

Abstract
The coordinated behavior of proteins is central to systems biology. However, the underlying mechanisms are poorly known and methods to analyze coordination by conventional quantitative proteomics are still lacking. We present the Systems Biology Triangle (SBT), a new algorithm that allows the study of protein coordination by pairwise quantitative proteomics. The Systems Biology Triangle detected statistically significant coordination in diverse biological models of very different nature and subjected to different kinds of perturbations. The Systems Biology Triangle also revealed with unprecedented molecular detail an array of coordinated, early protein responses in vascular smooth muscle cells treated at different times with angiotensin-II. These responses included activation of protein synthesis, folding, turnover, and muscle contraction – consistent with a differentiated phenotype—as well as the induction of migration and the repression of cell proliferation and secretion. Remarkably, the majority of the altered functional categories were protein complexes, interaction networks, or metabolic pathways. These changes could not be detected by other algorithms widely used by the proteomics community, and the vast majority of proteins involved have not been described before to be regulated by AngII. The unique capabilities of The Systems Biology Triangle to detect functional protein alterations produced by the coordinated action of proteins in pairwise quantitative proteomics experiments make this algorithm an attractive choice for the biological interpretation of results on a routine basis.
Cellular processes are executed by proteins working together in complexes or functional pathways, and recent evidence indicates that proteins carry out their functions in a coordinated manner. The concept of coordinated behavior of genes and proteins has been studied from different experimental and conceptual perspectives. High throughput mRNA analysis has revealed that genes that are functionally linked or are associated with the same metabolic pathway are often co-expressed (1–3), and that multiprotein complexes within the same functional class are regulated in the same direction (4). By combining gene expression information with data about protein interactions, growth phenotype, and transcription factor binding, it was possible to depict modules or groups of genes showing correlated behavior (5). Similarly, fluorescence techniques have revealed that transcript levels of temporally induced genes are highly correlated in individual yeast cells (6). At the protein level, high-throughput single-cell flow cytometry has been used to study biological noise (7), showing that proteins that are subunits of the same complex tend to attain similar levels in the cell, that fluctuations in protein levels tend to be smaller within large complexes (8), and that cell-to-cell variability in protein expression is similar for proteins sharing a similar function (9). Using a yeast fusion library for immunodetection and measurement of absolute expression levels (10), Carmi et al. showed that interacting proteins are expressed in significantly more similar cellular concentrations (11).
Recent advances in mass spectrometry (MS)-based proteomics allow the identification and relative quantification of thousands of proteins in a single study, making MS the technique of choice for the study of cellular processes on a proteome-wide scale. This technology has revealed that proteins with similar functions typically have similar expression levels (12). Similarly, a tendency of functionally related proteins to be coordinately regulated was shown by correlation analysis of protein abundance after density fractionation (13), over a time course (14), or in a large set of diverse conditions (15). Despite these efforts, the mechanisms by which cells coordinate the levels of functionally related proteins are still poorly understood. Furthermore, existing methods to analyze quantitative data obtained from conventional proteomics studies (for instance when only two different conditions are compared) are not designed to detect coordination and least of all to analyze its extent.
Here we present the Systems Biology Triangle (SBT)1, which to our knowledge is the first algorithm able to detect coordinated protein behavior in high-throughput pairwise quantitative proteomics experiments. We show the utility of the algorithm by revealing clearly coordinated protein behavior in biological models of varied origin subjected to different kinds of perturbations. We have also applied the model to characterize the molecular alterations that take place along time when vascular smooth muscle cells (VSMC) are treated with angiotensin-II (AngII). VSMC are a highly plastic cell type with a pivotal role in vascular wall remodeling, a pathophysiological process underlying prevalent cardiovascular diseases like hypertension and aneurism (16). Although numerous lines of evidence support the implication of AngII in this process (17–20), little is known about the signaling pathways and molecular mediators involved, particularly in the early phases of the process, where the responses can be more attributable to the direct action of AngII. In this work, we have performed the first proteome-wide, time-course study of the early response induced by AngII on VSMC. Application of the SBT algorithm to the quantitative data revealed that AngII produced on VSMC an array of increasingly coordinated protein responses that were reproducibly observed along time. Unlike other approaches widely used by the proteomics community, including enrichment, network analysis, and functional class scoring methods, SBT was able to detect the coordinated activation of protein synthesis, folding, turnover, and muscle contraction machineries, consistent with a differentiated phenotype, as well as the induction of migration and the repression of cell proliferation and secretion. Our work is the first to uncover the global picture of the changes that take place in VSMC in response to AngII stimulation at early times, and reveal the regulation by AngII of a considerable number of molecular pathways that have not been previously described. Thus, our work has important mechanistic implications in understanding the role of AngII in vascular remodeling. These results show that the new algorithm has unique capabilities to detect functional protein alterations produced by the coordinated action of proteins in relevant biological systems.
EXPERIMENTAL PROCEDURES
The Generic Integration Algorithm (GIA)
Rationale
During the analysis of quantitative proteomics data by mass spectrometry, the quantitative information is obtained from the spectra and this information is used to quantify the peptides from which the spectra are produced and the proteins that generate these peptides. In other words, the quantitative information is integrated from the spectrum level to the peptide level and then from the peptide level to the protein level. Here we first describe a generic integration algorithm (GIA) (Fig. 1A) that can be used to construct compact workflows containing sequential integration steps for the analysis of quantitative data (Fig. 1B) and the detection of significant protein abundance changes. Afterward, we show that GIA can be applied to construct workflows containing the Systems Biology Triangle (SBT) (Fig. 1C), a kind of integration that allows the detection of changes in functional categories produced by the coordinated behavior of their proteins.
Fig. 1.

Generic integration algorithm (GIA) for quantitative proteomics and extension to the Systems Biology Triangle (SBT). A, Scheme with the elements of the GIA, where log2-ratios of species in the lower level (xl) are rolled up taking into account their quantification weights (vl) to obtain the relative abundances of the species in the higher level (xh), (21). The relations table between the elements of the lower (l) and higher levels (h) is also schematized. B, Workflow for the analysis of a quantitative proteomics experiment using GIA. C, Workflow for the systems biology analysis of a quantitative proteomics experiment including the Systems Biology Triangle (SBT) (shaded in blue). Note that the variance obtained in the protein to category integration can be used as improved estimation of the experimental protein variance (green arrow). D, Scheme of the concept of proteome coordination according to the SBT, where the behavior of a hypothetical, completely coordinated proteome response (left panel) is compared with that of a completely noncoordinated response (right panel). The black curves represent the null hypothesis. E, Definition of the degree of coordination used in this work. The letters A and B represent the proteins outliers in the red and blue categories, whereas C and D represent the coordinated proteins in the green and blue categories.
Formulation of the GIA Model
Let us assume that the abundance of two samples is being compared and that during the process of quantification we are rolling up the quantitative information from a lower level (L) (e.g. peptide level) to calculate the quantitative values at a higher level (H) (e.g. protein level). We will use the scheme “L to H” to refer to this integration step (e.g. peptide to protein integration). We define the set of values xl as the relative abundances of the species in the lower level expressed as the 2-base logarithm of the ratio of abundance of the species present in each sample, and the set of values xh as the relative abundances of the species in the higher level (Fig. 1A). We express the relationships between the elements of the lower and higher levels by a relations table that indicates the correspondence between the indexes l and h of the elements of the lower and higher levels (Fig. 1A).
The model assumes that in the null hypothesis the quantification of any element l at the lower level may be expressed in the form of a mixed-effects model in relation to the quantification of the element h at the higher level it belongs to:
where ρhl is the random deviation between the element of the lower level and that from the higher level (e.g. the error introduced when peptides are prepared from their corresponding proteins), βl contains the random error carried by the element l of the lower level because of previous integrations from lower levels (e.g. the error carried by the peptide values as they are previously determined from the average of several spectra), and l∈h indicates that h is the element from which l is derived (e.g. that the peptide comes from a given protein).
The model assumes that the random errors are normallydistributed:
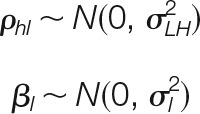 |
The parameter σLH2 is the general variance of the L to H integration, which is constant for all species upon which the integration is performed. Note that this assumption implies that all elements at the lower level are affected by the same variance in relation to the higher level. For instance, in the peptide to protein integration, all the peptides are assumed to be affected by the same error source when they are generated from the corresponding proteins.
The parameter σl2 is the prior local variance, and corresponds to the variance that affects to element l of the lower level because of previous integrations (e.g. the variance that affects to any peptide because of the quality of quantifications at the spectrum level). We define the prior weight, vl, as the inverse of σl2.
If we define the statistical weight wlh as the inverse of the total variance of xl, we will have
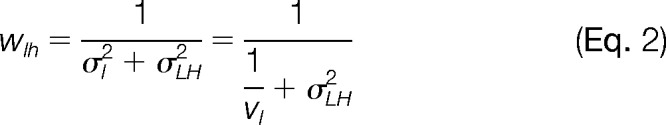 |
When the L to H integration is performed, the quantitative values at the higher level are calculated as weighted averages of the values at the lower level:
 |
According to error propagation theory, the inverse of the variance of the average is the sum of the inverses of the variances of the averaged elements. Therefore, the prior weights of the elements at the higher level, vh are the sum of statistical weights of the corresponding elements at the lower level:
Finally, the standardized variables of the elements of the lower level corresponding to the L to H integration are calculated as
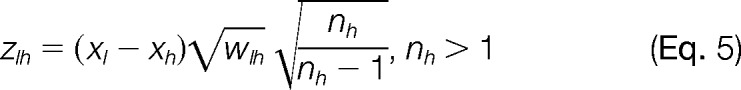 |
where nh is the number of elements from the lower level that are integrated to calculate xh. The values of zlh are expected to distribute according to the standard normal distribution N (0, 1). Note that although zlh is not defined when nh = 1, this does not preclude calculation of xh and vh.
Integrative Workflow and Estimation of the General Variance of the Integration
In the GIA integration the prior weights of the elements of the higher level are calculated from those in the lower level. However, the prior weights of the lower-level elements in the first integration step (e.g. the elements at the spectrum level) are not defined; these weights must be calculated in advance using other algorithms. In this work, we estimated the variances of the elements at the spectrum level using the method described in a previous work (21).
The input of the GIA algorithm is the table (idl, xl, vl) at the lower level, in plain tab-delimited text format, where idl is the identifier of the element l, and the relations table (idh, idl) that establishes the correspondence of the identifiers in the lower level with those in the upper level (Fig. 1A). The output is the table (idh, xh, vh) at the higher level and the general variance of the L to H integration, σLH2. The algorithm also computes the statistical weights at the lower level, wlh, and the standardized variables of the lower level corresponding to the L to H integration, zlh.
It is important to note here that the variables wlh and zlh, which fully describe the distribution of errors at the lower level, can only be calculated once the L to H integration is performed. In consequence, the output does not contain the complete information about the variance of the higher level. For instance, in the case of the peptide to protein integration, the total variance of each peptide, and therefore the standardized variable at the peptide level, cannot be estimated until peptides are integrated into proteins. In addition, this integration does not give information about total protein variance, which remains unknown until proteins are integrated at higher levels. Therefore, in this computational scheme the variances are indissolubly associated with the integrations.
In each integration, σLH2 is estimated using a robust, iterative method. An initial seed value of σLH2 = 0.001 is used by default, and the statistical weights at the lower level wlh are calculated using eq. 2. The integration is then performed to obtain the quantifications xh at the upper level using eq. 3, and the standardized variables of the integration zlh calculated using eq. 5. Once these parameters are calculated, the difference
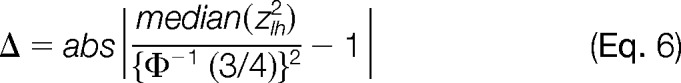 |
where Φ−1(x) is the inverse error function, is used as a robust parameter to determine the deviation of the zlh distribution from the standard normal distribution N(0,1). The process is iterated to minimize Δ using the Levenberg-Marquardt algorithm (22).
Determination of Confidence Intervals of the Variance
Confidence intervals of the variance were calculated using an algorithm that creates simulated experiments. As input, it used the original set of (xl, vl) values, and as output it produced an altered (xl,random, vl) data set, where xl is substituted by a random error generated according to the variance of xl:
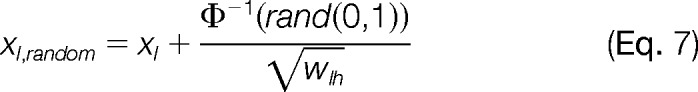 |
where rand(0, 1) is a random decimal number between 0 and 1. For each experiment, 21 simulated experiments were generated, and the variance σLH2 was calculated for each one by applying the GIA. After sorting the resulting list of variances, the median (11th element), along with the 3rd highest and 3rd lowest variances were taken to get the closest values to a one-sigma confidence for each side, in accordance to
where Φ(x) is the cumulative distribution function of the standard normal distribution.
Estimation of FDR and Detection and Elimination of Outliers
The false discovery rate (FDRlh) is calculated by ranking the data according to the absolute values of zlh (from high to low) and estimating the expected number of false deviations from the null hypothesis:
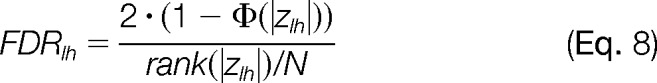 |
where N is the total number of zlh values. Note that in the plots presenting the distributions of zlh the values are ranked from low to high.
Outliers in the L to H integration are defined as the elements at the lower level whose FDRlh surpasses a given threshold (typically 1% or 5%). Outliers can be automatically eliminated by an iterative method. An initial GIA is executed to calculate FDRlh and σLH2. Then, for each element at the higher level, the most extreme lower level outlier is removed, and the GIA is repeated maintaining the original σLH2 variance. The procedure is repeated until no further outliers are detected.
The Systems Biology Triangle
In biological systems proteins can be classified as belonging to a functional class or category, meaning by that any general form of classification, including molecular function, cellular component, pathway, or interaction network. We hypothesized that if during the perturbation of a biological system all the proteins belonging to a category undergo the same abundance change, then the quantitative behavior of the set of proteins can be described by that of their category. We also hypothesized that the relative abundance of the category can be estimated by integrating the quantitative values of its protein components, using the GIA algorithm.
To develop an algorithm to detect significant changes in abundance of functional categories produced by the coordinated behavior of their protein components, we designed the Systems Biology Triangle (SBT). The SBT is constructed by applying the GIA algorithm to integrate from protein to category and then from category to all, as schematized in Fig. 1C (blue triangle). The variances calculated in the SBT analysis provide different levels of information. In the ideal case of complete coordination all the proteins distribute normally within each category, so that there are no protein outliers in the categories and therefore zqc follows a perfect N (0, 1) distribution (Fig. 1D, left). In this case, the variance σQC2 of the protein to category integration only reflects the experimental error by which the proteins are quantified around the corresponding category average, i.e. the experimental protein variance. In such a situation, the biological perturbation should only affect the abundance of the categories, and this effect would become evident in the category to all integration when the distribution of zca is confronted against the null hypothesis that the categories are not affected by the perturbation, i.e. σCA2 = 0 (Fig. 1D, left). In the extreme case of a noncoordinated behavior the perturbation affects proteins irrespective of their category, and therefore σQC2 would have a higher value than in the case of full coordination (Fig. 1D, right). Similarly, the perturbation should have no effect on the distribution of categories (Fig. 1D, right), which would distribute according to σCA2 = 0. In this situation we would not detect significantly changing categories. We speculated that in a real case the situation would be intermediate between these two extremes.
Therefore, we designed the SBT algorithm as follows: firstly, the protein to category integration is performed and the GIA-built in algorithm is applied to remove protein oultiers in each category. By eliminating the protein-category outliers we assured that the abundance change of the proteins remaining in the category did not significantly deviate from the category average; we defined such a situation by stating that these proteins follow a coordinated behavior. The categories that are significantly altered by the perturbation as a consequence of the coordinated behavior of their proteins are then detected as outliers in the category to all integration, by analyzing the distribution of zca values under the null hypothesis that σCA2 = 0. We also defined the degree of coordination of the biological response as the fraction of nonoutlier (i.e. coordinated) proteins belonging to the changing categories in the population of proteins that change individually or form part of changing categories (Fig. 1E).
SBT analysis can also improve the detection of individual protein abundance changes. These changes are detected through the analysis of the protein to all integration (Fig. 1B). However, the variance σQA2 obtained at this stage reflects the total deviations of protein quantifications around the grand mean, which includes not only the experimental uncertainties by which proteins are quantified, but also, to some extent, the effect of the perturbation on protein abundance. In proteomics publications it is generally assumed that the majority of the proteins do not change at the time of estimating protein variance; however, in the absence of a null hypothesis, it is impossible to determine whether this assumption is able to capture the experimental variance in a distribution influenced by a biological perturbation. We speculated that the variance σQC2, estimated through the SBT analysis (Fig. 1C) would be less influenced by the perturbation than σQA2, and therefore would constitute a more precise, conservative estimate of the experimental protein variance.
Clustering Algorithm
To facilitate the detection of similar categories (categories sharing many proteins), a clustering algorithm was applied. The set of changing categories was represented as a weighted directed graph, called similarity graph, where each vertex was associated to a category. Edges between two categories were weighted according to the number of proteins shared. A minimal threshold for the weights was calculated by a Durfee square-based optimization algorithm (23), and edges having a weight below the threshold were removed. This procedure left a number of isolated subgraphs containing categories that share similar sets of proteins, allowing the detection of the main category describing the behavior of the group.
Experimental Design and Statistical Rationale
The SBT model was tested by analyzing a total of eight different biological models subjected to quantitative proteomics by several stable isotope labeling methods. These included: 1) yeast cells treated with H2O2 or with vehicle (Yeast/H2O2); 2) untreated yeast cells (Yeast/-) (21); 3) human bone marrow mesensphere secretome obtained from cells cultured in the presence of human serum or chicken embryo extract (Human Secretome/HS versus CE) (24); 4) liver mitochondrial-associated membranes from Cav-1 knockout or wild type mice (Mouse MAM/Caveolin-1 KO) (25); 5) total fibroblast lysates from Zmpste24−/− or wild-type mice (Mouse Fibroblasts/Zmpste24 KO) (26); 6) total heart extracts from pigs after 1 h post-infarction reperfusion (27, 28), in these samples the infarcted and remote areas of the same animal were compared (Pig Heart/Infarct); 7) mouse lymphocytes obtained from HDAC6 KO and wild-type mouse spleen (Mouse Lymphocytes/HDAC6 KO); in these samples activated total spleen cells were cultured in 10% FBS-RPMI with IL-2 stimulation (29); and 8) primary mouse vascular smooth muscle cells (VSMC) treated with angiotensin-II (AngII) (see below); in these samples six independent biological preparations were used. Details about the null hypothesis used as a reference to determine significance, randomization and statistical tests used are described above. Stable isotope labeling and peptide identification and quantification methods are described below.
Vascular Smooth Muscle Cell Culture
Primary vascular smooth muscle cells were isolated from C57BL/6 mouse abdominal and thoracic aortas and cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 20% fetal bovine serum (FBS) (30). VSMC were rinsed thrice with PBS and starved for 48 h in serum-free DMEM. Cells were than treated with 1μΜ of AngII (Sigma-Aldrich) or vehicle in serum-free culture medium for 2, 4, 6, 8, and 10 h. Cells were lysed in RIPA buffer (NaCl 150 mm, 50 mm Tris pH 8.0, 1% Nonidet P-40, 0.5% sodium desoxycholate, 0.1% SDS) supplemented with 1 mm DTT, 3 mm EGTA, 1 mm PMSF, and protease inhibitor mixture (Sigma-Aldrich). Protein extracts were stored at −80 °C until use.
Sample Preparation, Peptide Labeling and Fractionation, and Mass Spectrometry Analysis
Yeast cells treated with H2O2 or with vehicle (Yeast/H2O2) and different technical replicates of untreated yeast cells (Yeast/-) were treated and labeled with SILAC, 18O or iTRAQ and the resulting data integrated as described (21). Trypsin digestion of all protein extracts was performed using a robust protocol described previously (31). VSMCs and total lymphocyte mouse lysates were labeled with iTRAQ8-plex and 4-plex, respectively, according to manufacturer's instructions. The secretome of human mesenspheres, murine liver MAMs, pig heart extracts, and mice total fibroblast lysates were subjected to 18O labeling (31).
Labeled peptides were subjected to peptide separation into six fractions using a Waters Oasis MCX cartridge (Waters Corp, Milford, MA). Briefly, desalted and dried peptides were taken up in 1 ml of 5 mm ammonium formate pH 3 (AF3). Each MCX cartridge was equilibrated by slowly passing 1 ml of 1:1 methanol/water across cartridge, followed by 3 ml of AF3 containing 25% acetonitrile (ACN) (v:v). Sample was applied at flow rate of 1 drop per second and cartridge was washed with 1 ml of AF3–25% ACN. The bound peptide was eluted into six fractions with 1 ml of freshly prepared buffers: (1) 500 mm AF3, 25% ACN, (2) 1 m AF3, 25% ACN, (3) 1.5 m AF3, 25% ACN, (4) 500 mm AF3, 1.5 m potassium chloride (KCl), 25% de ACN, (5) 1.25 m AF3, 37.5% ACN, and (6) 1 m AF3, 50% de ACN. Eluted peptides were dried in speed vac, desalted on HLB OASIS cartridges (32), and stored at −20 °C until MS analysis.
The samples were analyzed using LC-MS instrumentation consisting of an Easy nano-flow HPLC system (Thermo Fisher Scientific) coupled via a nanoelectrospray ion source (Thermo Fisher Scientific) to either an LTQ-Orbitrap Elite for 18O-labeled samples or to a Q Exactive mass spectrometer for iTRAQ-labeled samples (both Thermo Fisher Scientific). For LC, C18-based reverse phase separation was used with a 2-cm trap column and a 50-cm analytical column (EASY column, Thermo). Peptides were loaded in buffer A (0.1% formic acid (v/v)) and eluted with a 360 min linear gradient of buffer B (90% acetonitrile, 0.1% formic acid (v/v)) at 200 nL/min. Mass spectra were acquired in a data-dependent manner, with an automatic switch between MS and MS/MS using a top 20 method. MS spectra were acquired in the Orbitrap analyzer with a mass range of 390–1500 m/z and 120,000 resolution at m/z 400 (Orbitrap Elite) or 390–1500 m/z and 35,000 resolution at m/z 200 (Q Exactive). CID peptide fragments, acquired at 30 of normalized collision energy (Orbitrap Elite) or HCD peptide fragments obtained at 25 of normalized collision energy (Q Exactive), were analyzed at high resolution in the Orbitrap.
Peptide Identification and Quantification
The raw files were analyzed with Proteome Discoverer (version 1.4, Thermo Fisher Scientific), using a Uniprot database containing all human and chicken protein sequences (November 23th, 2011; 47,609 entries) for the human mesenspheres samples; all mouse protein sequences (April 28th, 2012; 122,974 entries) for the mouse samples; a joint Human+Yeast Swissprot database (Uniprot release 57.3 May 2009; 26,885 entries) for the yeast samples, and a joint Human+Pig Swissprot database (May 30th, 2012; 153,506 entries) for the pig samples. For database searching, parameters were selected as follows: trypsin digestion with two maximum missed cleavage sites, precursor mass tolerance of 800 ppm, fragment mass tolerance of 50 mmu. For 18O-labeled samples variable methionine oxidation, lysine and arginine modification of +4 Da and fixed cysteine carbamidomethylation were used. For iTRAQ, we allowed variable methionine oxidation and fixed cysteine carbamidomethylation, lysine and N-terminal modification of +144.1020 Da for iTRAQ 4-plex or + 304.2054 for iTRAQ 8-plex. The same collections of MS/MS spectra were also searched against inverted databases constructed from the same target databases. Peptide identification from MS/MS data was performed using the probability ratio method (33). False discovery rates (FDR) of peptide identifications were calculated using the refined method (34, 35); 1% FDR was used as criterion for peptide identification. Each peptide was assigned only to the best protein proposed by the Proteome Discoverer algorithm. Quantitative information was extracted from MS spectra, for 18O samples, or MS/MS spectra, for iTRAQ samples, using an in-house developed program (QuiXoT), as described (21), and protein abundance changes were analyzed using the Generic Integration Algorithm, as described above. Calculation of statistical weights of each quantitation at the spectrum level was performed according to the WSPP model (21). The validity of the null hypothesis at each one of the levels (spectrum, peptide, protein, and functional category) was carefully checked by plotting the cumulative distributions, as described (21). iBAQ parameter for all quantified proteins was calculated as in (36).
Protein Functional Annotation
Quantified proteins were functionally annotated using Ingenuity Knowledge Database (IPA) (37, 38), CORUM (39), and DAVID (40). The latter repository included 15 functional databases, such as KEGG, REACTOME, Gene Ontology, and Panther, among others. The Qiagen Transcription Factors Database was taken from http://www.sabiosciences.com/chipqpcrsearch.
Western Blotting and Immunofluorescence
Whole-cell lysates were prepared by lysing cells with RIPA buffer (50 mm Tris-HCl (pH 8.0), 150 mm NaCl, 3 mm EGTA and 1% Nonidet P-40, 0.5% sodium desoxycholate, 0.1%SDS) supplemented with 1 mm DTT, 1 mm PMSF, and a mixture of protease inhibitors (Sigma-Aldrich) on ice for 30 min and then boiling in 2× Laemmli buffer. Lysates were subjected to SDS-PAGE followed by immunoblotting with antibodies against various proteins, including calponin (Santa Cruz Biotechnology), tenascin (Millipore, AB19011), thrombospondin-1 (Thbs1) (Thermo Scientific, MS-421-B0), Prostaglandin G/H synthase 2 (PTGS-2) (Cayman, 160126) and methionine adenosyltransferase IIβ (Mat2β) (Santa Cruz Biotechnology, sc-390586). Tubulin-α (TUBA) (Sigma-Aldrich, T 6074) was used as protein loading control.
For immunofluorescence of cultured VSMCs, cells seeded onto coverslips were subjected to AngII treatment (10−6 m) for different times, fixed with 3% paraformaldehyde, permeabilized with 0.1% Triton-X-100, and then stained with anti-Calponin (1/100; Abcam 46794), or antitype III collagen (1/100, Abnova MAB1514), followed by Alexa Fluor 488-labeled secondary antibody. Nuclei were stained with Hoechst. Slides were mounted and visualized using an inverted confocal microscope (LSM700; Carl Zeiss) with 25× oil objective. Images were processed for presentation with Zen 2012 software (Carl Zeiss).
Data Access
The software needed to execute GIA workflows can be downloaded from ftp://ftp.cnic.es/ftpsvc/pub/SanXoT_package_Source_Code_Example.zip.
A readme.txt file is provided with basic instructions to install and execute the package. Help for each program is displayed by using the -h parameter.
The data set from the yeast experiment (21) is available in the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (41) under data set identifier PXD000325. The data set from the analysis of VSMC proteome (raw and msf files and excel table with identification and quantification data) is available in the PeptideAtlas repository (http://www.peptideatlas.org/PASS/PASS00690) that can be downloaded via ftp.peptideatlas.org, username: PASS00690, password: PU2454dpa.
RESULTS
A Generic Integration Algorithm to Analyze Quantitative Proteomics Data Allows the Development of the Systems Biology Triangle, a Functional Class Scoring Algorithm That Captures Protein Coordination
We firstly developed a generic integration algorithm (GIA) to integrate quantitative data from a lower to a higher level using error propagation theory (Fig. 1A) (see Experimental Procedures). By applying GIA sequentially, we developed automated workflows for conventional analysis of pairwise quantitative proteomics experiments (Fig. 1B), in which significant protein abundance changes were detected as outliers in the protein to all integration step.
We then applied GIA to construct the Systems Biology Triangle (SBT), a workflow that performs the protein to category integration, eliminates protein outliers within each category and detects significant category abundance changes produced by the coordinated behavior of their proteins (Fig. 1C). In parallel SBT detects significant protein abundance changes. The rationale of SBT design is explained under “Experimental Procedures.” In the most general case, biological systems will be somewhere between two extreme situations (Fig. 1D): a complete coordination (left), where the effect of the perturbation is noticed in the category to all integration (CA), and a completely noncoordinated behavior (right), where both the experimental error and the effect of the perturbation are reflected at the protein (QC) level. We defined the degree of coordination of the biological response as a measure of the fraction of proteins that are coordinated (Fig. 1E).
SBT Reveals Coordinated Protein Behavior in Real Biological Systems
To test the SBT model in practice, we first reanalyzed a published data set describing a quantitative comparison of untreated (A) and 0.5 mm H2O2-treated (B) Saccharomyces cerevisae cells (Yeast/H2O2), obtained by integrating the information from eight different replicate experiments using a variety of stable isotope labeling approaches (21). From that study, we also used a parallel integration of eight additional experiments where replicates from nontreated samples were compared as the null hypothesis (Yeast/-). Application of the SBT to the Yeast/- experiment produced z-variables that followed the N(0, 1) distribution in both the QC and CA integrations (Fig. 2A-1), with no significant category changes, indicating excellent agreement between experiment and theory. In contrast, clear evidence of coordination was found in the Yeast/H2O2 experiment, showing deviations from the null hypothesis at the category level (CA integration) with almost negligible deviations in proteins within categories (QC integration) (Fig. 2A-2). In addition, the proportion of protein-category outliers was remarkably low (1.6%, supplemental Table S1). To compare these results with those that would be obtained in a completely noncoordinated behavior, we randomly shuffled the quantitative data assigned to each protein without altering the protein to category relations table, so that the original relational structure and the protein redundancy in the categories were maintained; the process was repeated several times to determine average values and confidence intervals. Randomization increased the variance of proteins within their categories (compare σQR2 with σQC2) and caused category variance to disappear (compare σRA2 with σCA2) (Fig. 2B), showing that the coordination captured by the SBT was statistically significant. The pattern of coordinated protein alterations was clearly evident from analysis of protein changes within each category (Fig. 2C-1 and 2).
Fig. 2.

Evidence of coordinated protein behavior in real biological systems. A, The distribution of thestandardized variables describing the quantification variability among proteins within their category (zqc) and among different categories around the grand mean (zca), resemble more a coordinated than a noncoordinated behavior in all proteomes analyzed. The proteomes analyzed are described in Methods. B, Degree of coordination and effect of randomization on the variances. Randomization increases the protein-category variance and diminishes the category-all variance to values that are not significantly different from zero. Data are expressed as median ± standard deviation. C, Distribution curves of the standardized variable of coordinated proteins (zqa) plotted separately in the different changing categories.
SBT analysis of the category alterations in yeast revealed clear internal coherence in the coordinated protein responses induced by H2O2 (supplemental Fig. S1 and supplemental Table S2). Thus, there was a general repression of biosynthetic metabolic pathways, showing that oxidative stress did not selectively alter only a minority of metabolic enzymes (21), but rather produced a profound metabolic reconfiguration. Consistently, other processes central to cellular homeostasis were also decreased, such as sulfur compound biosynthesis, cell growth and protein trafficking, and proteasome activation was consistent with the induction of a cellular response for rapid elimination of misfolded oxidized proteins (42). Finally, the rest of activated categories where consistent with active mitochondrial adaptation to adverse conditions (43), cellular restoration of metal homeostasis (44, 45), and replacement of ribosomal proteins and rRNA damaged by oxidative stress (46).
SBT was then used to analyze a collection of five high-throughput quantitative experiments performed in several biological models in response to various stimuli and using different stable isotope labeling methods. As shown in Fig. 2A 3–7, in all cases the distributions of zqc and zca indicated the presence of a coordinated behavior, with clear deviations at the category level and almost negligible deviations of proteins within categories. Moreover, the fraction of proteins that were detected as outliers in their categories was remarkably low in all situations (supplemental Table S1). Similarly, randomization of protein values increased protein variance and almost eliminated category variance (Fig. 2B). Finally, the distributions of abundance changes (zqa values) of proteins within the most representative changing categories (Fig. 2C) revealed that in all the biological models most categories were displaced along the x axis while maintaining the same sigmoidal shape, as expected for a coordinated behavior.
In a further study, we analyzed whether the database used for ontological protein classification influenced the degree of coordination detected by our model. We selected three mouse experiments and repeated the SBT analysis using several functional classification databases. The degree of coordination and the fraction of coordinated categories tended to be higher when using curated databases like PANTHER and KEGG and had a tendency to diminish when using databases containing more general terms, such as GO and GO Slim (supplemental Fig. S2). However, the absolute number of coordinated categories tended to increase, because of the higher information content of the latter ones, suggesting that the optimum choice is a compromise between sensitivity and specificity.
Protein Coordination During VSMC Activation with AngII Builds Up Dynamically Over Time
We next analyzed whether the SBT model was able to detect coordinated protein abundance changes over time when primary VSMC were stimulated with AngII, a factor involved in vascular wall remodeling (47, 48). Cells were incubated with AngII for 0, 2, 4, 6, 8, and 10h and the quantitative changes were analyzed using multiplexed isobaric labeling followed by peptide fractionation to increase proteome coverage. This experiment also served to test if coordination of the same proteins could be reproducibly observed in different biological preparations from the same cell type.
Analysis of protein abundance changes was performed by applying the GIA-derived workflow (Fig. 1B), obtaining standardized values that in all the cases and integration levels followed normal distributions (supplemental Fig. S3 and supplemental Table S3A). The results revealed a reproducible pattern with a slight tendency to increase over time (Fig. 3A). We then analyzed coordinated protein changes using SBT (Fig. 1C). The quantitative protein information was integrated into functional categories using a database of more than 4500 categories constructed from IPA and DAVID repositories and also protein complexes from CORUM. 95% of the proteins could be annotated using these databases. Remarkably, the proportion of protein to category outliers was very low, and <3% in all cases (supplemental Table S3B), and no evidence of coordination was detected when the data were subjected to randomization (Fig. 3B and C). Interestingly, after removal of outliers, a clear pattern of coordinated category changes emerged, but only after 4 h of AngII treatment (Fig. 3B–E). Correlation analysis revealed that the categories altered at 4, 8, and 10 h, but not those at 2 h, were essentially the same that were altered at 6 h (Fig. 3E). Consistently, at 2 h practically all proteins changed in a noncoordinated way and hence were detected as category outliers, and these independent changes were similar along time (supplemental Fig. S4). These results indicate that proteins begin to change as early as 2h, being this change predominantly uncoordinated, and that coordinated behavior is gradually built up at later times. We also observed that the proportion of proteins changing in a coordinated manner tended to increase with abundance (Fig. 3F), and the majority of proteins in the quartiles of higher abundance were coordinated (Fig. 3G). These findings may suggest that a tight regulation of the most abundant proteins may be required upon stimulation by AngII to maintain cell homeostasis; however, this interpretation should be taken with caution, because the lower abundance proteins have a higher protein variance, making it more difficult to detect coordinated protein behavior.
Fig. 3.

Analysis of protein coordination in the VSMC proteome along the time course of AngII-treatment. A, Heat-map showing the time-course of the standardized protein quantifications (zqa) of the 1000 proteins having the most significant abundance change. The magnitude of the standardized variable is shaded according to the color scale at the bottom. B, Time-course of the number of categories changing in a coordinated manner. C, Time-course of the degree of coordination. D, Heat-map showing the time-course of the standardized category quantifications (zca) of the categories changing in at least one time-point. The magnitude of the standardized variable is shaded according to the color scale at the bottom. E, Correlation analysis between the standardized category quantifications at the different time-points in relation to those at 6h of AngII treatment. F, Distribution of coordinated proteins in the four iBAQ abundance quartiles at the different time points, in relation to the total number of proteins in each quartile. G, Distribution of protein coordination within the four iBAQ abundance quartiles. Coordination was calculated considering together all the proteins in the five time points.
AngII Promotes Coordinated Protein Changes Consistent with a Hypertrophic and Contractile VSMC Phenotype
Because the analysis used several functional category databases, and categories were often redundant in their protein content, to simplify interpretation we developed a clustering algorithm that assembled changing categories containing similar proteins into functional clusters, so that a representative category could be defined for each cluster (supplemental Fig. S5). This step simplified the outcome, reducing the total number of AngII-sensitive protein categories that changed in a coordinated manner to 25 (supplemental Table S4); after this process only the collagen and muscle protein categories needed a further classification (supplemental Fig. S6 and supplemental Table S5; see also below). Interestingly, more than one third of the categories corresponded to known protein complexes described in CORUM, in good agreement with the expected coordinated behavior of complex-forming proteins (11, 49, 50).
A subset of these complexes were involved in DNA packing and mRNA splicing, and included the nucleosome core, composed by histones, which constitute the basic scaffold for DNA packing (51), and small nuclear ribonucleoproteins, which form part of the spliceosome (Fig. 4). Other complexes were involved in ribosomal protein synthesis, including the 40S and 60S subunits of ribosome, and in protein folding and degradation, including the prefoldin complex, the 20S core particle of proteasome and cyclophilins, which have been reported to form part of spliceosomes (52). Together these categories depict a clear picture of coordinated activation of protein synthesis, folding and turnover machineries (Fig. 4).
Fig. 4.

AngII promotes a coordinated activation of protein synthesis, folding, and turnover in VSMC. A, Time course of protein changes belonging to categories implicated in protein synthesis, folding, and turnover machineries. All these proteins are known to form protein complexes. Although it is not clear which specific histone isoforms interact to each other to form the nucleosome, we represented them as a complex because it is well known that eight of these proteins (two copies of each histone protein, H2A, H2B, H3, and H4) bind together to form the core of the nucleosome (124). In addition, the proteins annotated as “Sm-like ribonucleoproteins” are actually from three different complexes LSm1–7, LSm2–8, and SMN (125, 126), and all of them are part of the spliceosome. B, Time-course of changes in the categories shown in panel A. For the sake of graph clarity, the zca of categories that increase in response to AngII are shown as positive. C, Distribution of the standardized protein quantifications (zqa) belonging to the categories indicated in the legend of panel B, showing coordinated protein changes.
We also detected a coordinated increase of a group of categories that were all associated with a contractile and differentiated phenotype (Fig. 5), including muscle proteins, tropomyosins and other well-known smooth muscle components (SM22/calponin-related proteins) (53). Among these, we observed a coordinated increase in proteins composing the B-Ksr1-MEK-MAPK-14-3-3 complex, which acts as a scaffold integrating the actions of diverse proteins including kinases, transcription factors, and apoptotic molecules (54). This complex translocates to the plasma membrane in response to growth factors, regulating Ras signaling, and producing several outcomes in VSMC, ranging from contraction to proliferation (55). We also detected an increase in abundance of the 5HT3 type receptor mediated signaling pathway, which promotes calcium release from intracellular stores, causing smooth muscle contraction (56). An increase in abundance of calcium-activated proteins involved in the regulation of PGC-1α was also in clear agreement with the augmented contractile capacity of VSMC, because PGC-1α promotes biogenesis of mitochondria (57), an important intracellular calcium reservoir and ATP source for muscle contraction. Accordingly, we observed an increase in the protein abundance of most of the components of mitochondrial F1F0-ATP synthase. Interestingly, a STRING-based interaction analysis revealed that all these groups of proteins formed highly interconnected networks, which sometimes joined together several of these categories (Fig. 5). This array of physical and functional protein interactions might be the cause of the high degree of coordination between the proteins contained in these categories, and explains why these categories were captured by the SBT model.
Fig. 5.

Evidences of coordinated protein changes consistent with a contractile VSMC phenotype upon AngII stimulation. A, B, and C, have the same meaning as in Fig. 4. Known protein complexes are represented as in Fig. 4; the rest of proteins are represented as an interaction network according to String.
AngII Induces Coordinated Protein Alterations Reflecting VSMC Migration and Repression of Cell Proliferation and Secretion
An additional group of coordinated categories was related to the induction of VSMC migration (Fig. 6). Among these, the cytoskeletal regulation by RhoGTPase and ARF/SAR proteins were both upregulated by AngII. RhoGTPases act as the primary Ca2+ sensitizers in smooth muscle, increasing the contractile output of these cells (58), and participate in the formation of focal adhesions, which are important for cell migration (59, 60). Proteins belonging to the ARF/SAR category have important roles actin remodeling (61, 62) and some of its members act in coordination with Rac1 and RhoA (63), which are implicated in cell migration. AngII-activated small GTPases have also been implicated in promoting migration in neuronal and epithelial cells (64, 65). Further evidence toward a migratory phenotype was provided by the coordinated upregulation of proteins from the Arp2/3 complex, a classical podosome component of smooth muscle cells (66), and the Upstream Regulator_MAP3K1 category. The components of this category include tenascin, a glycoprotein providing de-adhesive properties to the cells and enabling cell movement (67), and thrombospondin-1, a potent stimulus for VSMC migration (68). These changes were paralleled by downregulation of fibrillar collagen, which is known to suppress smooth muscle cell migration (69). Finally, the upregulated VSMC migration response is consistent with a progressive increase upon AngII treatment of the levels of PTGS-2 (supplemental Table S6), a well-known VSMC migration marker (70). Interestingly, the six most AngII-responsive proteins from the ARF/SAR category are regulated by the transcription factor PAX-4a (according to the Qiagen Transcription Factors Database), whereas the proteins of the rest of categories form complexes or densely interconnected networks (Fig. 6), providing a molecular basis for the detection of coordinated behavior by the SBT model.
Fig. 6.

AngII induces coordinated protein alterations reflecting VSMC migration. A, B, and C, have the same meaning as in Fig. 5. Proteins not known to form complexes or not forming a String interaction network are presented as squares.
Finally, we detected a coordinated alteration of several categories indicating a repression in cell proliferation and secretion (Fig. 7). Thus, we detected repression of the DNA-dependent ATPase MCM complex, which in eukaryotes is composed by six proteins (MCM2 to 7) and is required for initiation of chromosome replication (71, 72), and the glutamine amidotransferase category, containing mostly key enzymes involved in glutamine use for RNA and DNA synthesis (73), were both repressed (Fig. 7A). Homocysteine biosynthesis was also decreased (Fig. 7B). This pathway promotes smooth muscle cell proliferation (74) and is an important methylation route targeting DNA (75, 76), mRNA (75), and histones (77, 78). This methylation reaction is catalyzed in part by members of the protein arginine methyltransferase (Prmt) family, four of which were downregulated (Fig. 7). Histone methylation generally promotes tighter DNA packing around nucleosomes, resulting in heterochromatin formation and repression of gene expression (79). Repression of this route would thus promote more relaxed chromatin packing and consequent histone release from nucleosomes, which agrees well with the observed increased in histone levels (Fig. 4). The complement pathway, known for triggering inflammation (80) and VSMC switch to a proliferative phenotype (81), was also downregulated (Fig. 7D). In addition, antioxidant enzymes and gluconeogenesis were coordinately upregulated (Fig. 7A). The coordinated upregulation of gluconeogenesis, which was detected together with a concomitant repression of glycolytic enzymes (Fig. 7C), is consistent with nonproliferative phenotype of VSMC because proliferative VSMC display a highly glycolytic phenotype (82). Similarly, the increase in antioxidant enzymes would reflect the activation of defense mechanisms. This activation would counteract the described action of AngII as a promoter of oxidative stress in VSMC (83), which has been described as a mediator for VSMC proliferation (84).
Fig. 7.

AngII induces coordinated protein alterations reflecting VSMC repression of cell proliferation and secretion. A, E, and F, have the same meaning as in Fig. 5. B, C, and D, Schematic representation of protein changes in the homocysteine biosynthesis, gluconeogenesis, and complement pathways, respectively. Each protein is shown as a colored square, where each stripe represents the standardized protein quantification at each time point.
Remarkably, with the exception of MCM proteins, which were detected as a CORUM complex, within each category most proteins related to repressed cell proliferation and secretion were regulated by the same transcription factors. For example, the most responsive enzymes from the homocysteine pathway were regulated by NFkappa-B (85), a key mediator of AngII-driven proliferation and migration (86). Moreover, antioxidant enzymes are known to be regulated through antioxidant-response elements (87, 88) recognized by the nuclear factor (erythroid-derived 2)-related factor 2 (Nrf2) (89). Similarly, the complement system genes share common effectors, such as IL-1, IL-6, and interferon gamma (90). Gluconeogenesis enzymes also share common activating transcription factors, including the CREB-regulated transcription coactivator 2 and the glucocorticoid receptor (91). Together these results reveal that most coordinated categories that are discovered by the SBT model include proteins that share either physical or functional interconnections or common mechanisms of regulation.
Additional technical approaches were used to confirm the progressive acquisition of the contractile, nonproliferative, and migratory VSMC phenotype in response to AngII. Immunofluorescence staining detected increased levels of the contractile protein calponin (CNN) and decreased levels of type-III collagen (Fig. 8A), whereas Western blot revealed reduced levels of methionine adenosyltransferase 2β (Mat2β), the key regulatory enzyme of the homocysteine biosynthesis pathway (Fig. 8B). Western analysis also showed increased abundance several migration-associated factors, including tenascin (TnC), thrombospondin-1 (Thbs1), and prostaglandin-endoperoxide synthase 2 (PTGS-2) (70, 92–94) (Fig. 8C).
Fig. 8.

Immunological validation of the results obtained by the SBT. A, Representative calponin (CNN) and type-III collagen (col3) immunostaining performed with VSMC treated with AngII for 2, 6, and 8 h. Bar, 50 μm. B, and C, Western blots showing methionine adenosyltransferase IIβ (Mat2β) downregulation and tenascin (TnC), thrombospondin-1 (Thbs1) and prostaglandin-endoperoxide synthase 2 (PTGS-2) upregulation by AngII along time. Tubulin-α (TUBA) was used as protein loading control. The time-course of the standardized protein quantification (zqa) for these proteins are also indicated; for the sake of graph clarity, the zqa of categories that increase in response to AngII are shown as positive values.
Protein Outliers Maintain Their Noncoordinated Behavior Over Time and Tend to Play Differential Functional Roles
The functional category alterations detected by our model are not affected by the presence of isolated proteins (outliers) showing high changes in abundance, because these outliers are not considered to analyze the behavior of categories. This raises the question whether these outliers have specific biological roles that explain their abnormal behavior. To answer this question we first analyzed individual protein changes and separated those proteins belonging to a changing category (as described above). Remarkably, independent proteins (that change but do not belong to a changing category) were detected as early as 2h, when coordination was not yet detectable (compare Fig. 3C with supplemental Fig. S4). Moreover, these independent proteins consistently maintained their behavior over time, displaying in all cases a close resemblance to the behavior at 6h (supplemental Fig. S4B and C). Outlier proteins thus seem to respond to the stimulus differently to proteins following a coordinated pattern.
In a second analysis, we inspected the nature of the clearest protein outliers that belonged to the 25 categories altered by AngII (supplemental Table S7). Some of them belonged to a subset of proteins with a clearly separated functionality, suggesting inappropriate classification. This was the case of some collagens and muscle proteins (supplemental Table S5 and supplemental Fig. S6). Among the remaining proteins, PROF2 and PROF1, which were opposite outliers in the same category, were found to be differentially regulated; thus, PROF1, has been described to promote cell motility, whereas PROF-2 acts as a suppressor (95). MYH10, an outlier also assigned to this category, is a nonmuscle heavy chain myosin and is known to play specialized functions in cytokinesis, concentrating in microspikes at the tips of filopodia and retraction fibers (96–98). Similarly, the ribosomal phosphoprotein P0 (Rplp0), the only outlier detected in the category of 60S ribosomal proteins, participates in protein complexes with other cytosolic proteins (Grap2 and cyclin D interacting protein) (99), and its free form has been implicated in other extra-ribosomal functions (100). Finally, PAI-1, the clearest example of an outlier changing in the same direction as its category, is a major mediator of the effect of AngII on VSMC (101). Together these results indicate that the outliers detected by the model correspond to proteins that are either incorrectly classified or play specific roles that are different from those of the other proteins in the category.
Finally, in a third analysis, we inspected the independent proteins. From these, we selected a subset of 21 high-response (HR) proteins (supplemental Table S8). Interestingly, most of these proteins have been reported to play a relevant role in cell signaling, migration, and proliferation (supplemental Table S8), but none of them has been previously shown to be regulated by AngII in VSMC. Taken together, our results provide compelling evidence that the protein outliers detected by the SBT model are proteins with an independent behavior, probably because of their involvement in specific functions different from those of the proteins detected as having a coordinated behavior.
Results Obtained Using the SBT Model could not be Reproduced Using Other Representative Algorithms Widely Used by the Proteomics Community
To compare the performance of SBT with existing methods, we tried to interpret the quantitative proteomics results using several approaches widely used by the proteomics community. The proteins showing significant abundance changes were subjected to functional enrichment and network analysis using GOrilla (102) and STRING (103). Among the proteins that showed significant abundance changes (FDRqa < 5%) and were upregulated, we were only able to detected a significant enrichment in two functional categories: structural constituent of ribosome and calcium-dependent protein binding; similarly, only a significant network of functional interactions was evident among ribosomal and proteins related to calcium-binding. No significant enrichment or interaction network was detected among the proteins that had significant abundance changes and were downregulated. Similar results were obtained using GSEA (104), a functional class scoring algorithm that was not influenced by the threshold used to determine significant protein abundance changes. For GSEA analysis we used the same quantitative values at the protein level and the same functional category database used by the SBT model. Although SBT only detected coordinated categories, the sensitivity of GSEA was clearly inferior, being only able to detect 14% of the category changes detected by SBT at 5% FDRca (supplemental Fig. S7). The differences were even more remarkable for categories containing less than 100 proteins, where GSEA only detected 7% of the changes detected by SBT, whereas SBT practically detected all the category changes detected by GSEA. These results suggest that the SBT model is unique not only for its ability to capture protein coordination, but also for its sensitivity to detect functional category alterations.
DISCUSSION
In this study we present SBT, which to the best of our knowledge is the first algorithm that analyzes coordinated protein behavior in high-throughput quantitative pairwise proteomics experiments and is able to detect functional categories affected as a consequence of coordinated protein behavior. In contrast with other approaches used to study protein coordination, which are commented in the introduction, our approach is not based on correlation analysis, but takes information from ontological databases to classify proteins into functional categories. SBT directly addresses the question whether a protein has a coordinated behavior within a category by determining if it has the same relative abundance change as the other proteins in the category. Significant changes at the category level are then detected using only the population of proteins that have a coordinated behavior in each category. Consistently, we define the degree of coordination as the fraction of changing proteins that are coordinated, including in the calculation the proteins that belong to changing categories. This approach allows the analysis of protein coordination when only two conditions are compared, making it possible to measure the evolution and consistency of coordination over time. Here we show that this approach can capture coordinated protein behavior in seven different biological models subjected to various kinds of perturbations. However, in our extensive experience with the model we have successfully detected functional changes because of coordinated action of proteins in more than 40 comparative pairwise experiments. For this reason, we believe that the SBT model provides the proteomics community with a very useful tool for biological interpretation of pairwise quantitative proteomics experiments, obviating the need to calculate correlations in large sample data sets.
Because our model needs previous ontological knowledge about the quantified proteins, it is expected to be very sensitive to the quality and exhaustiveness of the classification. Indeed, we found a higher degree of coordination in databases using a more exhaustive classification method, such as PANTHER and KEGG, whereas larger, less curated databases, tended to detect a higher number of coordinated proteins and categories, but with a lower degree of coordination (supplemental Fig. S2). We also found that inclusion of information about protein complexes, such as the CORUM database, was particularly useful for detecting coordination, in agreement with the evidence that subunits of complexes have a remarkable tendency to be coregulated (8, 11, 14). Interestingly, we found that most of the outliers analyzed here seem to play a differential role, like regulatory or signaling functions, suggesting that our algorithm can be used to detect protein abundance changes of particular biological relevance. Our finding resembles results obtained in a previous report, showing that deviation from the correlation pattern by one or more proteins indicated a specific function during the biological process (14).
Quantitative transcriptomics data is usually interpreted in relation to biological knowledge stored in databases, a procedure known as gene set analysis or pathway analysis (105). These knowledge databases can contain ontological information about genes, like GO or Kyoto Encyclopedia of Genes and Genomes (KEGG), or provide information about gene/protein interactions and how and where these occur, like Reactome or STRING. Pathway analysis algorithms that use ontological information are classified into two major subtypes: over-representation (ORA) and functional class scoring (FCS) (105). ORA, also known as enrichment analysis, statistically evaluates whether the subset of genes showing significant expression changes relative to a given threshold is enriched in a given category (ontology) (106, 107). This approach is widely used to analyze quantitative proteomics data, but algorithms of this kind only consider significantly changing proteins and therefore ignore most of the acquired quantitative information; moreover, they do not consider protein abundance or fold-change information. In FCS methods, the quantitative values of all genes from a category are integrated to produce a category-level value (105), which is statistically analyzed to determine significant category changes (104, 108–110). Although these threshold-free methods have also been used in proteomics and take account of all the information obtained, they were originally designed to treat transcriptomics data and therefore do not take optimal account of specific characteristics of protein quantification by mass spectrometry. Particularly, they do not consider the large dynamic range of protein concentrations typical of biological systems, which makes MS-based quantification of proteins present in low amounts very challenging and, in general, less reliable. This problem is aggravated by undersampling, whereby the number of peptides used to quantify a protein is variable and cannot be controlled (21). Furthermore, current FCS methods are not designed to analyze the presence and extent of coordination in protein behavior.
The algorithm presented in this work is conceptually inspired on the WSPP model, an algorithm that we developed to treat quantitative proteomics data produced by mass spectrometry using stable isotope labeling techniques (21). The SBT model introduces a generic integration algorithm that rigorously integrates errors made at the inferior level with the error made at the superior level, allowing full control of the error associated with the integrated elements (i.e. proteins or categories) so that quantitative information may be analyzed using the standardized z variable (i.e. abundance changes expressed in units of standard deviations). The SBT model takes into account the dynamic range of protein abundances by introducing statistical weights when performing data integration, and also addresses the problem of undersampling. The model allows the detection of protein to category outliers, so that analysis of categories after outlier removal allows the detection of significant category changes that are produced by proteins acting in a coordinated manner, and hence can also be used as a robust functional class scoring algorithm. These properties of SBT make this model unique to treat quantitative proteomics data and to detect functional alterations that take place in a coordinated manner. These properties are not shared by other algorithms like GOrilla, STRING, or GSEA, and this fact explains why in this work they were not as sensitive as the SBT model to detect the functional categories affected by AngII in VSMC.
We should also note here that the SBT model is based in a null hypothesis that has been experimentally shown. In previous studies we showed that the z variable at different levels consistently follows the expected N (0, 1) distribution (21, 31, 111). Here, using two lines of evidence, we further show that, in the null hypothesis, the standardized variable at the category level (zca) also follows the standard normal distribution. First, we show the absence of category changes in a null hypothesis experiment comparing two preparations of untreated yeast cells. Second, we show the absence of category changes when protein to category assignations were randomized, a process that disrupts any underlying coordination at the category levels. Therefore, the category changes may be detected by robust estimates of statistical significance. Notably, our integration model assigns more weight to quantifications of higher quality, making it unnecessary to eliminate poor quantifications (21, 111).
In this report, we present the first quantitative, time-course proteomics study on the effects of a vascular remodeling factor, AngII, on VSMC. We show the discriminatory power of SBT by unraveling with unprecedented molecular detail the coordinated mechanisms taking place during AngII stimulation of VSMC. The robustness of the model is supported by remarkable maintenance of coordination in the same categories in different VSMC preparations over time. Although from our data we cannot judge whether this regulation occurs at transcriptional or posttranscriptional levels, our finding that the vast majority of coordinated categories are protein complexes, metabolic pathways or proteins forming interaction networks, suggests that protein-protein interactions play an important role in producing tight coregulation of protein levels. This conclusion is supported by detection of proteins with a seemingly uncoordinated behavior at the earliest time points, whereas the coordinated response elicited by AngII in VSMC builds up gradually, suggesting an adjustment of the regulatory protein machinery.
Some of the proteins described here to be affected by AngII have been previously observed as AngII sensitive in smooth muscle and other type of cells, including calponin (112), contractile proteins (113), small GTP-binding proteins (114), Rho-associated protein kinase (115), MAP kinases (116–118), and histones (119–122). However, the vast majority of the proteins detected here as being implicated in the coordinated action of AngII have not been described previously, and all of them together provide for the first time a global picture of the early phenotypic alterations taking place in VSMC. The increases in the protein synthesis, folding and turnover machineries suggest the induction of a hypertrophic phenotype where for the first time we show the ability of AngII to activate spliceosome and prefoldin subunits. This phenotype is consistent withthe observed reinforcement in contractility and migration, where we describe a novel effect of AngII as activator of the 5HT3 type receptor mediated signaling pathway, the complex-V of electron transport chain and the proteins of Arp2/3 complex. The decrease in fibrillar collagen during early times of AngII activation is also a completely new finding in the sense that it is opposite to the upregulatory effect produced by AngII on VSMC collagens at longer incubation times (48 h) (123). Finally, we also detect a general repression of proliferative and secretory protein categories, including the DNA-dependent ATPase MCM complex, the glutamine amidotransferase, and the complement, gluconeogenesis and homocysteine biosynthesis pathways, which have never been observed before to be downregulated by AngII in VSMC. These results therefore suggest that at early times AngII induces migration and inhibits proliferation in VSMC. This characterization of novel signaling pathways and effector molecules regulated by AngII in VSMCs during the early phase response might facilitate the identification of therapeutic targets for intervention in many cardiovascular diseases that occur with vascular wall remodeling.
Supplementary Material
Acknowledgments
We thank S. Bartlett for English editing.
Footnotes
Author contributions: J.R., E.B., and J.V. designed research; F.G., M.T., S. Martínez-Martínez, E.B., and J.V. performed research; F.G., M.T., E.B., and J.V. analyzed data; F.G., M.T., E.B., and J.V. wrote the paper; F.G., E.C., I.J., J.L., N.M., E.B., and J.V. performed the proteomics experiments; M.T. developed the software package; S. Méndez-Ferrer prepared biological samples; M.d., B.I., V.A., F.S., and J.R. prepared the biological samples.
* This work was supported by grant BIO2012–37926 from the Spanish Ministry of Economy and Competitiveness and grant PRB2 (IPT13/0001 - ISCIII-SGEFI/FEDER, ProteoRed) and RD12/0042/0056, RD12/0042/0054, RD12/0042/0028, and RD12/0042/0022 from RIC-Red Temática de Investigación Cooperativa en Salud (RETICS), Fondo de Investigaciones Sanitarias, Instituto de Salud Carlos III, and co-funding by Fondo Europeo de Desarrollo Regional (FEDER). SM is supported by the Fundación La Marató TV3 (122532). MTH was supported by a fellowship from the Spanish Ministry of Economy and Competitiveness. The CNIC is supported by the Spanish Ministry of Economy and Competitiveness (MINECO) and the Pro CNIC Foundation, and is a Severo Ochoa Center of Excellence (MINECO award SEV-2015-0505).
 This article contains supplemental Figs. S1 to S7 and Tables S1 to S8.
This article contains supplemental Figs. S1 to S7 and Tables S1 to S8.
Email addresses: Fernando García-Marqués fernando.garcia@externo.cnic.es; Marco Trevisan-Herraz mtrevisan@cnic.es; Sara Martínez-Martínez smartinez@cnic.es; Emilio Camafeita ecamafeita@cnic.es; Inmaculada Jorge Cerrudo inmaculada.jorge@cnic.es; Juan Antonio López jalopez@cnic.es; Nerea Méndez Barbero nmendez@cnic.es; Simón Méndez Ferrer simon.mendez-ferrer@cnic.es; Miguel Angel del Pozo madelpozo@cnic.es; Borja Ibáñez bibanez@cnic.es; Vicente Andrés García vandres@cnic.es; Francisco Sánchez-Madrid fsanchez-madrid@cnic.es; Juan Miguel Redondo jmredondo@cnic.es.
1 The abbreviations used are:
- SBT
- Systems Biology Triangle
- VSMC
- Vascular Smooth Muscle Cells
- AngII
- Angiotensin II
- GIA
- Generic integration algorithm
- QC
- Protein to category
- CA
- Category to all (or grand mean)
- ORA
- Over-representation
- FCS
- Functional class scoring.
REFERENCES
- 1. Ihmels J., Friedlander G., Bergmann S., Sarig O., Ziv Y., and Barkai N. (2002) Revealing modular organization in the yeast transcriptional network. Nat. Genet. 370–377 [DOI] [PubMed] [Google Scholar]
- 2. Ihmels J., Levy R., and Barkai N. (2004) Principles of transcriptional control in the metabolic network of Saccharomyces cerevisiae. Nat. Biotechnol. 22, 86–92 [DOI] [PubMed] [Google Scholar]
- 3. Wei H., Persson S., Mehta T., Srinivasasainagendra V., Chen L., Page G. P., Somerville C., and Loraine A. (2006) Transcriptional coordination of the metabolic network in Arabidopsis. Plant Physiol. 762–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sprinzak E., Cokus S. J., Yeates T. O., Eisenberg D., and Pellegrini M. (2009) Detecting coordinated regulation of multiprotein complexes using logic analysis of gene expression. BMC Syst. Biol. 3, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tanay A., Sharan R., Kupiec M., and Shamir R. (2004) Revealing modularity and organization in the yeast molecular network by integrated analysis of highly heterogeneous genomewide data. Proc. Natl. Acad. Sci. U.S.A. 2981–2986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gandhi S. J., Zenklusen D., Lionnet T., and Singer R. H. (2011) Transcription of functionally related constitutive genes is not coordinated. Nat. Struct. Mol. Biol. 18, 27–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Newman J., Ghaemmaghami S., and Ihmels J. (2006) Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature [DOI] [PubMed] [Google Scholar]
- 8. Carmi S., Levanon E. Y., and Eisenberg E. (2009) Efficiency of complex production in changing environment. BMC Syst. Biol. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bar-Even A., Paulsson J., Maheshri N., Carmi M., O'Shea E., Pilpel Y., and Barkai N. (2006) Noise in protein expression scales with natural protein abundance. Nat. Genet. 636–643 [DOI] [PubMed] [Google Scholar]
- 10. Ghaemmaghami S., Huh W.-K., Bower K., Howson R. W., Belle A., Dephoure N., O'Shea E. K., and Weissman J. S. (2003) Global analysis of protein expression in yeast. Nature 737–741 [DOI] [PubMed] [Google Scholar]
- 11. Carmi S., Levanon E. Y., Havlin S., and Eisenberg E. (2006) Connectivity and expression in protein networks: proteins in a complex are uniformly expressed. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 031909–031906 [DOI] [PubMed] [Google Scholar]
- 12. Marguerat S., Schmidt A., Codlin S., Chen W., Aebersold R., and Bähler J. (2012) Quantitative analysis of fission yeast transcriptomes and proteomes in proliferating and quiescent cells. Cell 671–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Foster L. J., de Hoog C. L., Zhang Y., Zhang Y., Xie X., Mootha V. K., and Mann M. (2006) A mammalian organelle map by protein correlation profiling. Cell 187–199 [DOI] [PubMed] [Google Scholar]
- 14. Hansson J., Rafiee M. R., Reiland S., Polo J. M., Gehring J., Okawa S., Huber W., Hochedlinger K., and Krijgsveld J. (2012) Highly coordinated proteome dynamics during reprogramming of somatic cells to pluripotency. Cell Rep. 1579–1592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wu Y., Williams E. G., Dubuis S., Mottis A., Jovaisaite V., Houten S. M., Argmann C. A., Faridi P., Wolski W., Kutalik Z., Zamboni N., Auwerx J., and Aebersold R. (2014) Multilayered genetic and omics dissection of mitochondrial activity in a mouse reference population. Cell 1415–1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lacolley P., Regnault V., Nicoletti A., Li Z., and Michel J. B. (2012) The vascular smooth muscle cell in arterial pathology: a cell that can take on multiple roles. Cardiovasc. Res. 95, 194–204 [DOI] [PubMed] [Google Scholar]
- 17. Touyz R. M. (2005) Intracellular mechanisms involved in vascular remodeling of resistance arteries in hypertension: role of angiotensin II. Exp. Physiol. 90, 449–455 [DOI] [PubMed] [Google Scholar]
- 18. Daugherty A., Manning M. W., and Cassis L. A. (2000) Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J. Clin. Invest. 105, 1605–1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Heeneman S., Sluimer J. C., and Daemen M. J. (2007) Angiotensin-converting enzyme and vascular remodeling. Circ. Res. 101, 441–454 [DOI] [PubMed] [Google Scholar]
- 20. Weintraub N. L. (2009) Understanding abdominal aortic aneurysm. N. Engl. J. Med. 361, 1114–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Navarro P., Trevisan-Herraz M., Bonzon-Kulichenko E., Nunez E., Martinez-Acedo P., Perez-Hernandez D., Jorge I., Mesa R., Calvo E., Carrascal M., Hernaez M. L., Garcia F., Barcena J. A., Ashman K., Abian J., Gil C., Redondo J. M., and Vazquez J. (2014) General statistical framework for quantitative proteomics by stable isotope labeling. J. Proteome Res. 13, 1234–1247 [DOI] [PubMed] [Google Scholar]
- 22. Marquardt D. W. (1963) An algorithm for least-squares estimation of nonlinear parameters. J. Soc. Indust. Appl. Math. 11, 431–441 [Google Scholar]
- 23. Anderson T., Hankin R., and Killworth P. (2008) Beyond the Durfee square: enhancing the h-index to score total publication output. Scientometrics 76, 577–588 [Google Scholar]
- 24. Isern J., Martin-Antonio B., Ghazanfari R., Martin A. M., Lopez J. A., del Toro R., Sanchez-Aguilera A., Arranz L., Martin-Perez D., Suarez-Lledo M., Marin P., Van Pel M., Fibbe W. E., Vazquez J., Scheding S., Urbano-Ispizua A., and Mendez-Ferrer S. (2013) Self-renewing human bone marrow mesenspheres promote hematopoietic stem cell expansion. Cell Rep. 3, 1714–1724 [DOI] [PubMed] [Google Scholar]
- 25. Wieckowski M. R., Giorgi C., Lebiedzinska M., Duszynski J., and Pinton P. (2009) Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc. 4, 1582–1590 [DOI] [PubMed] [Google Scholar]
- 26. Varela I., Cadinanos J., Pendas A. M., Gutierrez-Fernandez A., Folgueras A. R., Sanchez L. M., Zhou Z., Rodriguez F. J., Stewart C. L., Vega J. A., Tryggvason K., Freije J. M., and Lopez-Otin C. (2005) Accelerated aging in mice deficient in Zmpste24 protease is linked to p53 signaling activation. Nature 437, 564–568 [DOI] [PubMed] [Google Scholar]
- 27. Danielsen M., Hornshoj H., Siggers R. H., Jensen B. B., van Kessel A. G., and Bendixen E. (2007) Effects of bacterial colonization on the porcine intestinal proteome. J. Proteome Res. 6, 2596–2604 [DOI] [PubMed] [Google Scholar]
- 28. Garcia-Prieto J., Garcia-Ruiz J. M., Sanz-Rosa D., Pun A., Garcia-Alvarez A., Davidson S. M., Fernandez-Friera L., Nuno-Ayala M., Fernandez-Jimenez R., Bernal J. A., Izquierdo-Garcia J. L., Jimenez-Borreguero J., Pizarro G., Ruiz-Cabello J., Macaya C., Fuster V., Yellon D. M., and Ibanez B. (2014) beta3 adrenergic receptor selective stimulation during ischemia/reperfusion improves cardiac function in translational models through inhibition of mPTP opening in cardiomyocytes. Basic Res. Cardiol. 109, 422. [DOI] [PubMed] [Google Scholar]
- 29. Gonzalez-Granado J. M., Silvestre-Roig C., Rocha-Perugini V., Trigueros-Motos L., Cibrian D., Morlino G., Blanco-Berrocal M., Osorio F. G., Freije J. M., Lopez-Otin C., Sanchez-Madrid F., and Andres V. (2014) Nuclear envelope lamin-A couples actin dynamics with immunological synapse architecture and T cell activation. Sci. Signal. 7, ra37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ray J. L., Leach R., Herbert J. M., and Benson M. (2001) Isolation of vascular smooth muscle cells from a single murine aorta. Methods Cell Sci. 23, 185–188 [DOI] [PubMed] [Google Scholar]
- 31. Bonzon-Kulichenko E., Perez-Hernandez D., Nunez E., Martinez-Acedo P., Navarro P., Trevisan-Herraz M., Ramos Mdel C., Sierra S., Martinez-Martinez S., Ruiz-Meana M., Miro-Casas E., Garcia-Dorado D., Redondo J. M., Burgos J. S., and Vazquez J. (2011) A robust method for quantitative high-throughput analysis of proteomes by 18O labeling. Mol. Cell. Proteomics 10, M110 003335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Leyfer D., and Weng Z. (2005) Genome-wide decoding of hierarchical modular structure of transcriptional regulation by cis-element and expression clustering. Bioinformatics 2, ii197–203 [DOI] [PubMed] [Google Scholar]
- 33. Martinez-Bartolome S., Navarro P., Martin-Maroto F., Lopez-Ferrer D., Ramos-Fernandez A., Villar M., Garcia-Ruiz J. P., and Vazquez J. (2008) Properties of average score distributions of SEQUEST: the probability ratio method. Mol. Cell. Proteomics 7, 1135–1145 [DOI] [PubMed] [Google Scholar]
- 34. Navarro P., and Vazquez J. (2009) A refined method to calculate false discovery rates for peptide identification using decoy databases. J. Proteome Res. 8, 1792–1796 [DOI] [PubMed] [Google Scholar]
- 35. Bonzon-Kulichenko E., Garcia-Marques F., Trevisan-Herraz M., and Vazquez J. (2015) Revisiting peptide identification by high-accuracy mass spectrometry: problems associated with the use of narrow mass precursor windows. J. Proteome Res. 14, 700–710 [DOI] [PubMed] [Google Scholar]
- 36. Schwanhausser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., and Selbach M. (2011) Global quantification of mammalian gene expression control. Nature 473, 337–342 [DOI] [PubMed] [Google Scholar]
- 37. Ficenec D., Osborne M., Pradines J., Richards D., Felciano R., Cho R. J., Chen R. O., Liefeld T., Owen J., Ruttenberg A., Reich C., Horvath J., and Clark T. (2003) Computational knowledge integration in biopharmaceutical research. Brief. Bioinformatics 4, 260–278 [DOI] [PubMed] [Google Scholar]
- 38. Calvano S. E., Xiao W., Richards D. R., Felciano R. M., Baker H. V., Cho R. J., Chen R. O., Brownstein B. H., Cobb J. P., Tschoeke S. K., Miller-Graziano C., Moldawer L. L., Mindrinos M. N., Davis R. W., Tompkins R. G., Lowry S. F., Inflamm, and Host Response to Injury Large Scale Collab. Res P. (2005) A network-based analysis of systemic inflammation in humans. Nature 437, 1032–1037 [DOI] [PubMed] [Google Scholar]
- 39. Ruepp A., Brauner B., Dunger-Kaltenbach I., Frishman G., Montrone C., Stransky M., Waegele B., Schmidt T., Doudieu O. N., Stumpflen V., and Mewes H. W. (2008) CORUM: the comprehensive resource of mammalian protein complexes. Nucleic Acids Res. 36, D646–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Huang da W., Sherman B. T., Zheng X., Yang J., Imamichi T., Stephens R., and Lempicki R. A. (2009) Extracting biological meaning from large gene lists with DAVID. Current protocols in bioinformatics / editoral board, Andreas D. Baxevanis. [et al.] Chapter 13, Unit 13 11 [DOI] [PubMed] [Google Scholar]
- 41. Vizcaino J. A., Cote R. G., Csordas A., Dianes J. A., Fabregat A., Foster J. M., Griss J., Alpi E., Birim M., Contell J., O'Kelly G., Schoenegger A., Ovelleiro D., Perez-Riverol Y., Reisinger F., Rios D., Wang R., and Hermjakob H. (2013) The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 41, D1063–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Reinheckel T., Sitte N., Ullrich O., Kuckelkorn U., Davies K. J., and Grune T. (1998) Comparative resistance of the 20S and 26S proteasome to oxidative stress. Biochem. J. 335, 637–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Thorpe G. W., Fong C. S., Alic N., Higgins V. J., and Dawes I. W. (2004) Cells have distinct mechanisms to maintain protection against different reactive oxygen species: oxidative-stress-response genes. Proc. Natl. Acad. Sci. U.S.A. 101, 6564–6569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lapinskas P. J., Cunningham K. W., Liu X. F., Fink G. R., and Culotta V. C. (1995) Mutations in PMR1 suppress oxidative damage in yeast cells lacking superoxide dismutase. Mol. Cell. Biol. 15, 1382–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu X. F., and Culotta V. C. (1994) The requirement for yeast superoxide dismutase is bypassed through mutations in BSD2, a novel metal homeostasis gene. Mol. Cell. Biol. 14, 7037–7045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shenton D., Smirnova J. B., Selley J. N., Carroll K., Hubbard S. J., Pavitt G. D., Ashe M. P., and Grant C. M. (2006) Global translational responses to oxidative stress impact upon multiple levels of protein synthesis. J. Biol. Chem. 281, 29011–29021 [DOI] [PubMed] [Google Scholar]
- 47. Daemen M. J., Lombardi D. M., Bosman F. T., and Schwartz S. M. (1991) Angiotensin II induces smooth muscle cell proliferation in the normal and injured rat arterial wall. Circ. Res. 68, 450–456 [DOI] [PubMed] [Google Scholar]
- 48. Dubey R. K., Jackson E. K., and Luscher T. F. (1995) Nitric oxide inhibits angiotensin II-induced migration of rat aortic smooth muscle cell. Role of cyclic-nucleotides and angiotensin1 receptors. J. Clin. Invest. 96, 141–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sprinzak E., Cokus S. J., Yeates T. O., Eisenberg D., and Pellegrini M. (2009) Detecting coordinated regulation of multiprotein complexes using logic analysis of gene expression. BMC Syst. Biol. 3, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Simonis N., Gonze D., Orsi C., van Helden J., and Wodak S. J. (2006) Modularity of the transcriptional response of protein complexes in yeast. J. Mol. Biol. 363, 589–610 [DOI] [PubMed] [Google Scholar]
- 51. Kouzarides T. (2002) Histone methylation in transcriptional control. Curr. Opin. Genet. Dev. 12, 198–209 [DOI] [PubMed] [Google Scholar]
- 52. Chen Y. I., Moore R. E., Ge H. Y., Young M. K., Lee T. D., and Stevens S. W. (2007) Proteomic analysis of in vivo-assembled pre-mRNA splicing complexes expands the catalog of participating factors. Nucleic Acids Res. 35, 3928–3944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nishida W., Kitami Y., and Hiwada K. (1993) cDNA cloning and mRNA expression of calponin and SM22 in rat aorta smooth muscle cells. Gene 130, 297–302 [DOI] [PubMed] [Google Scholar]
- 54. Fu H., Subramanian R. R., and Masters S. C. (2000) 14–3-3 proteins: structure, function, and regulation. Annu. Rev. Pharmacol. Toxicol. 40, 617–647 [DOI] [PubMed] [Google Scholar]
- 55. McKay M. M., Ritt D. A., and Morrison D. K. (2009) Signaling dynamics of the KSR1 scaffold complex. Proc. Natl. Acad. Sci. U.S.A. 106, 11022–11027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Maricq A. V., Peterson A. S., Brake A. J., Myers R. M., and Julius D. (1991) Primary structure and functional expression of the 5HT3 receptor, a serotonin-gated ion channel. Science 254, 432–437 [DOI] [PubMed] [Google Scholar]
- 57. Koves T. R., Li P., An J., Akimoto T., Slentz D., Ilkayeva O., Dohm G. L., Yan Z., Newgard C. B., and Muoio D. M. (2005) Peroxisome proliferator-activated receptor-gamma coactivator 1alpha-mediated metabolic remodeling of skeletal myocytes mimics exercise training and reverses lipid-induced mitochondrial inefficiency. J. Biol. Chem. 280, 33588–33598 [DOI] [PubMed] [Google Scholar]
- 58. Somlyo A. P., and Somlyo A. V. (2003) Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiolog. Rev. 83, 1325–1358 [DOI] [PubMed] [Google Scholar]
- 59. Takai Y., Sasaki T., and Matozaki T. (2001) Small GTP-binding proteins. Physiolog. Rev. 81, 153–208 [DOI] [PubMed] [Google Scholar]
- 60. Ohtsu H., Mifune M., Frank G. D., Saito S., Inagami T., Kim-Mitsuyama S., Takuwa Y., Sasaki T., Rothstein J. D., Suzuki H., Nakashima H., Woolfolk E. A., Motley E. D., and Eguchi S. (2005) Signal-crosstalk between Rho/ROCK and c-Jun NH2-terminal kinase mediates migration of vascular smooth muscle cells stimulated by angiotensin II. Arterioscler. Thromb. Vasc. Biol. 25, 1831–1836 [DOI] [PubMed] [Google Scholar]
- 61. Frank S. R., Hatfield J. C., and Casanova J. E. (1998) Remodeling of the actin cytoskeleton is coordinately regulated by protein kinase C and the ADP-ribosylation factor nucleotide exchange factor ARNO. Mol. Biol. Cell 9, 3133–3146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Franco M., Peters P. J., Boretto J., van Donselaar E., Neri A., D'Souza-Schorey C., and Chavrier P. (1999) EFA6, a sec7 domain-containing exchange factor for ARF6, coordinates membrane recycling and actin cytoskeleton organization. EMBO J. 18, 1480–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Boshans R. L., Szanto S., van Aelst L., and D'Souza-Schorey C. (2000) ADP-ribosylation factor 6 regulates actin cytoskeleton remodeling in coordination with Rac1 and RhoA. Mol. Cell. Biol. 20, 3685–3694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Nishiya N., Kiosses W. B., Han J., and Ginsberg M. H. (2005) An alpha4 integrin-paxillin-Arf-GAP complex restricts Rac activation to the leading edge of migrating cells. Nat. Cell Biol. 7, 343–352 [DOI] [PubMed] [Google Scholar]
- 65. Santy L. C., and Casanova J. E. (2001) Activation of ARF6 by ARNO stimulates epithelial cell migration through downstream activation of both Rac1 and phospholipase D. J. Cell Biol. 154, 599–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gimona M., Kaverina I., Resch G. P., Vignal E., and Burgstaller G. (2003) Calponin repeats regulate actin filament stability and formation of podosomes in smooth muscle cells. Mol. Biol. Cell 14, 2482–2491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Fluck M., Mund S. I., Schittny J. C., Klossner S., Durieux A. C., and Giraud M. N. (2008) Mechano-regulated tenascin-C orchestrates muscle repair. Proc. Natl. Acad. Sci. U.S.A. 105, 13662–13667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yabkowitz R., Mansfield P. J., Ryan U. S., and Suchard S. J. (1993) Thrombospondin mediates migration and potentiates platelet-derived growth factor-dependent migration of calf pulmonary artery smooth muscle cells. J. Cell. Physiol. 157, 24–32 [DOI] [PubMed] [Google Scholar]
- 69. Tanaka S., Koyama H., Ichii T., Shioi A., Hosoi M., Raines E. W., and Nishizawa Y. (2002) Fibrillar collagen regulation of plasminogen activator inhibitor-1 is involved in altered smooth muscle cell migration. Arterioscler. Thromb. Vasc. Biol. 22, 1573–1578 [DOI] [PubMed] [Google Scholar]
- 70. Zhang J., Zou F., Tang J., Zhang Q., Gong Y., Wang Q., Shen Y., Xiong L., Breyer R. M., Lazarus M., Funk C. D., and Yu Y. (2013) Cyclooxygenase-2-derived prostaglandin E(2) promotes injury-induced vascular neointimal hyperplasia through the E-prostanoid 3 receptor. Circ. Res. 113, 104–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Coxon A., Maundrell K., and Kearsey S. E. (1992) Fission yeast cdc21+ belongs to a family of proteins involved in an early step of chromosome replication. Nucleic Acids Res. 20, 5571–5577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hu B., Burkhart R., Schulte D., Musahl C., and Knippers R. (1993) The P1 family: a new class of nuclear mammalian proteins related to the yeast Mcm replication proteins. Nucleic Acids Res. 21, 5289–5293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lacey J. M., and Wilmore D. W. (1990) Is glutamine a conditionally essential amino acid? Nutr. Rev. 48, 297–309 [DOI] [PubMed] [Google Scholar]
- 74. Chiang J. K., Sung M. L., Yu H. R., Chang H. I., Kuo H. C., Tsai T. C., Yen C. K., and Chen C. N. (2011) Homocysteine induces smooth muscle cell proliferation through differential regulation of cyclins A and D1 expression. J. Cell. Physiol. 226, 1017–1026 [DOI] [PubMed] [Google Scholar]
- 75. Chiang P. K., Gordon R. K., Tal J., Zeng G. C., Doctor B. P., Pardhasaradhi K., and McCann P. P. (1996) S-Adenosylmethionine and methylation. FASEB J. 10, 471–480 [PubMed] [Google Scholar]
- 76. Lindsay H., and Adams R. L. (1996) Spreading of methylation along DNA. Biochem. J. 320, 473–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Strahl B. D., Grant P. A., Briggs S. D., Sun Z. W., Bone J. R., Caldwell J. A., Mollah S., Cook R. G., Shabanowitz J., Hunt D. F., and Allis C. D. (2002) Set2 is a nucleosomal histone H3-selective methyltransferase that mediates transcriptional repression. Mol. Cell. Biol. 22, 1298–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Davis C. D., and Ross S. A. (2007) Dietary components impact histone modifications and cancer risk. Nutr. Rev. 65, 88–94 [DOI] [PubMed] [Google Scholar]
- 79. Rice J. C., Briggs S. D., Ueberheide B., Barber C. M., Shabanowitz J., Hunt D. F., Shinkai Y., and Allis C. D. (2003) Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol. Cell 12, 1591–1598 [DOI] [PubMed] [Google Scholar]
- 80. Gasque P., Neal J. W., Singhrao S. K., McGreal E. P., Dean Y. D., Van B. J., and Morgan B. P. (2002) Roles of the complement system in human neurodegenerative disorders: pro-inflammatory and tissue remodeling activities. Mol. Neurobiol. 25, 1–17 [DOI] [PubMed] [Google Scholar]
- 81. Lin Z. H., Fukuda N., Jin X. Q., Yao E. H., Ueno T., Endo M., Saito S., Matsumoto K., and Mugishima H. (2004) Complement 3 is involved in the synthetic phenotype and exaggerated growth of vascular smooth muscle cells from spontaneously hypertensive rats. Hypertension 44, 42–47 [DOI] [PubMed] [Google Scholar]
- 82. Paul R. J. (1989) Smooth muscle energetics. Annu. Rev. Physiol. 51, 331–349 [DOI] [PubMed] [Google Scholar]
- 83. Dzau V. J. (1998) Mechanism of protective effects of ACE inhibition on coronary artery disease. Eur. Heart J. 19, J2–6 [PubMed] [Google Scholar]
- 84. Gao P., Qian D. H., Li W., and Huang L. (2009) NPRA-mediated suppression of AngII-induced ROS production contribute to the antiproliferative effects of B-type natriuretic peptide in VSMC. Mol. Cell. Biochem. 324, 165–172 [DOI] [PubMed] [Google Scholar]
- 85. Yang H., Ara A. I., Magilnick N., Xia M., Ramani K., Chen H., Lee T. D., Mato J. M., and Lu S. C. (2008) Expression pattern, regulation, and functions of methionine adenosyltransferase 2beta splicing variants in hepatoma cells. Gastroenterology 134, 281–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Zahradka P., Werner J. P., Buhay S., Litchie B., Helwer G., and Thomas S. (2002) NF-kappaB activation is essential for angiotensin II-dependent proliferation and migration of vascular smooth muscle cells. J. Mol. Cell. Cardiol. 34, 1609–1621 [DOI] [PubMed] [Google Scholar]
- 87. Dai G., Vaughn S., Zhang Y., Wang E. T., Garcia-Cardena G., and Gimbrone M. A. Jr. (2007) Biomechanical forces in atherosclerosis-resistant vascular regions regulate endothelial redox balance via phosphoinositol 3-kinase/Akt-dependent activation of Nrf2. Circ. Res. 101, 723–733 [DOI] [PubMed] [Google Scholar]
- 88. Chen X. L., Varner S. E., Rao A. S., Grey J. Y., Thomas S., Cook C. K., Wasserman M. A., Medford R. M., Jaiswal A. K., and Kunsch C. (2003) Laminar flow induction of antioxidant response element-mediated genes in endothelial cells. A novel anti-inflammatory mechanism. J. Biol. Chem. 278, 703–711 [DOI] [PubMed] [Google Scholar]
- 89. Kim Y. J., Ahn J. Y., Liang P., Ip C., Zhang Y., and Park Y. M. (2007) Human prx1 gene is a target of Nrf2 and is upregulated by hypoxia/reoxygenation: implication to tumor biology. Cancer Res. 67, 546–554 [DOI] [PubMed] [Google Scholar]
- 90. Volanakis J. E. (1995) Transcriptional regulation of complement genes. Annu. Rev. Immunol. 13, 277–305 [DOI] [PubMed] [Google Scholar]
- 91. Jitrapakdee S. (2012) Transcription factors and coactivators controlling nutrient and hormonal regulation of hepatic gluconeogenesis. Int. J. Biochem. Cell Biol. 44, 33–45 [DOI] [PubMed] [Google Scholar]
- 92. Sharifi B. G., LaFleur D. W., Pirola C. J., Forrester J. S., and Fagin J. A. (1992) Angiotensin II regulates tenascin gene expression in vascular smooth muscle cells. J. Biol. Chem. 267, 23910–23915 [PubMed] [Google Scholar]
- 93. Majack R. A., Mildbrandt J., and Dixit V. M. (1987) Induction of thrombospondin messenger RNA levels occurs as an immediate primary response to platelet-derived growth factor. J. Biol. Chem. 262, 8821–8825 [PubMed] [Google Scholar]
- 94. Scott-Burden T., Resink T. J., Hahn A. W., and Buhler F. R. (1990) Induction of thrombospondin expression in vascular smooth muscle cells by angiotensin II. J. Cardiovasc. Pharmacol. 7, S17–20 [PubMed] [Google Scholar]
- 95. Mouneimne G., Hansen S. D., Selfors L. M., Petrak L., Hickey M. M., Gallegos L. L., Simpson K. J., Lim J., Gertler F. B., Hartwig J. H., Mullins R. D., and Brugge J. S. (2012) Differential remodeling of actin cytoskeleton architecture by profilin isoforms leads to distinct effects on cell migration and invasion. Cancer Cell 22, 615–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Tokuo H., and Ikebe M. (2004) Myosin X transports Mena/VASP to the tip of filopodia. Biochem. Biophys. Res. Commun. 319, 214–220 [DOI] [PubMed] [Google Scholar]
- 97. Zhang H., Berg J. S., Li Z., Wang Y., Lang P., Sousa A. D., Bhaskar A., Cheney R. E., and Stromblad S. (2004) Myosin-X provides a motor-based link between integrins and the cytoskeleton. Nat. Cell Biol. 6, 523–531 [DOI] [PubMed] [Google Scholar]
- 98. Kerber M. L., Jacobs D. T., Campagnola L., Dunn B. D., Yin T., Sousa A. D., Quintero O. A., and Cheney R. E. (2009) A novel form of motility in filopodia revealed by imaging myosin-X at the single-molecule level. Curr. Biol. 19, 967–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Xia C., Bao Z., Tabassam F., Ma W., Qiu M., Hua S., and Liu M. (2000) GCIP, a novel human grap2 and cyclin D interacting protein, regulates E2F-mediated transcriptional activity. J. Biol. Chem. 275, 20942–20948 [DOI] [PubMed] [Google Scholar]
- 100. Chang T. W., Chen C. C., Chen K. Y., Su J. H., Chang J. H., and Chang M. C. (2008) Ribosomal phosphoprotein P0 interacts with GCIP and overexpression of P0 is associated with cellular proliferation in breast and liver carcinoma cells. Oncogene 27, 332–338 [DOI] [PubMed] [Google Scholar]
- 101. Lee K. M., Seo H. Y., Kim M. K., Min A. K., Ryu S. Y., Kim Y. N., Park Y. J., Choi H. S., Lee K. U., Park W. J., Park K. G., and Lee I. K. (2010) Orphan nuclear receptor small heterodimer partner inhibits angiotensin II- stimulated PAI-1 expression in vascular smooth muscle cells. Exp. Mol. Med. 42, 21–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Eden E., Navon R., Steinfeld I., Lipson D., and Yakhini Z. (2009) GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J., Simonovic M., Roth A., Santos A., Tsafou K. P., Kuhn M., Bork P., Jensen L. J., and von Mering C. (2015) STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., and Mesirov J. P. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Khatri P., Sirota M., and Butte A. J. (2012) Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Comput. Biol. e1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Huang da W., Sherman B. T., and Lempicki R. A. (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic acids Res. 37, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Khatri P., and Draghici S. (2005) Ontological analysis of gene expression data: current tools, limitations, and open problems. Bioinformatics 21, 3587–3595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Mootha V. K., Lindgren C. M., Eriksson K. F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstrale M., Laurila E., Houstis N., Daly M. J., Patterson N., Mesirov J. P., Golub T. R., Tamayo P., Spiegelman B., Lander E. S., Hirschhorn J. N., Altshuler D., and Groop L. C. (2003) PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34, 267–273 [DOI] [PubMed] [Google Scholar]
- 109. Barry W. T., Nobel A. B., and Wright F. A. (2005) Significance analysis of functional categories in gene expression studies: a structured permutation approach. Bioinformatics 21, 1943–1949 [DOI] [PubMed] [Google Scholar]
- 110. Jiang Z., and Gentleman R. (2007) Extensions to gene set enrichment. Bioinformatics 23, 306–313 [DOI] [PubMed] [Google Scholar]
- 111. Jorge I., Navarro P., Martinez-Acedo P., Nunez E., Serrano H., Alfranca A., Redondo J. M., and Vazquez J. (2009) Statistical model to analyze quantitative proteomics data obtained by 18O/16O labeling and linear ion trap mass spectrometry: application to the study of vascular endothelial growth factor-induced angiogenesis in endothelial cells. Mol. Cell. Proteomics 8, 1130–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. di Gioia C. R., van de Greef W. M., Sperti G., Castoldi G., Todaro N., Ierardi C., Pieruzzi F., and Stella A. (2000) Angiotensin II increases calponin expression in cultured rat vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 279, 965–969 [DOI] [PubMed] [Google Scholar]
- 113. Turla M. B., Thompson M. M., Corjay M. H., and Owens G. K. (1991) Mechanisms of angiotensin II- and arginine vasopressin-induced increases in protein synthesis and content in cultured rat aortic smooth muscle cells. Evidence for selective increases in smooth muscle isoactin expression. Circ. Res. 68, 288–299 [DOI] [PubMed] [Google Scholar]
- 114. Ohtsu H., Suzuki H., Nakashima H., Dhobale S., Frank G. D., Motley E. D., and Eguchi S. (2006) Angiotensin II signal transduction through small GTP-binding proteins: mechanism and significance in vascular smooth muscle cells. Hypertension 48, 534–540 [DOI] [PubMed] [Google Scholar]
- 115. Rattan S., Puri R. N., and Fan Y. P. (2003) Involvement of rho and rho-associated kinase in sphincteric smooth muscle contraction by angiotensin II. Exp. Biol. Med. 228, 972–981 [DOI] [PubMed] [Google Scholar]
- 116. Duff J. L., Berk B. C., and Corson M. A. (1992) Angiotensin II stimulates the pp44 and pp42 mitogen-activated protein kinases in cultured rat aortic smooth muscle cells. Biochem. Biophys. Res. Commun. 188, 257–264 [DOI] [PubMed] [Google Scholar]
- 117. Ishida Y., Kawahara Y., Tsuda T., Koide M., and Yokoyama M. (1992) Involvement of MAP kinase activators in angiotensin II-induced activation of MAP kinases in cultured vascular smooth muscle cells. FEBS Letters 310, 41–45 [DOI] [PubMed] [Google Scholar]
- 118. Sadoshima J., Qiu Z., Morgan J. P., and Izumo S. (1995) Angiotensin II and other hypertrophic stimuli mediated by G protein-coupled receptors activate tyrosine kinase, mitogen-activated protein kinase, and 90-kD S6 kinase in cardiac myocytes. The critical role of Ca(2+)-dependent signaling. Circ. Res. 76, 1–15 [DOI] [PubMed] [Google Scholar]
- 119. Fukuda K., and Izumo S. (1998) Angiotensin II potentiates DNA synthesis in AT-1 transformed cardiomyocytes. J. Mol. Cell. Cardiol. 30, 2069–2080 [DOI] [PubMed] [Google Scholar]
- 120. Elliott M. E. (1990) Phosphorylation of adrenal histone H3 is affected by angiotensin, ACTH, dibutyryl cAMP, and atrial natriuretic peptide. Life Sci. 46, 1479–1488 [DOI] [PubMed] [Google Scholar]
- 121. Xu X., Ha C. H., Wong C., Wang W., Hausser A., Pfizenmaier K., Olson E. N., McKinsey T. A., and Jin Z. G. (2007) Angiotensin II stimulates protein kinase D-dependent histone deacetylase 5 phosphorylation and nuclear export leading to vascular smooth muscle cell hypertrophy. Arterioscler. Thromb. Vasc. Biol. 27, 2355–2362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Chu C. H., Lo J. F., Hu W. S., Lu R. B., Chang M. H., Tsai F. J., Tsai C. H., Weng Y. S., Tzang B. S., and Huang C. Y. (2012) Histone acetylation is essential for ANG-II-induced IGF-IIR gene expression in H9c2 cardiomyoblast cells and pathologically hypertensive rat heart. J. Cell. Physiol. 227, 259–268 [DOI] [PubMed] [Google Scholar]
- 123. Wang W., Huang X. R., Canlas E., Oka K., Truong L. D., Deng C., Bhowmick N. A., Ju W., Bottinger E. P., and Lan H. Y. (2006) Essential role of Smad3 in angiotensin II-induced vascular fibrosis. Circ. Res. 98, 1032–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Luger K., Mader A. W., Richmond R. K., Sargent D. F., and Richmond T. J. (1997) Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389, 251–260 [DOI] [PubMed] [Google Scholar]
- 125. Zaric B., Chami M., Remigy H., Engel A., Ballmer-Hofer K., Winkler F. K., and Kambach C. (2005) Reconstitution of two recombinant LSm protein complexes reveals aspects of their architecture, assembly, and function. J. Biol. Chem. 280, 16066–16075 [DOI] [PubMed] [Google Scholar]
- 126. Pillai R. S., Grimmler M., Meister G., Will C. L., Luhrmann R., Fischer U., and Schumperli D. (2003) Unique Sm core structure of U7 snRNPs: assembly by a specialized SMN complex and the role of a new component, Lsm11, in histone RNA processing. Genes Dev. 17, 2321–2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.