

Abstract
Human Ataxin-2 (ATXN2) gene locus variants have been associated with obesity, diabetes mellitus type 1,and hypertension in genome-wide association studies, whereas mouse studies showed the knock-out of Atxn2 to lead to obesity, insulin resistance, and dyslipidemia. Intriguingly, the deficiency of ATXN2 protein orthologs in yeast and flies rescues the neurodegeneration process triggered by TDP-43 and Ataxin-1 toxicity. To understand the molecular effects of ATXN2 deficiency by unbiased approaches, we quantified the global proteome and metabolome of Atxn2-knock-out mice with label-free mass spectrometry. In liver tissue, significant downregulations of the proteins ACADS, ALDH6A1, ALDH7A1, IVD, MCCC2, PCCA, OTC, together with bioinformatic enrichment of downregulated pathways for branched chain and other amino acid metabolism, fatty acids, and citric acid cycle were observed. Statistical trends in the cerebellar proteome and in the metabolomic profiles supported these findings. They are in good agreement with recent claims that PBP1, the yeast ortholog of ATXN2, sequestrates the nutrient sensor TORC1 in periods of cell stress. Overall, ATXN2 appears to modulate nutrition and metabolism, and its activity changes are determinants of growth excess or cell atrophy.
Ataxin-2 (ATXN2)1 is a stress-regulated protein of 124 kDa size, which is expressed in specific neuron populations, but also in hepatocytes (1–4). The structure of the human ATXN2 protein is characterized (1) by the N-terminal polyQ domain (5), (2) by dispersed proline-rich-domains that mediate association with various SH3-motif containing proteins of the tyrosine kinase receptor endocytosis machinery and thus modulate neuro-trophic signaling (6, 7), (3) by a C-terminal PAM2 motif that mediates interaction with the poly(A)-binding protein PABPC1 that is crucial for mRNA translation (8) and (4) by Lsm and Lsm-AD sequences that mediate the association with RNAs (9–11). ATXN2 is normally localized at the rough endoplasmic reticulum (12), but it relocalizes during periods of low cell energy together with PABPC1 to stress granules where the quality control of RNA occurs (3) and where fasting responses are modulated (13). The suppression of Ataxin-2 in mice and flies modulates mRNA translation and circadian clock (4, 14–16).
In human populations, several independent genome wide association studies (GWAS) have underlined the importance of variants at the genomic locus of ATXN2. They influence the risk for obesity, type 1 diabetes, hypertension, and cardiovascular infarction (17–26). Their impact is strong enough to stand out as genetic modifiers of human longevity (27, 28). Whether additive effects of coregulated neighbor genes at this locus play a causal role, has been discussed in a recent review (29).
In mice, two independent Atxn2-knock-out (KO) lines confirmed phenotypes of obesity (30, 31), with excessive liver fat and glycogen deposits, insulin resistance, and dyslipidemia also being noted by the age of 6 months. Thus, even in the absence of high-fat-diet and within the short lifespan of rodents, the monogenic deficiency of Ataxin-2 is sufficient to trigger important features of the human metabolic syndrome.
Conversely, the overactivity of Ataxin-2 because of polyglutamine (polyQ) expansions encoded by unstable (CAG)-repeat expansions in the human ATXN2 gene results in cell atrophy of specific neuronal populations. The polyQ-expansion of ATXN2 leads to a process of protein insolubility and aggregate formation with insidious toxicity throughout the nervous system, depending on the expansion size and polygenic interactions, possibly also on CAA-interruptions within the CAG-repeat. Neurodegenerative processes known as Spinocerebellar Ataxia type 2 (SCA2), the motor neuron degeneration Amyotrophic Lateral Sclerosis (ALS13), Frontotemporal dementia, Supranuclear palsy, or Levodopa-responsive Parkinsonism can be triggered by this mechanism (32–39). In Drosophila melanogaster flies, the Ataxin-2 ortholog dATX2 was shown to act as a generic modifier gene that affects multiple if not all neurodegenerative disorders (40). The protein interactions between ATXN2 and several other disease proteins of neurodegenerative disorders (41) and the similarity of atrophy patterns between these polyglutamine expansion diseases suggests a common molecular pathomechanism among these diverse neurodegenerative disorders (42).
Interestingly, the deficiency of ATXN2 orthologs was observed to play a beneficial role in these neurodegenerative processes, by alleviating or postponing them in microorganism and animal models (35, 43). Thus, the elucidation of the molecular effects of ATXN2 deficiency and of the physiological roles of Ataxin-2 may help to design neuroprotective approaches that are novel. Hence, we chose to document the global proteomic and metabolomic profile of Atxn2-KO mice in crucial tissues affected by Ataxin-2 loss- and gain-of-function, the liver and the cerebellum, respectively. This effort may provide insights on the target molecules and pathways of Ataxin-2 functions.
EXPERIMENTAL PROCEDURES
Mouse Breeding and Dissection
Animals were bred and aged in individually ventilated cages with continuous health monitoring, 4–6 animals per cage, under a 12 h light cycle with food (Ssniff M-Z, calories from protein 36%, fat 11%, and carbohydrates 53%) and water provided ad libitum. Mice were housed in accordance with the German Animal Welfare Act, the Council Directive of 24 November 1986 (86/609/EWG) with Annex II and the ETS123 (European Convention for the Protection of Vertebrate Animals) at the FELASA-certified Central Animal Facility (ZFE) of the Frankfurt University Medical School. All analyses were performed on male mice with a mixed C57BL/6 _ 129/Ola background. Genotyping was performed with tail biopsies by PCR with three sets of primers as previously described (30).
RNA Preparation and cDNA Synthesis
After cervical dislocation, cerebellum and liver were dissected from homozygous wild-type (Atxn2+/+) and knock-out (Atxn2−/−) mice at 4 months (for proteomics and metabolomics) or 6 months of age (for immunoblots). Total RNA was extracted from these tissues by homogenization in 1 ml of Trizol® Reagent per 50–100 mg of tissue using a Pellet Pestle® Motor tissue homogenizer (Kontes, The Glass Company Kontes, The Glass company, Vineland, NJ). One μg total RNA was digested with a DNase I Amplification Grade Kit (Invitrogen, Karlsruhe, Germany) in a reaction volume of 10 μl per tube to eliminate DNA during RNA purification prior to reverse transcription (RT-PCR) amplification. cDNA synthesis was performed with the Fermentas Life Sciences First Strand cDNA Synthesis Kit as instructed in the manual.
Validation of Quantitative Real-Time Reverse-Transcriptase Polymerase-Chain-Reaction
Quantitative Real-Time Reverse-Transcriptase Polymerase-Chain-Reaction (qPCR) was performed using a GeneAmp® 5700 Sequence Detection System (Applied Biosystems, Carlsbad, CA) with 96-well Optical Reaction Plates (Applied Biosystems, Carlsbad, CA). Twenty μl final reaction volume per well contained 25–30 ng cDNA, TaqMan® Universal PCR Master Mix, No AmpErase® UNG and primers and probes in predesigned TaqMan® Gene Expression Assays. All assays were run in triplicates. The following assays were used: Atxn2 (Mm 01199894_m1) to verify the KO genotype per tissue and Tbp (Mm 00446973_m1) as an endogenous control in all experiments, run in wells separate from the target gene assays. The PCR conditions were 50 °C for 2 min and 95 °C for 10 min followed by 40 cycles at 95 °C for 15 s and 60 °C for 40 s. Analysis of relative gene expression data was performed using the ΔΔCT method.
Metabolite Extraction for Metabolome Profiling
Tissues were dissected, washed in phosphate buffered saline (PBS, pH7.4) and shock frozen in liquid nitrogen. About 40–50 mg of each liver tissue and cerebellum was used for metabolite profiling, six WT versus six Atxn2-KO, respectively. Cold methanol (1.5 ml, −20 °C) and 0.1% ammonium acetate (0.2 ml) were added to the frozen tissues and subsequently homogenized by FastPrep (settings: 1 × 60 s; 4.5 m/s) with a steel ball. The first set of internal standards was added to each sample, containing chloramphenicol and C13-labeled l-glutamine, l-arginine, l-proline, l-valine, and uracil (3.5 μm final concentration). Lysates were transferred to a 15 ml tube, 5 ml methyl-tert-butyl ester (MTBE) was added and incubated at RT for 1 h on a rocking platform. Afterward, 1.25 ml MS grade water was added and incubated for additional 10 min. After centrifugation at 1000 × g at 4 °C for 10 min, organic and inorganic phases were separately transferred and combined in three tubes containing the second internal standard set (Avanti Polar Lipids, Alabaster, AL), dedicated for subsequent analysis by using methanol, acetonitrile, and water as alternative solvents for LC-MS runs. Metabolites were lyophilized and stored at −80 °C until needed. The residuals were suspended in 50 μl of ACN with 0.1% FA and 35 μl of MeOH with 0.1% FA for analysis by HILIC and in 50 μl of H2O with 0.1% FA for RPLC and were then sonicated for redissolving as needed and centrifuged at 17,500 × g for 5 min at 4 °C. The supernatants were transferred to microvolume inserts, and 5 μl per run was injected for LC-MS/MS analysis.
LC Conditions for Metabolomics
The target metabolites were selected to cover most of the important metabolic pathways in mammals, more than 400 metabolites in total. The chromatographic separations were performed on RP, RP-shield, and zicHILIC stationary phases. Following columns were used: Reprosil-PUR C18-AQ (1.9 μm, 120 Å, 150 × 2 mm ID; Dr. Maisch; Ammerbuch, Germany) column, BEH Shield RP18 (1.7 μm, 130 Å, 150 × 2.1 mm ID; Waters, Milford, MA) and a zicHILIC (3.5 μm, 100 Å, 150 × 2.1 mm ID; di2chrom; Marl, Germany) A 1290 series UHPLC (Agilent, Santa Clara, CA) with six different buffer conditions was used: (A1) 10 mm ammonium acetate in LC-MS grade H2O (adjusted with FA to pH 3.5), (A2) 10 mm ammonium acetate in LC-MS grade H2O (adjusted with ammonium hydroxide to pH 7.5), (A3) 20 mm ammonium formate in LC-MS grade H2O (adjusted with ammonium hydroxide to pH 5.0), (B1) LC-MS grade ACN with 0.1% FA, (B3) LC-MS grade MeOH, and (B2) LC-MS grade MeOH with 0.1% FA. Gradients and flow conditions are described in supplemental Table S1. Columns were run under controlled temperature of 30 °C, RP-shield with 80 °C. The columns were equilibrated with a blank run in the corresponding buffer system.
The LC was online coupled to a triple quadrupole hybrid ion trap mass spectrometer QTrap 6500 (ABSciex, Toronto, Canada). MS settings were reported previously (44), transition settings for the MRM's in supplemental Table S2.
Metabolome Data Analysis
Relative quantification was performed using MultiQuant software v.2.1.1 (AB Sciex, Foster City, CA), as described previously (44). Briefly, the identification of metabolites was based on the retention time, three co-occurring transitions and the ion ratio between the transitions, which had to match to MRM ion ratios of the corresponding tuned metabolites. Biological replicates were normalized for the weighted mg of tissue and internal standards.
Sample Preparation for Proteome Profiling
The same tissues were used as for metabolome profiling. About 10 mg of liver tissue and cerebellum was weighted under frozen condition. Samples were lysed and reduced in buffer containing 8% SDS, 0.1 m DTT, 0.1 m Tris, pH 8, and subsequently homogenized by FastPrep (settings: 3 × 60 s; 4 m/s) with a steel ball. Lysates were sonicated on ice for 1 min and boiled at 95 °C for 5 min at a rocking platform (750 rpm). Samples were prepared for LC-MS analysis as previously (44). In brief, lysates were centrifuged for 5 min at 15,000 × g, supernatants were reduced in 50 mm DTT for 1 h at 56 °C. Alkylation was done with a final concentration of 5.5 mm chloroacetamide for 30 min at RT in the dark. Samples were precipitated with 4-times excess of acetone overnight at −20 °C. Precipitates were lyophilized and dissolved in 8 m urea/10 mm Tris, pH 8. Lys-C digestion (1 μg/sample) was performed for 4 h at room temperature followed by a trypsin digestion (2 μg/sample) in 2 m urea overnight at 37 °C. Peptides were purified with C18 columns (Sep-Pak® Vac, 100 mg, Waters, Milford, MA). 1% of the total peptides were finally injected and analyzed by liquid chromatography online coupled to a nanoHPLC-MS/MS system, after dissolving in 5% ACN and 2% FA. Every sample was run as a technical replicate.
LC-MS Settings for Proteomics
LC-MS/MS was carried out by nanoflow reverse phase liquid chromatography (Dionex Ultimate 3000, Thermo Scientific, Waltham, MA) coupled online to a Q-Exactive Plus Orbitrap mass spectrometer (Thermo Scientific, Waltham, MA). Briefly, the LC separation was performed using a PicoFrit analytical column (75 μm ID × 25 cm long, 15 μm Tip ID (New Objectives, Woburn, MA) in-house packed with 3 μm C18 resin (Reprosil-AQ Pur, Dr. Maisch, Ammerbuch-Entringen, Germany). Peptides were eluted using a nonlinear gradient from 2 to 40% solvent B over 210 min at a flow rate of 266 nL/min (solvent A: 99.9% H2O, 0.1% formic acid; solvent B: 79.9% acetonitrile, 20% H2O, 0.1% formic acid). 3 kV were applied for nanoelectrospray generation. A cycle of one full FT scan mass spectrum (300–1750 m/z, resolution of 70,000 at m/z 200, AGC target 1e6) was followed by 12 data-dependent MS/MS scans (resolution of 35,000, AGC target 5e5) with normalized collision energy of 25 eV. To avoid repeated sequencing of the same peptides, a dynamic exclusion window of 30 s was used and only the peptide charge states between two to eight were allowed.
Label Free Proteomics Data Analysis
Raw MS data were processed with MaxQuant software (v1.5.0.0) (45) and searched against the Mus musculus (MOUSE) proteome database UniProtKB with 52,490 entries, released in 2014–11, respectively. Additionally, the “requantify” feature was implemented to increase the number of peptides which can be used for quantification. LC-MS runs of technical replicates were jointly analyzed. A false discovery rate (FDR) of 0.01 for proteins and peptides and a minimum peptide length of seven amino acids, a mass tolerance of 4.5 ppm for precursor and 20 ppm for fragment ions were required. A minimum Andromeda score of 0 and 40 (delta score 0 and 9) for unmodified peptides and modified peptides was applied. A maximum of two missed cleavages was allowed for the tryptic digest. Cysteine carbamidomethylation was set as fixed modification, whereas N-terminal protein acetylation and methionine oxidation were set as variable modifications.
The LFQ, a generic method for label-free quantification (46) within MaxQuant, was used for relative quantification. MaxQuant processed output files can be found in supplemental Tables S3–S4, showing peptide and protein identification, accession numbers, % sequence coverage of the protein, posterior error probability (PEP) values, log2 transformed LFQ intensities, and fold changes between KO/WT. Contaminants as well as proteins identified by site modification and proteins derived from the reversed part of the decoy database were strictly excluded from further analysis. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (47) with the dataset identifier PXD003155.
Statistical, Pathway, and PPI Network Analyses
Pathway and network analyses are novel methods to understand the complex data sets derived from deep omics studies. For the metabolome and proteome data, a two-sample t test was performed within Perseus software (v1.5.0.8, (45). Multiple test correction by Benjamini-Hochberg (BH) with a FDR of 0.05 was performed after the p value calculation. Significantly regulated metabolites and proteins were marked by an asterisk in the corresponding supplemental Tables S3–S4. Furthermore, volcano plots and Pearson correlation were created within Perseus. Only proteins with valid values in all replicates were used for the above mentioned tests.
For comprehensive proteome data analyses, gene set enrichment analysis (GSEA, v2.2.0) (48) was applied to see, if a priori defined sets of proteins show statistically significant, concordant differences between KO and WT state. Only proteins with at least two valid values in each cohort were used for GSEA analysis and the ratios were log2 transformed. GSEA default settings were used, except that the minimum size exclusion was set to five and KEGG v5.0 was used as gene set database. The cutoff for significantly regulated pathways was set to ≤ 0.05 p value and ≤ 0.05 FDR.
For protein–protein interaction (PPI) network analyses, the software tool String v.10 was used to visualize networks of significantly regulated proteins of the entire proteome with a confidence level of 0.7 (49). The cutoff for significantly regulated proteins was determined using one standard deviation from the log2 transformed median by using GraphPad Prism 5.03. Protein nodes which were not integrated into a network were removed.
Quantitative Immunoblot Analyses
The isolation of total proteins from liver of seven wild-type and seven Atxn2-KO mice at the age of 6 months was performed as described (50) with minor modifications: Prior to gel electrophoresis, 20 μg samples were heated at 90 °C for 2 min, using Precision Plus Protein™ Dual Color Standards as size marker. Transfer to nitrocellulose membranes (Protran, GE Healthcare, Little Chalfont, UK) was done at 50 V for 90 min, with blocking in 5% BSA solution in 1x TBS-T for 1 h at room temperature (RT). Primary antibodies against β-Actin (1:5000, A5441, Sigma-Aldrich, St. Louis, MO), ACADS (1:500, ab154823, Abcam, Cambridge, UK), ALDH6A1 (PA5–34691, Thermo Fischer, Waltham, MA), ALDH7A1 (PA5–29974, Thermo Fischer), IVD (1:250, PA5–22342, Thermo Fischer, Waltmam, MA), MCCC2 (PA5–27758, Thermo Fischer), OTC (1:500, PA5–28197, Thermo Fischer), and PCCA (1:250, ABIN518660, MaxPab®, Abnova, Taipei City, Taiwan) in 5% BSA solutions initially for 1 h at RT, and then overnight at 4 °C. Fluorescent-labeled α-mouse (1:15,000, IRDye 800CW, Li-Cor, Lincoln, NE), and α-rabbit (1:15,000, IRDye 680RD, Li-Cor) were the secondary antibodies. Fluorescence detection occurred on the Li-Cor Odyssey Classic Instrument. Statistical significance was assessed using unpaired t test with Welch's correction.
RESULTS
Proteomic Profiling
To determine the molecular effects of the Atxn2-KO in 4-month-old mouse liver and cerebellar tissue, we first performed global proteome quantifications by label-free mass spectrometry. Atxn2-KO was verified by genotyping of tail biopsies at the DNA level as previously described (30) and verified in the tissues under study by real-time qPCR at the mRNA level. Additionally, we looked at the proteome level for the evidence of ATXN2 presence in all samples, and observed six different ATXN2 specific peptides exclusively in wild-type (WT) samples, never in KO samples (data not shown).
Comparison of the proteome profiles of all six biological replicates of WT and Atxn2-KO mice was done by Pearson correlation. The Pearson correlation coefficients were highly similar, ranging from 0.766 to 0.981 in WT and KO liver tissue (supplemental Fig. S1), and from 0.928 to 0.992 in WT and KO cerebellar tissue (supplemental Fig. S2). These Pearson correlation coefficients suggested a very good quality of the proteome data sets.
The entire list of identified and quantified protein groups can be found in supplemental Tables S3–S4 for liver and cerebellar tissue, respectively. This resulted in 823 protein groups in liver versus 999 protein groups in cerebellar tissue, respectively. For pathway- and PPI network analyses, we accepted at least two valid values per sample cohort, resulting in 1609 protein groups in liver versus 1982 protein group ratios in cerebellar tissue, respectively.
Proteomic Profiling in Liver Tissue
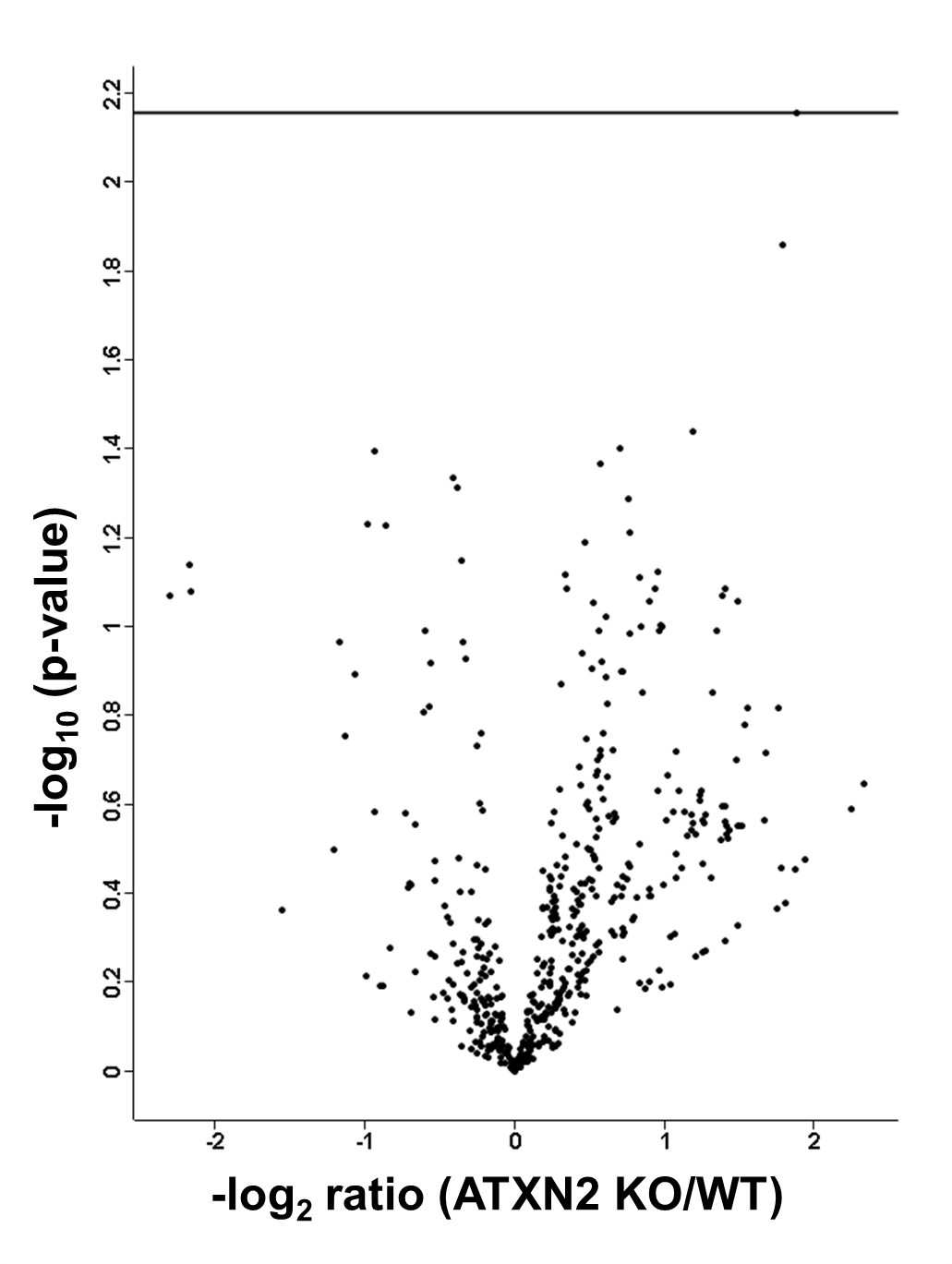
To identify significantly (after BH correction) regulated protein groups, the distribution between KO and WT cohorts was visualized in a volcano plot (Fig. 1).
Fig. 1.

Volcano plot featuring liver proteome data, with the mean difference of LFQ intensities between Atxn2 KO and WT tissues on the x axis versus statistical significance on the y axis (-log10 of the p value). Significant regulated proteins are identified in red.
The significantly dysregulated proteins were detailed in Table I. PCCA, ALDH6A1, IVD, and MCCC2 are involved in the valine, leucine, and isoleucine (branched chain amino acids, BCAA) degradation pathway. OTC is part of the arginine biosynthesis pathway, synthesizing l-citrulline from l-ornithine. ALDH7A1 is a key enzyme in the pathways lysine biosynthesis/lysine degradation and ACADS is involved in the pathway mitochondrial fatty acid beta-oxidation.
Table I. List of significant altered proteins in KO/WT liver tissue. Posterior error probability (PEP), MS2 spectral count frequency (MS/MS Count), Benjamini-Hochberg corrected p values and fold changes are shown.
| Protein names | Gene names | t-test Significant after BH | PEP | MS/MS Count | -LOG t-test p value | fold change KO/WT |
|---|---|---|---|---|---|---|
| Propionyl-CoA carboxylase alpha chain, mitochondrial | Pcca | + | 0 | 454 | 4,0698 | 0.55 |
| Isovaleryl-CoA dehydrogenase, mitochondrial | Ivd | + | 0 | 363 | 4,0434 | 0.48 |
| α-aminoadipic semialdehyde dehydrogenase | Aldh7a1 | + | 0 | 642 | 3,9197 | 0.67 |
| Short-chain specific acyl-CoA dehydrogenase, mitochondrial | Acads | + | 0 | 480 | 3,7971 | 0.57 |
| Ornithine carbamoyltransferase, mitochondrial | Otc | + | 0 | 1476 | 3,6804 | 0.61 |
| Methylcrotonoyl-CoA carboxylase beta chain, mitochondrial | Mccc2 | + | 0 | 308 | 3,6148 | 0.61 |
| Methylmalonate-semialdehyde dehydrogenase [acylating], mitochondrial | Aldh6a1 | + | 0 | 1085 | 3,4525 | 0.73 |
To illustrate the relative protein abundance and the variability of measurements as criteria for their relevance, the LFQ ion intensity distributions of every single individual mouse tissue was shown for the seven significant proteins (Fig. 2).
Fig. 2.

Log2 LFQ ion intensities of all six biological replicates in Atxn2-KO/WT liver tissues are shown. Red = KO; blue = WT. Error bars are mean values with S.D.
Applying the STRING software, we visualized the network of molecular interactions among downregulated proteins (at least one standard deviation from the mean) in Atxn2-KO/WT liver tissue (Fig. 3). The most striking pathway within this network was again the BCAA metabolism. Furthermore, protein–protein interaction networks involved in the ribosome and cytochrome P450 metabolism, fatty acid metabolism, and several amino acid metabolisms were downregulated as well (smaller networks in Fig. 3).
Fig. 3.

STRING PPI network and pathway analysis of proteins with downregulation at least one standard deviations from the log2 transformed median values (0.03227 ± 0.4819) in Atxn2-KO/WT liver tissue. Highlighted in red are proteins which belong to the KEGG pathway BCAA. Unconnected nodes were removed.
Even more sophisticated ways to analyze the entire proteomics dataset, taking all ratios into account without any ratio cutoffs, are assessment methods of pathway enrichment such as GSEA (Gene Set Enrichment Analysis). For liver tissue, no significantly upregulated KEGG pathway was identified with this approach, but several significantly downregulated KEGG pathways were identified. Most striking was the downregulation of the pathway branched chain amino acids (BCAA, that is leucine, isoleucine, and valine) and several related amino acid metabolism pathways. Furthermore, the pathways fatty acid metabolism and the citric acid cycle (tricarboxylic acid or TCA cycle) were significantly downregulated (Table II, supplemental Table S5).
Table II. Significantly (p value and q-value ≤0.05) downregulated KEGG pathways in KO/WT liver tissue according to GSEA. Size, number of identified proteins within the pathway; NES, normalized enrichment score.
| KEGG pathway | Size | NES | NOM p value | FDR q-value |
|---|---|---|---|---|
| Valine leucine and isoleucine degradation | 36 | −2.69 | 0 | 0 |
| Propanoate metabolism | 25 | −2.36 | 0 | 0 |
| Fatty acid metabolism | 25 | −2.31 | 0 | 0 |
| Glycine serine and threonine metabolism | 21 | −2.28 | 0 | 0 |
| Butanoate metabolism | 24 | −2.17 | 0 | 0 |
| Citric cycle TCA cycle | 23 | −2.07 | 0.0024 | 0.0014 |
| βalanine metabolism | 16 | −1.98 | 0 | 0.0041 |
| Alanine aspartate and glutamate metabolism | 19 | −1.88 | 0 | 0.0152 |
| Arginine and proline metabolism | 27 | −1.85 | 0.0022 | 0.0197 |
| Lysine degradation | 18 | −1.84 | 0.0044 | 0.0201 |
| Tryptophan metabolism | 25 | −1.79 | 0.0023 | 0.0294 |
| Limonene and pinene degradation | 8 | −1.76 | 0.0177 | 0.0389 |
| Pyruvate metabolism | 24 | −1.73 | 0.0024 | 0.0452 |
Proteomic Profiling in Cerebellar Tissue
For cerebellar tissue, we used identical approaches for data analysis as for liver tissue. On the protein level, no significantly regulated proteins were identified after BH correction (supplemental Table S4, volcano plot in supplemental Fig. S3). Compared with the PPI network generated for liver tissues, the BCAA pathway could still be identified as a network by applying at least one standard deviation (cerebellum 0.06144 ± 0.3175) of down regulated proteins, but to a much lesser degree (supplemental Fig. S4).
GSEA analyses revealed a nominal significance for upregulations of KEGG pathways proteasome, purine metabolism, amyotrophic lateral sclerosis, and VEGF signaling (supplemental Table S5), and nominal significance for downregulations of pathways ribosomes and valine, leucine, and isoleucine degradation again. In general, the fold changes in cerebellum were rather small.
Metabolomic Profiling
We applied a targeted LC-MS/MS methodology, based on multiple reaction monitoring (MRM) to identify and quantify relative differences in metabolite changes of selected metabolites between Atxn2-KO and WT mice in liver and cerebellar tissue. The correct retention time, three MRM's and a matching MRM ion ratio of tuned pure metabolites as a reference were used for correct identification of metabolites (44). In total, 176 metabolites could be identified and relatively quantified in liver tissue, 204 in cerebellum. The entire lists of all quantified metabolites can be found in supplemental Table S6.
Comparison of the metabolome profiles of all six biological replicates of WT and Atxn2-KO mice was done by Pearson correlation. The Pearson correlation coefficients were highly similar, ranging from 0.803 to 0.994 in KO and WT liver tissue (supplemental Fig. S5). In cerebellum, the range was from 0.83 to 0.985 (supplemental Fig. S6). These Pearson correlation coefficients suggested a very good quality of the metabolome data sets. Statistical analyses by a two sample t test with BH correction was visualized by volcano plots and revealed no significantly regulated metabolites (supplemental Fig. S7–S8), the fold ratios were too small in comparison to the variability.
Proteome- Metabolome Integration
Nevertheless, the fold-changes in the metabolome data fitted the proteome profile closely.
Within mitochondria, PCCA and PCCB control the conversion between propionyl-CoA and methyl-malonyl-CoA, MLYCD converts malonyl-CoA to acetyl-CoA in the β-alanine metabolism. Propionyl-CoA and malonyl-CoA are conjugated to carnitine for elimination from mitochondria.
Carnitine O-palmitoyltransferase 1 (liver isoform) and 2 (CPT1A, CTP2), catalyzing the transfer of the acyl group of long-chain fatty acid-CoA conjugates onto carnitine, an essential step for the mitochondrial uptake of long-chain fatty acids and their subsequent beta-oxidation in the mitochondrion, were 1.5- and 2.3-fold down regulated in Atxn2- KO liver tissue, respectively. Interestingly, acylcarnitines, such as propionylcarnitine, malonylcarnitine, and pimelylcarnitine were 2- to 2.5-fold increased. Malonylcarnitine is known to accumulate with specific disruption of fatty-acid oxidation caused by impaired entry of long-chain acylcarnitine esters into the mitochondria and failure of the mitochondrial respiratory chain (51). Furthermore, medium-chain acyl-CoA dehydrogenase deficiency is caused by mutation in the medium-chain acyl-CoA dehydrogenase gene (ACADM) (52), which was twofold decreased in Atxn2-KO liver tissue.
The previously reported accumulation of fat droplets in Atxn2-KO liver (30) is in excellent agreement with these metabolomics findings and with the proteomic observation of downregulated ACADS, which catalyzes the initial step of the mitochondrial fatty acid beta-oxidation. Furthermore, one of the strongest downregulations in the metabolomics profile of Atxn2-KO liver was for biotin (0.54-fold), an important cofactor for the BCAA pathway enzymes. The interplay of the key findings in Atxn2-KO liver were displayed in an integrative “omics” figure (Fig. 4).
Fig. 4.

Metabolomics and proteomics overview of the impact of an Atxn2-KO in liver tissue. Indicated are the fold changes of detected proteins and metabolites in KO/WT.
Candidate Protein Validation in Liver Tissue
So as to assess the main proteomics findings by an independent technique, RIPA-buffer extracted proteins from liver tissue of Atxn2-KO/WT mice at age 6 months were analyzed in quantitative immunoblots with commercially available antibodies (supplemental Fig. S9). A reduced abundance was confirmed for IVD (change to 0.06-fold ± 0.03, p = 0.0038), PCCA (to 0.25-fold ± 0.02, p < 0.0001), ACADS (to 0.16-fold ± 0.018, p < 0.0016), OTC (to 0.58-fold ± 0.03, p < 0.0042), ALDH6A1 (to 0.67-fold ± 0.03, p = 0.0006) and MCCC2 (to 0.44-fold ± 0.04, p < 0.0001). These studies confirm the mass spectrometry data on altered BCAA and fatty acid metabolism and should facilitate future mechanistic investigations in vitro.
DISCUSSION
Our global proteome quantification and biomathematical enrichment analysis by GSEA documented several significant effects. Less impressively, the metabolomics quantification showed several changes above twofold, but none of them was significant and sophisticated pathway enrichment software tools such as GSEA are unavailable for metabolome data. The proteome showed strong downregulations of the KEGG pathways for branched chain amino acid (BCAA) metabolism, fatty acid metabolisms, and citric acid cycle in liver, an observation that was supported with nominal significance and smaller fold-changes in cerebellar tissue for the BCAA pathway. Of course, fluctuations in food intake and exercise or stress perturb the leucine homeostasis and trophic signaling. Because our analyses started without prior hypothesis, we used animals that had been fed ad libitum and had been allowed to move freely or fight among several siblings per cage, without any standardization effort such as overnight fasting and individual cages. Thus, it is understandable that considerable variance was encountered for the short-lived compounds in the metabolomic survey, whereas the variance was milder for the long-lived proteins. The variance reduced the significance levels of our metabolomics findings, but the fold-changes of acylcarnitines and biotin should still be taken into account, given that it is well established that acylcarnitines will vary with food intake and exercise and stress (53). Biological systems are always balancing their homeostasis and compensating small anomalies, obscuring minor metabolic effects of the Atxn2 gene. That might be one reason why the citrate cycle appeared unchanged at the metabolome level while it was detectable as pathway enrichment of minor effects in the proteome. Furthermore, many metabolites from different pathways are joining or exiting the citrate cycle at different points that can flatten out Atxn2-KO specific effects.
In the proteomics survey, the significant downregulations of the individual proteins ACADS (involved in mitochondrial fatty acid beta-oxidation), ALDH6A1 (involved in mitochondrial valine and pyrimidine catabolism via malonate-methylmalonate decarboxylation to acetyl/propionyl-CoA), ALDH7A1 (involved in mitochondrial lysine catabolism and stress-protection by metabolizing lipid peroxidation-derived aldehydes), IVD (involved in mitochondrial leucine catabolism), MCCC2 (involved in mitochondrial leucine and isovaleric acid catabolism), PCCA (involved in mitochondrial valine/leucine/isoleucine catabolism and lipid degradation), and OTC (involved in mitochondrial glutamate degradation) converge to impair the degradation of amino acids and fatty acids while altering the generation of methyl-malonyl-CoA, propionyl-CoA, and malonyl-CoA (Fig. 4). The usage of malonyl-CoA can switch from: 1) serving as building block for the biosynthesis of larger fatty acids in a process that is insulin-dependent, via 2) regulating fatty acid oxidation and neuroprotective ketogenesis with glycogen breakdown in the liver during starvation periods, to 3) the conversion to acetyl-CoA as a substrate for the TCA cycle (54). A technically independent confirmation of these data, with commercially available antibodies being used in quantitative immunoblots, indicated that the strongest effect reduces the levels of IVD to less than 10% and suggests that leucine catabolism should be particularly affected.
Thus, our proteomic findings correspond well to the previous documentation of chronic accumulation of lipid droplets and glycogen in the liver of Atxn2-KO mice (30). It is known that elevated blood plasma levels of BCAAs serve as markers of obesity, insulin resistance and diabetes mellitus (55), so they are key indicators of metabolic excess. Conversely, depleted plasma levels of BCAA serve as biomarkers of the polyQ-expansion triggered neurodegenerative process and weight loss in Huntington's disease (56, 57). Thus, it will be interesting to test whether depleted plasma levels of BCAA are also associated with the neural atrophy caused by the polyQ-expansion of ATXN2 in SCA2 and ALS13. It is well established now that polyQ-expansions trigger an aggregation process, which is toxic through oligomer formation, but also through the longer half-life (35) of the disease protein with a gain in physiological functions. This process also sequestrates interaction molecules into insolubility leading to partial losses of physiological functions (8). Thus, polyQ-expanded proteins contribute to pathogenesis in the context of their cellular function (58–63). Not only polyQ-expansions in ATXN2, but also other variants in the ATXN2 gene are associated with ALS risk, presumably via altered physiological functions (37). Furthermore, the aggregation process of SCA2 is less conspicuous in cerebellar neurons than in motor neurons, despite cerebellar vulnerability to SCA2 (64, 65). Therefore, our understanding of the role of ATXN2 in metabolic excess syndromes and neural atrophies may be furthered by the identification of downstream effects such as BCAA metabolism.
The accumulation of the Ataxin-2 protein in neurons is thought to occur in insidious manner over decades, so the resulting neurotoxicity develops slowly and does not manifest until reaching a threshold at advanced ages. For this reason, apparently insignificant twofold changes in metabolites such as the BCAA pathway intermediates might have cumulative neurotoxic effects across the human lifespan. This is relevant, because strongly deficient degradation of BCAA is well known to result in childhood-onset diseases summarized under the term branched-chain organic acidurias, comprising maple syrup urine disease, isovaleric aciduria, propionic aciduria, and methylmalonic aciduria, which frequently have delayed neurodevelopmental stages or neurodegenerative processes (66). Of therapeutic value may be the observation that dietary restriction of BCAAs may mitigate some neurological complaints (67). Similarly, the twofold reduction in biotin in the metabolomics survey might be relevant, despite apparently being insignificant. Biotin levels are mostly derived from protein degradation in food uptake and thus vary considerably in animals fed ad libitum. Biotin is relevant as an essential factor for amino acid metabolism, gluconeogenesis, and fatty acid metabolism. Biotinidase (BTD) deficiency leads to ataxia and paresis, a biotin supplementation is used to normalize leucine and isoleucine metabolism, and a biotinidase knockout mouse activates AMP Kinase while inhibiting mTOR signaling (68). To obtain normal biotin levels from dietary protein, not only biotinidase is necessary, but also the transporters monocarboxylate transporter 1 (SLC16A1) and sodium-dependent multivitamin transporter (SLC5A6) as well as the protein ligase holocarboxylase synthetase (HLCS), see Fig. 4 (69). Thus, a mild reduction in biotin levels in cumulative manner might contribute to pathogenesis.
It is also noteworthy that the availability of branched chain amino acids such as leucine is a strong stimulus for mTORC1 signaling and the initiation or mRNA translation and protein biosynthesis, while uncoupling the insulin signals (55, 70–72). The mTOR phosphorylation pathway directly governs the transcription of ERRalpha-target genes involved in energy metabolism including lipogenesis and citric acid cycle (71). Indeed, ATXN2 was found to modulate the leucine/mTOR-driven phosphorylation of ribosomal protein S6 and also the incorporation rate of radioactive amino acids during mRNA translation (4). Therefore, the impairment of BCAA metabolism may be a direct marker of putative effects of ATXN2 deficiency on mTORC1 signaling. Recently, the yeast ortholog of ATXN2, PBP1, was shown to sequestrate yeast TORC1 to stress granules in times of low cellular energy in a process that is regulated through phosphorylation signals by SNF1 – the yeast ortholog of human AMPK1 – and PAS kinase (73–75). According to this scenario, deficient TORC1 sequestration in the absence of ATXN2/PBP1 would result in unhampered cell growth even in periods of low nutrients and energy, whereas overactive TORC1 sequestration because of gain-of-function mutations of ATXN2/PBP1 such as polyQ expansions would result in cellular atrophy despite abundant nutrients and energy. Thus, the observations of downregulated BCAA, fatty acid and citric acid cycle pathways in our global proteome profiling are credible in view of previous reports on ATXN2 orthologs and on the pathomechanism of obesity versus polyQ-triggered neurodegenerative diseases.
It is interesting to note that a recent report identifies the branched-chain amino acid catabolism to be a conserved regulator of physiological aging (76), whereas age is of course the best-established risk factor for the manifestation of many human diseases such as obesity and neurodegeneration.
Overall, the global proteome and metabolome profiling of Atxn2-KO mouse liver and cerebellum indicates that ATXN2 modulates nutrition and basal metabolism. The deficient or excessive activity of ATXN2 can plausibly drive growth excess or cell atrophy, respectively.
Note added in proof: A recently published global transcriptome profiling effort of Atxn2-KO liver and cerebellum also documented the downregulated levels of IVD mRNA as the second strongest effect, supporting the notion that IVD levels are controlled at least partially at the transcriptional level. Even bigger was the downregulation of RORA mRNA. This transcription factor that may be difficult to detect in proteome approaches because of very low levels and DNA-binding. This transcriptional downregulation extended to a cluster of calcium homeostasis modifiers (77). The encoded protein ROR alpha exerts control over such calcium regulators as well as lipid metabolism and circadian rhythm, similar to ATXN2. Mutation of RORA leads to congenital cerebellar ataxia because of Purkinje neuron loss (78). Thus, Ataxin-2 regulates numerous nutrient enzymes in the mitochondrial matrix as well as key factors that may respond to altered Ca2+ storage in mitochondria – in conclusion, both the proteome and the transcriptome profile are compatible with a profound mitochondrial dysfunction triggered by ATXN2 mutations.
Supplementary Material
Acknowledgments
We thank Beata Lukaszewska-McGreal for assistance during mass spectrometry sample preparation.
Footnotes
Author contributions: D.M. and G.A. designed research; D.M., M.H., N.S., and S.G. performed research; D.M. and G.A. analyzed data; D.M. and G.A. wrote the paper.
* This study was funded by the Max Planck Institute for Molecular Genetics, Berlin, Germany, by the Neurology Clinic at the Goethe University Frankfurt am Main, by the Deutsche Forschungsgemeinschaft (AU96/13-1 and AU96/14-1), and by the Prof. Dr. Dieter Platt Stiftung.
 This article contains supplemental materials.
This article contains supplemental materials.
1 The abbreviations used are:
- Atxn2
- Ataxin-2
- FA
- Formic acid
- FDR
- False discovery rate
- GSEA
- Gene Set Enrichment Analysis
- HILIC
- Hydrophilic interaction chromatography
- KO
- Knock out
- LFQ
- Label free quantification
- MeOH
- Methanol
- MRM
- Multiple Reaction Monitoring
- MTBE
- Methyl-tert-butyl ester
- PEP
- Posterior error probability
- PPI
- Protein–protein interaction
- RP
- Reversed phase
- STRING
- Search Tool for the Retrieval of Interacting Genes/Proteins.
REFERENCES
- 1. Scoles D. R., Pflieger L. T., Thai K. K., Hansen S. T., Dansithong W., and Pulst S. M. (2012) ETS1 regulates the expression of ATXN2. Hum. Mol. Genet. 21, 5048–5065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Huynh D. P., Del Bigio M. R., Ho D. H., and Pulst S. M. (1999) Expression of ataxin-2 in brains from normal individuals and patients with Alzheimer's disease and spinocerebellar ataxia 2. Ann. Neurol. 45, 232–241 [DOI] [PubMed] [Google Scholar]
- 3. Nonhoff U., Ralser M., Welzel F., Piccini I., Balzereit D., Yaspo M. L., Lehrach H., and Krobitsch S. (2007) Ataxin-2 interacts with the DEAD/H-box RNA helicase DDX6 and interferes with P-bodies and stress granules. Mol. Biol. Cell 18, 1385–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fittschen M., Lastres-Becker I., Halbach M. V., Damrath E., Gispert S., Azizov M., Walter M., Muller S., and Auburger G. (2015) Genetic ablation of ataxin-2 increases several global translation factors in their transcript abundance but decreases translation rate. Neurogenetics 16, 181–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sahba S., Nechiporuk A., Figueroa K. P., Nechiporuk T., and Pulst S. M. (1998) Genomic structure of the human gene for spinocerebellar ataxia type 2 (SCA2) on chromosome 12q24.1. Genomics 47, 359–364 [DOI] [PubMed] [Google Scholar]
- 6. Nonis D., Schmidt M. H., van de Loo S., Eich F., Dikic I., Nowock J., and Auburger G. (2008) Ataxin-2 associates with the endocytosis complex and affects EGF receptor trafficking. Cell Signal. 20, 1725–1739 [DOI] [PubMed] [Google Scholar]
- 7. Drost J., Nonis D., Eich F., Leske O., Damrath E., Brunt E. R., Lastres-Becker I., Heumann R., Nowock J., and Auburger G. (2013) Ataxin-2 modulates the levels of Grb2 and SRC but not ras signaling. J. Mol. Neurosci. 51, 68–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Damrath E., Heck M. V., Gispert S., Azizov M., Nowock J., Seifried C., Rub U., Walter M., and Auburger G. (2012) ATXN2-CAG42 sequesters PABPC1 into insolubility and induces FBXW8 in cerebellum of old ataxic knock-in mice. PLoS Genet. 8, e1002920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Satterfield T. F., and Pallanck L. J. (2006) Ataxin-2 and its Drosophila homolog, ATX2, physically assemble with polyribosomes. Hum. Mol. Genet. 15, 2523–2532 [DOI] [PubMed] [Google Scholar]
- 10. Yokoshi M., Li Q., Yamamoto M., Okada H., Suzuki Y., and Kawahara Y. (2014) Direct binding of Ataxin-2 to distinct elements in 3′ UTRs promotes mRNA stability and protein expression. Mol. Cell 55, 186–198 [DOI] [PubMed] [Google Scholar]
- 11. Castello A., Fischer B., Eichelbaum K., Horos R., Beckmann B. M., Strein C., Davey N. E., Humphreys D. T., Preiss T., Steinmetz L. M., Krijgsveld J., and Hentze M. W. (2012) Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 149, 1393–1406 [DOI] [PubMed] [Google Scholar]
- 12. van de Loo S., Eich F., Nonis D., Auburger G., and Nowock J. (2009) Ataxin-2 associates with rough endoplasmic reticulum. Exp. Neurol. 215, 110–118 [DOI] [PubMed] [Google Scholar]
- 13. Heck M. V., Azizov M., Stehning T., Walter M., Kedersha N., and Auburger G. (2014) Dysregulated expression of lipid storage and membrane dynamics factors in Tia1 knockout mouse nervous tissue. Neurogenetics 15, 135–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sudhakaran I. P., Hillebrand J., Dervan A., Das S., Holohan E. E., Hulsmeier J., Sarov M., Parker R., VijayRaghavan K., and Ramaswami M. (2014) FMRP and Ataxin-2 function together in long-term olfactory habituation and neuronal translational control. Proc. Natl. Acad. Sci. U.S.A. 111, E99–E108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang Y., Ling J., Yuan C., Dubruille R., and Emery P. (2013) A role for Drosophila ATX2 in activation of PER translation and circadian behavior. Science 340, 879–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lim C., and Allada R. (2013) ATAXIN-2 activates PERIOD translation to sustain circadian rhythms in Drosophila. Science 340, 875–879 [DOI] [PubMed] [Google Scholar]
- 17. Wellcome-Trust-Case-Control-Consortium (2007) Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447, 661–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Todd J. A., Walker N. M., Cooper J. D., Smyth D. J., Downes K., Plagnol V., Bailey R., Nejentsev S., Field S. F., Payne F., Lowe C. E., Szeszko J. S., Hafler J. P., Zeitels L., Yang J. H., Vella A., Nutland S., Stevens H. E., Schuilenburg H., Coleman G., Maisuria M., Meadows W., Smink L. J., Healy B., Burren O. S., Lam A. A., Ovington N. R., Allen J., Adlem E., Leung H. T., Wallace C., Howson J. M., Guja C., Ionescu-Tirgoviste C., Simmonds M. J., Heward J. M., Gough S. C., Dunger D. B., Wicker L. S., and Clayton D. G. (2007) Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat. Genet. 39, 857–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Smyth D. J., Plagnol V., Walker N. M., Cooper J. D., Downes K., Yang J. H., Howson J. M., Stevens H., McManus R., Wijmenga C., Heap G. A., Dubois P. C., Clayton D. G., Hunt K. A., van Heel D. A., and Todd J. A. (2008) Shared and distinct genetic variants in type 1 diabetes and celiac disease. N. Engl. J. Med. 359, 2767–2777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Barrett J. C., Clayton D. G., Concannon P., Akolkar B., Cooper J. D., Erlich H. A., Julier C., Morahan G., Nerup J., Nierras C., Plagnol V., Pociot F., Schuilenburg H., Smyth D. J., Stevens H., Todd J. A., Walker N. M., and Rich S. S. (2009) Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet. 41, 703–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Figueroa K. P., Farooqi S., Harrup K., Frank J., O'Rahilly S., and Pulst S. M. (2009) Genetic variance in the spinocerebellar ataxia type 2 (ATXN2) gene in children with severe early onset obesity. PLoS One 4, e8280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ganesh S. K., Tragante V., Guo W., Guo Y., Lanktree M. B., Smith E. N., Johnson T., Castillo B. A., Barnard J., Baumert J., Chang Y. P., Elbers C. C., Farrall M., Fischer M. E., Franceschini N., Gaunt T. R., Gho J. M., Gieger C., Gong Y., Isaacs A., Kleber M. E., Mateo Leach I., McDonough C. W., Meijs M. F., Mellander O., Molony C. M., Nolte I. M., Padmanabhan S., Price T. S., Rajagopalan R., Shaffer J., Shah S., Shen H., Soranzo N., van der Most P. J., Van Iperen E. P., Van Setten J., Vonk J. M., Zhang L., Beitelshees A. L., Berenson G. S., Bhatt D. L., Boer J. M., Boerwinkle E., Burkley B., Burt A., Chakravarti A., Chen W., Cooper-Dehoff R. M., Curtis S. P., Dreisbach A., Duggan D., Ehret G. B., Fabsitz R. R., Fornage M., Fox E., Furlong C. E., Gansevoort R. T., Hofker M. H., Hovingh G. K., Kirkland S. A., Kottke-Marchant K., Kutlar A., Lacroix A. Z., Langaee T. Y., Li Y. R., Lin H., Liu K., Maiwald S., Malik R., Murugesan G., Newton-Cheh C., O'Connell J. R., Onland-Moret N. C., Ouwehand W. H., Palmas W., Penninx B. W., Pepine C. J., Pettinger M., Polak J. F., Ramachandran V. S., Ranchalis J., Redline S., Ridker P. M., Rose L. M., Scharnag H., Schork N. J., Shimbo D., Shuldiner A. R., Srinivasan S. R., Stolk R. P., Taylor H. A., Thorand B., Trip M. D., van Duijn C. M., Verschuren W. M., Wijmenga C., Winkelmann B. R., Wyatt S., Young J. H., Boehm B. O., Caulfield M. J., Chasman D. I., Davidson K. W., Doevendans P. A., Fitzgerald G. A., Gums J. G., Hakonarson H., Hillege H. L., Illig T., Jarvik G. P., Johnson J. A., Kastelein J. J., Koenig W., Marz W., Mitchell B. D., Murray S. S., Oldehinkel A. J., Rader D. J., Reilly M. P., Reiner A. P., Schadt E. E., Silverstein R. L., Snieder H., Stanton A. V., Uitterlinden A. G., van der Harst P., van der Schouw Y. T., Samani N. J., Johnson A. D., Munroe P. B., de Bakker P. I., Zhu X., Levy D., Keating B. J., and Asselbergs F. W. (2013) Loci influencing blood pressure identified using a cardiovascular gene-centric array. Hum. Mol. Genet. 22, 1663–1678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gudbjartsson D. F., Bjornsdottir U. S., Halapi E., Helgadottir A., Sulem P., Jonsdottir G. M., Thorleifsson G., Helgadottir H., Steinthorsdottir V., Stefansson H., Williams C., Hui J., Beilby J., Warrington N. M., James A., Palmer L. J., Koppelman G. H., Heinzmann A., Krueger M., Boezen H. M., Wheatley A., Altmuller J., Shin H. D., Uh S. T., Cheong H. S., Jonsdottir B., Gislason D., Park C. S., Rasmussen L. M., Porsbjerg C., Hansen J. W., Backer V., Werge T., Janson C., Jonsson U. B., Ng M. C., Chan J., So W. Y., Ma R., Shah S. H., Granger C. B., Quyyumi A. A., Levey A. I., Vaccarino V., Reilly M. P., Rader D. J., Williams M. J., van Rij A. M., Jones G. T., Trabetti E., Malerba G., Pignatti P. F., Boner A., Pescollderungg L., Girelli D., Olivieri O., Martinelli N., Ludviksson B. R., Ludviksdottir D., Eyjolfsson G. I., Arnar D., Thorgeirsson G., Deichmann K., Thompson P. J., Wjst M., Hall I. P., Postma D. S., Gislason T., Gulcher J., Kong A., Jonsdottir I., Thorsteinsdottir U., and Stefansson K. (2009) Sequence variants affecting eosinophil numbers associate with asthma and myocardial infarction. Nat. Genet. 41, 342–347 [DOI] [PubMed] [Google Scholar]
- 24. Levy D., Ehret G. B., Rice K., Verwoert G. C., Launer L. J., Dehghan A., Glazer N. L., Morrison A. C., Johnson A. D., Aspelund T., Aulchenko Y., Lumley T., Kottgen A., Vasan R. S., Rivadeneira F., Eiriksdottir G., Guo X., Arking D. E., Mitchell G. F., Mattace-Raso F. U., Smith A. V., Taylor K., Scharpf R. B., Hwang S. J., Sijbrands E. J., Bis J., Harris T. B., Ganesh S. K., O'Donnell C. J., Hofman A., Rotter J. I., Coresh J., Benjamin E. J., Uitterlinden A. G., Heiss G., Fox C. S., Witteman J. C., Boerwinkle E., Wang T. J., Gudnason V., Larson M. G., Chakravarti A., Psaty B. M., and van Duijn C. M. (2009) Genome-wide association study of blood pressure and hypertension. Nat. Genet. 41, 677–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Newton-Cheh C., Johnson T., Gateva V., Tobin M. D., Bochud M., Coin L., Najjar S. S., Zhao J. H., Heath S. C., Eyheramendy S., Papadakis K., Voight B. F., Scott L. J., Zhang F., Farrall M., Tanaka T., Wallace C., Chambers J. C., Khaw K. T., Nilsson P., van der Harst P., Polidoro S., Grobbee D. E., Onland-Moret N. C., Bots M. L., Wain L. V., Elliott K. S., Teumer A., Luan J., Lucas G., Kuusisto J., Burton P. R., Hadley D., McArdle W. L., Brown M., Dominiczak A., Newhouse S. J., Samani N. J., Webster J., Zeggini E., Beckmann J. S., Bergmann S., Lim N., Song K., Vollenweider P., Waeber G., Waterworth D. M., Yuan X., Groop L., Orho-Melander M., Allione A., Di Gregorio A., Guarrera S., Panico S., Ricceri F., Romanazzi V., Sacerdote C., Vineis P., Barroso I., Sandhu M. S., Luben R. N., Crawford G. J., Jousilahti P., Perola M., Boehnke M., Bonnycastle L. L., Collins F. S., Jackson A. U., Mohlke K. L., Stringham H. M., Valle T. T., Willer C. J., Bergman R. N., Morken M. A., Doring A., Gieger C., Illig T., Meitinger T., Org E., Pfeufer A., Wichmann H. E., Kathiresan S., Marrugat J., O'Donnell C. J., Schwartz S. M., Siscovick D. S., Subirana I., Freimer N. B., Hartikainen A. L., McCarthy M. I., O'Reilly P. F., Peltonen L., Pouta A., de Jong P. E., Snieder H., van Gilst W. H., Clarke R., Goel A., Hamsten A., Peden J. F., Seedorf U., Syvanen A. C., Tognoni G., Lakatta E. G., Sanna S., Scheet P., Schlessinger D., Scuteri A., Dorr M., Ernst F., Felix S. B., Homuth G., Lorbeer R., Reffelmann T., Rettig R., Volker U., Galan P., Gut I. G., Hercberg S., Lathrop G. M., Zelenika D., Deloukas P., Soranzo N., Williams F. M., Zhai G., Salomaa V., Laakso M., Elosua R., Forouhi N. G., Volzke H., Uiterwaal C. S., van der Schouw Y. T., Numans M. E., Matullo G., Navis G., Berglund G., Bingham S. A., Kooner J. S., Connell J. M., Bandinelli S., Ferrucci L., Watkins H., Spector T. D., Tuomilehto J., Altshuler D., Strachan D. P., Laan M., Meneton P., Wareham N. J., Uda M., Jarvelin M. R., Mooser V., Melander O., Loos R. J., Elliott P., Abecasis G. R., Caulfield M., and Munroe P. B. (2009) Genome-wide association study identifies eight loci associated with blood pressure. Nat. Genet. 41, 666–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ehret G. B., Munroe P. B., Rice K. M., Bochud M., Johnson A. D., Chasman D. I., Smith A. V., Tobin M. D., Verwoert G. C., Hwang S. J., Pihur V., Vollenweider P., O'Reilly P. F., Amin N., Bragg-Gresham J. L., Teumer A., Glazer N. L., Launer L., Zhao J. H., Aulchenko Y., Heath S., Sober S., Parsa A., Luan J., Arora P., Dehghan A., Zhang F., Lucas G., Hicks A. A., Jackson A. U., Peden J. F., Tanaka T., Wild S. H., Rudan I., Igl W., Milaneschi Y., Parker A. N., Fava C., Chambers J. C., Fox E. R., Kumari M., Go M. J., van der Harst P., Kao W. H., Sjogren M., Vinay D. G., Alexander M., Tabara Y., Shaw-Hawkins S., Whincup P. H., Liu Y., Shi G., Kuusisto J., Tayo B., Seielstad M., Sim X., Nguyen K. D., Lehtimaki T., Matullo G., Wu Y., Gaunt T. R., Onland-Moret N. C., Cooper M. N., Platou C. G., Org E., Hardy R., Dahgam S., Palmen J., Vitart V., Braund P. S., Kuznetsova T., Uiterwaal C. S., Adeyemo A., Palmas W., Campbell H., Ludwig B., Tomaszewski M., Tzoulaki I., Palmer N. D., Aspelund T., Garcia M., Chang Y. P., O'Connell J. R., Steinle N. I., Grobbee D. E., Arking D. E., Kardia S. L., Morrison A. C., Hernandez D., Najjar S., McArdle W. L., Hadley D., Brown M. J., Connell J. M., Hingorani A. D., Day I. N., Lawlor D. A., Beilby J. P., Lawrence R. W., Clarke R., Hopewell J. C., Ongen H., Dreisbach A. W., Li Y., Young J. H., Bis J. C., Kahonen M., Viikari J., Adair L. S., Lee N. R., Chen M. H., Olden M., Pattaro C., Bolton J. A., Kottgen A., Bergmann S., Mooser V., Chaturvedi N., Frayling T. M., Islam M., Jafar T. H., Erdmann J., Kulkarni S. R., Bornstein S. R., Grassler J., Groop L., Voight B. F., Kettunen J., Howard P., Taylor A., Guarrera S., Ricceri F., Emilsson V., Plump A., Barroso I., Khaw K. T., Weder A. B., Hunt S. C., Sun Y. V., Bergman R. N., Collins F. S., Bonnycastle L. L., Scott L. J., Stringham H. M., Peltonen L., Perola M., Vartiainen E., Brand S. M., Staessen J. A., Wang T. J., Burton P. R., Soler Artigas M., Dong Y., Snieder H., Wang X., Zhu H., Lohman K. K., Rudock M. E., Heckbert S. R., Smith N. L., Wiggins K. L., Doumatey A., Shriner D., Veldre G., Viigimaa M., Kinra S., Prabhakaran D., Tripathy V., Langefeld C. D., Rosengren A., Thelle D. S., Corsi A. M., Singleton A., Forrester T., Hilton G., McKenzie C. A., Salako T., Iwai N., Kita Y., Ogihara T., Ohkubo T., Okamura T., Ueshima H., Umemura S., Eyheramendy S., Meitinger T., Wichmann H. E., Cho Y. S., Kim H. L., Lee J. Y., Scott J., Sehmi J. S., Zhang W., Hedblad B., Nilsson P., Smith G. D., Wong A., Narisu N., Stancakova A., Raffel L. J., Yao J., Kathiresan S., O'Donnell C. J., Schwartz S. M., Ikram M. A., Longstreth W. T. Jr., Mosley T. H., Seshadri S., Shrine N. R., Wain L. V., Morken M. A., Swift A. J., Laitinen J., Prokopenko I., Zitting P., Cooper J. A., Humphries S. E., Danesh J., Rasheed A., Goel A., Hamsten A., Watkins H., Bakker S. J., van Gilst W. H., Janipalli C. S., Mani K. R., Yajnik C. S., Hofman A., Mattace-Raso F. U., Oostra B. A., Demirkan A., Isaacs A., Rivadeneira F., Lakatta E. G., Orru M., Scuteri A., Ala-Korpela M., Kangas A. J., Lyytikainen L. P., Soininen P., Tukiainen T., Wurtz P., Ong R. T., Dorr M., Kroemer H. K., Volker U., Volzke H., Galan P., Hercberg S., Lathrop M., Zelenika D., Deloukas P., Mangino M., Spector T. D., Zhai G., Meschia J. F., Nalls M. A., Sharma P., Terzic J., Kumar M. V., Denniff M., Zukowska-Szczechowska E., Wagenknecht L. E., Fowkes F. G., Charchar F. J., Schwarz P. E., Hayward C., Guo X., Rotimi C., Bots M. L., Brand E., Samani N. J., Polasek O., Talmud P. J., Nyberg F., Kuh D., Laan M., Hveem K., Palmer L. J., van der Schouw Y. T., Casas J. P., Mohlke K. L., Vineis P., Raitakari O., Ganesh S. K., Wong T. Y., Tai E. S., Cooper R. S., Laakso M., Rao D. C., Harris T. B., Morris R. W., Dominiczak A. F., Kivimaki M., Marmot M. G., Miki T., Saleheen D., Chandak G. R., Coresh J., Navis G., Salomaa V., Han B. G., Zhu X., Kooner J. S., Melander O., Ridker P. M., Bandinelli S., Gyllensten U. B., Wright A. F., Wilson J. F., Ferrucci L., Farrall M., Tuomilehto J., Pramstaller P. P., Elosua R., Soranzo N., Sijbrands E. J., Altshuler D., Loos R. J., Shuldiner A. R., Gieger C., Meneton P., Uitterlinden A. G., Wareham N. J., Gudnason V., Rotter J. I., Rettig R., Uda M., Strachan D. P., Witteman J. C., Hartikainen A. L., Beckmann J. S., Boerwinkle E., Vasan R. S., Boehnke M., Larson M. G., Jarvelin M. R., Psaty B. M., Abecasis G. R., Chakravarti A., Elliott P., van Duijn C. M., Newton-Cheh C., Levy D., Caulfield M. J., and Johnson T. (2011) Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature 478, 103–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sebastiani P., Solovieff N., Puca A., Hartley S. W., Melista E., Andersen S., Dworkis D. A., Wilk J. B., Myers R. H., Steinberg M. H., Montano M., Baldwin C. T., and Perls T. T. (2010) Genetic signatures of exceptional longevity in humans. Science 2010 [DOI] [PubMed] [Google Scholar]
- 28. Sebastiani P., Solovieff N., Dewan A. T., Walsh K. M., Puca A., Hartley S. W., Melista E., Andersen S., Dworkis D. A., Wilk J. B., Myers R. H., Steinberg M. H., Montano M., Baldwin C. T., Hoh J., and Perls T. T. (2012) Genetic signatures of exceptional longevity in humans. PLoS One 7, e29848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Auburger G., Gispert S., Lahut S., Omur O., Damrath E., Heck M., and Basak N. (2014) 12q24 locus association with type 1 diabetes: SH2B3 or ATXN2? World J. Diabetes 5, 316–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lastres-Becker I., Brodesser S., Lutjohann D., Azizov M., Buchmann J., Hintermann E., Sandhoff K., Schurmann A., Nowock J., and Auburger G. (2008) Insulin receptor and lipid metabolism pathology in ataxin-2 knock-out mice. Hum. Mol. Genet. 17, 1465–1481 [DOI] [PubMed] [Google Scholar]
- 31. Kiehl T. R., Nechiporuk A., Figueroa K. P., Keating M. T., Huynh D. P., and Pulst S. M. (2006) Generation and characterization of Sca2 (ataxin-2) knockout mice. Biochem. Biophys. Res. Commun. 339, 17–24 [DOI] [PubMed] [Google Scholar]
- 32. Gwinn-Hardy K., Chen J. Y., Liu H. C., Liu T. Y., Boss M., Seltzer W., Adam A., Singleton A., Koroshetz W., Waters C., Hardy J., and Farrer M. (2000) Spinocerebellar ataxia type 2 with parkinsonism in ethnic Chinese. Neurology 55, 800–805 [DOI] [PubMed] [Google Scholar]
- 33. Ross O. A., Rutherford N. J., Baker M., Soto-Ortolaza A. I., Carrasquillo M. M., DeJesus-Hernandez M., Adamson J., Li M., Volkening K., Finger E., Seeley W. W., Hatanpaa K. J., Lomen-Hoerth C., Kertesz A., Bigio E. H., Lippa C., Woodruff B. K., Knopman D. S., White C. L. 3rd, Van Gerpen J. A., Meschia J. F., Mackenzie I. R., Boylan K., Boeve B. F., Miller B. L., Strong M. J., Uitti R. J., Younkin S. G., Graff-Radford N. R., Petersen R. C., Wszolek Z. K., Dickson D. W., and Rademakers R. (2011) Ataxin-2 repeat-length variation and neurodegeneration. Hum. Mol. Genet. 20, 3207–3212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lattante S., Millecamps S., Stevanin G., Rivaud-Pechoux S., Moigneu C., Camuzat A., Da Barroca S., Mundwiller E., Couarch P., Salachas F., Hannequin D., Meininger V., Pasquier F., Seilhean D., Couratier P., Danel-Brunaud V., Bonnet A. M., Tranchant C., LeGuern E., Brice A., Le Ber I., and Kabashi E. (2014) Contribution of ATXN2 intermediary polyQ expansions in a spectrum of neurodegenerative disorders. Neurology 83, 990–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Elden A. C., Kim H. J., Hart M. P., Chen-Plotkin A. S., Johnson B. S., Fang X., Armakola M., Geser F., Greene R., Lu M. M., Padmanabhan A., Clay-Falcone D., McCluskey L., Elman L., Juhr D., Gruber P. J., Rub U., Auburger G., Trojanowski J. Q., Lee V. M., Van Deerlin V. M., Bonini N. M., and Gitler A. D. (2010) Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466, 1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pulst S. M., Nechiporuk A., Nechiporuk T., Gispert S., Chen X. N., Lopes-Cendes I., Pearlman S., Starkman S., Orozco-Diaz G., Lunkes A., DeJong P., Rouleau G. A., Auburger G., Korenberg J. R., Figueroa C., and Sahba S. (1996) Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat. Genet. 14, 269–276 [DOI] [PubMed] [Google Scholar]
- 37. Lahut S., Omur O., Uyan O., Agim Z. S., Ozoguz A., Parman Y., Deymeer F., Oflazer P., Koc F., Ozcelik H., Auburger G., and Basak A. N. (2012) ATXN2 and its neighbouring gene SH2B3 are associated with increased ALS risk in the Turkish population. PLoS One 7, e42956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gispert S., Kurz A., Waibel S., Bauer P., Liepelt I., Geisen C., Gitler A. D., Becker T., Weber M., Berg D., Andersen P. M., Kruger R., Riess O., Ludolph A. C., and Auburger G. (2012) The modulation of Amyotrophic Lateral Sclerosis risk by ataxin-2 intermediate polyglutamine expansions is a specific effect. Neurobiol. Dis. 45, 356–361 [DOI] [PubMed] [Google Scholar]
- 39. Lee T., Li Y. R., Ingre C., Weber M., Grehl T., Gredal O., de Carvalho M., Meyer T., Tysnes O. B., Auburger G., Gispert S., Bonini N. M., Andersen P. M., and Gitler A. D. (2011) Ataxin-2 intermediate-length polyglutamine expansions in European ALS patients. Hum. Mol. Genet. 20, 1697–1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Na D., Rouf M., O'Kane C. J., Rubinsztein D. C., and Gsponer J. (2013) NeuroGeM, a knowledgebase of genetic modifiers in neurodegenerative diseases. BMC Med. Genomics 6, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lim J., Hao T., Shaw C., Patel A. J., Szabo G., Rual J. F., Fisk C. J., Li N., Smolyar A., Hill D. E., Barabasi A. L., Vidal M., and Zoghbi H. Y. (2006) A protein–protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell 125, 801–814 [DOI] [PubMed] [Google Scholar]
- 42. Rub U., Schols L., Paulson H., Auburger G., Kermer P., Jen J. C., Seidel K., Korf H. W., and Deller T. (2013) Clinical features, neurogenetics, and neuropathology of the polyglutamine spinocerebellar ataxias type 1, 2, 3, 6 and 7. Prog. Neurobiol. 104, 38–66 [DOI] [PubMed] [Google Scholar]
- 43. Al-Ramahi I., Perez A. M., Lim J., Zhang M., Sorensen R., de Haro M., Branco J., Pulst S. M., Zoghbi H. Y., and Botas J. (2007) dAtaxin-2 mediates expanded Ataxin-1-induced neurodegeneration in a Drosophila model of SCA1. PLoS Genet. 3, e234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gielisch I., and Meierhofer D. (2015) Metabolome and proteome profiling of complex I deficiency induced by rotenone. J. Proteome Res. 14, 224–235 [DOI] [PubMed] [Google Scholar]
- 45. Tyanova S., Mann M., and Cox J. (2014) MaxQuant for in-depth analysis of large SILAC datasets. Methods Mol. Biol. 1188, 351–364 [DOI] [PubMed] [Google Scholar]
- 46. Cox J., Hein M. Y., Luber C. A., Paron I., Nagaraj N., and Mann M. (2014) Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 13, 2513–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vizcaino J. A., Cote R. G., Csordas A., Dianes J. A., Fabregat A., Foster J. M., Griss J., Alpi E., Birim M., Contell J., O'Kelly G., Schoenegger A., Ovelleiro D., Perez-Riverol Y., Reisinger F., Rios D., Wang R., and Hermjakob H. (2013) The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 41, D1063–D1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., and Mesirov J. P. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Franceschini A., Szklarczyk D., Frankild S., Kuhn M., Simonovic M., Roth A., Lin J., Minguez P., Bork P., von Mering C., and Jensen L. J. (2013) STRING v9.1: protein–protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 41, D808–D815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Halbach M. V., Stehning T., Damrath E., Jendrach M., Sen N. E., Basak A. N., and Auburger G. (2015) Both ubiquitin ligases FBXW8 and PARK2 are sequestrated into insolubility by ATXN2 PolyQ expansions, but only FBXW8 expression is dysregulated. PLoS One 10, e0121089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wolf A., Weir P., Segar P., Stone J., and Shield J. (2001) Impaired fatty acid oxidation in propofol infusion syndrome. Lancet 357, 606–607 [DOI] [PubMed] [Google Scholar]
- 52. Leal J., Ades A. E., Wordsworth S., and Dezateux C. (2014) Regional differences in the frequency of the c.985A>G ACADM mutation: findings from a meta-regression of genotyping and screening studies. Clin. Genet. 85, 253–259 [DOI] [PubMed] [Google Scholar]
- 53. Thompson D. K., Sloane R., Bain J. R., Stevens R. D., Newgard C. B., Pieper C. F., and Kraus V. B. (2012) Daily variation of serum acylcarnitines and amino acids. Metabolomics 8, 556–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Foster D. W. (2012) Malonyl-CoA: the regulator of fatty acid synthesis and oxidation. J. Clin. Invest. 122, 1958–1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lynch C. J., and Adams S. H. (2014) Branched-chain amino acids in metabolic signaling and insulin resistance. Nat. Rev. Endocrinol. 10, 723–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mochel F., Charles P., Seguin F., Barritault J., Coussieu C., Perin L., Le Bouc Y., Gervais C., Carcelain G., Vassault A., Feingold J., Rabier D., and Durr A. (2007) Early energy deficit in Huntington disease: identification of a plasma biomarker traceable during disease progression. PLoS One 2, e647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mochel F., Benaich S., Rabier D., and Durr A. (2011) Validation of plasma branched chain amino acids as biomarkers in Huntington disease. Arch. Neurol. 68, 265–267 [DOI] [PubMed] [Google Scholar]
- 58. Orr H. T. (2012) Cell biology of spinocerebellar ataxia. J. Cell Biol. 197, 167–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cary G. A., and La Spada A. R. (2008) Androgen receptor function in motor neuron survival and degeneration. Phys. Med. Rehabil. Clin. N. Am. 19, 479–494, viii [DOI] [PubMed] [Google Scholar]
- 60. Di Prospero N. A., and Fischbeck K. H. (2005) Therapeutics development for triplet repeat expansion diseases. Nat. Rev. Genet. 6, 756–765 [DOI] [PubMed] [Google Scholar]
- 61. Schulte J., and Littleton J. T. (2011) The biological function of the Huntingtin protein and its relevance to Huntington's Disease pathology. Curr. Trends Neurol. 5, 65–78 [PMC free article] [PubMed] [Google Scholar]
- 62. Cattaneo E., Rigamonti D., Goffredo D., Zuccato C., Squitieri F., and Sipione S. (2001) Loss of normal huntingtin function: new developments in Huntington's disease research. Trends Neurosci. 24, 182–188 [DOI] [PubMed] [Google Scholar]
- 63. Lim J., Crespo-Barreto J., Jafar-Nejad P., Bowman A. B., Richman R., Hill D. E., Orr H. T., and Zoghbi H. Y. (2008) Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature 452, 713–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Huynh D. P., Figueroa K., Hoang N., and Pulst S. M. (2000) Nuclear localization or inclusion body formation of ataxin-2 are not necessary for SCA2 pathogenesis in mouse or human. Nat. Genet. 26, 44–50 [DOI] [PubMed] [Google Scholar]
- 65. Koyano S., Iwabuchi K., Yagishita S., Kuroiwa Y., and Uchihara T. (2002) Paradoxical absence of nuclear inclusion in cerebellar Purkinje cells of hereditary ataxias linked to CAG expansion. J. Neurol. Neurosurg. Psychiatry 73, 450–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ogier de Baulny H., and Saudubray J. M. (2002) Branched-chain organic acidurias. Semin. Neonatol. 7, 65–74 [DOI] [PubMed] [Google Scholar]
- 67. Chuang D. T., Chuang J. L., and Wynn R. M. (2006) Lessons from genetic disorders of branched-chain amino acid metabolism. J. Nutr. 136, 243S–249S [DOI] [PubMed] [Google Scholar]
- 68. Hernandez-Vazquez A., Wolf B., Pindolia K., Ortega-Cuellar D., Hernandez-Gonzalez R., Heredia-Antunez A., Ibarra-Gonzalez I., and Velazquez-Arellano A. (2013) Biotinidase knockout mice show cellular energy deficit and altered carbon metabolism gene expression similar to that of nutritional biotin deprivation: clues for the pathogenesis in the human inherited disorder. Mol. Genet. Metab. 110, 248–254 [DOI] [PubMed] [Google Scholar]
- 69. Zempleni J. (2005) Uptake, localization, and noncarboxylase roles of biotin. Annu. Rev. Nutr. 25, 175–196 [DOI] [PubMed] [Google Scholar]
- 70. Kimball S. R., and Jefferson L. S. (2006) Signaling pathways and molecular mechanisms through which branched-chain amino acids mediate translational control of protein synthesis. J. Nutr. 136, 227S–231S [DOI] [PubMed] [Google Scholar]
- 71. Morita M., Gravel S. P., Hulea L., Larsson O., Pollak M., St-Pierre J., and Topisirovic I. (2015) mTOR coordinates protein synthesis, mitochondrial activity, and proliferation. Cell Cycle 14, 473–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kim S. G., Buel G. R., and Blenis J. (2013) Nutrient regulation of the mTOR complex 1 signaling pathway. Mol. Cells 35, 463–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Takahara T., and Maeda T. (2012) Transient sequestration of TORC1 into stress granules during heat stress. Mol. Cell 47, 242–252 [DOI] [PubMed] [Google Scholar]
- 74. Buchan J. R., Capaldi A. P., and Parker R. (2012) TOR-tured yeast find a new way to stand the heat. Mol. Cell 47, 155–157 [DOI] [PubMed] [Google Scholar]
- 75. DeMille D., Badal B. D., Evans J. B., Mathis A. D., Anderson J. F., and Grose J. H. (2015) PAS kinase is activated by direct SNF1-dependent phosphorylation and mediates inhibition of TORC1 through the phosphorylation and activation of Pbp1. Mol. Biol. Cell 26, 569–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Mansfeld J., Urban N., Priebe S., Groth M., Frahm C., Hartmann N., Gebauer J., Ravichandran M., Dommaschk A., Schmeisser S., Kuhlow D., Monajembashi S., Bremer-Streck S., Hemmerich P., Kiehntopf M., Zamboni N., Englert C., Guthke R., Kaleta C., Platzer M., Suhnel J., Witte O. W., Zarse K., and Ristow M. (2015) Branched-chain amino acid catabolism is a conserved regulator of physiological ageing. Nat. Commun. 6, 10043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Halbach M. V., Gispert S., Stehning T., Damrath E., Walter M., and Auburger G. (2016) Atxn2 knockout and CAG42-knock-in cerebellum shows similarly dysregulated expression in calcium homeostasis pathway. Cerebellum accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gold D. A., Gent P. M., and Hamilton B. A. (2007) ROR alpha in genetic control of cerebellum development: 50 staggering years. Brain Res. 1140, 19–25 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}