

Abstract
Numerous affinity purification-mass spectrometry (AP-MS) and yeast two-hybrid screens have each defined thousands of pairwise protein-protein interactions (PPIs), most of which are between functionally unrelated proteins. The accuracy of these networks, however, is under debate. Here, we present an AP-MS survey of the bacterium Desulfovibrio vulgaris together with a critical reanalysis of nine published bacterial yeast two-hybrid and AP-MS screens. We have identified 459 high confidence PPIs from D. vulgaris and 391 from Escherichia coli. Compared with the nine published interactomes, our two networks are smaller, are much less highly connected, and have significantly lower false discovery rates. In addition, our interactomes are much more enriched in protein pairs that are encoded in the same operon, have similar functions, and are reproducibly detected in other physical interaction assays than the pairs reported in prior studies. Our work establishes more stringent benchmarks for the properties of protein interactomes and suggests that bona fide PPIs much more frequently involve protein partners that are annotated with similar functions or that can be validated in independent assays than earlier studies suggested.
Proteins often function by interacting with partner proteins to form complexes, which range from heterodimers to large macromolecular assemblies (1, 2). If we can accurately learn the heteromeric interactions that each protein makes, it will greatly aid the modeling of all aspects of cellular biochemistry and physiology.
Over the last 15 years, protein-protein “interactomes” have been characterized on a genome-wide scale in bacteria and eukaryotes by yeast 2-hybrid (Y2H)1 and affinity purification-mass spectrometry (AP-MS) screens (1, 3–18). Y2H screens detect binary physical interactions between pairs of proteins expressed in a non-native host, whereas AP-MS detects proteins that co-purify with a tagged protein expressed in the native organism. The Y2H method can detect transient, lower affinity interactions of as little as micromolar affinity (19), whereas AP-MS is better suited to identify stable protein complexes (5). The resulting networks generally comprise thousands of pairwise interactions between proteins in which hub proteins are highly connected to functionally diverse arrays of other proteins (1, 20), with the total interactome being estimated to contain ∼10,000 protein pairs in Escherichia coli (4) to ∼130,000 in humans (21).
As part of a large interdisciplinary project (enigma.lbl.gov), we are conducting detailed system-wide analyses of the model sulfate-reducing bacterium Desulfovibrio vulgaris, a Deltaproteobacteria and obligate anaerobe (22). D. vulgaris has been extensively characterized by functional genomic studies of its response to environmentally relevant conditions (23, 24), but relatively few protein complexes have been analyzed to date (25–27). Therefore, we have performed a global AP-MS screen to characterize its interactome and have also critically reexamined nine published AP-MS and Y2H screens. We have developed a rigorous data analysis strategy that has identified 459 high confidence protein-protein interactions (PPIs) for D. vulgaris and 391 PPIs from an existing AP-MS dataset for E. coli, many of which are supported by low throughput data from the literature. Importantly, compared with the protein-protein networks proposed previously, our two interactomes are smaller, less interconnected. and more strongly enriched in protein partners that share similar function or whose interactions have been validated in independent high throughput assays. We also show that the ∼3% of PPIs from the earlier Y2H and AP-MS screens that were reciprocally confirmed as both bait-prey and prey-bait pairs, and thus are more confidently detected, share very similar characteristics with our two high confidence interactomes. The remaining ∼97% of protein pairs from the earlier screens, in contrast, do not. Our work provides more stringent criteria for assessing the quality of protein interactomes and suggests that the number of bona fide interactions from the earlier screens that are supportable by independent evidence is limited to hundreds and not the thousands claimed.
EXPERIMENTAL PROCEDURES
Recombinant Strain Construction and Affinity Purification
D. vulgaris Hildenborough wild-type ATCC29579 was genetically engineered to encode locus-specific affinity purification (AP)-tagged fusion proteins using electroporation of non-replicating “suicide constructs” (28). Of the 3525 predicted D. vulgaris protein-coding genes, we attempted to create tagged strains for a priority list of 2086 genes. These genes were selected based on several criteria, including detection of the proteins they encode in fractionated cell-free extracts by MS,2 expected complexes based on E. coli interologs, and functional interest, such as energy generation. We constructed plasmids for generating chromosomal AP-tagged alleles for 1963 of the priority genes, 1681 of which were successfully integrated into the D. vulgaris chromosome. From this set, 1498 strains expressing an AP-tagged fusion protein were verified by Western blot, of which 1415 were constructed using Sequence and Ligation Independent Cloning, 77 using Gatewayn and 6 using recombineering procedures, supplemental Dataset S1. The primary AP tag utilized was Strep-TEV-FLAG (1231 strains); however, Strep-TEV-FLAG-His6 (237 strains) and Sequential Peptide Affinity tag (30 strains) (29) were also used. A non-redundant total of 1401 unique genes are represented as AP-tagged alleles in the 1498 strains constructed. All affinity purifications were performed as described previously (28). In all cases, Strep-TEV-FLAG-His6 strains were treated exactly as Strep-TEV-FLAG strains for the purposes of affinity purification of protein complexes.
Sample Preparation for Mass Spectrometry and LC MS Data Acquisition
The majority of AP samples were analyzed by parallel gel-free and gel-based workflows. In a gel-free approach, AP-isolated proteins were digested with trypsin utilizing a 96-well PVDF membrane-based protocol and analyzed by LC MS/MS using either LTQ XL linear ion trap mass spectrometer (Thermo Scientific, Fremont, CA) or LTQ Velos Orbitrap mass spectrometer (Thermo Scientific), essentially as described by Chhabra et al. and Roan et al., respectively (28, 30). Five sample sets, however, used a QSTAR XL mass spectrometer (AB Sciex, Framingham, MA) as described by Chiu et al. (31). In all cases, an additional wash-run (5-μl injection of 50% isopropyl alcohol to clean the trap cartridge; 30-min gradient over analytical column, including two 5-min ramps from 3% acetonitrile to 97% acetonitrile) and a protein standards run consisting of bovine 6-protein mix (Michrom Bioresources, Auburn, CA) (10-fmol injection; 15-min gradient 3% acetonitrile to 40% acetonitrile) were incorporated between AP samples to minimize carry-over of D. vulgaris proteins. The final 6-protein mix standard-run was used to assess carry-over between samples (i.e. represents the “background-run” described below). In the gel-based workflow, proteins were fractionated by SDS-PAGE (12%) and bands visualized by silver staining (13). Selected bands were excised from the gel; proteins were in-gel digested with trypsin using a ProGest robot (Genomics Solutions, Ann Arbor, MI), and proteolytic peptides were analyzed by LC MS/MS using LTQ XL linear ion trap mass spectrometer (Thermo Scientific), as described in Walian et al. (27). For the in-gel workflow, additional wash and standard runs were introduced between samples that resulted from processing of gel slices from a single lane of the SDS-polyacrylamide gel, i.e. between different AP purifications. Wash runs consisted of a blank injection followed by a 30-min gradient containing two 5-min ramps from 2% acetonitrile to 97% acetonitrile. Protein standard runs consisted of a 25-fmol injection of bovine 6-protein mix via a 10-ml metered injection followed by a 14-min gradient from 2 to 50% acetonitrile. The 6-protein mix standard runs were used to assess carryover between samples (i.e. represents the background-run described below).
Identifying Peptides from Mass Spectrometry Data
Peak lists were extracted from .raw files using the Mascot Distiller 2.3.2.0 software (Matrix Science, London, United Kingdom). Data were searched with an in-house Mascot version 2.2.04 search engine (Matrix Science) against a custom protein database containing all potential protein products generated via 6-frame translation of the D. vulgaris genome supplemented by frequently observed contaminants and concatenated with the decoy database generated by reversing all D. vulgaris protein sequences (102,572 sequences; 9,848,210 residues) (32). Search was limited to doubly and triply charged precursors. The following search parameters were utilized for most searches: precursor mass tolerance of 0.8 Da for the LTQ XL-generated and 3 ppm for the LTQ Velos Orbitrap-generated data, respectively; fragment mass tolerance of 0.8 Da for both instruments; tryptic digestion allowing for cleavages before Pro; 1 missed cleavage; fixed modification, Cys-carbamidomethyl; variable modifications, deamidation (Asn and Gln), Met-sulfoxide, and Pyro-Glu (N-terminal Gln). A limited number of searches were performed with a precursor mass tolerance of 1.5 Da and 50 ppm for LTQ XL-generated and LTQ Velos-generated data, respectively. Precursor and product ion mass tolerances for analysis of the QSTAR-generated data were 100 ppm and 0.15 Da, respectively. Significance threshold was set to a p value of ≤0.05. Protein acceptance required the presence of at least one distinct peptide with expectation value of ≤0.05. >90.5% of peptide identifications met the “bold red” Mascot match quality criteria, i.e. (i) peptide assignment to a protein with the highest score (rank) within the potential homologs with overlapping sequences, and (ii) a top scoring match for the spectrum. <0.5% of peptide identifications had rank two scores. 97% of these, however, were the only identification supporting a protein and as such were filtered out at a later step, as described below. The remaining 25 cases supported a protein identified by at least one peptide that met the bold red criteria and were thus retained. All peptide matches with expectation value of ≤0.05 were used for spectral counting (33, 34).
Initial MS Data Filtering
High abundance or “sticky” proteins were observed in some cases in subsequent unrelated protein samples even after extensive washing of the LC column between samples (background-run). Proteins identified based on the presence of these peptides were designated “carry-over” and removed from subsequent analysis if the Mascot score for the protein in the sample in question was lower than its Mascot score from the immediately preceding background-run. This automatic procedure was augmented in 21 cases by manual removal of a single protein that appeared to be a contaminant from other samples processed the same day. In addition, peptides that cannot be unambiguously mapped to a single D. vulgaris protein were removed from the analysis and not used to assign the proteins' identification. Some ambiguous peptides were retained for spectral counting, however, but only if the identified protein was also supported by at least one unambiguous peptide. Peptide level mass spectrometry data for the resulting partially filtered dataset are provided in supplemental Dataset S2.
For our final high confidence interactome, we additionally filtered out low signal proteins and overly abundant proteins by removing prey proteins identified with a single-peptide hit from the results of a given purification: ribosomal proteins and protein chaperones (DnaK, DVU0811; GrpE, DVU0812; GroEL, DVU1976; and GroES, DVU1977); and the following top nine frequent fliers: PpaC (DVU1636); Mrp (DVU2109); GroEL; DVU2405; ApsA (DVU0847); Sat (DVU1295); Pyc (DVU1834); DnaK; and Tuf (DVU2920) (supplemental Dataset S3). The 31 instances of a bait being detected by a single peptide were retained at this stage, however (see supplemental Dataset S2). Skyline software (35) was used to generate a spectral library for these baits, and the spectra have been deposited at Panoromaweb. After this series of filtering steps, 53,506 protein pairs remained for a Matrix model (supplemental Dataset S4) and 5177 for a Spoke model (20).
Definition of Homologs and Interologs
To compare protein-protein networks from various species as well as to project the EcoCyc reference set onto species other than E. coli, we mapped homologs between all nine studied species using bi-directional best Basic Local Alignment Search Tool (BLAST) searches (36). All predicted protein sequences encoded by one genome were queried against a database of protein sequences encoded by another genome using BLASTP 2.2.9 with default options, and then the search direction was switched. Pairs in which each protein was the most significant hit for a query from the other genome and for which both E-values were at least as significant as 10−4 were mapped to each other. The supplemental Table S1 lists the number of mapped homologs between all nine species. A pair of PPIs (a and b) and (a′ and b′) from two different species is called an interolog if a is th emapped homolog of a′ and b is the mapped homolog of b′.
Defining Gold Standard Positive and Negative Interactions
Computational analysis was performed using curated gold standard sets of interacting and non-interacting pairs of proteins (supplemental Dataset S5). Because of the lack of truly known interacting and non-interacting proteins in D. vulgaris, our gold standard sets should be considered as imperfect gold standards. 38 of the gold standard positive set are pairs of D. vulgaris proteins that have been shown to interact in stable complexes in low throughput experiments in this organism. The remaining 500 gold-positives were E. coli interologs, i.e. D. vulgaris proteins mapped to homologous E. coli proteins (as described above), of either PPIs from EcoCyc version 12.0 (supplemental Dataset S6) (37) or reciprocally confirmed PPIs from recent AP-MS experiments in E. coli (17). This dataset was curated to account for expected differences between E. coli and D. vulgaris complexes (e.g. a classical RNA degradosome complex configuration was not expected to be found in D. vulgaris due to the truncation of a scaffold protein (38)). We also excluded all interactions with ribosomal proteins, as this complex is atypical due to the RNA component as well as highly abundant. This resulted in a set of 536 pairs, of which 69 were observed among the 53,506 protein pairs for our matrix model.
Because the number of potential pairwise interactions between all proteins vastly exceeds the number of bona fide PPIs, the number of protein pairs in a gold-negative set should approximate the square of the number of proteins in the gold-positive set divided by two to capture the large number of potential pairwise combinations. Therefore, a negative gold standard set of non-interacting protein pairs was prepared by randomizing pairs of proteins from the positive gold standard set. We included all pairs of D. vulgaris proteins mapping to E. coli proteins that (a) were present in a heteromeric complex in EcoCyc but not observed to interact with each other in either EcoCyc or AP experiments (both reciprocal and non-reciprocal interactions from (17)), and (b) an interaction should have been possible to detect via AP because both bait and prey were identified in other AP pulldowns. We excluded pairs made between ribosomal proteins and other proteins, as well as pairs in which one partner was annotated as a protein chaperone or protease, because the latter functional categories are expected to form nonspecific complexes with a variety of partners. This resulted in 27,542 protein pairs. We observed a total of 1171 pairs among the 53,506 protein pairs for the matrix model.
To compute the FDRs for our high confidence interactome and the published bacterial Y2H and AP-MS interactomes, a different gold standard set was used to allow a common approach for multiple species. This gold standard set was solely based on EcoCyc complexes without any additional correction for D. vulgaris biology or any additional AP-MS data. Only ribosomal proteins were excluded from the EcoCyc dataset. The gold standard positive set was constructed out of all EcoCyc interacting pairs, and the negative set was constructed by taking all possible pairs of proteins in the positive set that do not appear in the positive set of interactions. The gold standard set for species other than E. coli was created by mapping the E. coli gold standard set to interologs in other species (supplemental Table S2).
Scoring Protein-Protein Interactions
To measure the likelihood of two proteins being involved in a physical interaction, each protein pair is assigned values for four scoring functions that each separate bona fide interactions from background noise.
Dice score indicates the number of times two proteins are observed together in a purification divided by the sum of individual appearances of the two proteins in all purifications (39). This feature reduces the problem of “frequent fliers,” either “sticky” proteins that bind non-specifically to many other proteins or abundant proteins found in many fractions. For frequent fliers this value is close to zero, and for proteins that form specific interactions, the value is higher.
The Completeness score measures the consistency of purifications. For each purification, we define a directed graph that includes edges from the bait to each prey and, in addition, includes edges from prey a to prey b if b was also observed as a prey in a different purification in which a was used as bait. The completeness score is defined as a ratio of the number of edges in the graph to the total number of possible edges in the graph. This score is computed for each purification, and each pair of proteins is assigned the maximum value over all the purifications in which both proteins appeared. The completeness score is a generalization of the clustering coefficient (20).
- The normalized spectral abundance factor (NSAF) filters out prey observed at low abundance. For a given purification, p, NSAF for protein v is defined as shown in Equation 1 (40),
where SCv is the number of spectral counts for protein v; Lv is the length of protein v, and index i iterates over all proteins in the given purification. For the matrix model, the score for a pair of proteins (v,u) is extended to Model 1,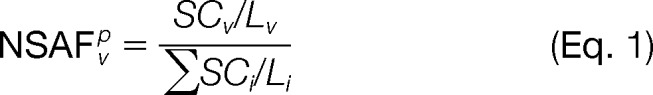
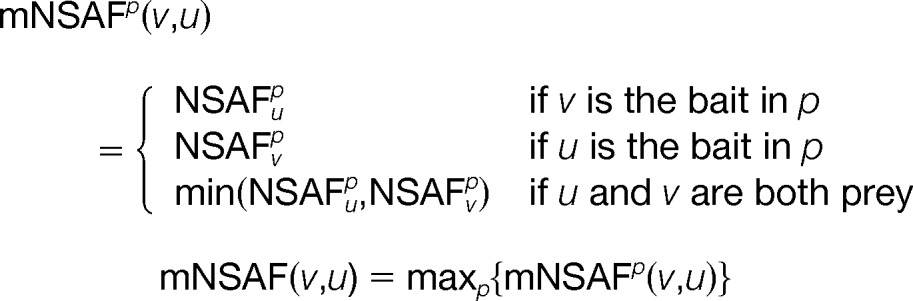
- For absolute normalized abundance (ANA), this metric is similar and complementary to NSAF. Unlike NSAF, it is not normalized with respect to the sum of spectral counts per purification, and therefore it estimates the absolute abundance across all purifications as shown in Model 2,
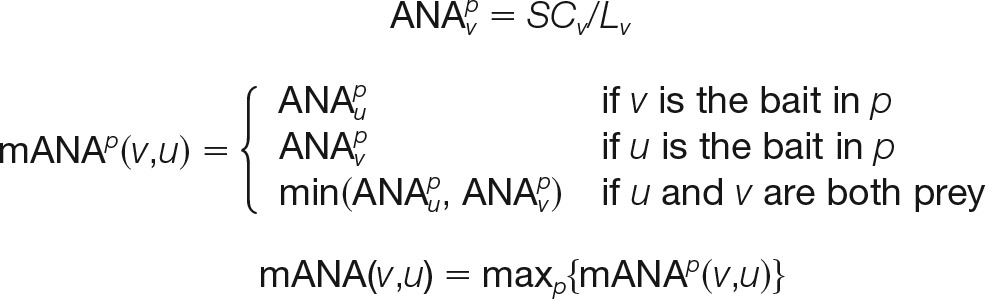
Inference of High Confidence Protein-Protein Interactions
Using the gold standard set, we trained a logistic regression to compute the likelihood scores for all pairs of proteins observed together in all the purifications. Such a strategy allows relative weights for our four scoring function to be derived and the result to be summarized with a single likelihood score. Similar approaches have been previously proposed using either spectral counts (40–42) or co-purification statistics for pairs of proteins (14, 43), although here we use a combination of these two approaches and somewhat modified metrics. We tested three types of cross-validation to reduce learning bias (44) as follows: 1) 10-fold cross-validation; 2) leave-one-out cross-validation; and 3) leave-one-operon-out cross-validation, in which at each iteration of cross-validation all proteins from a given operon are selected and all their intra-operon and inter-operon interactions are used for validation and the rest are used for training. We observed only small differences in the results of these three cross-validations, and the third was used for all the results presented here as it is the more stringent.
Despite joining the four scoring functions described above into a single classifier, we still observed that (probably due to the limitations of our gold standard set and the scoring functions) some fraction of PPIs received high combined scores even though some of the most important individual feature scores were very low. Because of this, we applied the following filtering steps.
(i) Feature-based Filtering
(a) Remove pairs of proteins that co-appeared together in fewer than 10% of the purifications in which either of the proteins appeared individually, i.e. the dice score has to be higher than 0.1. (b) Remove pairs of proteins for which mNSAF score (defined above) is less than 5% for at least of one of them. This mostly eliminates proteins from very large pulldowns, which were likely the result of experimental error, where some proteins appear at very low abundance. This resulted in 1136 PPIs with an FDR of ∼35% (supplemental Dataset S7).
(ii) Classification Based Filtering
To obtain a higher confidence set of PPIs, we applied a threshold to the confidence scores, as computed by logistic regression that gives 459 matrix model PPIs at 17% FDR and 352 spoke model PPIs at 10% FDR upon cross-validation (supplemental Dataset S8).
Experimental Design and Statistical Rationale
This project determines high confidence PPIs using a logistic regression that combines four different features from the mass spectrometry data, as described above. For this reason, no single aspect of the mass spectrometry data, such as reproducibility between technical or biological replicas, provides the most telling measure of accuracy. Instead, the fundamental criteria for judging the accuracy of our high confidence PPIs are the FDRs calculated using gold standard and gold-negative protein pairs, see above, and the additional quality metrics shown under “Results.” Our analysis indicates that the PPIs in our two high confidence interactomes are comparable in accuracy to those in three benchmark sets of validated PPIs: the EcoCyc dataset and AP-MS and Y2H PPIs that have been reciprocally confirmed in biological replicas as bait prey and prey bait pairs. In contrast, by the same suite of criteria, nine previously proposed bacterial interactomes are much less accurate.
The following suggests that many protein pairs that are highly reproducible between replica affinity purifications are nonetheless false positives, likely because at least one of the pairs is a highly abundant contaminant in many purifications. In our unfiltered data, we observed 140 reciprocal interactions, including 35 pairs that are encoded in the same operon (25%). However, only 82 of these reciprocal pairs are present in our final set of 459 high confidence PPIs. Of the remaining 58, only one is a same-operon pair, suggesting that these 58 reciprocal pairs are mostly false positives. Conversely, of the 377 high confidence PPIs that were not reciprocally confirmed 17% are same operon pairs, and therefore the non-reciprocal high confidence PPIs are likely almost as accurate a set as the 140 reciprocally confirmed PPIs. Reciprocally confirmed PPIs are thus a useful but by no means decisive indication of bona fide interactions. Instead, by using logistic regression to consider multiple features of the mass spectrometry data at once, frequent fliers and other false positives can be identified more effectively.
That said, we have determined the reproducibility of our AP-MS assay at several steps. There are two measures of the technical reproducibility between different mass spectrometry assays of the same affinity-purified sample of eluted proteins. 73% of the 1628 instances where a protein was detected after elution from an SDS-polyacrylamide gel were also detected after in-solution digestion from the same affinity purification. For the 99 cases where in-solution digests were performed in replicas on the same set of 46 eluted proteins, the mean overlap in proteins identified between replicas was 71%.
There also two measures for reproducibility between biological replicas. The first is that for the 178 instances where the same bait was affinity-purified two or more times (76 different bait proteins), the mean overlap in proteins identified between replicas was 65%. The second is given by the 34% reciprocal confirmation percent among high confidence PPIs, which is considerably higher than the 0.3–9.8% reciprocal confirmation percent seen in the nine previously published AP-MS and Y2H interactomes, see under “Results.”
Identifying Low Confidence Protein Pair Sets
Single Peptide Hit
All identified proteins, including those whose identification was based on a single unique peptide, were included in the analysis. The same pipeline and criteria for selecting high confidence set were used otherwise.
Ribo-other
PPIs involving ribosomal proteins and having regression score satisfying the selection of the high confidence set defined above were selected. Among these pairs only those that have one ribosomal protein and the other non-ribosomal protein were finally considered for this set.
No Thresholds
We did not apply the threshold on the dice and mNSAF regression features (see under “Feature-based Filtering”) and selected a subset of high scoring PPIs based on the regression scores that resulted in 17% FDR, similar to our high confidence set.
High FDR
Pairs that were part of the partially filtered set of 1136 PPIs described above were excluded from the high confidence set by the 20% FDR threshold.
Gene and Protein Annotations
Gene names, protein names, protein function annotation, and gene to operon assignment are taken from the MicrobesOnline database (45). Cellular localization for D. vulgaris and E. coli proteins was taken from PSORTb (46, 47).
Other Bacterial Interactome Data
We have identified nine bacterial AP-MS or Y2H screens in the literature. Where a screen is reported in more than one paper by the same group, we have taken the interactome data from the latest, most complete report of the screen. One AP-MS network for E. coli was taken from supplemental Table S6 of Hu et al. (17), and the reciprocal PPIs from this study were identified using supplemental Table S4 from Hu et al. (17) (supplemental Dataset S9). In addition, from private correspondence with Hu et al. (17), we obtained their complete LC MS “in-solution” set of identified peptides, which was used for peptide statistics as well as to obtain our revised high confidence interactome for E. coli. A second AP-MS interactome of E. coli generated by a different group was taken from supplemental Table S1 of Arifuzzaman et al. (18), including a list of reciprocal PPIs (supplemental Dataset S10). The AP-MS interactome of Mycoplasma pneumoniae was taken from supplemental Table S3of Kuhner et al. (16) (supplemental Dataset S11), and reciprocal PPIs were taken from their supplemental Table S2 and reduced to those pairs that appear in supplemental Table S3. The Treponema pallidum Y2H interactome and its reciprocals were taken from supplemental Table S1 of Titz et al. (9), subset TPA-HCA (supplemental Dataset S12). Campylobacter jejuni Y2H PPIs having a score above 0.5, i.e. the high confidence set as defined by the authors, and the set of reciprocal PPIs were taken from supplemental Table S13 of Parrish et al. (7) (supplemental Dataset S13). The Y2H interactome of Bacillus subtilis was taken from supplemental Table S6 of Marchadier et al. (6) (supplemental Dataset S14). No information on reciprocal PPIs was available for this study. The E. coli Y2H interactome and its reciprocal PPIs were taken from supplemental Table S3 of Rajagopala et al. (4) (supplemental Dataset S15). The list of reciprocal PPIs was extended with pairs that were confirmed with two types of vector of fusion to an activator domain. The Helicobacter pylori Y2H interactome was taken from supplemental Table S2 of Hauser et al. (3), with reciprocal PPIs taken as those having an overlap value of at least two (supplemental Dataset S16). The Synechocystis sp. Y2H interactome, PPIs having interaction category “A,” and its reciprocals were taken from supplemental Table S2 of Sato et al. (8) (supplemental Dataset S17). For all data sets, we kept only heterologous interactions; no homomeric pairs were considered.
Calculating Overlap between Two PPI Networks
Because different studies could target different subsets of the proteome, we calculated percent of overlap between two networks relative to the common proteins only. Given two sets of PPIs, each of them is reduced to pairs for which each protein appears in the other set. Then the overlap is defined as a fraction of common PPIs relative to the reduced sets.
Calculating the Number of Proteins Detected Per Bait in Non-filtered MS Data
To calculate the number of preys detected per bait in the previously published AP-MS screens, we used data that were as close as possible to the raw peptide data identified as significant hits by an MS data search engine, i.e. the datasets used had not been filtered based on criteria or features such as a co-purification score (17) or the machine learning approach used in our study. For data from Arifuzzaman et al. (18), we used their supplemental Table S1, even though some frequent fliers identified by the authors from the control experiments were not present in that table. For data from Kuhner et al. (16), we used their supplemental Table II. For the Hu et al. (17), we obtained the data on all peptides detected by “in-solution” LC-MS from personal correspondence with the authors.
Revised High Confidence Set of E. coli PPIs
To the in-solution LC MS data collected for E. coli (17), we applied our pipeline in the same manner used to obtain our high confidence PPIs for D. vulgaris. Single peptide hits as well as ribosomal and chaperons (DnaK, b0014; GrpE, b2614; GroEL, b4143; and GroS, b4142) were removed. We used the same feature-based filtering and thresholds. Then the regression score cutoff was selected at 20% FDR to obtain 391 matrix model PPIs (supplemental Dataset S18).
Calculating False Negative Rates
The false negative (FN) rate was estimated using the EcoCyc gold-positive set from which both ribosomal and flagella related PPIs were excluded. There are 301 D. vulgaris interologs corresponding to this set. Among these 301 pairs, 79 had at least one protein tagged as a bait in our experiments and the second protein detected in at least one purification. Out these 79, 41 are found among the 53,406 potentially interacting pairs in the unfiltered matrix model, giving an estimated experimental FN rate of 100·(1 − 41/79) = 48%. Of the 41 potential protein pairs, 24 are reported in our high confidence set, implying an analysis pipeline FN rate of 41% and an overall FN rate of 69%. Given that our high confidence set of 459 PPIs has ∼20% false positives, 1196 PPIs should have been detected from the 957 tagged bait purifications we conducted. Using similar calculations for as for the Hu et al. (17) in-solution E. coli AP-MS dataset, the experimental and computational FN rates are 4 and 17.6%, respectively. Assuming our revised set of 391 PPIs contains 20% false positives, the number of E. coli PPIs that are potentially detectable in this experiment is 716.
To extrapolate to the number of PPIs detectable if all proteins were tagged as baits, we observe that for both the D. vulgaris and E. coli data, the relation between the number of newly observed proteins with each new purification correlates linearly with the number of high confidence PPIs confirmed by these purifications, see under “Results.” Thus, for all 3399 D. vulgaris and 4151 E. coli proteins, and assuming a 20% FDR and the appropriate FN rate as described above, we estimated there are 3067 and 3002 PPIs for D. vulgaris and E. coli, respectively.
Estimating the FDR Using Gold Standards from Rajagopala et al. (4)
Rajagopala et al. (4) tested 212 gold-positive and 500 gold-negative protein pairs, detecting 44 and 4, respectively, as interacting in their Y2H assay. If one assumes a similar false positive rate for an appropriately sized gold-negative set of ∼90,000 pairs (∼(4242/2)-212), this implies an FDR for the Rajagopala et al. (4) interactome of >80%.
Visualization of Biotin Binding Proteins
Wild-type D. vulgaris (ATCC 29579) and D. vulgaris CAT400742 (DVU2224-STF-His6) were grown at 30 °C in an anaerobic growth chamber (Coy Laboratory Product, Inc., Grass Lake, MI) in rich liquid MOYLS4 medium for 20 h (10% inoculum) (48). G418 was added to a final concentration of 400 μg/ml for CAT400742. E. coli BW25113 cells were grown overnight in LB media. Cells from 1-liter cultures were harvested by centrifugation. For affinity blotting, cells were lysed at room temperature with 10 ml of Bacteria Protein Extraction Reagent (Thermo Scientific) containing 200 μl of 50× protease inhibitor and 2 μl of Benzonase nuclease (Sigma). Cell lysate was cleared by centrifugation for 30 min at 16,000 × g. 250 μl of a 50% slurry of anti-FLAG agarose beads was added to 10 ml of lysate, and the mixture was incubated for 3 h at 4 °C. Beads were separated by centrifugation and washed in 5× 1 ml of 50 mm Tris, pH 8.0, 150 mm NaCl buffer. 50 μl of beads were boiled in 1× SDS, and proteins were separated on 15% Tris glycine gel. Proteins were transferred on PVDF membrane and blocked overnight in 5% milk. Membrane was washed five times in PBS with 0.05% Tween 20 buffer (PBST), and membrane was incubated with streptavidin-horseradish peroxidase polymer conjugate (Sigma) diluted 1:2500 in PBST at room temperature for 1 h (49). Membrane was washed five times with PBST and incubated for 5 min in SuperSignal West Substrate working solution (Thermo Scientific). Membrane was exposed to x-ray film to visualize biotinylated proteins.
Data Reporting
The protein interactions from this publication have been submitted to the IMEx consortium through IntAct and assigned the identifier IM-22740 (50). All raw MS files and associated Mascot search engine results files wereuploaded at the UCSD Center for Computational MassSpectrometry, MassIVE, and can be downloaded on line (MassIVE identifiers: MSV000079275, MSV000079276, MSV000079277). A spectral library containing annotated MS/MS spectra for baits and virtually all preys identified on the basis of a single peptide has been deposited at the Panoramaweb site (51). These data can be viewed at Panorama Public, project title: “Bacterial interactomes: interacting protein partners share similar function and are validated in independent assays more frequently than previously reported”. A small minority of single peptide hit spectra that could not be uploaded to Panoramaweb are provided in supplemental Dataset S19.
RESULTS
AP-MS Screen of D. vulgaris
D. vulgaris is an obligate anaerobe that had previously been challenging to use to purify affinity-tagged proteins. Therefore, we first tailored high throughput genetic modification, immunological screening, cell culture, and affinity purification protocols for this bacteria (Fig. 1) (28). Out of a set of genes prioritized for screening (see “Experimental Procedures”), 1401 were successfully fused with a tandem affinity tag (supplemental Dataset S1), and 957 of the resulting fusion proteins were detected as baits in AP-MS experiments along with an average of 7.7 co-purifying prey proteins per purification (supplemental Dataset S2 and Fig. S1). The proteins detected tend to be among those translated from highly expressed mRNAs (supplemental Figs. S2 and S3) and included members of all but one of the 17 functional categories defined by The Institute of Genome Research (TIGR roles) (supplemental Fig. S4) (52).
Fig. 1.

High throughput affinity purification and mass spectrometry pipeline in D. vulgaris. Schematic diagram showing the stages of the high throughput pipeline, including suicide vector construction, recombinant strain generation, affinity purification, mass spectrometry, and data analysis to annotate interactions along with corresponding statistics.
Inferring High Confidence Protein-Protein Interactions from MS Data
In AP-MS experiments, many proteins that are not genuine interacting partners will nonetheless co-purify with a tagged bait protein (16, 17). To overcome this contamination problem, we first filtered the data to remove prey proteins detected by only one peptide, the nine proteins most frequently observed by MS in purifications (supplemental Dataset S3), and ribosomal proteins and chaperonins. The spoke model for interpreting AP-MS data assigns a potential interaction between each bait and all of the prey proteins that co-purify with it (Fig. 2A) (20). The matrix model (20) extends the spoke model by including all pairwise interactions between the prey identified in a given purification (Fig. 2A). After our initial filtering, we obtained 53,406 potentially interacting pairs for the matrix model (supplemental Dataset S4) and 5177 for the spoke model.
Fig. 2.

Evaluation of PPI inference for two AP-MS representation models. A, schematic representation of two possible models, spoke and matrix, used to annotate PPIs from AP-MS experiments. B, precision/recall curve as measured from the cross-validation procedure. True positives (TP) (correctly inferred gold standard positive interactions), false positives (incorrectly inferred as interacting gold standard negative interactions), and FN (not inferred as interacting gold standard positive interactions). C, precision as the function of the number of PPIs inferred by the logistic regression classifier.
Each of the 53,406 putative protein pairs was described using four features based on the MS data (see “Experimental Procedures”). Two features, NSAF and absolute normalized abundance, use MS spectral counts normalized by polypeptide length to down-weight instances where one or both partners are present at lower relative or absolute abundance. The other two features, dice and completeness, are used to favor protein pairs that co-occur in the same purifications more commonly than they are found in separate purifications. In addition, based on published evidence, a set of 69 “gold standard-positive” protein pairs were identified that are likely bona fide PPIs and a set of 1171 “gold standard-negative” protein pairs that likely do not interact (see “Experimental Procedures” and supplemental Dataset S5). The four features each give partially separated distributions for the gold standard-positive and -negative protein pairs (supplemental Figs. S5–S8) and were combined using a logistic regression to give a single score for each protein pair.
After imposing thresholds on the features NSAF and dice, a set of 1136 protein pairs was identified (see “Experimental Procedures” and supplemental Dataset S7). The accuracy of this set of pairs at different points down the logistic regression rank list was then estimated using the gold standard-positive and -negative sets with cross-validation, see “Experimental Procedures” (Figs. 2B and supplemental Fig. S9). For the matrix model, we identified 459 PPIs involving 469 proteins at 17% FDR (Fig. 2C and supplemental Dataset S8), and for the spoke model, we identified 352 PPIs at 10% FDR. Because the spoke and matrix models are similar (supplemental Fig. S10), we define the PPIs from the matrix model as our high confidence set. The resulting network of interactions is shown in Fig. 3.
Fig. 3.

D. vulgaris protein interaction network. All 459 interactions present in the high confidence interaction network are shown. Spoke model (bait-to-prey) interactions are indicated with solid arrows, and matrix model (prey-prey) interactions are shown with dashed lines. Gold-positive interactions inferred from 10-fold cross-validation are shown in purple color. All proteins are colored according to their assigned TIGR role as indicated. Gray areas enclose complexes discussed in the supplemental text associated with sulfate reduction and energy conservation (a1–a3), cofactor biosynthesis (b1–b3), RNA synthesis and degradation (c1–c3), motility (d1–d3), megaplasmid encoded complexes (e1–e5), and novel and uncharacterized complexes (f1–f2).
An FDR, however, is necessarily an approximation as it assumes that the gold standard sets used contain no errors and that they have properties typical of the set of bona fide PPIs present in the original unfiltered dataset. Given that we do not know the true set of PPIs in any organism, there is no guarantee that these conditions are met. Consequently, we have adopted the following five additional metrics to assess the quality of our sets and other sets of proposed PPIs.
Percentage of Same Operon PPIs
This measures the fraction of protein pairs whose members are encoded in the same operon.
Fold Enrichment in the Same TIGR Role
This measures the fraction of protein pairs, both of whose members share the same TIGR role annotation, were divided by the fraction of the same TIGR role pairs that are found in gene pairs chosen at random from the genome.
Percentage of PPIs in AP-MS
This measures the fraction of protein pairs that are found in at least one AP-MS interactome other than the dataset being assessed.
Percentage of PPIs in Y2H
This measures the fraction of PPIs in a given set that are found in at least one Y2H interactome other than the dataset being assessed.
Reciprocal Confirmation Percent
This measures the protein pairs for which interaction was reciprocally confirmed within the same AP-MS or Y2H study as both bait-prey and prey-bait pairs, as a percent of pairs in the interactome for which both members were tested as baits.
To provide a benchmark for the properties of well characterized bacterial complexes, we identified three “benchmark” PPI datasets. One comprises the complexes in the EcoCyc database, a manually curated dataset based on low throughput experiments from the literature (supplemental Dataset S6) (53). The other two are for the ∼3% of protein pairs that have been confirmed as reciprocal PPIs in either bacterial Y2H or AP-MS interactomes (Fig. 4 and supplemental Table S3). Encouragingly, our high confidence PPIs score within the same range as the benchmark datasets for our first four metrics, never having a lower value (Fig. 4), although the fifth metric is not applicable to the benchmark sets.
Fig. 4.

PPI quality metrics for benchmark datasets and high and low confidence D. vulgaris protein pair sets. The top three rows show metrics for benchmark bacterial datasets: the EcoCyc complexes (53) and protein pairs that have been reciprocally confirmed in either four AP-MS studies, including ours, or in six Y2H studies (supplemental Table S3). The remaining rows show metrics for our high confidence set and the four low confidence sets of protein pairs described in the text. The numbers of protein pairs in each set are given in parentheses. The columns shows from left to right are as follows: percent of pairs whose members are encoded in the same operon; reciprocal PPI confirmation percent; fold enrichment of pairs for which both members have the same TIGR role over that expected among randomly chosen pairs; percent overlap with a combined set of interologs from the six bacterial Y2H interactomes; and percent overlap with a combined set of interologs from the three published bacterial AP-MS interactomes (see “Experimental Procedures” for more details).
The accuracy of our high confidence PPIs is further supported by the fact that they include many previously characterized interactions or are consistent with other data (see supplemental text and supplemental Figs. S11–S13 for further discussion). For example, interactions previously identified in low throughput studies of D. vulgaris or other bacteria are observed among components of dissimilatory sulfite reductase, quinone-interacting membrane-bound oxidoreductase, flavin oxidoreductase, and RNA polymerase. Novel high confidence PPIs supported by other data include associations between an uncharacterized protein, DsrD and DsrAB; FlxABCD and heterodisulfide reductase A; two proteins of unknown function and RNA polymerase; and an uncharacterized protein, a carbon storage regulator, and flagellin proteins. In addition, of the 11 high confidence PPIs that include a protein encoded on the native 200-kb D. vulgaris plasmid pDV1, only one includes a protein encoded on the much larger bacterial chromosome, a result consistent with a low FDR.
Lower Confidence Sets of Protein Pairs
We next explored whether it is possible to detect additional PPIs by using alternative criteria to interrogate the raw mass spectrometry data. Four sets of “low confidence” protein pairs were identified (Fig. 4 and “Experimental Procedures”). The first three were captured as matrix models at ∼20% FDR and included many PPIs from our high confidence set, but here we only consider those unique pairs that were found in addition to the high confidence PPIs. The four sets are (i) “single hit” prey proteins that are detected by only one peptide; (ii) ribosomal and chaperonin proteins, but limited only to “ribo-other” protein pairs between a ribosomal or a chaperonin protein and another protein; (iii) “no threshold” pairs that were excluded by the thresholds placed on the features NSAF and dice; and (iv) the 677 “high FDR” protein pairs excluded from our high confidence set at local FDRs between 20 and 61%.
Compared with our high confidence PPIs and the three benchmark datasets, the low confidence sets are each less enriched in protein pairs that are encoded in the same operon, reciprocally confirmed, or found in Y2H interactomes or, with one exception, in AP-MS interactomes (Fig. 4). The single hit and ribo-other sets are likely to be largely false positives as they also show no enrichment for pairs with similar functions (Fig. 4). This is not unexpected as single peptide hits are present at lower abundance in purified material and thus are more likely to be incompletely removed contaminants, and ribosomal proteins are highly expressed in cells (supplemental Fig. S3) and thus also likely to be difficult to completely remove during purification. The “no threshold” and “high FDR” sets, in contrast, may contain many functional but lower affinity interactions. The no threshold pairs are almost as strongly enriched in the same TIGR role protein pairs as our high confidence set (Fig. 4). Given that this strong enrichment is unlikely to occur by chance (p value ≤ 0.05 after correction for multiple testing), it is reasonable that most no threshold pairs are functional. The lower reproducibility with which these PPIs are observed either in other interactomes or as reciprocal PPIs could be because low affinity interactions are harder to observe. Similarly for the high FDR set, pairs between 500 and 1000 on the regression score rank list remain enriched in same TIGR role pairs, but they show little enrichment in the same operon pairs, pairs found in other interactomes, or reciprocally confirmed pairs (Fig. 5), suggesting again that there is a class of functional associations among proteins that differ from those that predominate our high confidence set.
Fig. 5.

PPI quality metrics for regression score cohorts. Predicted interactions are sorted by the logistic regression score (x axis) for the 4000 highest scoring matrix model interactions in the absence of any regression feature threshold. The y axis shows the relative enrichment of protein pairs that are encoded in the same operon (maroon) that is reciprocally confirmed (green), is found in Y2H studies (red) and in other AP-MS studies (blue), and is shared the same TIGR role (gold). Enrichment is expressed as a ratio over the frequency expected for pairs randomly chosen from the genome, with the horizontal dashed line indicating the background frequency ratio of 1. The values are calculated for a series of windows of 500 protein pairs, except that the first n <250 points the window size is n +250.
Estimating a False Negative Rate
Although we cannot identify additional PPIs with properties similar to those in our high confidence set, it is possible to estimate the percent of such PPIs that we have failed to detect by calculating the false negative rate using gold standard positive PPIs (see “Experimental Procedures”). From the estimated false negative rate of 69%, the number of PPIs that should have been detected in our screen of 957 tagged baits is ∼1196 versus the 459 we report. Extrapolating to a screen in which all genes are tagged, we estimate that one might detect ∼3000 PPIs with characteristics similar to both our high confidence set of PPIs and the EcoCyc PPIs (see “Experimental Procedures”; supplemental Fig. S14).
Comparison with Other Bacterial AP-MS Interactomes
We next compared our high confidence interactome for D. vulgaris with the three previously published bacterial AP-MS networks, two for E. coli (17, 18) and one for M. pneumoniae (16) (see “Experimental Procedures”). Our interactome is dramatically less connected than these other networks (Fig. 6 and supplemental Table S4). For example, the number of protein pairs per bait and the size of linked complexes are both >8-fold lower in our interactome compared with the other three (supplemental Table S4). In addition, the protein pairs in our interactome have very different qualities compared with the other AP-MS networks (Fig. 7). For example, using a common gold standard, the FDR of our interactomes is 29% versus 66–91% for the other networks. In addition, same operon protein pairs, same TIGR role pairs, reciprocal PPIs, and pairs found in other bacterial interactomes are, with one partial exception, substantially more enriched in our high confidence set than in the other interactomes (Fig. 7). The partial exception is for M. pneumoniae. These protein pairs, however, were chosen using genomic proximity data (16), which could well have introduced a selection bias.
Fig. 6.

Interactome connectivity of EcoCyc PPIs and proposed bacterial AP-MS interactomes. Visualization of interactomes for manually curated protein complexes in the EcoCyc database (A), the high confidence AP-MS interactions inferred in this study for D. vulgaris (B) and E. coli (C), the published AP-MS interactomes for M. pneumoniae (16) (D), and the E. coli (E and F) (17, 18).
Fig. 7.

PPI quality metrics for benchmark datasets and proposed bacterial interactomes. The top three rows show metrics for the three benchmark datasets described in Fig. 4. The remaining rows show our high confidence interactomes for D. vulgaris and E. coli (supplemental Datasets S8 and S18), the three previously published AP-MS interactomes (supplemental Datasets S9–S11), and the six Y2H interactomes (supplemental Datasets S12–S17). The numbers of protein pairs in each set are given in parentheses. The left most column shows the FDR estimated using gold standard positive and negative sets based only on complexes from the EcoCyc dataset or, in the case of the non E. coli studies, their interologs. The remaining columns are as in Fig. 4.
The different properties of our interactomes versus those proposed previously could result from differences in data quality or how the data was analyzed. The following suggests that differences in data analysis methods are chiefly responsible.
23% of PPIs in the three previous AP-MS interactomes include a ribosomal protein paired with a non-ribosomal protein. Only 0.4% of these pairs, however, were reciprocally confirmed in a given study; only 1.1% are encoded in the same operon; only 1.1% are present in at least one bacterial Y2H interactome; and they are only 1.03-fold more frequently annotated with the same TIGR role than expected for randomly selected pairs of genes. The inclusion of ribo-other pairs in the published bacterial AP-MS interactomes thus significantly affects the quality metrics for the complete interactome.
An “in solution” MS dataset of 3361 PPIs from the Hu et al. (17) AP-MS study of E. coli used biochemical and MS methods closest to ours and thus was most appropriate for complete reanalysis. Using our data filtering and analysis methods to identify high confidence PPIs from this dataset, only 391 matrix model PPIs are identified at 20% FDR (supplemental Dataset S18). These PPIs have network connectivity and PPI quality metrics scores similar to those of our high confidence D. vulgaris interactome and the three benchmark datasets (Figs. 6 and 7 and supplemental Table S4). In contrast, the 2970 protein pairs from the Hu et al. (17) dataset that were not selected by our reanalysis have metric scores similar to protein pairs from the three published bacterial AP-MS interactomes (supplemental Table S5). In addition, several hundred pairs that were excluded just below the 20% FDR threshold in our reanalysis tend to share the same TIGR role but are not to be encoded in the same operon, found in other interactomes, or reciprocally confirmed (Fig. 8, regression score ranks 500–1,500). Finally, based on our estimate for the false negative rate, ∼716 PPIs could in principle be detected from the Hu et al. (17) in solution dataset, versus the 3361 PPIs reported by Hu et al. (17) and the 391 identified in our re-analysis of these data (see under “Experimental Procedures”). Thus, by reanalyzing the Hu et al. (17) MS data, we can achieve results very similar to those obtained using our D. vulgaris data.
Although Hu et al. (17) used a gold-positive set that, similar to ours, was based on low throughput experiments, their gold-negative set was built differently. It consisted of pairs comprising a cytoplasmic protein (CY) and either a periplasmic (PE) or an outer membrane (OM) protein. There are, however, seven times fewer OM and PE proteins than CY proteins, and OM and PE proteins are detected by mass spectrometry with half the efficiency of CY proteins (see supplemental Fig.S1b in Ref.17). Gold standard negatives estimate the frequency of contaminating proteins that are incompletely removed during purification. If a member of each negative pair is, on average, of lower abundance or less detectable than the positive proteins, this frequency will be underestimated. To reduce the possibility of such a bias, our gold-negative set comprised non-interacting pairings between proteins from our gold-positive set. To test the impact of the difference between ours and the negative sets of Hu et al. (17), we constructed a negative set identical in size to our original one, but used pairs between D. vulgaris CY proteins and either PE or OM proteins. Using this set together with our usual gold standard positive set, the recalculated FDR of our high confidence interactome of 459 PPIs is 10% (versus 17% previously), and the FDR for the set of 1136 protein pairs input to the regression is 22% (versus 35% previously). Thus, a gold-negative set built of CY pairs underestimates the FDR.
Fig. 8.

PPI quality metrics for regression confidence score cohorts for the Hu et al. (17) revised dataset. The values plotted were determined as described in Fig. 5 except that information from our reanalysis of Hu et al. (17) data are shown.
Comparison with Bacterial Y2H Interactomes
We have also compared our D. vulgaris high confidence interactome and our revised version of the Hu et al. (17) E. coli interactome to Y2H networks from six bacteria: E. coli (4), H. pylori (3), T. pallidum (9), C. jejuni (7), B. subtilis (6), and Synechocystis sp. (Fig. 7 and supplemental Fig. S15 and Table S6 and “Experimental Procedures”) (8). These six Y2H interactomes are at least severalfold less well enriched in same operon, same TIGR role, and reciprocal protein pairs than are our two high confidence networks or the benchmark datasets and have substantially higher FDRs (Fig. 7). By these four PPI quality metrics, the Y2H networks instead closely resemble the three published AP-MS interactomes. Two other quality metrics, however, reveal differences among the Y2H interactomes. The Y2H networks of E. coli, H. pylori, and Synechocystis are more similar to our high confidence sets when judged by the percent of protein pairs found in other interactomes and overall network connectivity (Fig. 7 and supplemental Table S6), although the other Y2H interactomes resemble the published AP-MS networks. Thus, there is some heterogeneity in the quality of the protein pairs between the Y2H datasets, but despite this, all differ in important regard to our high confidence AP-MS networks and the benchmark sets.
DISCUSSION
Accurately defining the spectrum of PPIs in an organism is challenging, in part because there is a poor overlap between PPIs proposed in different studies (Fig. 7) (3, 10). For example, even when overlap is computed only for proteins present in both an E. coli AP-MS (17) and an E. coli Y2H (4) interactome, the overlaps are only 13 and 24%, respectively. Such poor agreement could result from the different specificities of each method for detecting classes of PPIs (3, 54), high false positive rates (55, 56), or high false negative rates. A further complication is that there are many non-functional PPIs that are not under evolutionary constraint but that result because short amino acid sequences that bind at low affinity evolve frequently by chance (57). Such interactions are hard to distinguish from genuine false positives because both fail to display characteristics helpful in validating functional PPIs, such as having related functions.
With the above challenges in mind, we have conducted an AP-MS survey of the sulfate-reducing bacterium D. vulgaris and have reanalyzed nine Y2H and AP-MS screens for other bacteria. Using a more stringent data analysis strategy than employed previously, we have identified 391 and 456 high confidence PPIs for E. coli and D. vulgaris, respectively, many of which are supported by low throughput data from the literature (supplemental text and Figs. S11–S13). The PPIs we identified have dramatically different properties from previous Y2H and AP-MS interactomes. Compared with these other interactomes, our two high confidence networks are smaller, much less connected, and have a higher fraction of PPIs homologous to those in other interactomes (Figs. 6 and 7 and supplemental Fig. S15). In addition, the protein pairs in our networks have lower FDRs, are more frequently confirmed as reciprocal PPIs, and are more commonly comprised of partners encoded in the same operon or sharing the same TIGR role (Fig. 7). By these same metrics, three benchmark datasets (PPIs in the EcoCyc database and the ∼3% of PPIs that have been reciprocally confirmed in AP-MS or Y2H studies) are much more similar to our high confidence interactomes than to the complete versions of the published bacterial PPI networks (Fig. 7).
These striking differences suggest that only a minority of protein pairs in the other interactomes are stable interactions of the sort captured in our high confidence sets and the benchmark sets. Of the remaining pairs, several hundred may be lower affinity, functional interactions because some lower confidence protein pairs excluded from our interactomes tend to share the same TIGR role, while being rarely found in other interactomes, reciprocally confirmed or encoded in the same operon (e.g. Figs. 4, 5, and 8). Our interpretation is that these pairs interact, but at lower affinity, and thus the interactions are harder to detect. To explain the low frequency of the same operon PPIs in these low confidence sets, we assume that bona fide PPIs not encoded in the same operon must be generally of weaker affinity than the bona fide same operon PPIs. Beyond the high and low affinity functional pairs, however, several lines of evidence suggest that most pairs in the previously published interactomes are false positives that do not interact and/or are non-functional interactions.
Our FDR estimates for the previous nine interactomes are 42–91% when gold standards based on protein complexes from E. coli are employed (Fig. 7). Although our estimates for the non-E. coli interactomes are necessarily limited to well conserved protein pairs, and thus the high FDRs could be explained if there is a rapid evolution of bona fide PPIs between species, this concern does not apply to the published E. coli networks. Because the published non-E. coli interactomes are by other metrics similar in quality to the three published E. coli interactomes (Fig. 7), the proportion of false positives among non-conserved protein pairs is probably similar to that for conserved pairs in all interactomes. Therefore, the high FDRs we have estimated for prior interactomes do not result from a rapid evolution of PPIs.
Four of the earlier AP-MS and Y2H studies (17) estimated FDRs. These prior FDR estimates are all lower than ours for the same datasets (9–28% versus 66–81%), and the following suggest that this is due to the earlier studies underestimating the percent of false positives. Hu et al. (17), for example, employed gold standard negatives that were biased toward proteins that are harder to detect by MS than typical proteins (see “Results”). Two of the other studies used gold-negative sets that were the same size as the gold-positive sets (7, 9), whereas negative sets should approximate the square of the number of proteins in the positive set divided by two (see “Experimental Procedures”). As a result, the FDR would be underestimated by ∼4-fold. Finally, although Rajagopala et al. (4) did not estimate an FDR, this can be derived from their gold standard control data, implying an FDR of >80% (see “Experimental Procedures”).
Most of the published interactomes have far fewer protein pairs that share the same TIGR role than either our high confidence networks or the benchmark datasets. Low affinity functional interactors, however, should share the same TIGR role as often as high affinity pairs. Thus, the low prevalence of same TIGR role pairs suggests that most pairs in the published interactomes have no functional relevance.
In AP-MS datasets, pairs between a ribosomal protein and a non-ribosomal protein lack the hallmarks of bona fide PPIs (see “Results” and Fig. 4). Such pairs constitute 19–34% of PPIs in the previously proposed AP-MS interactomes, although they are excluded from our high confidence sets, suggesting that these pairs contribute significantly to high FDRs.
Our results also suggest that the poor overlap between Y2H and AP-MS interactomes cannot be due solely to differences in the types of interaction that the two methods detect. When PPIs are confined to only those that have been reciprocally confirmed within each study, the agreement between the Y2H and AP-MS methods increases 5–10-fold (Fig. 7). Furthermore, when Y2H and AP-MS interactomes from the same species, E. coli, are directly compared, the overlaps are 2–3-fold higher using our AP-MS interactome than using the interactome proposed by Hu et al. (17) that was based on the same initial MS dataset (13 and 24% versus 39 and 49%).
The AP-MS and Y2H interactomes for eukaryotes also have a much higher connectivity than our high confidence D. vulgaris and E. coli interactomes or the EcoCyc PPIs (5, 10–12, 14, 15, 58, 59). These interactomes could genuinely have quite different connectivities due to fundamental differences in the biology of bacteria and eukaryotes. The eukaryotic studies, however, used methods similar to those employed in bacteria. For example, several did not calculate an FDR (10, 15, 59); one used a gold-negative set that contained only pairs whose members are localized differently within cells (5), which our analysis suggests can lead to underestimated FDRs, see “Results”; and two performed single step affinity purifications (58, 59), which are in principle less accurate than tandem affinity purifications and produce the least accurate of the three published bacterial AP-MS interactomes (Fig. 7) (compare Arifuzzaman et al. (18) with both Hu et al. (17) and Kuhner et al. (16)). Thus, many of the proposed protein pairs in the eukaryotic networks may be either false positives or low affinity non-functional interactions.
Conversely, our more selective analysis strategy fails to detect many bona fide PPIs. For example, we estimate that our D. vulgaris study should have detected ∼1196 high confidence PPIs from the 957 bait proteins tested versus the 459 PPIs we reported (see “Results”). We also estimate that if all proteins were tested as baits ∼3000 PPIs should be detectable in both E. coli and in D. vulgaris. These estimates, however, are for PPIs that have similar properties to the protein pairs in the EcoCyc database and in our high confidence sets. Our work also shows that there are additional lower affinity PPIs that are not well represented in the EcoCyc dataset. The proportion of such transient yet functional interactions is unknown as is the number of interactions involving membrane proteins because of technological challenges in surveying membrane complexes. Future efforts should thus focus on identifying a larger proportion of bona fide PPIs, both high and low affinity, while maintaining an acceptable FDR.
Supplementary Material
Acknowledgments
We acknowledge Dr. Rich Niles for support of data management, and we thank Sahni Phase and Prof. Andrew Emili for providing MS data from their Hu et al. (17) study. We are indebted to Dr. Birgit Schilling for generous help with data deposition to Massive and Panorama repositories.
Footnotes
Author contributions: S.C.H., S.J.F., S.R.C., T.C.H., J.D.W., H.E.W., M.D.B., J.C., and G.B. designed research; S.A., B.L.G., N.L.L., T.R.J., S.A.R., D.A.E., R.P., J.H., W.Y., E.D.S., H.L., M.E.S., J.T.G., B.R.L., A.S., V.V.T., S.R.C., and H.E.W. performed research; M.S., S.E.B., H.E.W., M.D.B., J.C., and G.B. analyzed data; M.S., S.A., H.E.W., M.D.B., J.C., and G.B. wrote the paper.
* This work was initiated by the Protein Complex Analysis Project and later conducted as part of ENIGMA (Ecosystems and Networks Integrated with Genes and Molecular Assemblies (enigma.lbl.gov)), a Scientific Focus Area Program at Lawrence Berkeley National Laboratory, both supported by the Office of Science, Office of Biological and Environmental Research, of the United States Department of Energy under Contract DE-AC02-05CH11231, and mass spectrometry analyses were performed by the UCSF Sandler-Moore Mass spectrometry Core Facility, which acknowledges support from the Sandler Family Foundation, the Gordon and Betty Moore Foundation, the Canary Foundation and National Institutes of Health Cancer Center Support Grant P30 CA08210 from NCI. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
 This article contains supplemental text, Datasets S1–S19, Tables S1–S6, and Figs. S1–S15.
This article contains supplemental text, Datasets S1–S19, Tables S1–S6, and Figs. S1–S15.
2 M. Dong, H. Liu, J. JIn, H.E. Witkowska and M.D. Biggin., unpublished observations.
1 The abbreviations used are:
- Y2H
- yeast two-hybrid
- AP-MS
- affinity purification-mass spectrometry
- BLAST
- Basic Local Alignment Search Tool
- CY
- cytoplasmic protein
- FDR
- false discovery rate
- NSAF
- normalized spectral abundance factor
- OM
- outer membrane protein
- PE
- periplasmic protein
- PPI
- protein-protein interaction
- TIGR
- The Institute of Genome Research
- FN
- false negative.
REFERENCES
- 1. Kristensen A. R., and Foster L. J. (2013) High throughput strategies for probing the different organizational levels of protein interaction networks. Mol. Biosyst. 9, 2201–2212 [DOI] [PubMed] [Google Scholar]
- 2. Vidal M., Cusick M. E., and Barabási A. L. (2011) Interactome networks and human disease. Cell 144, 986–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Häuser R., Ceol A., Rajagopala S. V., Mosca R., Siszler G., Wermke N., Sikorski P., Schwarz F., Schick M., Wuchty S., Aloy P., and Uetz P. (2014) A second-generation protein-protein interaction network of Helicobacter pylori. Mol. Cell. Proteomics 13, 1318–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rajagopala S. V., Sikorski P., Kumar A., Mosca R., Vlasblom J., Arnold R., Franca-Koh J., Pakala S. B., Phanse S., Ceol A., Häuser R., Siszler G., Wuchty S., Emili A., Babu M., et al. (2014) The binary protein-protein interaction landscape of Escherichia coli. Nat. Biotechnol. 32, 285–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yu H., Braun P., Yildirim M. A., Lemmens I., Venkatesan K., Sahalie J., Hirozane-Kishikawa T., Gebreab F., Li N., Simonis N., Hao T., Rual J. F., Dricot A., Vazquez A., Murray R. R., et al. (2008) High quality binary protein interaction map of the yeast interactome network. Science 322, 104–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marchadier E., Carballido-López R., Brinster S., Fabret C., Mervelet P., Bessières P., Noirot-Gros M. F., Fromion V., and Noirot P. (2011) An expanded protein-protein interaction network in Bacillus subtilis reveals a group of hubs: exploration by an integrative approach. Proteomics 11, 2981–2991 [DOI] [PubMed] [Google Scholar]
- 7. Parrish J. R., Yu J., Liu G., Hines J. A., Chan J. E., Mangiola B. A., Zhang H., Pacifico S., Fotouhi F., DiRita V. J., Ideker T., Andrews P., and Finley R. L. Jr. (2007) A proteome-wide protein interaction map for Campylobacter jejuni. Genome Biol. 8, R130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sato S., Shimoda Y., Muraki A., Kohara M., Nakamura Y., and Tabata S. (2007) A large-scale protein-protein interaction analysis in Synechocystis sp. PCC6803. DNA Res. 14, 207–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Titz B., Rajagopala S. V., Goll J., Häuser R., McKevitt M. T., Palzkill T., and Uetz P. (2008) The binary protein interactome of Treponema pallidum–the syphilis spirochete. PLoS ONE 3, e2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rolland T., Tașan M., Charloteaux B., Pevzner S. J., Zhong Q., Sahni N., Yi S., Lemmens I., Fontanillo C., Mosca R., Kamburov A., Ghiassian S. D., Yang X., Ghamsari L., Balcha D., et al. (2014) A proteome-scale map of the human interactome network. Cell 159, 1212–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Murali T., Pacifico S., Yu J., Guest S., Roberts G. G. 3rd, and Finley R. L. Jr. (2011) DroID 2011: a comprehensive, integrated resource for protein, transcription factor, RNA and gene interactions for Drosophila. Nucleic Acids Res. 39, D736–D743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Simonis N., Rual J. F., Carvunis A. R., Tasan M., Lemmens I., Hirozane-Kishikawa T., Hao T., Sahalie J. M., Venkatesan K., Gebreab F., Cevik S., Klitgord N., Fan C., Braun P., Li N., et al. (2009) Empirically controlled mapping of the Caenorhabditis elegans protein-protein interactome network. Nat. Methods 6, 47–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Butland G., Peregrín-Alvarez J. M., Li J., Yang W., Yang X., Canadien V., Starostine A., Richards D., Beattie B., Krogan N., Davey M., Parkinson J., Greenblatt J., and Emili A. (2005) Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature 433, 531–537 [DOI] [PubMed] [Google Scholar]
- 14. Gavin A. C., Aloy P., Grandi P., Krause R., Boesche M., Marzioch M., Rau C., Jensen L. J., Bastuck S., Dümpelfeld B., Edelmann A., Heurtier M. A., Hoffman V., Hoefert C., Klein K., et al. (2006) Proteome survey reveals modularity of the yeast cell machinery. Nature 440, 631–636 [DOI] [PubMed] [Google Scholar]
- 15. Krogan N. J., Cagney G., Yu H., Zhong G., Guo X., Ignatchenko A., Li J., Pu S., Datta N., Tikuisis A. P., Punna T., Peregrín-Alvarez J. M., Shales M., Zhang X., Davey M., et al. (2006) Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440, 637–643 [DOI] [PubMed] [Google Scholar]
- 16. Kühner S., van Noort V., Betts M. J., Leo-Macias A., Batisse C., Rode M., Yamada T., Maier T., Bader S., Beltran-Alvarez P., Castaño-Diez D., Chen W. H., Devos D., Güell M., Norambuena T., et al. (2009) Proteome organization in a genome-reduced bacterium. Science 326, 1235–1240 [DOI] [PubMed] [Google Scholar]
- 17. Hu P., Janga S. C., Babu M., Díaz-Mejía J. J., Butland G., Yang W., Pogoutse O., Guo X., Phanse S., Wong P., Chandran S., Christopoulos C., Nazarians-Armavil A., Nasseri N. K., Musso G., et al. (2009) Global functional atlas of Escherichia coli encompassing previously uncharacterized proteins. PLos Biol. 7, e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Arifuzzaman M., Maeda M., Itoh A., Nishikata K., Takita C., Saito R., Ara T., Nakahigashi K., Huang H. C., Hirai A., Tsuzuki K., Nakamura S., Altaf-Ul-Amin M., Oshima T., Baba T., et al. (2006) Large-scale identification of protein-protein interaction of Escherichia coli K-12. Genome Res. 16, 686–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Estojak J., Brent R., and Golemis E. A. (1995) Correlation of two-hybrid affinity data with in vitro measurements. Mol. Cell. Biol. 15, 5820–5829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Seebacher J., and Gavin A. C. (2011) SnapShot: protein-protein interaction networks. Cell 144, 1000, 1000 e1001 doi: 10.1016/j.cell.2011.02.025. [DOI] [PubMed] [Google Scholar]
- 21. Venkatesan K., Rual J. F., Vazquez A., Stelzl U., Lemmens I., Hirozane-Kishikawa T., Hao T., Zenkner M., Xin X., Goh K. I., Yildirim M. A., Simonis N., Heinzmann K., Gebreab F., Sahalie J. M., et al. (2009) An empirical framework for binary interactome mapping. Nat. Methods 6, 83–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhou J., He Q., Hemme C. L., Mukhopadhyay A., Hillesland K., Zhou A., He Z., Van Nostrand J. D., Hazen T. C., Stahl D. A., Wall J. D., and Arkin A. P. (2011) How sulphate-reducing microorganisms cope with stress: lessons from systems biology. Nat. Rev. Microbiol. 9, 452–466 [DOI] [PubMed] [Google Scholar]
- 23. He Q., He Z., Joyner D. C., Joachimiak M., Price M. N., Yang Z. K., Yen H. C., Hemme C. L., Chen W., Fields M. M., Stahl D. A., Keasling J. D., Keller M., Arkin A. P., Hazen T. C., et al. (2010) Impact of elevated nitrate on sulfate-reducing bacteria: a comparative study of Desulfovibrio vulgaris. ISME J. 4, 1386–1397 [DOI] [PubMed] [Google Scholar]
- 24. Mukhopadhyay A., Redding A. M., Joachimiak M. P., Arkin A. P., Borglin S. E., Dehal P. S., Chakraborty R., Geller J. T., Hazen T. C., He Q., Joyner D. C., Martin V. J., Wall J. D., Yang Z. K., Zhou J., and Keasling J. D. (2007) Cell-wide responses to low-oxygen exposure in Desulfovibrio vulgaris Hildenborough. J. Bacteriol. 189, 5996–6010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Han B. G., Dong M., Liu H., Camp L., Geller J., Singer M., Hazen T. C., Choi M., Witkowska H. E., Ball D. A., Typke D., Downing K. H., Shatsky M., Brenner S. E., Chandonia J. M., et al. (2009) Survey of large protein complexes in D. vulgaris reveals great structural diversity. Proc. Natl. Acad. Sci. U.S.A. 106, 16580–16585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Oliveira T. F., Vonrhein C., Matias P. M., Venceslau S. S., Pereira I. A., and Archer M. (2008) The crystal structure of Desulfovibrio vulgaris dissimilatory sulfite reductase bound to DsrC provides novel insights into the mechanism of sulfate respiration. J. Biol. Chem. 283, 34141–34149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Walian P. J., Allen S., Shatsky M., Zeng L., Szakal E. D., Liu H., Hall S. C., Fisher S. J., Lam B. R., Singer M. E., Geller J. T., Brenner S. E., Chandonia J. M., Hazen T. C., Witkowska H. E., et al. (2012) High throughput isolation and characterization of untagged membrane protein complexes: outer membrane complexes of Desulfovibrio vulgaris. J. Proteome Res. 11, 5720–5735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chhabra S. R., Butland G., Elias D. A., Chandonia J. M., Fok O. Y., Juba T. R., Gorur A., Allen S., Leung C. M., Keller K. L., Reveco S., Zane G. M., Semkiw E., Prathapam R., Gold B., et al. (2011) Generalized schemes for high throughput manipulation of the Desulfovibrio vulgaris genome. Appl. Environ. Microbiol. 77, 7595–7604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zeghouf M., Li J., Butland G., Borkowska A., Canadien V., Richards D., Beattie B., Emili A., and Greenblatt J. F. (2004) Sequential peptide affinity (SPA) system for the identification of mammalian and bacterial protein complexes. J. Proteome Res. 3, 463–468 [DOI] [PubMed] [Google Scholar]
- 30. Roan N. R., Chu S., Liu H., Neidleman J., Witkowska H. E., and Greene W. C. (2014) Interaction of fibronectin with semen amyloids synergistically enhances HIV infection. J. Infect. Dis. 210, 1062–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chiu Y. L., Witkowska H. E., Hall S. C., Santiago M., Soros V. B., Esnault C., Heidmann T., and Greene W. C. (2006) High molecular-mass APOBEC3G complexes restrict Alu retrotransposition. Proc. Natl. Acad. Sci. U.S.A. 103, 15588–15593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Elias J. E., and Gygi S. P. (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 [DOI] [PubMed] [Google Scholar]
- 33. Liu H., Sadygov R. G., and Yates J. R. 3rd. (2004) A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 76, 4193–4201 [DOI] [PubMed] [Google Scholar]
- 34. Lundgren D. H., Hwang S. I., Wu L., and Han D. K. (2010) Role of spectral counting in quantitative proteomics. Expert Rev. Proteomics 7, 39–53 [DOI] [PubMed] [Google Scholar]
- 35. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., and MacCoss M. J. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Altschul S. F., Gish W., Miller W., Myers E. W., and Lipman D. J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410 [DOI] [PubMed] [Google Scholar]
- 37. Karp P. D., Riley M., Saier M., Paulsen I. T., Collado-Vides J., Paley S. M., Pellegrini-Toole A., Bonavides C., and Gama-Castro S. (2002) The EcoCyc Database. Nucleic Acids Res. 30, 56–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chhabra S. R., Joachimiak M. P., Petzold C. J., Zane G. M., Price M. N., Reveco S. A., Fok V., Johanson A. R., Batth T. S., Singer M., Chandonia J. M., Joyner D., Hazen T. C., Arkin A. P., Wall J. D., et al. (2011) Towards a rigorous network of protein-protein interactions of the model sulfate reducer Desulfovibrio vulgaris Hildenborough. PLoS One 6, e21470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wodak S. J., Vlasblom J., Turinsky A. L., and Pu S. (2013) Protein-protein interaction networks: the puzzling riches. Curr. Opin. Struct. Biol. 23, 941–953 [DOI] [PubMed] [Google Scholar]
- 40. Zybailov B., Mosley A. L., Sardiu M. E., Coleman M. K., Florens L., and Washburn M. P. (2006) Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J. Proteome Res. 5, 2339–2347 [DOI] [PubMed] [Google Scholar]
- 41. Choi H., Larsen B., Lin Z. Y., Breitkreutz A., Mellacheruvu D., Fermin D., Qin Z. S., Tyers M., Gingras A. C., and Nesvizhskii A. I. (2011) SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat. Methods 8, 70–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Breitkreutz A., Choi H., Sharom J. R., Boucher L., Neduva V., Larsen B., Lin Z. Y., Breitkreutz B. J., Stark C., Liu G., Ahn J., Dewar-Darch D., Reguly T., Tang X., Almeida R., et al. (2010) A global protein kinase and phosphatase interaction network in yeast. Science 328, 1043–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Collins S. R., Kemmeren P., Zhao X. C., Greenblatt J. F., Spencer F., Holstege F. C., Weissman J. S., and Krogan N. J. (2007) Toward a comprehensive atlas of the physical interactome of Saccharomyces cerevisiae. Mol. Cell. Proteomics 6, 439–450 [DOI] [PubMed] [Google Scholar]
- 44. Park Y., and Marcotte E. M. (2012) Flaws in evaluation schemes for pair-input computational predictions. Nat. Methods 9, 1134–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dehal P. S., Joachimiak M. P., Price M. N., Bates J. T., Baumohl J. K., Chivian D., Friedland G. D., Huang K. H., Keller K., Novichkov P. S., Dubchak I. L., Alm E. J., and Arkin A. P. (2010) MicrobesOnline: an integrated portal for comparative and functional genomics. Nucleic Acids Res. 38, D396–D400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Díaz-Mejía J. J., Babu M., and Emili A. (2009) Computational and experimental approaches to chart the Escherichia coli cell-envelope-associated proteome and interactome. FEMS Microbiol. Rev. 33, 66–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gardy J. L., Laird M. R., Chen F., Rey S., Walsh C. J., Ester M., and Brinkman F. S. (2005) PSORTb v.2.0: expanded prediction of bacterial protein subcellular localization and insights gained from comparative proteome analysis. Bioinformatics 21, 617–623 [DOI] [PubMed] [Google Scholar]
- 48. Zane G. M., Yen H. C., and Wall J. D. (2010) Effect of the deletion of qmoABC and the promoter-distal gene encoding a hypothetical protein on sulfate reduction in Desulfovibrio vulgaris Hildenborough. Appl. Environ. Microbiol. 76, 5500–5509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Delli-Bovi T. A., Spalding M. D., and Prigge S. T. (2010) Overexpression of biotin synthase and biotin ligase is required for efficient generation of sulfur-35 labeled biotin in E. coli. BMC Biotechnol. 10, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Orchard S., Ammari M., Aranda B., Breuza L., Briganti L., Broackes-Carter F., Campbell N. H., Chavali G., Chen C., del-Toro N., Duesbury M., Dumousseau M., Galeota E., Hinz U., Iannuccelli M., et al. (2014) The MIntAct project–IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 42, D358–D363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sharma V., Eckels J., Taylor G. K., Shulman N. J., Stergachis A. B., Joyner S. A., Yan P., Whiteaker J. R., Halusa G. N., Schilling B., Gibson B. W., Colangelo C. M., Paulovich A. G., Carr S. A., Jaffe J. D., et al. (2014) Panorama: a targeted proteomics knowledge base. J. Proteome Res. 13, 4205–4210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Davidsen T., Beck E., Ganapathy A., Montgomery R., Zafar N., Yang Q., Madupu R., Goetz P., Galinsky K., White O., and Sutton G. (2010) The comprehensive microbial resource. Nucleic Acids Res. 38, D340–D345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Keseler I. M., Mackie A., Peralta-Gil M., Santos-Zavaleta A., Gama-Castro S., Bonavides-Martínez C., Fulcher C., Huerta A. M., Kothari A., Krummenacker M., Latendresse M., Muñiz-Rascado L., Ong Q., Paley S., Schroder I., et al. (2013) EcoCyc: fusing model organism databases with systems biology. Nucleic Acids Res. 41, D605–D612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Braun P., Tasan M., Dreze M., Barrios-Rodiles M., Lemmens I., Yu H., Sahalie J. M., Murray R. R., Roncari L., de Smet A. S., Venkatesan K., Rual J. F., Vandenhaute J., Cusick M. E., Pawson T., et al. (2009) An experimentally derived confidence score for binary protein-protein interactions. Nat. Methods 6, 91–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. D'Haeseleer P., and Church G. M. (2004) Estimating and improving protein interaction error rates. Proc. IEEE Comput. Syst. Bioinform Conf. 2004 216–223 [DOI] [PubMed] [Google Scholar]
- 56. von Mering C., Krause R., Snel B., Cornell M., Oliver S. G., Fields S., and Bork P. (2002) Comparative assessment of large-scale data sets of protein-protein interactions. Nature 417, 399–403 [DOI] [PubMed] [Google Scholar]
- 57. Landry C. R., Levy E. D., Abd Rabbo D., Tarassov K., and Michnick S. W. (2013) Extracting insight from noisy cellular networks. Cell 155, 983–989 [DOI] [PubMed] [Google Scholar]
- 58. Guruharsha K. G., Rual J. F., Zhai B., Mintseris J., Vaidya P., Vaidya N., Beekman C., Wong C., Rhee D. Y., Cenaj O., McKillip E., Shah S., Stapleton M., Wan K. H., Yu C., et al. (2011) A protein complex network of Drosophila melanogaster. Cell 147, 690–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Malovannaya A., Lanz R. B., Jung S. Y., Bulynko Y., Le N. T., Chan D. W., Ding C., Shi Y., Yucer N., Krenciute G., Kim B. J., Li C., Chen R., Li W., Wang Y., et al. (2011) Analysis of the human endogenous coregulator complexome. Cell 145, 787–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.