

Abstract
During mammalian lung development, the morphological transition from respiratory tree branching morphogenesis to a predominantly saccular architecture, capable of air-breathing at birth, is dependent on physical forces as well as molecular signaling by a range of transcription factors including the cAMP response element binding protein 1 (Creb1). Creb1−/− mutant mice exhibit complete neonatal lethality consistent with a lack of lung maturation beyond the branching phase. To further define its role in the developing mouse lung, we deleted Creb1 separately in the respiratory epithelium and mesenchyme. Surprisingly, we found no evidence of a morphological lung defect nor compromised neonatal survival in either conditional Creb1 mutant. Interestingly however, loss of mesenchymal Creb1 on a genetic background lacking the related Crem protein showed normal lung development but poor neonatal survival. To investigate the underlying requirement for Creb1 for normal lung development, Creb1−/− mice were re-examined for defects in both respiratory muscles and glucocorticoid hormone signaling, which are also required for late stage lung maturation. However, these systems appeared normal in Creb1−/− mice. Together our results suggest that the requirement of Creb1 for normal mammalian lung morphogenesis is not dependent upon its expression in lung epithelium or mesenchyme, nor its role in musculoskeletal development.
Development of the mammalian lung is a highly intricate, multiphase process which is regulated largely by inter-germ layer molecular signaling, and by physical forces. Initially, epithelial buds emerge from the foregut endoderm, then branch extensively into the surrounding mesenchyme to produce the respiratory tree. In mice, these events begin during the embryonic phase (~E9.5-E10.5) and are essentially complete by the end of the “pseudoglandular” phase (~E10.5-E16.5)1. Subsequently, during the “cannalicular” (~16.5-E17.5) and “saccular” (~E17.5 till postnatal day 0.5) phases the mesenchymal content is considerably reduced and un-expanded distal epithelial tubules undergo extensive remodeling to form a fluid-filled dilated structure, with clefts (or primary septae) emerging to subdivide terminal airways into primitive alveolar sacs2. Concurrently, epithelial progenitors within distal tubules differentiate into type-I and –II alveolar epithelial cells (AECs) which then populate these sacs, mediating gas exchange and surfactant biosynthesis, respectively. To survive birth, which in rodents occurs during the saccular phase, proper sacculation is critical to ensure sufficient oxygen delivery to the blood once in a gaseous environment1. Although numerous studies have elegantly described the complex signaling pathways which regulate early lung budding and branching events, the molecular mechanisms which mediate the morphological transition from a pseudoglandular to a saccular structure are poorly understood.
Among the few factors implicated to have an important function during the lung saccular stage is the cAMP response element binding protein (Creb1). Creb1 is a member of the Creb/Atf subfamily of cAMP-responsive basic region-leucine zipper (bZIP) transcription factors which includes the cAMP response element modulatory protein (Crem) and activating transcription factor 1 (Atf1)3. In the nucleus, Creb1 is bound to DNA at specific gene promoter regions termed cAMP response elements (CREs) and transactivation normally only occurs when Creb1 is activated by upstream Ser/Thr-kinases, including cAMP-dependent PKA, which phosphorylate Ser-1334. Due to a high similarity in transactivation domain and bZIP (DNA binding domain) sequence identity between Creb1, Crem and Atf1, all can be activated by the same kinases and bind to target gene promoters as homodimers or as heterodimers with each other, thus providing a complex level of gene regulatory diversity5.
The role of Creb1/Atf family members in developmental (and other) processes has, in part, been revealed by gene targeted mutations in mice. Inactivation of Atf1 or Crem does not appear to affect gross development, although Crem−/− males become sterile due to impaired spermatogenesis6,7. Mice lacking Creb1 isoforms α and β (Creb1αΔ) are found at a reduced Mendelian frequency indicating a developmental disadvantage, but are otherwise viable and healthy8. However, complete inactivation of Creb1 function via loss of isoforms α, β and Δ (Creb1−/−) suffer multiple defects including reduced birth weight, brain deformities, impaired T cell development and cessation of lung sacculation causing neonatal lethality9. Our investigation of a similar Creb1 mutant revealed an almost identical whole body and respiratory phenotype, and further showed that Creb1−/− lungs had not developed beyond the pseudoglandular stage10. Importantly, analysis of this latter mutant demonstrated that complete loss of Creb1 could not be rescued by the activities of other Creb/Atf members, a compensatory mechanism described in studies using both compound Creb/Atf1 knockouts7 as well as conditional Creb1 mutants on a genetic background lacking either Crem or Atf111,12.
Although the phenotype of total Creb1-mutants indicates a requirement for Creb1 signaling during lung saccular stage development, the underlying molecular mechanisms remain unclear. Localization of transcriptionally active Creb1 in the fetal mouse lung using Ser133 phosphorylated Creb1 (pCreb1) immunostaining strongly suggests a role in the distal epithelium10,13. This is also consistent with the known stimulatory effect of cAMP on surfactant protein family gene transcription (Reviewed in14). Furthermore, total Creb1-mutants display a severe lack of AEC, and to a lesser extent, proximal epithelial cell marker expression while mesenchymal-derived vasculature and smooth muscle development appear unaffected10. Despite this, persistent Creb1 and pCreb1 expression in mesenchymal cell subsets, particularly during the saccular phase10, also suggest a function for Creb1 in the developing lung mesenchyme.
In this study, we have investigated the functional role of Creb1 in the developing respiratory epithelium and mesenchyme by conditional Creb1 deletion within these germ layers. We initially explored the importance of Creb1 for neonatal survival, morphological lung development and also epithelial cell differentiation during the saccular phase. Surprisingly, in these analyses we did not observe any requirement for Creb1 in any specific lung germ layer. To test for possible compensation by Creb/Atf members in conditional Creb1 knockout lungs, we assessed relative Crem and Atf1 gene expression levels and also lung morphology in conditional knockouts on a Crem deficient background. Ruling out these effects, we then re-examined total Creb1−/− fetal mice for defects in extra-pulmonary systems which are known to indirectly influence saccular lung development. Together our results indicate that loss of either respiratory epithelial or mesenchymal Creb1 expression is dispensable for lung development and neonatal survival, and is not compensated for by other Creb/Atf1 members. Lastly, we find that the respiratory phenotype in Creb1−/− mice is likely not due to deficiencies in either glucocorticoid (GC) signaling or muscular-driven physical forces which are also essential for normal saccular lung development.
Results
Conditional loss of Creb1 in lung epithelium and mesenchyme does not impair survival at birth
We first assessed the impact of lung epithelial Creb1 deletion on neonatal survival. Lung epithelial deletion was achieved using a doxycycline-inducible Cre/LoxP system. In this approach, Creb1fl/fl mice were bred to SPCrtTAtg/− and TetO-cretg/− strains to generate triple transgenic pups bearing all three alleles. This allows for Creb1 deletion in the respiratory epithelium due to the restricted expression of reverse tetracycline transactivator (rtTA) from the hSFTPC promoter. The rtTA, together with externally provided doxycycline in rodent food, then drive expression of Cre recombinase via the Tet(O7)CMV promoter to completely remove Creb1 expression in the lung epithelium (Fig. 1a). Therefore, maternal doxycycline-fed triple transgenic mice (Creb1fl/fl; SPCrtTAtg/−; TetO-cretg/−) are hereafter referred to as ‘Creb1EpKO’ mice. To account for known deleterious effects associated with doxycycline and rtTA toxicity in embryonic mouse lungs15, Creb1fl/fl; SPCrtTAtg/− animals were used as controls for Creb1EpKO mice in all analyses. Surprisingly, genotyping analysis of litters at two weeks of age showed no significant deviations from the expected 25% Mendelian frequency in Creb1EpKO mice (Table 1). Furthermore, we did not observe any obvious abnormalities in these mice throughout adulthood.
Figure 1. Creb1 deletion in developing lung epithelium and mesenchyme.

(a) Mechanism of respiratory-epithelial Creb1 deletion using a doxycyline-inducible triple transgenic mouse system. In the presence of Creb1fl, SPCrtTAtg and TetO-cretg alleles, doxycycline treatment induces lung-epithelial Cre recombinase expression. (b–d) Immunohistochemistry for Creb1 in Dox-treated E17.5 control (Creb1fl/fl; SPCrtTAtg/−) and Creb1EpKO lungs. Creb1 expression is virtually ubiquitous in control lungs (b) while lung epithelial Creb1 expression is mostly absent in Creb1EpKO lungs (c). Boxed area of (c) is magnified in (d) to show sporadic Creb1+ cells (arrows) in the Creb1EpKO proximal lung epithelium. (e–h) Creb1 immunohistochemistry in E17.5 (non Dox-treated) control, Creb1MesKO, and Creb1fl−/− lungs. Control lungs show almost ubiquitous Creb1 expression (e) while mesenchymal Creb1 expression is mostly lost in Creb1MesKO lungs (f). Boxed area of (f) is magnified in (g) to show rare Creb1+ cells (arrows) in the Creb1MesKO lung mesenchyme. Creb1 expression is completely absent in Creb1fl−/− lungs (h). Dotted lines in (D) and (G) indicate the epithelial-mesenchymal boundary. ‘P’ indicates proximal airway lumen; ‘D’ indicates distal airway lumen. All images are representative of at least three animals per genotype. Scale bars: b,c,e,f,h; 90 μm.
Table 1. Analysis of survival at two weeks of age in Creb1 EpKO mice.
| Cross | Litters | Pups | Average litter size | Creb1fl/fl | Creb1fl/fl; TetO-cretg/− | Creb1fl/fl; SPCrtTAtg/− | Creb1fl/fl; SPCrtTAtg/−; TetO-cretg/− | Loss of Creb1EpKO pups |
|---|---|---|---|---|---|---|---|---|
| Creb1fl/fl; TetO-cretg/− × Creb1fl/fl; SPCrtTAtg/− +Doxycycline | 8 | 50 | 6.25 | 20 (12.5) | 10 (12.5) | 11 (12.5) | 9 (12.5) | Non-significant |
We then investigated the requirement for mesenchymal Creb1 in the developing lung via mesenchymal-specific Creb1 deletion. This was achieved using a Dermo1-cre strain which express Cre recombinase as early as E13.5 within a variety of mesodermal-derived tissues including lung mesenchyme16,17,18. However, due to the very close proximity of the Creb1 and Dermo1 (Twist2) genes on chromosome 1 (~29 Mb apart), matings between Creb1fl/+;Dermo1Cre/+ and Creb1fl/fl mice resulted in a highly unequal frequency of progeny genotypes, (distinct from the knockout phenotype) and was therefore not an appropriate means of assessing survival. Nevertheless, the few Creb1fl/fl;Dermo1Cre/+ mice (hereafter referred to as Creb1MesKO) that were produced appeared healthy and were fertile. In addition, when Creb1MesKO mice were mated with Creb1fl/fl mice, we found no significant deviations from the expected 50% Creb1MesKO frequency when litters were genotyped at two weeks of age (Table 2). We also investigated Creb1 deletion in the endothelium using Tie2-cre mice which express Cre recombinase as early as E7.5 in the vascular endothelium19. However, in the two litters analysed (15 pups) we did not observe any loss of survival or other defects in Creb1fl/fl;Tie2Cre/+ mice (hereafter referred to as “Creb1EndoKO”). Total Creb1 knockout mice were also generated by mating Creb1fl/fl to CMV-Cre mice, which show Cre activity in all tissues before embryo implantation20. Complete neonatal lethality in a separate strain of Creb1−/− mice (bearing an exon 10 disruption) has previously been shown9, and although our Creb1fl/fl;CMVCre/+ mice (hereafter referred to as “Creb1fl−/−”) represent a slightly different Creb1 mutation, we have previously demonstrated the same morphological (whole body dwarfism) and respiratory phenotype (loss of sacculation) in the mice described herein10, as that exhibited in the previous strain. Therefore, we assumed an outcome of neonatal lethality in Creb1fl−/− mice and we did not investigate this further.
Table 2. Analysis of survival at two weeks of age in Creb1 MesKO mice.
| Cross | Litters | Pups | Average litter size | Creb1fl/f; Dermo1+/+ | Creb1fl/fl; Dermo1Cre/+ | Loss of Creb1MesKO pups |
|---|---|---|---|---|---|---|
| Creb1fl/fl; Dermo1+/+; × Creb1fl/fl; Dermo1Cre/+ | 11 | 65 | 5.9 | 40 (32.5) | 24 (32.5) | Non-significant |
Efficacy of respiratory epithelial and mesenchymal Creb1 deletion
To determine the degree of Creb1 deletion in respective lung germ layers, we performed immunohistochemistry for Creb1 at E17.5 where the mesenchymal tissue between developing epithelial buds is clearly defined. In all controls examined, Creb1 expression was highly expressed in all germ layers (Fig. 1b,e). Respiratory epithelial-specific Creb1 deletion was variable in Creb1EpKO mice, with some animals showing almost complete loss of Creb1 expression in lung epithelium (as shown in Fig. 1c,d) while others showed greatly reduced, but incomplete deletion in lung epithelium. The extent of Creb1 deletion in Creb1EpKO mice appeared particularly variable in the proximal lung epithelium, with distinct clusters of Creb1+ cells often observed in the conducting airway (Supplementary Fig. S1). This was also evident as early as E15.5, although at these earlier stages Creb1+ cells were less frequently observed (Supplementary Fig. S1). In comparison, the distal lung epithelium showed a much more complete and consistent absence of Creb1 expression in Creb1EpKO mice (Fig. 1d and Supplementary Fig. S1). In Creb1MesKO mice, lung mesenchymal Creb1 expression was almost completely abolished, although a few rare interstitial Creb1+ cells were observed (Fig. 1f,g). We also observed loss of Creb1 in chondrocytes in Creb1MesKO mice, consistent with the known spatial expression of Dermo1Cre in several non-respiratory tissues16 (Supplementary Fig. S1). Efficient Creb1 deletion in endothelial cells was also observed in lungs of Creb1EndoKO mice (Supplementary Fig. S1). As expected, total loss of Creb1 expression was always observed in the lung of Creb1fl−/− mice (Fig. 1h).
The Creb1−/− respiratory phenotype is not recapitulated by loss of Creb1 in lung epithelium and mesenchyme
Histological analysis was used to investigate potential alterations to lung morphology in E18.5 Creb1EpKO, Creb1MesKO and Creb1EndoKO mice. Unexpectedly, we found no consistent morphological differences in E18.5 Creb1EpKO lungs when compared to controls (Fig. 2a,b). As we had often found variable lung epithelial Creb1 deletion in fetal Creb1EpKO mice, we looked to see whether the extent of Creb1 deletion correlated to any degree of abnormal lung morphology. However, even in Creb1EpKO mice with the most complete level of lung epithelial Creb1 deletion we noted no difference in morphology compared with littermate controls. A tissue-to-airspace measurement also revealed no difference in overall cellularity in E17.5 Creb1EpKO lungs (Supplementary Fig. S2). Similarly, loss of mesenchymal or endothelial Creb1 in Creb1MesKO and Creb1EndoKO mice respectively, did not affect lung morphology (Fig. 2c–e). Due to the lack of significant pCreb1 expression in lung endothelial cells relative to other mesenchymal lineages and the epithelium10, from herein our analysis does not include Creb1EndoKO mice. Adult lung structure in Creb1EpKO and Creb1MesKO mice also appeared similar to controls (Supplementary Fig. S3). In comparison, lungs of E18.5 Creb1fl−/− mice displayed almost no expansion of airways and retained a morphology similar to that of late pseudoglandular-stage lungs, consistent with previous analysis of the Creb1−/− respiratory phenotype10 (Fig. 2f). Together these results suggest that Creb1 expression in lung epithelium, mesenchyme or endothelium separately is not required for normal lung development.
Figure 2. Normal lung development in Creb1EpKO, Creb1MesKO and Creb1EndoKO mice.

(a–f) Haematoxylin and eosin-stained tissue sections from E18.5 Creb1EpKO, Creb1MesKO, Creb1EndoKO and Creb1fl−/− lungs. Loss of Creb1 separately in lung epithelium (b), mesenchyme (d) and endothelium (e) produces no phenotype compared to controls (a,c). Lungs of Creb1fl−/− lungs mice exhibit a severe defect in sacculation, with little or no expansion of proximal and distal airways (f). All images are representative of at least three animals per genotype. Scale bar: 180 μm.
Epithelial differentiation is unaffected in Creb1 EpKO and Creb1 MesKO fetal lungs
Although neonatal survival and fetal lung morphology appeared to be unaffected in Creb1EpKO and Creb1MesKO mice, we reasoned that abnormalities in lung epithelial cell differentiation may still be present based on our previous findings showing altered levels of proximal and distal epithelial cell markers in Creb1fl−/− lungs10. We therefore used immunohistochemistry to investigate the differentiation of neuroendocrine, ciliated and secretory (Clara) cells in E18.5 Creb1EpKO and Creb1MesKO lungs using the proximal lung epithelial cell markers CGRP (also known as Calca), β-tubulin IV and CC10 (also known as Scgb1a1), respectively. However, we did not observe any difference in relative levels or spatial localization of these markers in Creb1EpKO and Creb1MesKO lungs (Fig. 3a–f,i–n). Distal epithelial cell differentiation also appeared unaffected in E18.5 Creb1EpKO and Creb1MesKO lungs as shown by immunohistochemistry for the type-II AEC marker, ProSPC (Fig. 3g,h,o,p). Using qPCR, we also examined gene expression of proximal and distal lung epithelial cell markers in E18.5 Creb1EpKO and Creb1MesKO lungs using qPCR. Consistent with our immunostaining results, no prominent alterations in mRNA levels were observed, though a reduction in levels for FoxJ1 (in Creb1EpKO lungs) and Aqp5 (in Creb1MesKO lungs) was noted (Supplementary Fig. S4).
Figure 3. Normal epithelial differentiation in Creb1EpKO lungs.

Immunohistochemistry for the proximal epithelial markers CGRP (a,b, i,j), β-tubulin IV (c,d,k,l), and CC10 (e,f,m,n) in E18.5 Creb1EpKO and Creb1MesKO lungs. Immunohistochemistry for the distal epithelial (type-II AEC) marker ProSPC (g,h,o,p) in E17.5 Creb1EpKO and E18.5 Creb1MesKO lungs. No differences in expression or localization was observed for proximal or epithelial markers. Boxed areas are magnified either in insets (CGRP, ProSPC) or below the main image (β-tubulin IV, CC10), while arrowheads indicate marker-positive cells. All images are representative of at least three animals per genotype. Scale bars: a-f, i-n; 180 μm. g,h,o,p; 90 μm.
Loss of respiratory epithelial or mesenchymal Creb1 in fetal lung is not rescued by Crem or Atf1 compensation
To test for possible compensatory up-regulation by the Creb/Atf family members Crem and Atf1, we firstly examined mRNA levels for these factors in E18.5 Creb1EpKO and Creb1MesKO lungs using qPCR. We found no changes in Atf1 mRNA levels in Creb1EpKO or Creb1MesKO lungs (Fig. 4a). However, Crem mRNA levels increased in Creb1EpKO lungs (1.4 fold, p < 0.05), and a greater increase was observed in Creb1MesKO lungs (2.7 fold, p < 0.001) (Fig. 4a). Despite this, immunohistochemical analysis using either an N-terminal or a C-terminal Crem antibody found no detectable levels of Crem protein in either control, Creb1MesKO or Creb1EpKO fetal lungs at E18.5 (Supplementary Fig. S5). Furthermore, we could not detect Crem protein in the embryonic lung at any developmental stage examined (E13.5, E15.5, PN6.5, Adult) (Supplementary Fig. S5). Normal Crem immunoreactivity (from both N- and C-terminal antibodies) however, was seen in round spermatid cell nuclei of the adult mouse testis, consistent with known sites of Crem expression6 (Supplementary Fig. S5). To further investigate potential Crem involvement in Creb1MesKO lung development we bred Creb1MesKO mice onto a Crem+/− or Crem−/− genetic background, and assessed fetal lung morphology at E18.5 as well as neonatal survival at 2 weeks postnatally. Fetal lungs from E18.5 Creb1MesKO; Crem−/− mice appeared normally developed relative to Creb1MesKO; Crem+/− controls (Fig. 4b,c), and immunohistochemical analysis of Creb1MesKO; Crem−/− lungs did not exhibit any defect in epithelial differentiation (Supplementary Fig. S6). Interestingly however, numbers of Creb1MesKO; Crem+/− and Creb1MesKO; Crem−/− mice at 2 weeks was significantly lower than the expected 25% Mendelian frequency (Table 3). Creb1MesKO; Crem−/− mice were never found, although due to extremely low progeny numbers and a high rate of maternal cannibalization there was insufficient statistical power to determine a causal link from this genotype to loss of survival (Supplementary Table S3). We did not attempt to generate Creb1EpKO; Crem−/− mice due to several breeding limitations including Crem−/− male sterility and also the inability to breed Creb1fl/fl mice on a homozygote SPCrtTAtg/tg background21. Together, these observations support the view that the normal lung phenotype in Creb1EpKO and Creb1MesKO mice is not due to compensatory up-regulation by Creb/Atf family members.
Figure 4. Lack of compensatory Crem/Atf1 upregulation in Creb1EpKO and Creb1MesKO lungs.
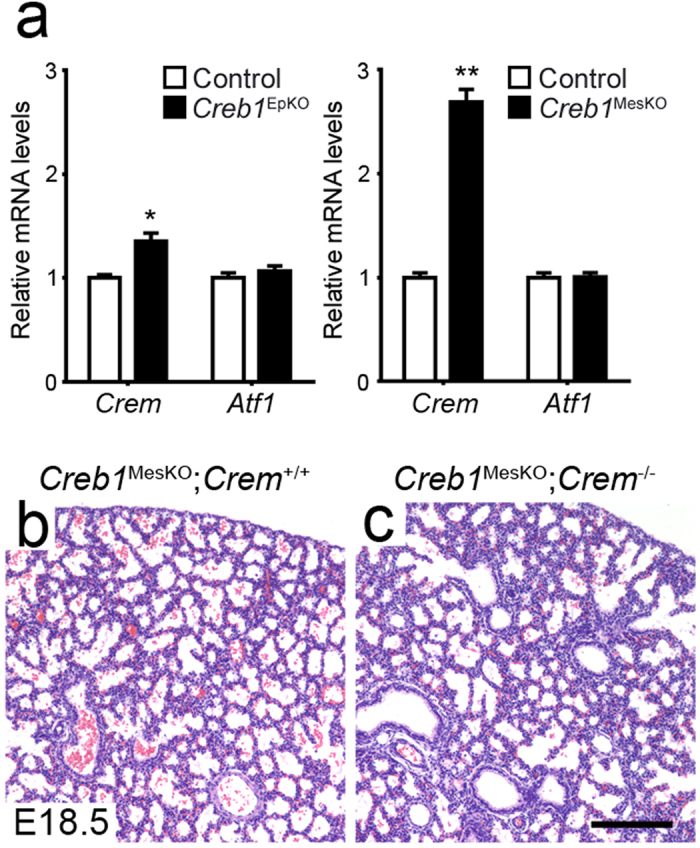
(a) qPCR analysis of Crem and Atf1 in E18.5 E18.5 Creb1EpKO (n = 7) and Creb1MesKO lungs (n = 6). (b,c) Haematoxylin and eosin-stained tissue sections from E18.5 control (Creb1MesKO; Crem+/+) and Creb1MesKO; Crem−/− lungs. No major difference in lung morphology was observed. Error bars represent SEM. Single asterisk (*) indicates p < 0.05, while double asterisk (**) indicates p < 0.01. White bars: Controls, Black bars: Conditional Creb1 deletions. All images are representative of at least three animals per genotype. Scale bars: 180 μm.
Table 3. Analysis of survival at two weeks of age in Creb1 MesKO; Crem +/− mice.
| Cross | Litters | Pups | Average litter size | Creb1fl/fl; Dermo1+/+; Crem+/+ | Creb1fl/fl; Dermo1+/+; Crem+/− | Creb1fl/fl; Dermo1Cre/+; Crem+/+ | Creb1fl/fl; Dermo1Cre/+; Crem+/− | Loss of Creb1MesKO; Crem+/− pups |
|---|---|---|---|---|---|---|---|---|
| Creb1fl/fl; Dermo1Cre/+; Crem+/− × Creb1fl/fl; Dermo1+/+; Crem+/+ | 23 | 129 | 5.6 | 45 (28.5) | 38 (28.5) | 33 (28.5) | 13 (28.5) | Significant loss p = 0.0005 |
The genotype of the progeny was determined as described in Materials and Methods at two weeks of age. The expected number of each genotype is indicated in brackets, and was calculated according to a predicted Mendelian allele inheritance ratio. Statistically significant deviations from the expected frequency was determined using a goodness-of-fit test with significance set a p < 0.05.
Glucocorticoid hormone synthesis is unaffected in in Creb1 fl−/− mice
As we had not observed any respiratory phenotype in Creb1EpKO or Creb1MesKO mice, we re-examined Creb1fl−/− fetal mice and tested for disruption of the GC signaling pathway, which is required for late stage lung maturation via the GC Receptor (GR, Nr3c1)22. We examined relative adrenal gland proportions, plasma corticosterone levels as well as pulmonary GR expression in E18.5 control and Creb1fl−/− fetal mice. However, transverse sections through lumbar vertebrae showed that adrenal glands in Creb1fl−/− fetal mice appeared similar to controls (Fig. 5a,b). Corticosterone levels were slightly increased in Creb1fl−/− fetal mice, though this did not reach statistical significance (Fig. 5e). Lastly, the level of GR immunoreactivity in Creb1fl−/− fetal lungs was comparable to controls, and appeared to be localized primarily to the nucleus indicative of normal GC ligand-bound GR activation (Fig. 5c,d). Together, these results suggest that the respiratory phenotype in Creb1fl−/− fetal mice is not caused by loss of GC signaling.
Figure 5. Loss of Creb1 does not impair GC signaling in the fetal lung.

(a,b) Haematoxylin and eosin-stained tissue in transverse sections through lower thoracic vertebrae in E18.5 control and Creb1fl−/− mice. Adrenal gland proportions appear comparable between control and Creb1fl−/− mice. (c,d) Immunohistochemistry for GR in E18.5 control and Creb1fl−/− lungs. (e) Measurement of plasma corticosterone in E18.5 Creb1+/+ (white bars), Creb1+/− (grey bars) and Creb1fl−/− (black bars) mice. Error bars represent SEM. All images are representative of at least three animals per genotype. Ad: adrenal gland, Li: liver, Sp: spleen, St: stomach. Scale bars: a, b: 360 μm, c, d: 90 μm.
Diaphragm and intercostal musculature development are unaffected in Creb1 fl−/− mice
We also re-examined Creb1fl−/− fetal mice for defects in muscular elements known to be important for late stage lung development such as the diaphragm and intercostal muscle. We firstly inspected E18.5 Creb1fl−/− fetal mice for possible diaphragm herniation, however careful examination of the thoracic cavity (in 3 separate knockout animals) did not reveal any noticeable defects in diaphragm muscle tissue. Secondly, we tested whether FBM-associated muscle fibres were properly formed in Creb1fl−/− fetal mice at E18.5 using immunohistochemistry for the mature muscle marker myosin heavy chain (MyHC). In transverse sections through thoracic vertebrae, we noticed no difference in MyHC-positive diaphragm or skeletal intercostal muscle fibres in Creb1fl−/− mice compared with controls (Fig. 6a–f). Another Creb1-regulated structural muscle protein, desmin23, similarly appeared at normal levels in all skeletal and diaphragm musculature examined in E18.5 Creb1fl−/− fetal mice (Fig. 6g,h). In addition, CD31 immunohistochemistry showed that these muscular elements were properly vascularized (Fig. 6i–l). Lastly, we tested for evidence of lung hypoplasia in E18.5 Creb1fl−/− mice via analysis of lung wet weight/body weight ratios (Fig. 6m). No difference was detected between Creb1fl−/− (3.8 ± 0.2%, n = 3) and Creb1fl+/− littermate controls (3.4 ± 0.1%, n = 3). Thus, it is unlikely that the respiratory phenotype in Creb1fl−/− mice is due to loss of in utero FBM-associated muscular activity.
Figure 6. Diaphragm and intercostal musculature are normally developed in Creb1fl−/− fetal mice.

Immunohistochemistry for the muscle markers MyHC (a–f), desmin (g,h) and CD31 (i–l) in transverse sections through thoracic vertebrae in E18.5 control and Creb1fl−/− fetal mice. Magnified images of MyHC-positive intercostal (c,d) and diaphragm (e,f) musculature. Intercostal and diaphragm musculature appear normally developed in Creb1fl−/− fetal mice. (m) Scatterplot showing the percentage fetal lung wet weight/body weight in E18.5 control and Creb1fl−/− littermates (n = 3). Error bars represent SEM. All images are representative of at least three animals per genotype. Scale bars: a,b,g,h; 900 μm, c,d,i-l; 180 μm, e,f; 90 μm. Lu: lung, Li: liver, D: diaphragm, Im: intercostal muscle, “*”indicates thoracic cavity.
Discussion
In this study we initially show that conditional Creb1 deletion in the respiratory epithelium, endothelium or mesenchyme surprisingly has no major impact on lung sacculation, suggesting that lung-localized Creb1 has no essential role in mammalian lung development. While Creb1 is widely expressed in the developing lung, the primary site of Creb1 transcriptional function is believed to be the distal epithelium, based on the expression of Ser133 pCreb1 in the pseudoglandular phase10,13. Our Creb1 immunohistochemistry in Creb1EpKO mice showed that a subset of epithelial cells, primarily from proximal regions, often escaped deletion. It is therefore possible that sufficient Creb1 activity allowed normal lung development to proceed in Creb1EpKO mice. While it is expected that a small subset of proximal epithelial cells will not undergo Creb1 deletion due to lack of SPCrtTA transgene activation (such as the neuroepithelial lineage24), it is also conceivable that the Crebfl locus is somewhat resistant to Cre-mediated recombination. This latter possibility appears more likely given that we observe no expansion of neuroepithelial (or other) proximal epithelial lineages in Creb1EpKO lungs. Nevertheless, our finding of largely complete Creb1 deletion in the distal epithelium of Creb1EpKO lungs argue against a normal phenotype in these mice resulting from residual epithelial Creb1 signaling. On the other hand, strong Ser133 pCreb1 expression in mesenchymal cells during the saccular phase also argues for a role for Creb1 in the mesenchyme10, yet while Dermo1Cre-mediated deletion appeared mostly complete in Creb1MesKO lungs, we also did not observe any respiratory phenotype in these mice.
Interestingly, mice with a Ser133 to Alanine residue mutation (Creb1 Ser133A) in the Creb1 gene are viable and healthy, albeit born at a reduced Mendelian frequency when bred on a C57BL/6J background25. This suggests that distal epithelial Ser133 pCreb1 expression is not an indicator of important Creb1 activity in the lung, and/or that a partial functional redundancy exists between other phosphorylation sites for Creb1 transcriptional activation and normal embryonic development. It would be valuable to generate mice with compound Creb1 phosphorylation site mutations to pursue this hypothesis. In addition, it would be worthwhile to generate compound lung epithelial/mesenchymal Creb1 mouse mutants to investigate the potential combined requirement of Creb1 in these germ layers for normal lung development.
We next investigated several alternate mechanisms in order to explain the severe lung phenotype observed in total Creb1 mouse mutants. Firstly, we provide evidence that the lack of a respiratory phenotype in conditional Creb1 deletions is not caused by compensatory activities of Crem or Atf1. This functional redundancy within the Creb/Atf1 subfamily is believed to occur via up-regulation of these factors’ expression in the absence of Creb18,26,27. Our investigation shows that while upregulation of Crem, and not Atf1, gene expression takes place in both E18.5 Creb1EpKO and Creb1MesKO fetal lungs, the lack of Crem immunoreactivity in both controls and conditional knockout lungs indicates that Crem protein is neither synthesized in the wildtype fetal lung, nor upregulated to detectable levels in the absence of Creb1. Although we did not test all Crem isoforms in our qPCR and immunohistochemcial analyses, more compelling is our finding that lungs of Creb1MesKO mice, which showed a higher upregulation of Crem than Creb1EpKO lungs, displayed no lung phenotype even when bred on a Crem deficient genetic background. It is therefore possible that in most tissues, Crem mRNA upregulation in the absence of Creb1 has little or no functional outcome, with the exception of perhaps the brain where Crem compensation protects against neuronal cell death11. Interestingly however, our finding of reduced neonatal survival in Creb1MesKO; Crem+/− and Creb1MesKO; Crem−/− mice point to a compensatory role for Crem in Creb1-deficient mesodermal tissues during early postnatal life. This phenotype would be interesting to investigate, however given the wide range of non-respiratory organs also targeted by the Dermo1Cre allele including bone16, kidney28 and heart29, isolating the affected system(s) may be a challenging task.
Tracing the origin of respiratory defects in mouse mutants can be difficult given that a loss of gene function in a separate tissue can potentially impair lung development via an indirect (or non-autonomous) mechanism. For example, mice lacking the myogenic factor Myogenin (Myog) exhibit defective lung sacculation due to poorly developed diaphragm muscle30, though Myog is not expressed in the lung. Similarly, loss of the skeletal muscle-specific factor Myf5, also not expressed in the lung, prevents normal lung sacculation31. More difficult to interpret are lung phenotypes from total mouse mutants in which the deleted gene is expressed in multiple tissues including lung, yet other musculoskeletal defects are also present which may contribute or represent the sole cause of the lung phenotype. For example, the insulin-like growth factor-1 (Igf1r) is widely expressed during embryonic development, and Igf1r total knockout mice exhibit both fetal lung and diaphragmatic defects32.
Advantageously, the multiple systems targeted by the Dermo1Cre allele also help to explore whether certain non-respiratory mechanisms potentially underpin the Creb1−/− lung phenotype. For example, it could be argued that impaired development of the ribcage or vertebrae may prevent normal expansion of the underlying lung or provide inadequate structural support to respiratory muscles, thus arresting lung development prior to sacculation33. This suggestion has significant merit given the vertebral fusions occasionally observed in Creb1−/− mice34 but is also supported by previous findings in mice which utilize tissue-specific overexpression of a dominant negative Creb1 transgene known as A-CREB, which specifically blocks DNA binding of the Creb/Atf1 subfamily35. A-CREB overexpression in chondrocytes results in a reduced rib-cage circumference and neonatal lethality36, while mice overexpressing A-CREB in the mesoderm also die at birth, presumably due to respiratory failure and exhibit profound ribcage, as well as other skeletal defects34. Conversely, we did not observe any lung or skeletal phenotype in Creb1MesKO or Creb1MesKO; Crem−/− fetal mice, although a lack of Creb1 expression in ribcage chondrocytes due to Dermo1Cre-mediated deletion was apparent in these mutants. We speculate that these discrepancies are partially due to differences in promoter-specific Dermo1Cre versus A-CREB expression in mesodermal cell lineages, although it is also conceivable that Atf1 can compensate for lack of Creb1/Crem for correct skeletal development. Another possibility is that the lack of CRE occupancy in A-CREB mutants leads to aberrant promoter binding by other factors which may produce off-target developmental outcomes via abnormal target gene regulation. We therefore suggest a level of caution when interpreting the phenotype in A-CREB mouse mutants.
We also investigated, but excluded, the possibility that the Creb1−/− lung phenotype is secondary to muscular defects. Late stage lung development is known to be critically dependent on mechanical influences from in utero ‘fetal breathing movements’ (FBMs), which are driven by contractile activities of the diaphragm and skeletal intercostal musculature. Absence of skeletal muscles results in lung hypoplasia during late in utero development as seen in compound knockout mice which lack both of the myotomal factors Myf5 and MyoD37. Abnormalities in diaphragm development can similarly cause lung hypoplasia, most commonly due to diaphragmatic herniation whereby abdominal contents invade the thoracic space and prevent fetal lung expansion38. Intriguingly, a previous study using Creb1−/− mice found PKA-Creb1 signaling to be required for important myotomal factors including Pax3, MyoD and Myf5 during early development39. However, our MyHC immunohistochemistry show that Creb1fl−/− mice do not exhibit any defects in FBM-associated musculature that would be consistent with a loss of saccular lung development. Furthermore, previous and current findings from our group have not found any evidence of lung hypoplasia in Creb1fl−/− mice, which would be expected in the event of impaired FBM-associated musculature. For example, in our current study the lung wet weight/body weight ratios in Creb1fl−/− fetal mice were comparable to controls, though this is normally reduced in lung hypoplasia. Additionally, our previous work in E18.5 Creb1fl−/− lungs showed no significant difference in cell apoptosis and a slightly higher rate of cell proliferation10, though these are normally increased and reduced, respectively, in lung hypoplasia.
Another potential mechanism explored was the possibility that global loss of Creb1 disrupts components of the GC-GR signaling pathway, which is required for late stage lung maturation. On one hand, insufficient levels of corticosterone (the active rodent GC ligand) prevent GR transcriptional signaling as seen in corticotrophin releasing hormone (Crh) knockout mice40. On the other hand, GR mouse mutants exhibit elevated corticosterone levels secondary to marked adrenal hypertrophy22. Interestingly, Creb1 has a recognized role in promoting gene expression of Crh41, supporting a role for Creb1 in GC production. However, free corticosterone levels appeared relatively unchanged in Creb1fl−/− mice, consistent with normally-sized adrenal glands. In addition, our finding of nuclear GR localization in Creb1fl−/− lungs implies that sufficient GC ligand is available to promote cytoplasmic to nuclear GR translocation. The normal levels of corticosterone in Creb1fl−/− mice are perhaps unsurprising given that a fetal loss of GCs in various GC signaling deficient mouse mutants can be compensated for by a maternal transfer of corticosterone across the placenta42. Therefore, while our results do not preclude an absence of Crh expression in Creb1fl−/− mice, this is unlikely to be the cause of the Creb1fl−/− respiratory phenotype. Normal GR expression in Creb1fl−/− lungs also suggests Creb1 is not required for lung GR expression.
In summary, we find that the requirement of Creb1 for normal mammalian lung development is not dependent upon its expression in the respiratory epithelium or mesenchyme. Furthermore, we show that this requirement is not indirectly due to known or suspected Creb1 involvement in musculoskeletal development, nor the GC signaling pathway. Therefore, the underlying cause behind the respiratory phenotype in total Creb1 mouse mutants remains unknown. One further possibility not explored in this study is whether the recognized involvement of Creb1 in the peripheral nervous system43 indirectly stimulates lung sacculation via innervation of FBM-associated muscles. However, while innervation of muscular elements such as the diaphragm is known to be required for lung function at birth44, it is uncertain whether it is also essential for the saccular process of lung development. Conditional Creb1 deletion in a wider range of neuronal cell lineages would therefore prove a valuable exercise to address this potential cause of the lung phenotype observed in Creb1fl−/− mice.
Methods
Mouse models
SPCrtTA and TetO-cre mice were purchased from the Jackson Laboratory (Bar Harbor, Maine, USA). Dermo1-cre, CMV-cre and Tie2-cre mice were generously provided by Dr. Brandon Wainwright (University of Queensland, Brisbane, Australia), Dr. Christina Mitchell (Monash University, Melbourne, Australia) and Dr. Jinhua Li (Monash University) respectively. All mouse strains were maintained on a C57BL/6 background. To generate the Creb1EpKO mice, SPCrtTA and TetO-cre lines were both bred to Creb1fl/fl mice to produce transgenic (tg) Creb1fl/fl;SPCrtTAtg/− and Creb1fl/fl;TetO-cretg/− mice, respectively. These two lines were then time-mated, and rodent food was altered at E6.5 till E14.5 to contain doxycycline (600 mg/kg, Specialty Feeds, WA, Australia) to induce cre expression in pups bearing all three transgenic alleles (Creb1EpKO). Genotyping primer sequences are shown in Supplementary Table S1. All animal experimentation was approved and carried out according to the guidelines established by the School of Biomedical Sciences Animal Ethics Committee, Monash University (Ethics No. 2011/147).
Histology, Immunohistochemistry and Lung Tissue morphometry
Whole fetal mouse torsos were immersion-fixed in 4% paraformaldehyde (PFA) overnight at 4 °C with agitation then embedded in paraffin. 5μm sections were cut and mounted on slides, then used for histological or immunostaining analyses according to a standard protocol as previously described10. Lung tissue morphometry was performed according to previously published methods45. Primary antibodies used are shown in Supplementary Table S2.
RNA Extraction, cDNA synthesis and quantitative PCR
Total RNA and cDNA was obtained from fetal lungs using TRIzol reagent (Invitrogen, Carlsbad, CA) and M-MLV Reverse Transcriptase (Promega, Madison WI), respectively, as per the manufacturer’s instructions. The integrity of 28S and 18S rRNA from total RNA was always assessed on agarose gels prior to further analysis. qPCR analysis was performed in triplicate for each biological replicate using at least two housekeeping genes per analysis: 18S rRNA and Rps29. Other qPCR primers were designed to include at least one oligo overlapping an exon-exon boundary, using the web-based software Primer346. Primer efficiency was then calculated using a standard curve and fetal lung cDNA template. Sequences for these primers can be found in a previous publication10. Cycling was performed on a CFX384 Touch™ Real-Time PCR Detection System (Bio-Rad, Richmond, CA). Relative differential expression was then determined using CFX Manager™ software (Bio-Rad).
Corticosterone measurement
Trunk blood (5–10 μl) was obtained from fetal wildtype and Creb1fl−/− mice and assayed for free corticosterone levels using a Corticosterone Double Antibody RIA Kit (MP Biomedicals, Solon, OH), as per the manufacturer’s instructions. Radioactivity was measured using a 1470 Perkin Elmer automated gamma counter (Perkin Elmer, Waltham, MA).
Statistical Analysis
GraphPad Prism software was used to analyze the results of all experiments with a statistical significance set at p < 0.05. Statistically significant deviations in neonatal mouse survival at two weeks of age was calculated using a goodness-of-fit test.
Additional Information
How to cite this article: Antony, N. et al. Creb1 regulates late stage mammalian lung development via respiratory epithelial and mesenchymal-independent mechanisms. Sci. Rep. 6, 25569; doi: 10.1038/srep25569 (2016).
Supplementary Material
Acknowledgments
This work was funded by a Program Grant 606789 from the Australian National Health & Medical Research Council. The authors acknowledge the facilities and scientific and technical assistance of Monash Histology Platform, Department of Anatomy and Developmental Biology, Monash University. The authors also acknowledge the facilities and mouse husbandry of the Monash Animal Research Platform, Monash University. Lastly, we thank Dr. Rob Bryson-Richardson for muscle marker antibodies as well expertise regarding developmental muscle biology.
Footnotes
Author Contributions C.T.J. obtained the funding. M.T. provided the Crebfl/fl and Crem+/− mouse strains. A.N., M.R.A. and B.A.D. performed the experiments. B.A.D. wrote the main manuscript text. All authors reviewed the manuscript.
References
- Herriges M. & Morrisey E. E. Lung development: orchestrating the generation and regeneration of a complex organ. Development 141, 502–513, 10.1242/dev.098186 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J. & Chen J. Developmental programs of lung epithelial progenitors: a balanced progenitor model. Wiley interdisciplinary reviews. Developmental biology 3, 331–347, 10.1002/wdev.141 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr B. & Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol 2, 599–609, 10.1038/35085068 (2001). [DOI] [PubMed] [Google Scholar]
- Johannessen M., Delghandi M. P. & Moens U. What turns CREB on? Cell Signal 16, 1211–1227 (2004). [DOI] [PubMed] [Google Scholar]
- Shaywitz A. J. & Greenberg M. E. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem 68, 821–861 (1999). [DOI] [PubMed] [Google Scholar]
- Blendy J. A., Kaestner K. H., Weinbauer G. F., Nieschlag E. & Schutz G. Severe impairment of spermatogenesis in mice lacking the CREM gene. Nature 380, 162–165 (1996). [DOI] [PubMed] [Google Scholar]
- Bleckmann S. C. et al. Activating transcription factor 1 and CREB are important for cell survival during early mouse development. Mol Cell Biol 22, 1919–1925 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummler E. et al. Targeted mutation of the CREB gene: compensation within the CREB/ATF family of transcription factors. Proc Natl Acad Sci USA 91, 5647–5651 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph D. et al. Impaired fetal T cell development and perinatal lethality in mice lacking the cAMP response element binding protein. Proc Natl Acad Sci USA 95, 4481–4486 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A. D. et al. cAMP response element binding protein is required for differentiation of respiratory epithelium during murine development. Plos One 6, e17843, 10.1371/journal.pone.0017843 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantamadiotis T. et al. Disruption of CREB function in brain leads to neurodegeneration. Nat Genet 31, 47–54 (2002). [DOI] [PubMed] [Google Scholar]
- Baumann S. et al. CREB function is required for normal thymic cellularity and post-irradiation recovery. Eur J Immunol 34, 1961–1971, 10.1002/eji.200324826 (2004). [DOI] [PubMed] [Google Scholar]
- Xu J., Tian J., Grumelli S. M., Haley K. J. & Shapiro S. D. Stage-specific effects of cAMP signaling during distal lung epithelial development. J Biol Chem 281, 38894–38904 (2006). [DOI] [PubMed] [Google Scholar]
- Mendelson C. R. Role of transcription factors in fetal lung development and surfactant protein gene expression. Annu Rev Physiol 62, 875–915 (2000). [DOI] [PubMed] [Google Scholar]
- Morimoto M. & Kopan R. rtTA toxicity limits the usefulness of the SP-C-rtTA transgenic mouse. Developmental Biology 325, 171–178 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K. et al. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development 130, 3063–3074 (2003). [DOI] [PubMed] [Google Scholar]
- Chen H. et al. TGF-beta receptor II in epithelia versus mesenchyme plays distinct roles in the developing lung. Eur Respir J 32, 285–295, 10.1183/09031936.00165407 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- White A. C. et al. FGF9 and SHH signaling coordinate lung growth and development through regulation of distinct mesenchymal domains. Development 133, 1507–1517, 10.1242/dev.02313 (2006). [DOI] [PubMed] [Google Scholar]
- Kisanuki Y. Y. et al. Tie2-Cre Transgenic Mice: A New Model for Endothelial Cell-Lineage Analysis in Vivo. Developmental Biology 230, 230–242 (2001). [DOI] [PubMed] [Google Scholar]
- Schwenk F., Baron U. & Rajewsky K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res 23, 5080–5081 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perl A.-K., Zhang L. & Whitsett J. A. Conditional Expression of Genes in the Respiratory Epithelium in Transgenic Mice: Cautionary Notes and Toward Building a Better Mouse Trap. Am J Respir Cell Mol Biol 40, 1–3 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole T. J. et al. Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev 9, 1608–1621 (1995). [DOI] [PubMed] [Google Scholar]
- Berdeaux R. et al. SIK1 is a class II HDAC kinase that promotes survival of skeletal myocytes. Nat Med 13, 597–603, 10.1038/nm1573 (2007). [DOI] [PubMed] [Google Scholar]
- Perl A. K., Wert S. E., Nagy A., Lobe C. G. & Whitsett J. A. Early restriction of peripheral and proximal cell lineages during formation of the lung. Proc Natl Acad Sci USA 99, 10482–10487 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naqvi S., Martin K. J. & Arthur J. S. CREB phosphorylation at Ser133 regulates transcription via distinct mechanisms downstream of cAMP and MAPK signalling. Biochem J 458, 469–479, 10.1042/BJ20131115 (2014). [DOI] [PubMed] [Google Scholar]
- Vogt M. A. et al. Inducible forebrain-specific ablation of the transcription factor Creb during adulthood induces anxiety but no spatial/contextual learning deficits. Front Behav Neurosci 8, 407, 10.3389/fnbeh.2014.00407 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundersen B. B. et al. Increased hippocampal neurogenesis and accelerated response to antidepressants in mice with specific deletion of CREB in the hippocampus: role of cAMP response-element modulator tau. J Neurosci 33, 13673–13685, 10.1523/JNEUROSCI.1669-13.2013 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolpakova-Hart E. et al. Growth of cranial synchondroses and sutures requires polycystin-1. Dev Biol 321, 407–419, 10.1016/j.ydbio.2008.07.005 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavine K. J., Long F., Choi K., Smith C. & Ornitz D. M. Hedgehog signaling to distinct cell types differentially regulates coronary artery and vein development. Development 135, 3161–3171, 10.1242/dev.019919 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng B. S. et al. Pulmonary hypoplasia in the myogenin null mouse embryo. Am J Respir Cell Mol Biol 22, 304–315, 10.1165/ajrcmb.22.3.3708 (2000). [DOI] [PubMed] [Google Scholar]
- Inanlou M. R. & Kablar B. Abnormal development of the intercostal muscles and the rib cage in Myf5 −/− embryos leads to pulmonary hypoplasia. Developmental dynamics: an official publication of the American Association of Anatomists 232, 43–54, 10.1002/dvdy.20202 (2005). [DOI] [PubMed] [Google Scholar]
- Epaud R. et al. Knockout of insulin-like growth factor-1 receptor impairs distal lung morphogenesis. Plos One 7, e48071, 10.1371/journal.pone.0048071 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turgeon B. & Meloche S. Interpreting neonatal lethal phenotypes in mouse mutants: insights into gene function and human diseases. Physiol Rev 89, 1–26, 10.1152/physrev.00040.2007 (2009). [DOI] [PubMed] [Google Scholar]
- Lopez T. P. & Fan C. M. Dynamic CREB family activity drives segmentation and posterior polarity specification in mammalian somitogenesis. Proc Natl Acad Sci USA 110, E2019–2027, 10.1073/pnas.1222115110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn S. et al. A dominant-negative inhibitor of CREB reveals that it is a general mediator of stimulus-dependent transcription of c-fos. Mol Cell Biol 18, 967–977 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long F., Schipani E., Asahara H., Kronenberg H. & Montminy M. The CREB family of activators is required for endochondral bone development. Development 128, 541–550 (2001). [DOI] [PubMed] [Google Scholar]
- Inanlou M. R. & Kablar B. Contractile activity of skeletal musculature involved in breathing is essential for normal lung cell differentiation, as revealed in Myf5 −/−:MyoD −/− embryos. Developmental dynamics: an official publication of the American Association of Anatomists 233, 772–782, 10.1002/dvdy.20381 (2005). [DOI] [Google Scholar]
- Merrell A. J. & Kardon G. Development of the diaphragm–a skeletal muscle essential for mammalian respiration. FEBS J 280, 4026–4035, 10.1111/febs.12274 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A. E., Ginty D. D. & Fan C. M. Protein kinase A signalling via CREB controls myogenesis induced by Wnt proteins. Nature 433, 317–322 (2005). [DOI] [PubMed] [Google Scholar]
- Muglia L., Jacobson L., Dikkes P. & Majzoub J. A. Corticotropin-releasing hormone deficiency reveals major fetal but not adult glucocorticoid need. Nature 373, 427–432 (1995). [DOI] [PubMed] [Google Scholar]
- Liu Y., Coello A. G., Grinevich V. & Aguilera G. Involvement of transducer of regulated cAMP response element-binding protein activity on corticotropin releasing hormone transcription. Endocrinology 151, 1109–1118, 10.1210/en.2009-0963 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A. D., McDougall A. R., Seow B., Hooper S. B. & Cole T. J. Glucocorticoid regulation of lung development: lessons learned from conditional GR knockout mice. Mol Endocrinol 29, 158–171, 10.1210/me.2014-1362 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonze B. E. & Ginty D. D. Function and regulation of CREB family transcription factors in the nervous system. Neuron 35, 605–623 (2002). [DOI] [PubMed] [Google Scholar]
- Witzemann V., Chevessier F., Pacifici P. G. & Yampolsky P. The neuromuscular junction: selective remodeling of synaptic regulators at the nerve/muscle interface. Mech Dev 130, 402–411, 10.1016/j.mod.2012.09.004 (2013). [DOI] [PubMed] [Google Scholar]
- Bird A. D., Choo Y. L., Hooper S. B., McDougall A. R. & Cole T. J. Mesenchymal glucocorticoid receptor regulates the development of multiple cell layers of the mouse lung. American Journal of Respiratory Cell and Molecular Biology 50, 419–428, 10.1165/rcmb.2013-0169OC (2014). [DOI] [PubMed] [Google Scholar]
- Untergasser A. et al. Primer3–new capabilities and interfaces. Nucleic Acids Res 40, e115, 10.1093/nar/gks596 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.