

Abstract
Parthenolide (PTL), a major active sesquiterpene lactone from the herbal plant Tanacetum parthenium, has been applied in traditional Chinese medicine for centuries. Although PTL demonstrates potent anticancer efficacy in numerous types of malignant cells, the cellular targets of PTL have not been well defined. We reported here that PTL interacts with both cytosolic thioredoxin reductase (TrxR1) and mitochondrial thioredoxin reductase (TrxR2), two ubiquitous selenocysteine-containing antioxidant enzymes, to elicit reactive oxygen species-mediated apoptosis in HeLa cells. PTL selectively targets the selenocysteine residue in TrxR1 to inhibit the enzyme function, and further shifts the enzyme to an NADPH oxidase to generate superoxide anions, leading to reactive oxygen species accumulation and oxidized thioredoxin. Under the conditions of inhibition of TrxRs in cells, PTL does not cause significant alteration of cellular thiol homeostasis, supporting selective target of TrxRs by PTL. Importantly, overexpression of functional TrxR1 or Trx1 confers protection, whereas knockdown of the enzymes sensitizes cells to PTL treatment. Targeting TrxRs by PTL thus discloses an unprecedented mechanism underlying the biological activity of PTL, and provides deep insights to understand the action of PTL in treatment of cancer.
Keywords: apoptosis, oxidation-reduction (redox), oxidative stress, reactive oxygen species (ROS), thioredoxin reductase, parthenolide
Introduction
The highly conserved and ubiquitous thioredoxin system, composed of thioredoxin reductase (TrxR),2 thioredoxin (Trx), and NADPH, plays pivotal roles in maintaining intracellular redox homeostasis and regulating multiple redox signaling pathways (1–3). Two major isoforms of TrxR/Trx distribute in different cellular organelles: TrxR1/Trx1 are predominant in cytosol and nucleus, whereas TrxR2/Trx2 mainly localize within mitochondrion. TrxR1 and TrxR2 have similar overall structures and share the same catalytic mechanism. TrxRs catalyze the NADPH-dependent reduction of disulfide bonds in oxidized Trxs to generate reduced Trxs, which interact with a broad spectrum of downstream targets to regulate diverse cellular redox events during cell proliferation, differentiation, and death (1, 4). Mammalian TrxRs, compared with those from bacteria, are large selenocysteine (Sec)-containing flavoenzymes with a unique C-terminal -Gly-Cys-Sec-Gly active motif. The physiological significance of TrxRs is to catalyze the reduction of Trxs, and hence the function of the thioredoxin system desperately relies on the activity of the selenoenzymes. The thioredoxin system is often overexpressed in many cancer cells (5, 6), and TrxR deficiency or transfection with dominant-negative mutant Trx leads to a retardation in tumor progression and metastasis (7–9). In addition, high Trx expression is associated with the resistance to tumor chemotherapy (10, 11) and results in increased tumor angiogenesis (12). Supported by these observations, the thioredoxin system has been emerging as an important target for cancer treatment. Consequently, various small molecules targeting the thioredoxin system have been discovered and developed as potential cancer chemotherapeutic agents in past years (13–20).
Herb plants have been a source of medical agents since ancient time. With the development of modern molecular medicine, an increasing number of active components from herbal plants have been identified and their putative cellular targets have been disclosed. Parthenolide (PTL, Fig. 1) is a sesquiterpene lactone and principal active molecule present in the medicinal plant feverfew (Tanacetum parthenium), whose extract has been used in traditional Chinese medicine for centuries (21). PTL has been applied in treatment of inflammation through inhibiting NF-κB activation (22–24). In recent years, increasing attention has been drawn to its potent anticancer activity, and PTL is currently being tested in cancer clinical trials (21). Among various mechanisms in accounting for the anticancer activity of PTL (25), such as targeting EGF receptor (26) as well as AP-1 and mitogen-activated protein kinases (27), modulating the Akt/NF-κB pathway (28, 29) and protein kinase C (30), inhibiting STAT3 (31) and tubulin carboxypeptidase (32), activating Bcl-2 members (33) and p53 (34), depleting histone deacetylase 1 (35), and inducing autophagy (36), accumulating evidence supports that induction of reactive oxygen species (ROS) is critical for the cellular action of PTL (28, 29, 31, 36–40). However, the mechanism(s) of ROS induction by PTL remains poorly defined, and the primary cellular target and mode of action of PTL are still in debate.
FIGURE 1.
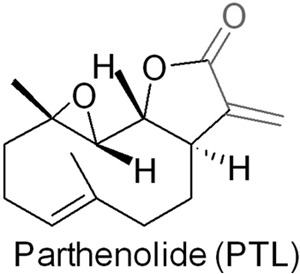
Chemical structure of PTL.
As our continuous efforts in discovering and developing novel small molecules manipulating the cellular redox system as potential therapeutic agents, we reported here that both TrxR1 and TrxR2 are novel cellular targets of PTL in human cervical carcinoma HeLa cells. PTL appears specifically binding to the Sec residue of TrxRs to suppress their Trx-reduction ability, but further elicits a new function to generate ROS, leading to disruption of cellular redox homeostasis, and eventual induction of apoptosis. Overexpression of the functional TrxR1 or Trx1 attenuates the cytotoxicity of PTL, whereas knockdown of TrxR1 or TrxR2 enhances the cytotoxicity, supporting the physiological significance of interaction of TrxRs with PTL. Targeting TrxRs by PTL thus reveals an unprecedented mechanism underlying the biological action of PTL, and would shed light on the potential application of PTL in the treatment of cancer.
Experimental Procedures
Materials
Recombinant rat TrxR1 was prepared as described (41), and was a gift from Prof. Arne Holmgren, Karolinska Institute, Sweden. The recombinant U498C TrxR1 mutant (Sec→Cys) was produced as described (15, 42). The recombinant TrxR1 and U498C TrxR1 have a specific activity of ∼50 and ∼5% of the wild type (WT) TrxR1 with the 5,5′-dithiobis-2-nitrobenzoic acid (DTNB) assay, respectively. The recombinant Escherichia coli Trx and PAO-Sepharose were prepared as described (15). Dulbecco's modified Eagle's medium (DMEM), l-buthionine-(S,R)-sulfoximine (BSO), NADPH, N-acetyl-l-cysteine (NAC), dl-dithiothreitol (DTT), G418, 2,3-dimercapto-1-propanesulfonic acid, Sephadex G-25, DTNB, N-acetyl-Asp-Glu-Val-Asp-p-nitroanilide (Ac-DEVD-pNA), reduced and oxidized glutathione (GSH and GSSG), bovine insulin, yeast glutathione reductase (GR), superoxide dismutase (SOD), tris(2-carboxyethyl)phosphine, Hoechst 33342, 2′,7′-dichlorfluorescein diacetate (DCFH-DA), and dihydroethidium (DHE) were from Sigma. Polyclonal rabbit anti-Trx1 (AB61173a, 1:1000 dilution for Western blots), cytochrome c, and the plasmid for overexpressing Trx1 (pEGFP-Trx1) and the control vector (pEGFP) were obtained from Sangon Biotech (Shanghai, China). PTL, monoclonal mouse anti-TrxR1 (sc-28321, 1:1000 dilution for Western blots), polyclonal goat anti-TrxR2 (sc-46279, 1:500 dilution for Western blots), and HRP-conjugated secondary antibodies (sc-2031, sc-2004 and sc-2020, 1:4000 dilution for Western blots) were from Santa Cruz (Santa Cruz, CA). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), penicillin, and streptomycin were products of Amresco (Solon, OH). Fetal bovine serum (FBS) was from Sijiqing (Hangzhou, China). Bovine serum albumin (BSA), phenylmethylsulfonyl fluoride (PMSF), and monoclonal mouse anti-actin (AA128-1, 1:1000 dilution for Western blots) were from Beyotime (Nantong, China). HeLa cells, HL-60 cells, A549 cells, and HepG2 cells were from the Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences. HEK cells stably overexpressing TrxR1 (HEK-TrxR1) and those stably transfected with a vector (HEK-IRES), short hairpin RNA (shRNA) plasmids targeting TrxR1 (shTrxR1), and nontargeting control (shNT) were gifts of Professor Constantinos Koumenis from the University of Pennsylvania School of Medicine (43, 44). The shRNA plasmids targeting coding regions of the human TrxR2 gene (shTrxR2) and the control nontargeting shRNA (shNT) were purchased from GenePharma (Shanghai, China). GeneTran III transfection reagent was obtained from Biomiga (San Diego, CA). Mitochondrial superoxide indicator MitoSOX was a product of Invitrogen. All other reagents were of analytical grade. A 100 mm solution of PTL was prepared in dimethyl sulfoxide and stored at −20 °C.
Cell Cultures
Cells were cultured in DMEM with 10% FBS, 2 mm glutamine, and 100 units ml−1 of penicillin/streptomycin and maintained in a humidified atmosphere of 5% CO2 at 37 °C (standard culture conditions). HEK-TrxR1 and HEK-IRES were cultured under the standard culture conditions supplemented with 0.1 μm sodium selenite and 0.4 mg/ml of G418. HeLa cells stably transfected with the shTrxR1 plasmid (HeLa-shTrxR1), shTrxR2 (HeLa-shTrxR2), or shNT plasmid (HeLa-shNT) were kept under the standard culture conditions supplemented with 1 μg/ml of puromycin.
MTT Assay
Unless otherwise noted, 8 × 103 cells were incubated with PTL or other agents in triplicate in 96-well plates for the indicated times at 37 °C in a final volume of 100 μl. Cells treated with DMSO alone were set as controls. At the end of the treatment, 10 μl of MTT (5 mg/ml) was added to each well and incubated for an additional 4 h. An extraction buffer (100 μl, 10% SDS, 5% isobutanol, 0.1% HCl) was added, and the cells were incubated overnight at 37 °C. The viability was calculated by measuring the absorbance at 570 nm using a microplate reader (Multiskan GO, Finland).
DTNB Reduction Assay
TrxR activity was determined at room temperature using a microplate reader. NADPH-reduced TrxR (170 nm) or U498C TrxR (700 nm) was incubated with different concentrations of PTL for the indicated times at room temperature (final volume of the mixture was 50 μl). A master mixture in 50 μl of TE buffer (50 mm Tris-HCl, pH 7.5, 1 mm EDTA) containing DTNB (4 mm) and NADPH (0.4 mm) was added, and the linear increase of absorbance at 412 nm during the initial 3 min was recorded for calculating the relative enzyme activity.
End Point Insulin Reduction
NADPH-reduced TrxR (170 nm) was incubated with different concentrations of PTL for 2 h at room temperature in a final volume of 50 μl. A master mixture in TE buffer (50 μl) containing 4 μm E. coli Trx, 0.4 mm NADPH, and 0.32 mm insulin was added to the solution, and the incubation continued at room temperature for 0.5 h. The reaction was terminated by addition of 1 mm DTNB in 6 m guanidine hydrochloride (100 μl, pH 8.0) and the absorbance at 412 nm was measured using a microplate reader. The activity was expressed as the percentage of the control.
GR Assay
NADPH-reduced GR (0.25 units/ml) in TE buffer was incubated with different concentrations of PTL for 2 h at room temperature in a total volume of 100 μl. Reactions were initiated by the addition of GSSG and NADPH (50 μl, final concentration: 1 mm and 400 μm, respectively). The GR activity was determined by measuring the decrease of absorbance at 340 nm during the initial 3 min. The activity was expressed as the percentage of the control.
In Vitro Trx Assay
Reduced E. coli Trx was prepared by incubation of the stock protein with DTT (100 mm). Excessive DTT was subsequently removed by a Sephadex G-25 desalting column. The reduced Trx (5 μm) was incubated with different concentrations of PTL for 2 h at 37 °C in a final volume of 50 μl. A master mixture in TE buffer (30 μl) containing DTT and insulin was added (final concentration: 2 and 0.16 mm, respectively). The Trx activity was measured at room temperature by monitoring the increase of absorbance at 650 nm.
Trx Activity in Cells
HeLa-Trx1 and HeLa-EGFP-Trx1 cells were harvested. Total cellular proteins were extracted by RIPA buffer (50 mm Tris-HCl, pH 7.5, 2 mm EDTA, 0.5% deoxycholate, 150 mm NaCl, 1% Triton X-100, 0.1% SDS, 1 mm Na3VO4 and 1 mm PMSF), and quantified using the Bradford procedure. Trx activity in cell lysates was measured by the end point insulin reduction assay (45, 46), and the activity was expressed as the percentage of the control.
TrxR Activity in Cells
After HeLa cells were treated with 5 or 10 μm PTL for 12 h, the cells were harvested. Total cellular proteins were extracted by RIPA buffer, and quantified using the Bradford procedure. TrxR activity in cell lysates was measured by the end point insulin reduction assay according to our previously published protocol (18, 19).
Imaging TrxR Activity
TRFS-green is a cell membrane permeable dye to specifically detect TrxR activity in cultured cells (47). HeLa cells (2 × 105 cells/well) were seeded into 12-well plates and allowed to grow overnight. After the cells were treated with PTL for 8 h, TRFS-green was added to a final concentration of 10 μm, and the incubation was continued for 4 h at 37 °C. The cells were visualized and photographed by a FLoid Cell Imaging Station (Life Technologies, USA). The intensity of the green fluorescence reflects the relative activity of TrxR in cells.
Determination of Trx Redox States
Reduced Trx was distinguished from the oxidized forms based on the binding of the vicinal dithiols to phenylarsine oxide (PAO) (15, 48, 49). PAO-Sepharose was prepared by immobilization of PAO on Sepharose 4B according to our published procedures (15). The reduced Trx was pulled down on the beads, whereas the oxidized forms were retained in the solution. The cells were treated with PTL (20 μm) or vehicle for 24 h. The cellular proteins were extracted by RIPA buffer and quantified by the Bradford procedure. The fully oxidized and reduced controls were prepared by treating the non-treated cell lysate with diamide (5 mm) and tris(2-carboxyethyl)phosphine (5 mm) for 30 min at 37 °C, respectively. After incubating the samples with PAO-Sepharose on a rotating shaker at room temperature for 30 min, the supernatant was collected and the beads were washed with RIPA buffer. Reduced Trx was eluted by the addition of RIPA buffer containing 2,3-dimercapto-1-propanesulfonic acid (20 mm). All samples were separated by 15% SDS-PAGE and electroblotted onto a PVDF membrane for Western blots analysis.
Induction of Superoxides
NADPH-reduced TrxR (1.3 μm) was incubated with PTL (50 μm) at room temperature for 2 h in TE buffer. The remaining enzyme activity was less than 10% of the control as monitored by the DTNB reduction assay. After removal of excessive PTL by a Sephadex G-25 desalting column, the modified enzyme (44 μl) was added to 256 μl of TE buffer containing 200 μm NADPH. Superoxides production was determined by the cytochrome c reduction assay. The control enzyme without incubation with PTL was treated in the same manner. Briefly, to the above reaction mixture was added 34 μl of 0.82 mm cytochrome c, and the absorbance spectra from 500 to 600 nm were recorded. After the indicated time, SOD was added to reach a final amount of 300 units. The inhibition of the increment of the absorbance at 550 nm after addition of SOD indicates the production of superoxides.
Assessment of Intracellular ROS
HeLa cells were plated in 12-well plates and allowed to grow for 24 h. The cells were incubated with PTL with or without NAC (1 mm) for 6 h. After removal of the medium, the ROS indicator DCFH-DA (10 μm) or DHE (10 μm) in fresh FBS-free medium was added and incubated for an additional 30 min at 37 °C. For monitoring the mitochondrial superoxides, MitoSOX (5 μm) in fresh FBS-free medium was added and incubated for an additional 20 min at 37 °C. Phase-contrast and fluorescence images were acquired by a FLoid Cell Imaging Station.
Imaging TrxR2 Activity
Mito-TRFS, a selective TrxR2 probe, was used to image the TrxR2 activity in live HeLa cells (50). Briefly, the cells were treated with the indicated concentrations of PTL for 10 h followed by further incubating with Mito-TRFS (2 μm) for 2 h. The bright field and fluorescence images were acquired by a FLoid Cell Imaging Station.
Trx1 Overexpression and TrxR2 Knockdown
For generation of the Trx1-overexpressing cell line, HeLa cells were transfected with pEGFP-Trx1 or the control vector (p-EGFP) using GeneTran III transfection reagent according to the manufacturer's instructions. To generate a TrxR2-knockdown cell line, HeLa cells were transfected with sh-TrxR2 or sh-NT plasmids using the GeneTran III transfection reagent. Overexpression of Trx1 was confirmed by measuring the total Trx activity in cells, and knockdown of TrxR2 was analyzed by Western blots.
Hoechst 33342 Staining
HeLa cells were plated in 12-well plates and incubated with the indicated concentrations of PTL and NAC (0.1 mm) for 24 h followed by addition of 5 μg/ml of Hoechst 33342. Phase-contrast and fluorescence images were acquired by a FLoid Cell Imaging Station.
Measurement of Caspase 3 Activity
HeLa cells were treated with different concentrations of PTL for 12 or 24 h. The cells were collected and then lysed with RIPA buffer. The protein content was quantified using the Bradford procedure. The caspase 3 activity was determined by a colorimetric assay using the Ac-DEVD-pNA as a substrate according to the published protocols (18, 19).
Annexin V/PI Staining
HeLa cells were treated with the indicated concentrations of PTL and NAC (0.1 mm) for 24 and 48 h in 12-well plates. The cells were harvested and washed with PBS. Apoptotic cells were identified by double staining with fluorescein 5-isothiocyanate-conjugated Annexin V and PI according to the manufacturer's instructions (Zoman Biotech, Beijing, China). Data were obtained and analyzed using a FACSCantoTM flow cytometer (BD Biosciences, USA) with the Cell Quest software.
Statistics
Data were presented as mean ± S.E. Statistical differences between two groups were assessed by the Student's t test. Comparisons among multiple groups were performed using one-way analysis of variance followed by a post hoc Scheffe test. p < 0.05 was set as the criterion for statistical significance.
Results
Inhibition of TrxR in Vitro
PTL contains an α-methylene-γ-lactone moiety (Fig. 1), which is reminiscent of many known TrxR inhibitors, such as curcumin and its analogues (20, 51, 52), cinnamaldehydes (53, 54), adenanthin (14), gambogic acid (18), and xanthohumol analogues (13). Thus we speculated that PTL might be also an inhibitor of TrxR. Initially, we determined the inhibition of TrxR by PTL in vitro. Preincubation of PTL with the reduced recombinant rat TrxR1 (WT TrxR1) elicits a dose-dependent inhibition of the enzyme with an IC50 value of ∼3 μm (Fig. 2A). The enzyme activity could not be recovered after removal of PTL by a Sephadex G-25 desalting column (data not shown), indicating an irreversible inhibition occurs. Next, we examined the inhibition potency of PTL toward GR and U498C TrxR1. As shown in Fig. 2A, PTL exhibits very weak inhibition to GR or U498C TrxR1 (IC50 > 20 μm). Selective inhibition of WT TrxR1 but not U498C TrxR1 or GR suggests the Sec residue is specifically targeted by PTL. Also, PTL shows a marginal effect on the activity of Trx (Fig. 2B). The potency of TrxR inhibition is dependent on incubation times (Fig. 2C). As DTNB is an artificial substrate of TrxR, we further measured the ability of PTL to inhibit the reduction of Trx, the physiological substrate of TrxR, by TrxR. PTL dose-dependently inhibits TrxR to reduce Trx determined by the Trx-mediated insulin reduction assay (Fig. 2D). Taken together, PTL selectively inhibits TrxR in vitro, and this inhibition appears primarily targeting the Sec residue of the enzyme.
FIGURE 2.
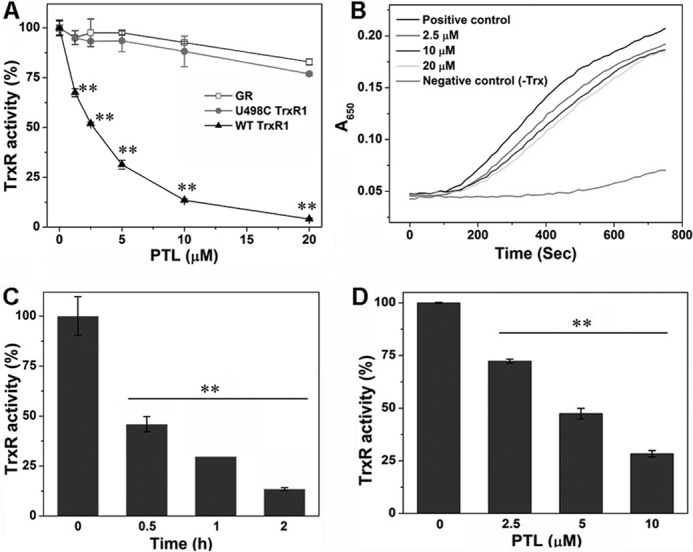
Inhibition of TrxR in vitro. A, dose-dependent inhibition of TrxR, U498C TrxR, and GR by PTL. NADPH-reduced WT TrxR1, U498C TrxR1, and GR were incubated with the indicated concentrations of PTL for 2 h at room temperature, and their activity was determined by the DTNB assay. B, no significant inhibition of Trx by PTL. Representative results are shown from 3 independent experiments. C, time-dependent inhibition of TrxR by PTL. NADPH-reduced WT TrxR1 was incubated with PTL (5 μm) for the indicated times at room temperature, and the activity was determined by the DTNB assay. D, inhibition of TrxR by PTL detected by the end point insulin reduction assay. Data in A, C, and D are from 3 independent experiments done in triplicate, and all the activity was expressed as the percentage of the control. **, p < 0.01 versus the control groups.
Cytotoxicity and Inhibition of TrxR in Cells
To extend the in vitro observation of TrxR inhibition by PTL, we then determined the action of PTL in cells. PTL inhibits the growth of HeLa cells in both dose- and time-dependent manners (Fig. 3A), and an IC50 value of ∼5 μm could be obtained after a 72-h treatment. We further examined the cytotoxicity of PTL to other cancer cell lines. As shown in Fig. 3B, PTL also potently inhibits the proliferation of A549 (human lung carcinoma cell), HL-60 (human promyelocytic leukemia cell), and HepG2 cells (human hepatocellular carcinoma cell). Next, we determined the inhibition of TrxR in HeLa cells. First, we employed the TRFS-green, a specific TrxR probe developed by our group (47), to image the cellular TrxR activity. As illustrated in Fig. 3C, the control cells display bright fluorescence. However, treatment of the cells with PTL causes dose-dependent decline of the fluorescence, indicating the inhibition of TrxR. Quantification of the fluorescence intensity in individual cells was shown in Fig. 3D. We then performed the classic Trx-mediated insulin reduction assay to confirm the TrxR inhibition by PTL. Again, treatment of the cells with PTL impairs the cellular TrxR activity dose-dependently (Fig. 3E), consistent with the observation from the live cell imaging experiment. PTL contains the α,β-unsaturated ketone structure (Fig. 1), a potentially thiol-reactive motif and key pharmacophore of the sesquiterpene lactones. We then assayed the cellular thiol homeostasis after PTL treatment. As shown in Fig. 3F, the total thiol level in cells treated with 10 μm PTL remains similar as that in the control cells. It should be noted here that at this concentration, cellular TrxR activity was severely inhibited (Fig. 3, C and E). Stimulation of the cells with a low concentration of PTL (5 μm) causes up-regulation (∼30% elevation) of total cellular thiols. This is likely due to activation of the ARE-Nrf2 cytoprotective pathways as molecules with the α,β-unsaturated ketone structure are usually potent Nrf2 activators (45, 46, 55). Collectively, PTL inhibits TrxR in HeLa cells and displays remarkable cytotoxicity to a variety of cancer cells.
FIGURE 3.
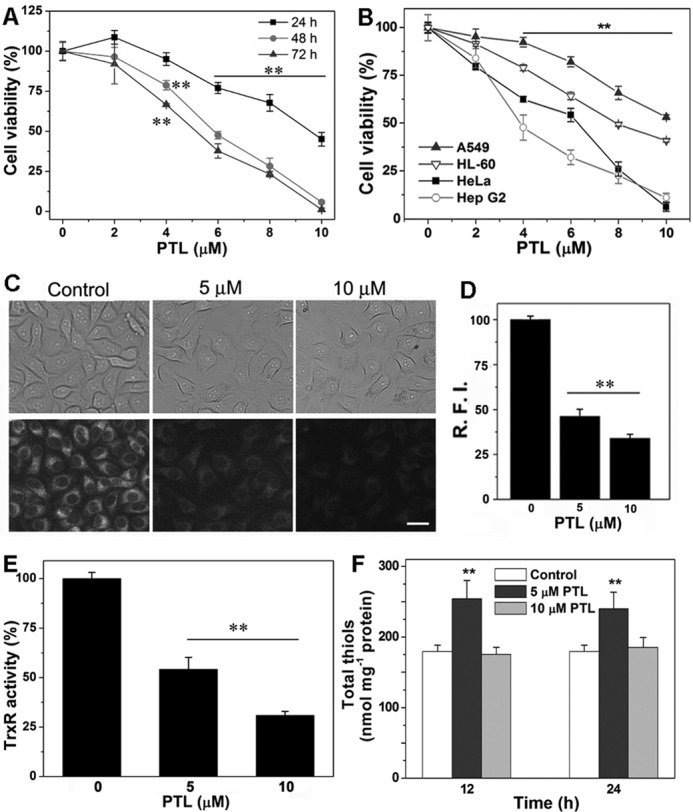
Cytotoxicity of PTL and inhibition of TrxR in cells. A, cytotoxicity of PTL to HeLa cells. The cells were treated with varying concentrations of PTL for 24, 48, or 72 h, and the viability was determined by the MTT assay. B, cytotoxicity of PTL to different types of cancer cells. The cells were treated with the indicated concentrations of PTL for 48 h, and the viability was determined by the MTT assay. C, imaging TrxR activity by TRFS-green in live HeLa cells. Images are representative of 3 independent experiments done in triplicate. Scale bar: 15 μm. D, 10 cells were randomly selected and relative fluorescence intensity (R.F.I.) was quantified in individual cells from C by ImageJ. E, inhibition of TrxR in HeLa cells determined by the end point insulin reduction assay. F, alteration of cellular thiol levels after PTL treatment. HeLa cells were treated with the indicated concentrations of PTL for 12 or 24 h, and total cellular thiols were determined by the DTNB titration. Data in A, B, E, and F are from 3 independent experiments done in triplicate. **, p < 0.01 versus the control groups.
Accumulation of Oxidized Trx in Cells
Because TrxR plays a critical role to catalyze the NADPH-dependent reduction of disulfide bond(s) in oxidized Trx in vivo, we next determined the ratio of reduced Trx to oxidized Trx in HeLa cells after PTL treatment. We prepared PAO-Sepharose to pull down the reduced Trx and retain the oxidized Trx in the solution. After addition of 2,3-dimercapto-1-propanesulfonic acid, the reduced Trx was released from the PAO-Sepharose (49). To validate this “capture and release” assay, we treated the cell lysate with diamide or tris(2-carboxyethyl)phosphine to generate the fully oxidized Trx or reduced Trx, respectively. As illustrated in Fig. 4A, Trx from the diamide-treated lysate (oxidized Trx, O) is only present in the supernatant (S), whereas that from the tris(2-carboxyethyl)phosphine-treated lysate (reduced Trx, R) is fully captured by the PAO-Sepharose (P), demonstrating this assay is suitable to distinguish the reduced Trx from the oxidized one. As shown in Fig. 4A, the majority of Trx was found in the reduced form in the vehicle-treated cells. However, the oxidized Trx was elevated notably upon treatment of the cells with PTL. The reduced Trx and oxidized Trx were quantified by measuring the bands intensity, and the ratio of the reduced Trx to the oxidized one was shown in Fig. 4B. There is no apparent alteration of the total Trx level after PTL treatment (Fig. 4C).
FIGURE 4.
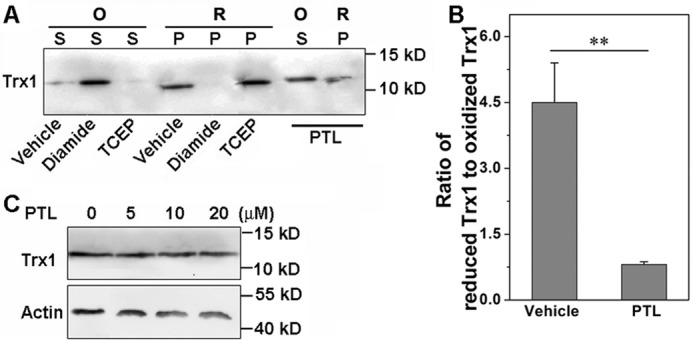
Accumulation of oxidized Trx in HeLa cells. A, HeLa cells were treated with PTL (20 μm) for 24 h. The oxidized and reduced Trx1 were determined by the PAO-Sepharose pulldown assay. Relative protein level was quantified by densitometry using ImageJ, and the ratio shown in B. C, no significant alteration of Trx1 expression in HeLa cells after PTL treatment. The cells were treated with varying concentrations of PTL for 24 h, and the Trx1 level was determined by Western blot analysis. Data in A and C are representative of 3 independent experiments. **, p < 0.01 versus the control groups.
Inflation of ROS in Cells
Many TrxR inhibitors, such as DNCB (56), curcumin (20, 52), and shikonin (19) could modify the enzyme to inhibit its reduction of the oxidized Trx, but further shift the enzyme to an NADPH oxidase to generate ROS. Thus we determined whether PTL has a similar effect. As shown in Fig. 5A, the PTL-modified TrxR1 displayed steady cytochrome c reduction activity, which was inhibited by the addition of SOD, suggesting production of superoxides in such a process. The kinetic change of A550 was determined to be 6.8 × 10−3/min before addition of SOD. Under the same conditions, the change of A550 caused by the native non-inhibited enzyme was less than 2.0 × 10−3/min. One of the outstanding functions of the thioredoxin system is to maintain intracellular redox homeostasis and defend against oxidative stress. PTL inhibits TrxR, and further shifts the enzyme to a ROS generator. We next determined the ROS level in HeLa cells after PTL treatment. DCFH-DA is a general ROS probe. DCFH-DA staining indicates the burst of ROS after stimulation of the cells with PTL (Fig. 5B). Pretreatment of the cells with antioxidant NAC blocks ROS accumulation. DCFH-DA-based ROS assay is prone to artifacts as extensively discussed in the literature (57). We next employed another dye, DHE, to confirm the ROS production in the cells (Fig. 5C). DHE readily penetrates plasma membrane to intercept ROS, especially the superoxides, to form a product that intercalates with nucleic acids and emits red fluorescence. Again, the PTL-treated cells evoke the fluorescence, which is blocked by pretreatment of the cells with NAC. We further applied the mitochondrial-specific ROS dye MitoSox to determine the mitochondrial ROS level. As shown in Fig. 5D, MitoSox staining also gives a bright signal after PTL insult, indicating the elevation of ROS in mitochondrial. Altogether, PTL promotes ROS accumulation in HeLa cells.
FIGURE 5.
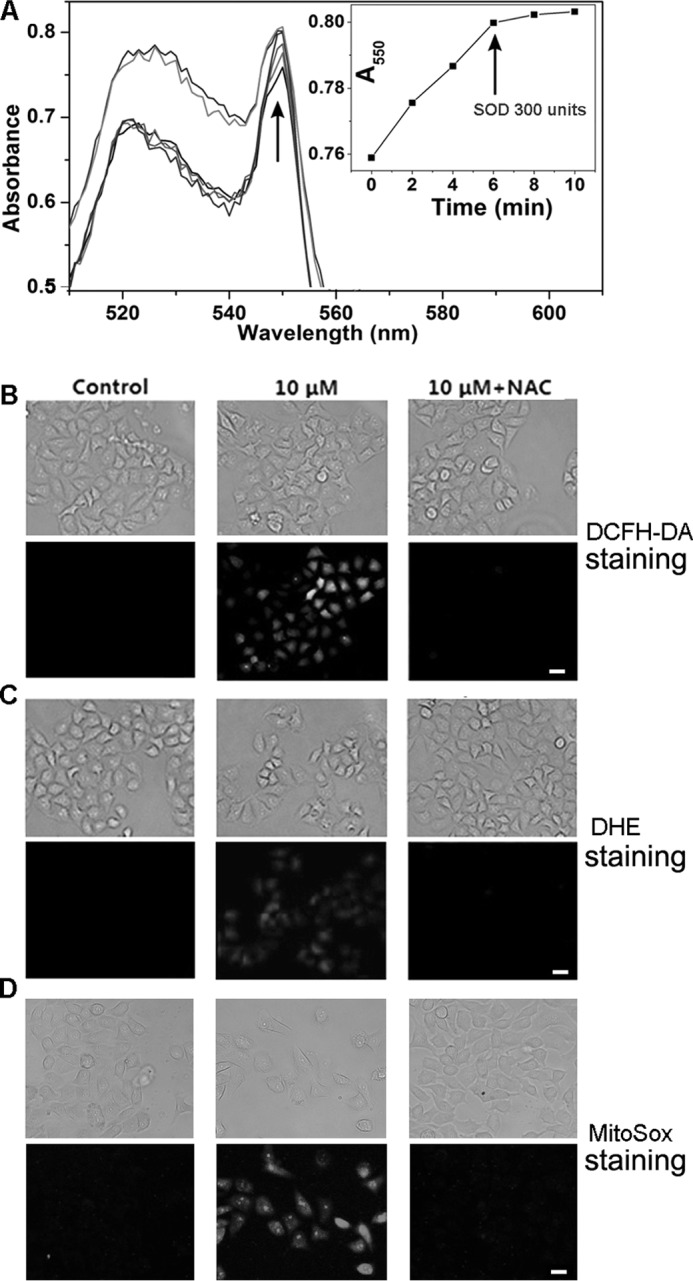
Induction of ROS by PTL. A, induction of superoxides by the PTL-modified TrxR. Superoxides production was monitored by the cytochrome c reduction assay. The inset shows the change of absorbance at 550 nm after addition of cytochrome c and SOD. SOD was added as indicated by the arrow in the inset. Data are representative of 3 independent experiments done in duplicate. ROS accumulation in cells determined by DCFH-DA staining (B), DHE staining (C), and MitoSox staining (D). Top panels, bright field; bottom panels, fluorescence field (green channel for DCFH-DA staining, and red channel for DHE and MitoSox staining). Images in B–D are representative of 3 independent experiments done in triplicate. Scale bar: 20 μm.
Protection of Cell Death by NAC and Promotion of Cell Death by GSH Depletion
NAC is a known antioxidant and precursor for biosynthesis of GSH. Pretreatment of HeLa cells with NAC impairs the cytotoxicity of PTL (Fig. 6A), consistent with the prevention of ROS accumulation by NAC (Fig. 5, B–D). Next, we determined the effect of GSH on the cytotoxicity of PTL. In line with the protective role of NAC, depletion of cellular GSH by pretreatment of the cells with BSO enhances the cytotoxicity of PTL (Fig. 6B). Under our experimental conditions, pretreatment of HeLa cells with 100 μm BSO for 24 h has no apparent cytotoxicity but decreases the intracellular GSH level to less than 20% of the control. GSH is a pivotal component of the glutathione system, which is another redox regulation network in cells besides the thioredoxin system and also acts as a backup of the thioredoxin system (58). Sensitizing the cells to PTL treatment by depletion of cellular GSH suggests the involvement of the thioredoxin system in the cellular action of PTL.
FIGURE 6.
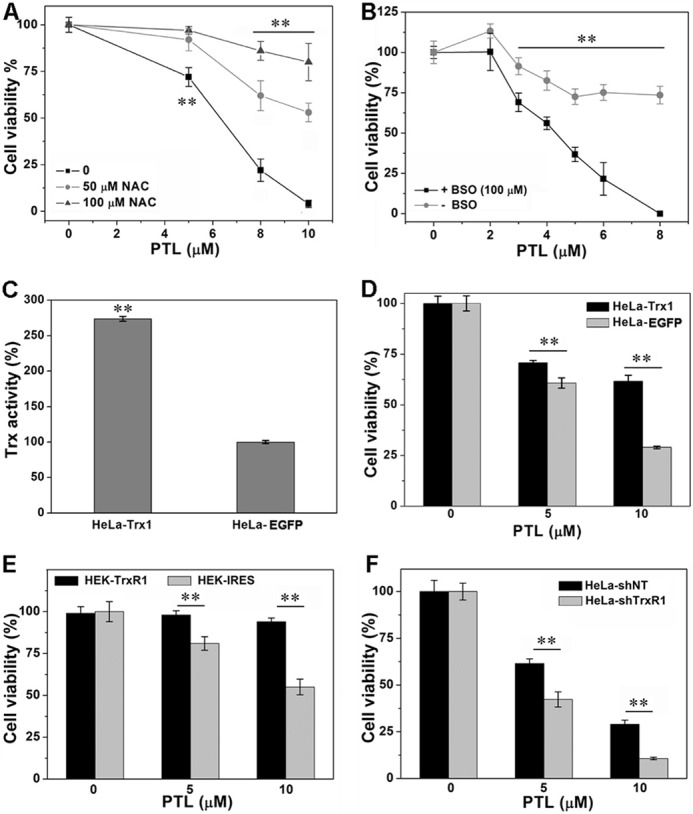
The role of TrxR in PTL-induced cell death. A, protection of the cells by NAC. HeLa cells were incubated with the indicated concentrations of NAC and PTL for 48 h. B, augmentation of cytotoxicity by GSH depletion. HeLa cells were treated with BSO for 24 h to lower the intracellular GSH level, followed by PTL treatment for an additional 48 h. C, determination of Trx activity in HeLa-EGFP and HeLa-EGFP-Trx1 cells. D, protection of cells by Trx1 overexpression. The cells were treated with the indicated concentrations of PTL for 48 h. E, cytotoxic effects of PTL on HEK-IRES and HEK-TrxR1 cells. The cells were treated with the indicated concentrations of PTL for 48 h. F, cytotoxic effects of PTL on HeLa-shNT and HeLa-shTrxR1 cells. The cells were treated with the indicated concentrations of PTL for 48 h. The cell viability was determined by the MTT assay in all experiments. All data are from 3 independent experiments done in triplicate. **, p < 0.01 versus the control groups.
Involvement of TrxR for PTL Cellular Action
PTL inhibits TrxR both in vitro and in HeLa cells, and causes ROS accumulation and Trx oxidation. To further address whether the cytotoxicity of PTL is related to its interaction with TrxR, we then compared the sensitivity of HEK cells stably overexpressing TrxR1 (HEK-TrxR1) and the cells that stably transfected with a vector (HEK-IRES) to PTL treatment. The transfection efficiency was confirmed by determining protein expression and enzyme activity in our previous publications (18, 19). As shown in Fig. 6E, PTL exhibits significantly higher cytotoxicity to HEK-IRES cells. Next, we overexpressed Trx1 in HeLa cells (HeLa-Trx1), and compared the cytotoxicity of PTL to HeLa-Trx1 cells and those transfected with a control vector (HeLa-EGFP). The overexpression of Trx1 was confirmed by measuring the cellular total Trx activity (Fig. 6C). Overexpression of Trx1 in HeLa cells (HeLa-Trx1) protects the cells from PTL-induced cell death (Fig. 6D). To further address the physiological relevance of TrxR-mediated PTL cytotoxicity, we transfected shRNA plasmid specifically targeting TrxR1 to generate HeLa cells stably knocking down the expression of TrxR1 (HeLa-shTrxR1). The non-targeting shRNA plasmid was transfected for the control cells (HeLa-shNT). Knockdown of TrxR1 in HeLa cells was fully evaluated in our previous publications (18, 19). TrxR knockdown could yield drug-specific alteration of cytotoxicity of therapeutic small molecules (59). In our case, PTL shows elevating potency to HeLa-shTrxR1 cells (Fig. 6F).
Because the ROS level in mitochondrial increases after PTL treatment (Fig. 5D), we next asked whether the mitochondrial TrxR (TrxR2) is also involved in the cellular actions of PTL. First, we determined the TrxR2 activity after the cells were treated with PTL. We employed our recently developed TrxR2 probe Mito-TRFS to stain TrxR2 activity in live HeLa cells (50). As shown in Fig. 7A, the cellular TrxR2 activity was severely inhibited by PTL. This is not surprising as mammalian TrxR1 and TrxR2 have similar structures and share the same catalytic mechanism. Second, we generated TrxR2-knockdown HeLa cells (HeLa-shTrxR2), and compared the cytotoxicity of PTL to the HeLa-shTrxR2 cells and those transfected with a control shNT plasmid (HeLa-shNT2). After evaluating the knockdown efficiency by different shRNAs, we noticed that shTrxR2–1356 is the most potent one to lower the TrxR2 expression (Fig. 7B). Thus we chose this shRNA to generate HeLa-shTrxR2 cells for the following experiment. Consistent with the inhibition of TrxR2 by PTL, knockdown of TrxR2 also enhances the cytotoxicity of PTL (Fig. 7C). Collectively, these results indicated that cytotoxicity of PTL to HeLa cells is related to its interaction with both TrxR1 and TrxR2.
FIGURE 7.
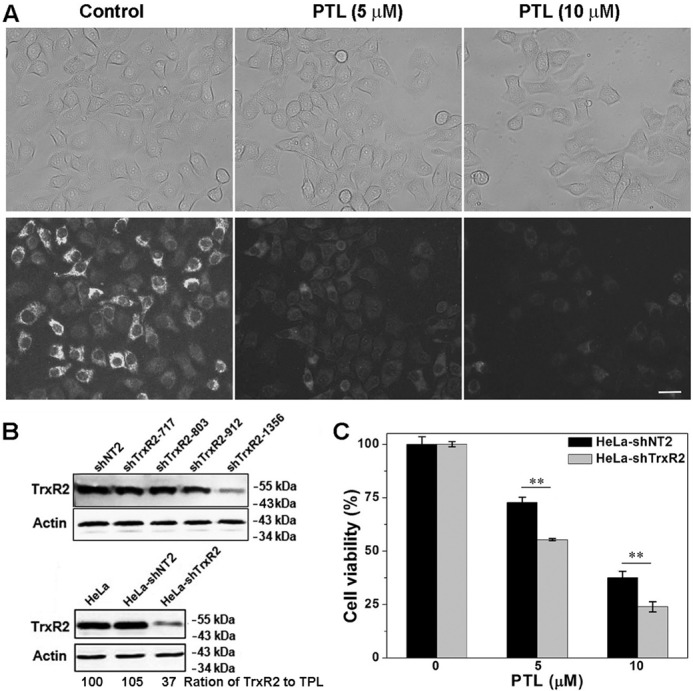
Involvement of TrxR2 for PTL cellular actions. A, inhibition of TrxR2 by PTL determined by the Mito-TRFS staining. Representative images are shown from 3 independent experiments done in triplicate. Scale bar: 20 μm. B, evaluation of TrxR2 knockdown efficiency by different shRNAs. Representative results are shown from 3 independent experiments. The ratio of Western blot samples was normalized to total protein loading (TPL). C, TrxR2 knockdown enhances the cytotoxicity of PTL. Cells were treated with the indicated concentrations of PTL for 48 h, and their viability was determined by the MTT assay from 3 independent experiments done in triplicate. **, p < 0.01 versus the control groups.
Induction of Apoptosis
Cell apoptosis is mediated by a series of precisely controlled events that are frequently altered in malignant cells. Abrogation of apoptotic pathways is often found in cancer cells arising from a complex interplay of genetic aberrations and misregulated pathways (60). We demonstrated here that cytotoxicity of PTL to HeLa cells is predominantly through the induction of apoptosis (Fig. 8). Multiple assays were employed to confirm the apoptosis. First, the morphological changes of the nuclei were characterized by Hoechst staining. Hoechst 33342 is a cell membrane-penetrating dye that specifically binds to double strand DNA and gives blue fluorescence. As shown in Fig. 8A, the control cells displayed normal, round nuclei with faint and even fluorescence. However, after PTL treatment, cells exhibited condensed and highly fluorescent nuclei, a characteristic morphology of cells undergoing apoptosis. Pretreatment of the cells with NAC alleviates nuclear morphological changes. Next, we determined activation of caspase 3 after PTL treatment. Caspase 3 is an essential component of the apoptotic machinery, and activation of caspase 3 is a central event in the process of apoptosis. PTL activates caspase 3 in both concentration- and time-dependent manners (Fig. 8B). Finally, we employed the Annexin V-fluorescein 5-isothiocyanate/PI double staining assay to quantify the apoptotic population by flow cytometry. The scattergrams were shown in Fig. 8C, and the corresponding quantification results were shown in Fig. 8D. Treatment of the cells with 10 μm PTL for 24 h causes ∼35% cell apoptosis. Longer time treatments (48 h) elicits more prominent apoptosis. The apoptotic population increases to ∼40 and ∼90% by 5 and 10 μm PTL treatments, respectively. Again, NAC antagonizes the apoptosis. A slight increase of the necrotic cells was observed after the cells were treated with PTL for 48 h. This could be due to the excessive oxidative stress caused by PTL. Thus, we concluded that PTL predominantly triggers the apoptotic cell death in HeLa cells.
FIGURE 8.
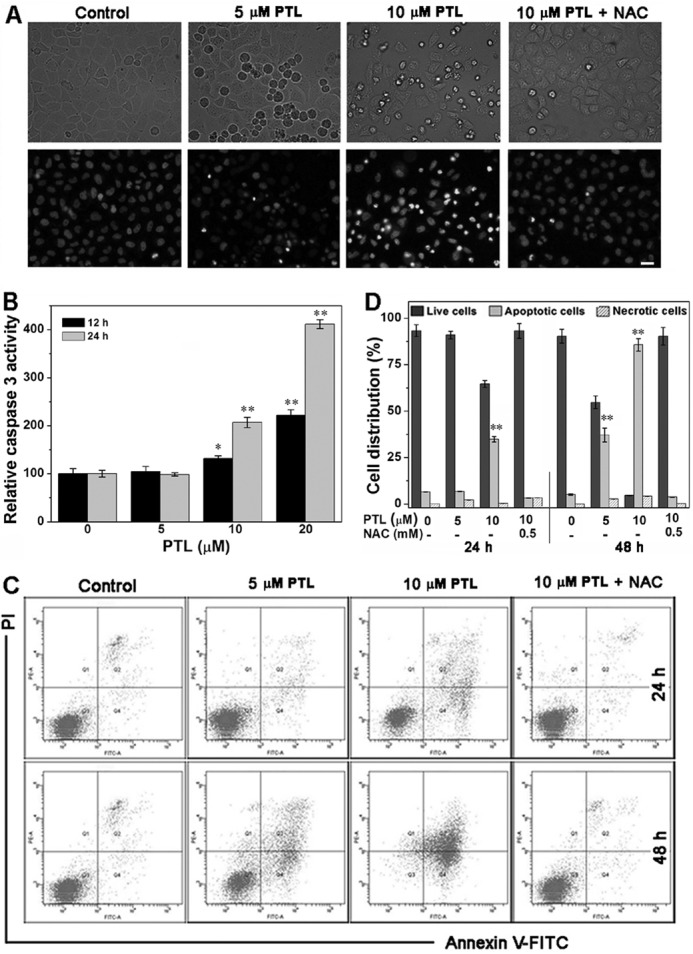
Induction of apoptosis of HeLa cells. A, nuclear morphological changes by Hoechst 33342 staining. Representative images are shown from 3 independent experiments done in triplicate. Scale bar: 20 μm. B, activation of caspase 3 by PTL. Caspase 3 activity in cell extracts was determined from 3 independent experiments done in triplicate. C, analysis of apoptosis by the annexin V/PI double-staining assay. HeLa cells were treated with varying concentrations of PTL for 24 or 48 h, and representative FACS analysis scattergrams for 10,000 cells from 3 independent experiments were shown. The cells show four different cell populations designated as the follows: double negative (unstained) cells showing the live cell population (lower left, Q3), annexin V positively and PI negatively stained cells showing early apoptosis (lower right, Q4), annexin V/PI double stained cells showing late apoptosis (upper right, Q2), and finally PI positively and annexin V negatively stained cells showing necrotic cells (upper left, Q1). D, the quantification of the necrotic cells (Q1), live cells (Q3), and apoptotic cells (Q2 and Q4) is illustrated. *, p < 0.05; and **, p < 0.01 versus the control groups.
Discussion
Accumulating evidence supports that ROS are double-edged swords in cellular processes: low level ROS are indispensable for redox signaling, whereas high dose ROS generally induce oxidative stress. ROS and oxidative stress have long been associated with cancer. Two key features of malignant cells are their unlimited proliferation and hyperactive metabolism (60). As generation of ROS is a byproduct of cell growth and metabolism, tumor cells usually have increased ROS levels compared with normal cells (61, 62). Therefore, elevation of ROS production or inhibition of the cellular antioxidant systems is increasingly considered as a therapeutic strategy to the treatment of cancers (63, 64). We disclosed that PTL, a sesquiterpene lactone from the medical plant feverfew, selectively targets the Sec residue to inhibit the physiological function of the antioxidant enzyme TrxR. Furthermore, PTL-modified TrxR gains a new function to keep producing ROS. Among various putative mechanisms, induction of ROS has been rationally adopted in accounting for the cellular action of PTL. Nevertheless, how the ROS are formed is not clear. We demonstrated that PTL-modified TrxR directly generates ROS, thus providing a mechanistic interpretation for the previous observations. Our attempt to link the ROS amount induced by PTL to the TrxR level in HeLa-shNT and HeLa-shTrxR1 cells failed (data not shown). We reasoned that this might be due to dual roles of TrxR in cells: antioxidant function of the non-modified TrxR and prooxidant function of the PTL-modified TrxR. Knockdown of TrxR decreases both active TrxR (for ROS scavenging) and PTL-modified TrxR (for ROS production). The two counteracting interactions might contribute to the nonsignificant change of the ROS level in HeLa-shNT and HeLa-shTrxR1 cells after PTL treatment. As a consequence of inhibition of TrxR and induction of ROS, PTL causes accumulation of oxidized Trx, and eventually elicits apoptosis in HeLa cells. The physiological significance of targeting TrxR by PTL was further demonstrated as overexpression of functional TrxR lowers the cytotoxicity of PTL, whereas knockdown of the enzyme expression sensitizes cells to PTL treatment.
Sesquiterpene lactones are a class of natural compounds distributed predominantly in the flowers of plants belonging to the Asteraceae family, most of which are widely used in folk medicine. Sesquiterpene lactones exhibit a broad spectrum of biological effects, including cytotoxic, anti-inflammatory, and anti-tumor activity. Among various sesquiterpene lactones, PTL, commonly extracted from the feverfew herb, is actively being investigated as an anticancer agent (25). The biological activity of PTL is thought to be mediated through its α-methylene-γ-lactone moiety (Fig. 1), which was supposed to covalently bind to sulfhydryl groups within proteins, thereby modulating their functions (22–24, 35). Mammalian TrxRs contain a selenol side chain (from the Sec residue) at their C-terminal, and this Sec residue determines the function of TrxR (42). Sec, compared with its counterpart Cys, is more reactive and prone to be modified by various electrophiles (13, 14, 16–20, 49, 52). Thus, PTL likely follows the same mechanism to bind to the Sec residue in TrxR to modify the enzyme (52).
The specific interaction of PTL with TrxR was demonstrated by the following evidence. First, we measured the in vitro inhibition potency of PTL to TrxR1, U498C TrxR1, GR, and Trx (Fig. 2, A and B). Single mutation of Sec to Cys sharply decreased the sensitivity of the enzyme to PTL, indicating that the Sec residue is a primary target of PTL. The structure of GR is closely related to TrxR, however, very weak inhibition of GR was observed under our experimental conditions. The small redox protein Trx is also little affected by PTL. The selective inhibition of WT TrxR1 but not U498C TrxR1, GR, or Trx suggests a specific interaction of PTL and TrxR1. In addition, PTL does not cause significant alteration of cellular thiol homeostasis under the conditions of TrxR inhibition, further underpinning the selective target of TrxR by PTL. Second, we provided evidence to support the unique role of TrxR for the biological action of PTL in cellular contexts. Cells overexpressing TrxR1/Trx1 show less sensitivity to PTL compared with the cells only transfected with vectors (Fig. 6, D and E). More physiologically relevant evidence that genetic knockdown of TrxR1/TrxR2 elevates the cytotoxicity of PTL further supports that TrxRs are critically involved in the biological effects of PTL (Figs. 6F and 7C). Third, the glutathione system and thioredoxin system are two major networks that work independently but with a few cross-talks to serve as mutual backup in regulating cellular redox events (58, 65). Our observation that depletion of GSH by BSO enhances the cytotoxicity of PTL, whereas up-regulation of GSH by NAC alleviates the cytotoxicity (Fig. 6, A and B) also suggests that TrxR is involved in the cellular action of PTL. Taken together, our data indicate that PTL targets TrxR in HeLa cells with high specificity.
Evading apoptosis, arising from a complex interplay of genetic aberrations and misregulated signaling pathways, is one of the hallmarks of malignant cells (60). Activation of apoptotic pathways in cancer cells is thus critical for cancer therapy. Multiple assays demonstrated that the cytotoxicity of PTL is through induction of apoptosis (Fig. 8), hence potentiating the clinical use of PTL in treatment of tumors. The apoptosis-inducing ability of PTL in HeLa cells could be attributed to its inhibition of TrxR. First, inhibition of TrxR by PTL directly promotes ROS production and restricts the availability of the reduced Trx. Because the reduced Trx is an electron donor for many antioxidant systems, TrxR inhibition may lead to the collapse of cellular antioxidant defense. Thus, inhibition of TrxR could elicit oxidative stress-mediated apoptosis. Second, the reduced Trx, but not the oxidized Trx, directly interacts with various apoptosis-related enzymes, such as ASK1 (66), procaspase 3 (67), and apoptosis inducing factor (68), to suppress apoptosis. TrxR inhibition causes accumulation of oxidized Trx, which would be expected to promote apoptosis. Third, the PTL-modified TrxR resembles the selenium compromised thioredoxin reductase-derived apoptotic proteins (SecTRAPs), where the Sec residue is modified by electrophiles. These SecTRAPs may trigger rapid cell apoptosis (69). Collectively, targeting TrxR by PTL elicits apoptosis eventually.
In conclusion, we have disclosed TrxR as a novel target of PTL, and demonstrated that PTL induces apoptosis of tumor cells through a previously unrecognized mechanism. The elucidation of PTL-TrxR interaction in cells may shed light to understand how this sesquiterpene lactone acts in vivo, and this novel targeting mechanism could lead to development of small molecule inhibitors of TrxR as potential cancer chemotherapeutic agents.
Author Contributions
D. D. and J. Z. conducted most of the experiments and analyzed the results. J. Y. and Y. L. conducted experiments on the preparation of PAO-Sepharose and determination of the redox states of Trx. J. F. conceived the idea for the project, analyzed the results, and wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We appreciate Prof. Arne Holmgren (Karolinska Institute) for the recombinant rat TrxR1 and Prof. Constantinos Koumenis (School of Medicine, University of Pennsylvania) for cells and plasmids.
This work was supported by Natural Science Foundation of China Grant 21572093 and Natural Science Foundation of Gansu Province Grant 145RJZA225. The authors declare they have no conflicts of interest with the contents of this article.
- TrxR
- thioredoxin reductase
- Trx
- thioredoxin
- BSO
- l-buthionine-(S,R)-sulfoximine
- DCFH-DA
- 2′,7′-dichlorofluorescein diacetate
- DHE
- dihydroethidium
- DTNB
- 5,5′-dithiobis-2-nitrobenzoic acid
- GR
- glutathione reductase
- HEK-TrxR1
- HEK cells overexpressing TrxR1
- HEK-IRES
- HEK cells transfected with a vector
- HeLa-shTrxR
- HeLa cells transfected with shTrxR plasmids
- HeLa-shNT
- HeLa cells transfected with shNT plasmid
- NAC
- N-acetyl-l-cysteine
- PAO
- phenylarsine oxide
- PTL
- parthenolide
- ROS
- reactive oxygen species
- SOD
- superoxide dismutase
- Sec
- selenocysteine
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PI
- propidium iodide.
References
- 1. Lu J., and Holmgren A. (2014) The thioredoxin antioxidant system. Free Radic. Biol. Med. 66, 75–87 [DOI] [PubMed] [Google Scholar]
- 2. Bindoli A., and Rigobello M. P. (2013) Principles in redox signaling: from chemistry to functional significance. Antioxid. Redox Signal. 18, 1557–1593 [DOI] [PubMed] [Google Scholar]
- 3. Arnér E. S. (2009) Focus on mammalian thioredoxin reductases: important selenoproteins with versatile functions. Biochim. Biophys. Acta 1790, 495–526 [DOI] [PubMed] [Google Scholar]
- 4. Hanschmann E. M., Godoy J. R., Berndt C., Hudemann C., and Lillig C. H. (2013) Thioredoxins, glutaredoxins, and peroxiredoxins: molecular mechanisms and health significance: from cofactors to antioxidants to redox signaling. Antioxid. Redox Signal. 19, 1539–1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lincoln D. T., Ali Emadi E. M., Tonissen K. F., and Clarke F. M. (2003) The thioredoxin-thioredoxin reductase system: over-expression in human cancer. Anticancer Res. 23, 2425–2433 [PubMed] [Google Scholar]
- 6. Berggren M., Gallegos A., Gasdaska J. R., Gasdaska P. Y., Warneke J., and Powis G. (1996) Thioredoxin and thioredoxin reductase gene expression in human tumors and cell lines, and the effects of serum stimulation and hypoxia. Anticancer Res. 16, 3459–3466 [PubMed] [Google Scholar]
- 7. Hellfritsch J., Kirsch J., Schneider M., Fluege T., Wortmann M., Frijhoff J., Dagnell M., Fey T., Esposito I., Kölle P., Pogoda K., Angeli J. P., Ingold I., Kuhlencordt P., et al. (2015) Knockout of mitochondrial thioredoxin reductase stabilizes prolyl hydroxylase 2 and inhibits tumor growth and tumor-derived angiogenesis. Antioxid. Redox Signal. 22, 938–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yoo M. H., Xu X. M., Carlson B. A., Gladyshev V. N., and Hatfield D. L. (2006) Thioredoxin reductase 1 deficiency reverses tumor phenotype and tumorigenicity of lung carcinoma cells. J. Biol. Chem. 281, 13005–13008 [DOI] [PubMed] [Google Scholar]
- 9. Gallegos A., Gasdaska J. R., Taylor C. W., Paine-Murrieta G. D., Goodman D., Gasdaska P. Y., Berggren M., Briehl M. M., and Powis G. (1996) Transfection with human thioredoxin increases cell proliferation and a dominant-negative mutant thioredoxin reverses the transformed phenotype of human breast cancer cells. Cancer Res. 56, 5765–5770 [PubMed] [Google Scholar]
- 10. Zhao L., Li W., Zhou Y., Zhang Y., Huang S., Xu X., Li Z., and Guo Q. (2015) The overexpression and nuclear translocation of Trx-1 during hypoxia confers on HepG2 cells resistance to DDP, and GL-V9 reverses the resistance by suppressing the Trx-1/Ref-1 axis. Free Radic. Biol. Med. 82, 29–41 [DOI] [PubMed] [Google Scholar]
- 11. Kim S. J., Miyoshi Y., Taguchi T., Tamaki Y., Nakamura H., Yodoi J., Kato K., and Noguchi S. (2005) High thioredoxin expression is associated with resistance to docetaxel in primary breast cancer. Clin. Cancer Res. 11, 8425–8430 [DOI] [PubMed] [Google Scholar]
- 12. Welsh S. J., Bellamy W. T., Briehl M. M., and Powis G. (2002) The redox protein thioredoxin-1 (Trx-1) increases hypoxia-inducible factor 1α protein expression: Trx-1 overexpression results in increased vascular endothelial growth factor production and enhanced tumor angiogenesis. Cancer Res. 62, 5089–5095 [PubMed] [Google Scholar]
- 13. Zhang B., Duan D., Ge C., Yao J., Liu Y., Li X., and Fang J. (2015) Synthesis of xanthohumol analogues and discovery of potent thioredoxin reductase inhibitor as potential anticancer agent. J. Med. Chem. 58, 1795–1805 [DOI] [PubMed] [Google Scholar]
- 14. Soethoudt M., Peskin A. V., Dickerhof N., Paton L. N., Pace P. E., and Winterbourn C. C. (2014) Interaction of adenanthin with glutathione and thiol enzymes: selectivity for thioredoxin reductase and inhibition of peroxiredoxin recycling. Free Radic. Biol. Med. 77, 331–339 [DOI] [PubMed] [Google Scholar]
- 15. Liu Y., Duan D., Yao J., Zhang B., Peng S., Ma H., Song Y., and Fang J. (2014) Dithiaarsanes induce oxidative stress-mediated apoptosis in HL-60 cells by selectively targeting thioredoxin reductase. J. Med. Chem. 57, 5203–5211 [DOI] [PubMed] [Google Scholar]
- 16. Citta A., Folda A., Bindoli A., Pigeon P., Top S., Vessières A., Salmain M., Jaouen G., and Rigobello M. P. (2014) Evidence for targeting thioredoxin reductases with ferrocenyl quinone methides: a possible molecular basis for the antiproliferative effect of hydroxyferrocifens on cancer cells. J. Med. Chem. 57, 8849–8859 [DOI] [PubMed] [Google Scholar]
- 17. Citta A., Schuh E., Mohr F., Folda A., Massimino M. L., Bindoli A., Casini A., and Rigobello M. P. (2013) Fluorescent silver(I) and gold(I)-N-heterocyclic carbene complexes with cytotoxic properties: mechanistic insights. Metallomics 5, 1006–1015 [DOI] [PubMed] [Google Scholar]
- 18. Duan D., Zhang B., Yao J., Liu Y., Sun J., Ge C., Peng S., and Fang J. (2014) Gambogic acid induces apoptosis in hepatocellular carcinoma SMMC-7721 cells by targeting cytosolic thioredoxin reductase. Free Radic. Biol. Med. 69, 15–25 [DOI] [PubMed] [Google Scholar]
- 19. Duan D., Zhang B., Yao J., Liu Y., and Fang J. (2014) Shikonin targets cytosolic thioredoxin reductase to induce ROS-mediated apoptosis in human promyelocytic leukemia HL-60 cells. Free Radic. Biol. Med. 70, 182–193 [DOI] [PubMed] [Google Scholar]
- 20. Cai W., Zhang B., Duan D., Wu J., and Fang J. (2012) Curcumin targeting the thioredoxin system elevates oxidative stress in HeLa cells. Toxicol. Appl. Pharmacol. 262, 341–348 [DOI] [PubMed] [Google Scholar]
- 21. Ghantous A., Sinjab A., Herceg Z., and Darwiche N. (2013) Parthenolide: from plant shoots to cancer roots. Drug Discov. Today 18, 894–905 [DOI] [PubMed] [Google Scholar]
- 22. Kwok B. H., Koh B., Ndubuisi M. I., Elofsson M., and Crews C. M. (2001) The anti-inflammatory natural product parthenolide from the medicinal herb Feverfew directly binds to and inhibits IκB kinase. Chem. Biol. 8, 759–766 [DOI] [PubMed] [Google Scholar]
- 23. García-Piñeres A. J., Castro V., Mora G., Schmidt T. J., Strunck E., Pahl H. L., and Merfort I. (2001) Cysteine 38 in p65/NF-κB plays a crucial role in DNA binding inhibition by sesquiterpene lactones. J. Biol. Chem. 276, 39713–39720 [DOI] [PubMed] [Google Scholar]
- 24. Hehner S. P., Hofmann T. G., Dröge W., and Schmitz M. L. (1999) The antiinflammatory sesquiterpene lactone parthenolide inhibits NF-κB by targeting the IκB kinase complex. J. Immunol. 163, 5617–5623 [PubMed] [Google Scholar]
- 25. Kreuger M. R., Grootjans S., Biavatti M. W., Vandenabeele P., and D'Herde K. (2012) Sesquiterpene lactones as drugs with multiple targets in cancer treatment: focus on parthenolide. Anticancer Drugs 23, 883–896 [DOI] [PubMed] [Google Scholar]
- 26. D'Anneo A., Carlisi D., Emanuele S., Buttitta G., Di Fiore R., Vento R., Tesoriere G., and Lauricella M. (2013) Parthenolide induces superoxide anion production by stimulating EGF receptor in MDA-MB-231 breast cancer cells. Int. J. Oncol. 43, 1895–1900 [DOI] [PubMed] [Google Scholar]
- 27. Won Y. K., Ong C. N., Shi X., and Shen H. M. (2004) Chemopreventive activity of parthenolide against UVB-induced skin cancer and its mechanisms. Carcinogenesis 25, 1449–1458 [DOI] [PubMed] [Google Scholar]
- 28. Steele A. J., Jones D. T., Ganeshaguru K., Duke V. M., Yogashangary B. C., North J. M., Lowdell M. W., Kottaridis P. D., Mehta A. B., Prentice A. G., Hoffbrand A. V., and Wickremasinghe R. G. (2006) The sesquiterpene lactone parthenolide induces selective apoptosis of B-chronic lymphocytic leukemia cells in vitro. Leukemia 20, 1073–1079 [DOI] [PubMed] [Google Scholar]
- 29. Guzman M. L., Rossi R. M., Karnischky L., Li X., Peterson D. R., Howard D. S., and Jordan C. T. (2005) The sesquiterpene lactone parthenolide induces apoptosis of human acute myelogenous leukemia stem and progenitor cells. Blood 105, 4163–4169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Won Y. K., Ong C. N., and Shen H. M. (2005) Parthenolide sensitizes ultraviolet (UV)-B-induced apoptosis via protein kinase C-dependent pathways. Carcinogenesis 26, 2149–2156 [DOI] [PubMed] [Google Scholar]
- 31. Kurdi M., and Booz G. W. (2007) Evidence that IL-6-type cytokine signaling in cardiomyocytes is inhibited by oxidative stress: parthenolide targets JAK1 activation by generating ROS. J. Cell. Physiol. 212, 424–431 [DOI] [PubMed] [Google Scholar]
- 32. Fonrose X., Ausseil F., Soleilhac E., Masson V., David B., Pouny I., Cintrat J. C., Rousseau B., Barette C., Massiot G., and Lafanechère L. (2007) Parthenolide inhibits tubulin carboxypeptidase activity. Cancer Res. 67, 3371–3378 [DOI] [PubMed] [Google Scholar]
- 33. Zhang S., Ong C. N., and Shen H. M. (2004) Involvement of proapoptotic Bcl-2 family members in parthenolide-induced mitochondrial dysfunction and apoptosis. Cancer Lett. 211, 175–188 [DOI] [PubMed] [Google Scholar]
- 34. Gopal Y. N., Chanchorn E., and Van Dyke M. W. (2009) Parthenolide promotes the ubiquitination of MDM2 and activates p53 cellular functions. Mol. Cancer Ther. 8, 552–562 [DOI] [PubMed] [Google Scholar]
- 35. Gopal Y. N., Arora T. S., and Van Dyke M. W. (2007) Parthenolide specifically depletes histone deacetylase 1 protein and induces cell death through ataxia telangiectasia mutated. Chem. Biol. 14, 813–823 [DOI] [PubMed] [Google Scholar]
- 36. Lan B., Wan Y. J., Pan S., Wang Y., Yang Y., Leng Q. L., Jia H., Liu Y. H., Zhang C. Z., and Cao Y. (2015) Parthenolide induces autophagy via the depletion of 4E-BP1. Biochem. Biophys. Res. Commun. 456, 434–439 [DOI] [PubMed] [Google Scholar]
- 37. Xu Y., Fang F., Miriyala S., Crooks P. A., Oberley T. D., Chaiswing L., Noel T., Holley A. K., Zhao Y., Kiningham K. K., Clair D. K., and Clair W. H. (2013) KEAP1 is a redox sensitive target that arbitrates the opposing radiosensitive effects of parthenolide in normal and cancer cells. Cancer Res. 73, 4406–4417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sun Y., St Clair D. K., Xu Y., Crooks P. A., and St Clair W. H. (2010) A NADPH oxidase-dependent redox signaling pathway mediates the selective radiosensitization effect of parthenolide in prostate cancer cells. Cancer Res. 70, 2880–2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim J. H., Liu L., Lee S. O., Kim Y. T., You K. R., and Kim D. G. (2005) Susceptibility of cholangiocarcinoma cells to parthenolide-induced apoptosis. Cancer Res. 65, 6312–6320 [DOI] [PubMed] [Google Scholar]
- 40. Wen J., You K. R., Lee S. Y., Song C. H., and Kim D. G. (2002) Oxidative stress-mediated apoptosis: the anticancer effect of the sesquiterpene lactone parthenolide. J. Biol. Chem. 277, 38954–38964 [DOI] [PubMed] [Google Scholar]
- 41. Arnér E. S., Sarioglu H., Lottspeich F., Holmgren A., and Böck A. (1999) High-level expression in Escherichia coli of selenocysteine-containing rat thioredoxin reductase utilizing gene fusions with engineered bacterial-type SECIS elements and co-expression with the selA, selB and selC genes. J. Mol. Biol. 292, 1003–1016 [DOI] [PubMed] [Google Scholar]
- 42. Zhong L., and Holmgren A. (2000) Essential role of selenium in the catalytic activities of mammalian thioredoxin reductase revealed by characterization of recombinant enzymes with selenocysteine mutations. J. Biol. Chem. 275, 18121–18128 [DOI] [PubMed] [Google Scholar]
- 43. Javvadi P., Hertan L., Kosoff R., Datta T., Kolev J., Mick R., Tuttle S. W., and Koumenis C. (2010) Thioredoxin reductase-1 mediates curcumin-induced radiosensitization of squamous carcinoma cells. Cancer Res. 70, 1941–1950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nalvarte I., Damdimopoulos A. E., Nystöm C., Nordman T., Miranda-Vizuete A., Olsson J. M., Eriksson L., Björnstedt M., Arnér E. S., and Spyrou G. (2004) Overexpression of enzymatically active human cytosolic and mitochondrial thioredoxin reductase in HEK-293 cells: effect on cell growth and differentiation. J. Biol. Chem. 279, 54510–54517 [DOI] [PubMed] [Google Scholar]
- 45. Yao J., Zhang B., Ge C., Peng S., and Fang J. (2015) Xanthohumol, a polyphenol chalcone present in hops, activating Nrf2 enzymes to confer protection against oxidative damage in PC12 cells. J. Agric. Food Chem. 63, 1521–1531 [DOI] [PubMed] [Google Scholar]
- 46. Peng S., Zhang B., Meng X., Yao J., and Fang J. (2015) Synthesis of piperlongumine analogues and discovery of nuclear factor erythroid 2-related factor 2 (Nrf2) activators as potential neuroprotective agents. J. Med. Chem. 58, 5242–5255 [DOI] [PubMed] [Google Scholar]
- 47. Zhang L., Duan D., Liu Y., Ge C., Cui X., Sun J., and Fang J. (2014) Highly selective off-on fluorescent probe for imaging thioredoxin reductase in living cells. J. Am. Chem. Soc. 136, 226–233 [DOI] [PubMed] [Google Scholar]
- 48. Johansson L., Chen C., Thorell J. O., Fredriksson A., Stone-Elander S., Gafvelin G., and Arnér E. S. (2004) Exploiting the 21st amino acid-purifying and labeling proteins by selenolate targeting. Nat. Methods 1, 61–66 [DOI] [PubMed] [Google Scholar]
- 49. Zhang J., Li Y., Duan D., Yao J., Gao K., and Fang J. (2016) Inhibition of thioredoxin reductase by alantolactone prompts oxidative stress-mediated apoptosis of HeLa cells. Biochem. Pharmacol. 102, 34–44 [DOI] [PubMed] [Google Scholar]
- 50. Liu Y., Ma H., Zhang L., Cui Y., Liu X., and Fang J. (2016) A small molecule probe reveals declined mitochondrial thioredoxin reductase activity in a Parkinson's disease model. Chem. Commun. (Camb) 52, 2296–2299 [DOI] [PubMed] [Google Scholar]
- 51. Qiu X., Liu Z., Shao W. Y., Liu X., Jing D. P., Yu Y. J., An L. K., Huang S. L., Bu X. Z., Huang Z. S., and Gu L. Q. (2008) Synthesis and evaluation of curcumin analogues as potential thioredoxin reductase inhibitors. Bioorg. Med. Chem. 16, 8035–8041 [DOI] [PubMed] [Google Scholar]
- 52. Fang J., Lu J., and Holmgren A. (2005) Thioredoxin reductase is irreversibly modified by curcumin: a novel molecular mechanism for its anticancer activity. J. Biol. Chem. 280, 25284–25290 [DOI] [PubMed] [Google Scholar]
- 53. Gan F. F., Kaminska K. K., Yang H., Liew C. Y., Leow P. C., So C. L., Tu L. N., Roy A., Yap C. W., Kang T. S., Chui W. K., and Chew E. H. (2013) Identification of Michael acceptor-centric pharmacophores with substituents that yield strong thioredoxin reductase inhibitory character correlated to antiproliferative activity. Antioxid. Redox Signal. 19, 1149–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chew E. H., Nagle A. A., Zhang Y., Scarmagnani S., Palaniappan P., Bradshaw T. D., Holmgren A., and Westwell A. D. (2010) Cinnamaldehydes inhibit thioredoxin reductase and induce Nrf2: potential candidates for cancer therapy and chemoprevention. Free Radic. Biol. Med. 48, 98–111 [DOI] [PubMed] [Google Scholar]
- 55. Peng S., Yao J., Liu Y., Duan D., Zhang X., and Fang J. (2015) Activation of Nrf2 target enzymes conferring protection against oxidative stress in PC12 cells by ginger principal constituent 6-shogaol. Food Funct. 6, 2813–2823 [DOI] [PubMed] [Google Scholar]
- 56. Arnér E. S., Björnstedt M., and Holmgren A. (1995) 1-Chloro-2,4-dinitrobenzene is an irreversible inhibitor of human thioredoxin reductase: loss of thioredoxin disulfide reductase activity is accompanied by a large increase in NADPH oxidase activity. J. Biol. Chem. 270, 3479–3482 [DOI] [PubMed] [Google Scholar]
- 57. Kalyanaraman B., Darley-Usmar V., Davies K. J., Dennery P. A., Forman H. J., Grisham M. B., Mann G. E., Moore K., Roberts L. J. 2nd, and Ischiropoulos H. (2012) Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic. Biol. Med. 52, 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Du Y., Zhang H., Lu J., and Holmgren A. (2012) Glutathione and glutaredoxin act as a backup of human thioredoxin reductase 1 to reduce thioredoxin 1 preventing cell death by aurothioglucose. J. Biol. Chem. 287, 38210–38219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Eriksson S. E., Prast-Nielsen S., Flaberg E., Szekely L., and Arnér E. S. (2009) High levels of thioredoxin reductase 1 modulate drug-specific cytotoxic efficacy. Free Radic. Biol. Med. 47, 1661–1671 [DOI] [PubMed] [Google Scholar]
- 60. Hanahan D., and Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 61. Nogueira V., and Hay N. (2013) Molecular pathways: reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin. Cancer Res. 19, 4309–4314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Trachootham D., Zhou Y., Zhang H., Demizu Y., Chen Z., Pelicano H., Chiao P. J., Achanta G., Arlinghaus R. B., Liu J., and Huang P. (2006) Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell 10, 241–252 [DOI] [PubMed] [Google Scholar]
- 63. Harris I. S., Treloar A. E., Inoue S., Sasaki M., Gorrini C., Lee K. C., Yung K. Y., Brenner D., Knobbe-Thomsen C. B., Cox M. A., Elia A., Berger T., Cescon D. W., Adeoye A., Brüstle A., et al. (2015) Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 27, 211–222 [DOI] [PubMed] [Google Scholar]
- 64. Gorrini C., Harris I. S., and Mak T. W. (2013) Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 12, 931–947 [DOI] [PubMed] [Google Scholar]
- 65. Kanzok S. M., Fechner A., Bauer H., Ulschmid J. K., Müller H. M., Botella-Munoz J., Schneuwly S., Schirmer R., and Becker K. (2001) Substitution of the thioredoxin system for glutathione reductase in Drosophila melanogaster. Science 291, 643–646 [DOI] [PubMed] [Google Scholar]
- 66. Saitoh M., Nishitoh H., Fujii M., Takeda K., Tobiume K., Sawada Y., Kawabata M., Miyazono K., and Ichijo H. (1998) Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 17, 2596–2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mitchell D. A., Morton S. U., Fernhoff N. B., and Marletta M. A. (2007) Thioredoxin is required for S-nitrosation of procaspase-3 and the inhibition of apoptosis in Jurkat cells. Proc. Natl. Acad. Sci. U.S.A. 104, 11609–11614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Shelar S. B., Kaminska K. K., Reddy S. A., Kumar D., Tan C. T., Yu V. C., Lu J., Holmgren A., Hagen T., and Chew E. H. (2015) Thioredoxin-dependent regulation of AIF-mediated DNA damage. Free Radic. Biol. Med. 87, 125–136 [DOI] [PubMed] [Google Scholar]
- 69. Anestål K., and Arnér E. S. (2003) Rapid induction of cell death by selenium-compromised thioredoxin reductase 1 but not by the fully active enzyme containing selenocysteine. J. Biol. Chem. 278, 15966–15972 [DOI] [PubMed] [Google Scholar]