

Abstract
Integrin-mediated rolling and firm cell adhesion are two critical steps in leukocyte trafficking. Integrin α4β1 mediates a mixture of rolling and firm cell adhesion on vascular cell adhesion molecule-1 (VCAM-1) when in its resting state but only supports firm cell adhesion upon activation. The transition from rolling to firm cell adhesion is controlled by integrin activation. Kindlin-3 has been shown to bind to integrin β tails and trigger integrin activation via inside-out signaling. However, the role of kindlin-3 in regulating resting α4β1-mediated cell adhesion is not well characterized. Herein we demonstrate that kindlin-3 was required for the resting α4β1-mediated firm cell adhesion but not rolling adhesion. Knockdown of kindlin-3 significantly decreased the binding of kindlin-3 to β1 and down-regulated the binding affinity of the resting α4β1 to soluble VCAM-1. Notably, it converted the resting α4β1-mediated firm cell adhesion to rolling adhesion on VCAM-1 substrates, increased cell rolling velocity, and impaired the stability of cell adhesion. By contrast, firm cell adhesion mediated by Mn2+-activated α4β1 was barely affected by knockdown of kindlin-3. Structurally, lack of kindlin-3 led to a more bent conformation of the resting α4β1. Thus, kindlin-3 plays an important role in maintaining a proper conformation of the resting α4β1 to mediate both rolling and firm cell adhesion. Defective kindlin-3 binding to the resting α4β1 leads to a transition from firm to rolling cell adhesion on VCAM-1, implying its potential role in regulating the transition between integrin-mediated rolling and firm cell adhesion.
Keywords: cell adhesion, conformational change, integrin, kindlin, shear stress, kindlin-3, conformation, firm cell adhesion, integrin α4β1, resting state
Introduction
Integrins are a large family of α/β heterodimeric cell adhesion molecules that mediate cell/cell, cell/extracellular matrix, and cell/pathogen interactions (1). Different from most integrins that mediate only firm cell adhesion upon activation, the resting integrin α4β1 can mediate a mixture of rolling and firm leukocyte adhesion to vascular cell adhesion molecule-1 (VCAM-1)3 but only supports firm cell adhesion postactivation, playing an important role in leukocyte trafficking and immune homeostasis (2–5). Integrin-mediated cell adhesion is regulated by the dynamic shift between low and high affinity conformations of integrin for ligand binding (6). In the resting state, integrin has a low affinity bent conformation with the headpiece facing down toward the cell membrane; upon activation, integrin undergoes a series of conformational rearrangements and extends upward in a switchblade-like opening motion, leading to the increased integrin affinity (7–9). This process is commonly controlled by inside-out signals from the cytoplasm that are dependent on specific interactions between intracellular effector molecules, such as talin and kindlins, and the integrin cytoplasmic tail (10–12). Binding of talin to integrin β tails is a final common element of cellular signaling cascades that control integrin activation (13, 14), and kindlins are thought to be coactivators (15–17). It is also reported that distinct kindlin-3 binding patterns can lead to distinct binding affinities of mucosal vascular addressin cell adhesion molecule-1 and VCAM-1 to integrin α4β7 (18). In addition to inside-out signaling, extracellular metal ions can also regulate integrin affinity via a cluster of three divalent cation-binding sites in integrin β I domain (19). Compared with the low affinity state in Ca2+/Mg2+, addition of Mn2+ or removal of Ca2+ strikingly increases the affinity and adhesiveness of almost all integrins (20–22).
Kindlins are a family of band 4.1-ezrin-radixin-moesin-containing intracellular proteins, including kindlin-1, -2, and -3 in mammals (23). Kindlin-3 is primarily expressed in hematopoietic cells (24) and has been shown to bind integrin β tails to induce integrin activation (25, 26). Kindlin-3-deficient platelets showed defective activation of αIIbβ3 and α2β1 integrins in response to chemokine stimulation (27). In addition, loss of kindlin-3 expression accounts for the rare autosomal human disease named leukocyte adhesion deficiency type III (28, 29). Lymphocytes derived from leukocyte adhesion deficiency type III patients showed defective activation of αLβ2 and α4β1 integrins upon phorbol 12-myristate 13-acetate stimulation and showed impaired cell spreading and migration (26, 30, 31). Besides the critical role of kindlin-3 in inducing integrin activation through inside-out signaling, it is also reported that kindlin-3 binds the resting integrin α4β7 at a high level, and the dissociation of kindlin-3 from β7 tail specifically increases the binding affinity of α4β7 to mucosal vascular addressin cell adhesion molecule-1 but suppresses VCAM-1 binding (18). Thus, kindlin-3 may have distinct functions in regulating the resting and activated integrins.
Although numerous studies have revealed the role of kindlin-3 in inside-out activation of integrin, little is known regarding its function in regulating the resting α4β1-mediated cell adhesion. Herein we report that kindlin-3 was required for the resting α4β1-mediated firm cell adhesion but not rolling adhesion under shear flow conditions. Silencing of kindlin-3 in K562 cells stably expressing human α4β1 (K562-α4β1) significantly decreased the binding of kindlin-3 to β1 and thus down-regulated the binding affinity of the resting α4β1 to soluble VCAM-1. Furthermore, knockdown of kindlin-3 converted the resting α4β1-mediated firm cell adhesion to rolling adhesion on VCAM-1 substrates, increased cell rolling velocity, and impaired the stability of cell adhesion under flow. By contrast, firm cell adhesion mediated by Mn2+-activated α4β1 was barely affected. Moreover, lack of kindlin-3 resulted in a more bent conformation of the resting α4β1. Re-expression of knockdown-resistant wild-type (WT) kindlin-3 but not integrin binding-deficient kindlin-3 mutant could rescue the observed defects in integrin α4β1-mediated cell adhesion and α4β1 conformation in kindlin-3 knockdown cells, suggesting that the observed defects were due to the deficient kindlin-3/integrin binding induced by kindlin-3 knockdown. Thus, kindlin-3 has an important role in maintaining a proper conformation of the resting α4β1 and its ability to mediate firm cell adhesion before activation.
Experimental Procedures
cDNA Construction and Cell Transfection
cDNA of human α4 integrin subunit was constructed in vector pcDNA3.1/Hygro(−) (Invitrogen). Kindlin-3 shRNA and luciferase shRNA were constructed in vector pLKO.1 (Invitrogen). The shRNA-resistant point mutation in WT kindlin-3 was generated using QuikChange (Stratagene); human kindlin-3 cDNA in vector pCDH-puro (Invitrogen) was used as the template. The kindlin-3 W596A mutation was generated using shRNA-resistant WT kindlin-3 construct as the template. All constructs were confirmed by DNA sequencing.
Transient transfection of 293T cells was performed as described (21). K562 cells stably expressing human α4β1 (K562-α4β1) were established by transfection of human α4 (32).
Antibodies and Reagents
Alexa Fluor 647-conjugated goat anti-mouse IgG, Cy3-conjugated goat anti-rat IgG, and Alexa Fluor 647-conjugated goat anti-human IgG were from Invitrogen. mAb to kindlin-3 was from Santa Cruz Biotechnology (N-12). mAb to β1 was from Abcam (EP1041Y). mAbs 9F10 and AIIB2 against human α4 and β1 integrin, respectively, were prepared from hybridomas (Developmental Studies Hybridoma Bank). mAb to CD45 was from Sino Biological (10086-H02H). mAb Act-1 against human β7 integrin was as described previously (33). Fab fragments were produced as described (34), and direct labeling of antibodies with Alexa Fluor 488 was performed using a protein labeling kit according to the manufacturer's instructions (Invitrogen). Human VCAM-1/Fc fusion protein containing Ig domains 1–7 of human VCAM-1 fused to the hinge and Fc region of human IgG1 was generated as described (35). Complete protease inhibitor mixture tablets were from Roche Applied Science.
Silencing and Rescue of Kindlin-3 Expression
Kindlin-3 knockdown was performed by transduction of K562-α4β1 cells with the recombinant lentivirus, which expressed the shRNA that annealed to kindlin-3 (5′-CCGAAUUGUACACGAGUAU-3′) (26). Stable knockdown level of kindlin-3 was confirmed by Western blotting. To rescue kindlin-3 expression in kindlin-3 knockdown K562-α4β1 cells, shRNA-resistant WT kindlin-3 or kindlin-3 W596A mutant was transiently expressed by recombinant lentivirus. Kindlin-3 expression was confirmed by Western blotting. Lentiviruses were generated and cells were transduced as described (36).
Immunoprecipitation and Western Blotting
Cells were treated with 5 mm dimethyl 3,3′-dithiopropionimidate dihydrochloride (Thermo) for 45 min and then lysed with lysis buffer (TBS containing 1% Triton X-100, 0.05% Nonidet P-40, Complete protease inhibitor mixture, 1 mm Ca2+/Mg2+ or 1 mm Mn2+) for 30 min on ice. Cell lysates were then immunoprecipitated with AIIB2 antibody. Kindlin-3, integrin β1, and β-actin were detected by immunoblotting. Mouse IgG was used as a control.
Flow Cytometry
Flow cytometry was done as described (37). Cell surface expression of integrin α4β1 on K562-α4β1 transfectants was determined by staining with mAbs 9F10 and AIIB2. Stained cells were then measured using a FACSCalibur (BD Biosciences) and analyzed using FlowJo software.
Soluble Ligand Binding Assay
The soluble ligand binding assay was performed as described (35, 37). Briefly, 20 μg/ml VCAM-1/Fc fusion protein was preincubated with Alexa Fluor 647-conjugated goat anti-human IgG in 50 μl of Hepes-buffered saline (20 mm Hepes, pH 7.4) containing either 1 mm Ca2+/Mg2+ or 1 mm Mn2+ and then incubated with cells for 30 min at room temperature. Next, cells were washed twice, measured using a FACSCalibur, and analyzed using FlowJo software. As a control, cells were preincubated with 20 μg/ml α4β1 blocking mAb AIIB2 for 5 min at 37 °C before addition of VCAM-1/Fc complexes.
Flow Chamber Assay
The flow chamber assay was performed as described (21, 38). A polystyrene Petri dish was coated with a 5-mm diameter, 20-μl spot of 5 μg/ml purified VCAM-1/Fc in coating buffer (PBS, 10 mm NaHCO3, pH 9.0) for 1 h at 37 °C followed by 2% BSA in coating buffer for 1 h at 37 °C to block nonspecific binding sites. Cells were diluted to 1 × 106/ml in Buffer A (Hepes-buffered saline, 0.5% BSA) containing the indicated divalent cations immediately before infusion in the flow chamber. Cells were allowed to accumulate for 30 s at 0.3 dyne/cm2 and 10 s at 0.4 dyne/cm2. Then shear stress was increased every 10 s from 1 dyne/cm2 up to 32 dynes/cm2 in 2-fold increments. The number of cells remaining bound at the end of each 10-s interval was determined.
Rolling velocity at each shear stress was calculated from the average distance traveled by rolling cells in 3 s. A velocity of 1 μm/s, which corresponds to a movement of ½ cell diameter during the 3-s measurement interval, was the minimum velocity required to define a cell as rolling instead of firmly adherent. For integrin α4β1 blocking, cells were preincubated with 20 μg/ml AIIB2 for 5 min at 37 °C.
Cell Detachment Assay
Cells were prepared as described in the flow chamber assay and then infused in the flow chamber. Cells were allowed to accumulate for 0.3 dyne/cm2 and 10 s at 0.4 dyne/cm2. Then shear stress was increased every 10 s from 1 dyne/cm2 up to 16 dynes/cm2 in 2-fold increments. The cells remaining bound to VCAM-1 substrates (5 μg/ml) at each wall shear stress were determined as a percentage of initial adherent cells at 1 dyne/cm2.
Fluorescence Resonance Energy Transfer (FRET) Assay
FRET was measured as described (33, 39). For detecting the orientation of integrin ectodomain relative to cell membrane, cells were seeded on a poly-l-lysine (100 μg/ml)-coated surface in serum-free DMEM with the indicated divalent cation and incubated for 30 min at 37 °C. Adherent cells were fixed with 3.7% paraformaldehyde for 15 min at room temperature, and nonspecific sites were blocked by incubation with 10% serum-rich medium for 10 min at room temperature. Then cells were stained with 20 μg/ml Alexa Fluor 488-conjugated AIIB2 Fab, Alexa Fluor 488-conjugated anti-CD45 Fab, or Alexa Fluor 488-conjugated Act-1 Fab for 40 min at 37 °C. After two washes, cells were labeled with 10 μm FM4-64 FX (Invitrogen) for 4 min on ice, washed once, and immediately mounted with Mowiol® 4-88 (Polysciences Inc.) mounting solution under a coverslip. The mounted slides were kept in the dark and subjected to photobleach FRET acquisition by a confocal microscope (TCS SP8, Leica). FRET efficiency (E) was calculated as E = 1 − (Fdonor(d)Pre/Fdonor(d)Post) where Fdonor(d)Pre and Fdonor(d)Post are the mean donor emission intensity of pre- and postphotobleaching.
Results
Kindlin-3 Knockdown Reduces Kindlin-3 Binding to β1 Integrin
To investigate the role of kindlin-3 in α4β1-mediated cell adhesion, we knocked down the expression of kindlin-3 in K562-α4β1 cells. Cells were transduced with lentivirus-based control shRNA (luciferase shRNA) or kindlin-3-targeting shRNA (kindlin-3 shRNA). Reduced expression of kindlin-3 was verified by Western blotting analysis. Compared with the untransduced control cells, cells transduced with kindlin-3 shRNA showed an approximately 80% decrease in kindlin-3 expression (Fig. 1A). As a control, luciferase shRNA did not influence the kindlin-3 expression in the transfectant. To examine the off-target effect of kindlin-3 shRNA and the effect of kindlin-3 silencing-induced deficient kindlin-3/β1 integrin binding, we included two controls by re-expressing shRNA-resistant WT kindlin-3 or shRNA-resistant kindlin-3 mutant containing a tryptophan 596 to alanine point mutation (kindlin-3 W596A), which impairs β1 integrin binding (40, 41), in kindlin-3 knockdown cells, respectively. The expression level of WT kindlin-3 and kindlin-3 W596A mutant in kindlin-3 knockdown K562-α4β1 stable cells was comparable with that of control cells (Fig. 1A). Flow cytometry analyses showed comparable levels of cell surface expression of α4β1 in control, kindlin-3-silenced, and kindlin-3-re-expressing cells (Fig. 1B), indicating that kindlin-3 expression does not affect the cell surface expression of α4β1 in K562-α4β1 cells.
FIGURE 1.

Knockdown of kindlin-3 expression in K562-α4β1 stable cells. A, left panel, expression levels of kindlin-3 were determined by immunoblotting in K562-α4β1 (Control), K562-α4β1 expressing luciferase shRNA, K562-α4β1 expressing kindlin-3 shRNA, K562-α4β1 expressing kindlin-3 shRNA and shRNA-resistant WT kindlin-3, and K562-α4β1 expressing kindlin-3 shRNA and shRNA-resistant kindlin-3 W596A mutant. A representative result of three independent experiments is shown. Right panel, quantification of three independent blots for kindlin-3 expression as a ratio relative to the expression level of control cells. B, cell surface expression of integrin α4β1 was determined by flow cytometry. Open histogram, mock control; filled histogram, integrin α4 (upper panels) and β1 (lower panels). C, co-immunoprecipitation (IP) of kindlin-3 with β1 integrin in 1 mm Ca2+/Mg2+ or 1 mm Mn2+. The precipitates were blotted with anti-β1 and anti-kindlin-3 antibodies. A representative result of three independent experiments is shown. Error bars represent ±S.D. (n = 3). ***, p < 0.001; NS, not significant (two-tailed Student's t test).
Next, we examined the effect of kindlin-3 knockdown on the association of kindlin-3 with the resting β1 integrin in 1 mm Ca2+/Mg2+ or with the activated β1 integrin in 1 mm Mn2+. A co-immunoprecipitation assay showed that knockdown of kindlin-3 significantly reduced the binding of kindlin-3 to both the resting and Mn2+-activated β1 integrins (Fig. 1C). As expected, re-expression of WT kindlin-3 in kindlin-3 knockdown cells restored the binding of kindlin-3 to β1 integrin to the level in control cells. However, re-expression of kindlin-3 W596A mutant did not rescue the kindlin-3 binding (Fig. 1C).
Kindlin-3 Knockdown Inhibits soluble VCAM-1 Binding to α4β1
Kindlins are coactivators of integrins (15–17); therefore we next investigated the effect of reduced kindlin-3 expression on the ligand binding affinity of α4β1 by examining the binding of soluble VCAM-1/Fc to K562-α4β1 cells. The binding of soluble VCAM-1/Fc complexed with Alexa Fluor 647-conjugated goat anti-human IgG to K562-α4β1 transfectants in different divalent cations was measured by flow cytometry. Compared with the binding of VCAM-1/Fc complexes to K562-α4β1 in 1 mm Ca2+/Mg2+, VCAM-1/Fc binding was greatly enhanced in 1 mm Mn2+, indicating the increased ligand binding affinity after integrin activation by Mn2+ (Fig. 2). In the presence of 1 mm Ca2+/Mg2+, kindlin-3 knockdown cells showed a 53% decrease in VCAM-1/Fc binding compared with control cells. However, those cells showed only a 36% decrease in VCAM-1/Fc binding in the presence of 1 mm Mn2+. These data suggest that knockdown of kindlin-3 induces a greater decrease in the ligand binding affinity of the resting α4β1 than of Mn2+-activated α4β1. Moreover, re-expression of WT kindlin-3 rescued the observed defects in VCAM-1/Fc binding to integrin α4β1 in kindlin-3 knockdown cells, whereas re-expression of kindlin-3 W596A mutant showed no rescue effect (Fig. 2). These data suggest that the observed defects in α4β1-VCAM-1 binding are due to the deficient kindlin-3/β1 integrin binding induced by kindlin-3 knockdown. As a control, the binding of VCAM-1 was completely blocked by β1 integrin-blocking antibody AIIB2, indicating that VCAM-1 binding is β1 integrin-dependent.
FIGURE 2.

Effect of kindlin-3 knockdown on soluble VCAM-1 binding to α4β1. Binding of soluble VCAM-1/Fc to K562-α4β1 control cells or transfectants in 1 mm Ca2+/Mg2+ or 1 mm Mn2+ was analyzed. As a control, the function of α4β1 was blocked by 20 μg/ml AIIB2. Mean fluorescence intensity of VCAM-1/Fc binding was calculated. Soluble ligand binding was expressed as the specific mean fluorescence intensity and quantified as a percentage of total α4β1 expression defined by staining with mAb 9F10 against α4. Error bars represent ±S.D. (n = 3). ***, p < 0.001; NS, not significant (two-tailed Student's t test).
Kindlin-3 Is Essential for Firm Cell Adhesion Mediated by the Resting α4β1
Integrin α4β1 mediates a mixture of rolling and firm cell adhesion in shear flow on VCAM-1 substrates when in its resting state and only supports firm cell adhesion upon activation (2). We next investigated the role of kindlin-3 in regulating the cell adhesion mediated by α4β1 pre- and postactivation. The adhesive behaviors of the K562-α4β1 transfectants in shear flow were characterized in a parallel wall flow chamber with human VCAM-1/Fc absorbed to its lower wall. The shear stress was incrementally increased, and the velocity of the cells remaining bound at each increment was determined (42). In 1 mm Ca2+/Mg2+, the control and luciferase shRNA-treated K562-α4β1 cells showed a mixture of about 30% of rolling events and 70% of firmly adherent events in the total adherent cells (Fig. 3, A and B). In contrast, kindlin-3 knockdown cells showed a similar number of adherent cells, but the percentage of firmly adherent cells decreased from 70 to 28% (Fig. 3, A and B), indicating that reduced kindlin-3 expression results in a transition from firm adhesion to rolling adhesion mediated by the resting α4β1. In addition, kindlin-3 knockdown cells showed significantly faster rolling compared with control cells (Fig. 3C). The addition of Mn2+ strikingly increased the adhesiveness of K562-α4β1 cells to VCAM-1, leading to significantly increased adherent cells with nearly 100% firmly adherent events (Fig. 3, A and B). Knockdown of kindlin-3 led to a slight decrease in the number of adherent cells but did not affect the percentage of firmly adherent events. These data indicate that kindlin-3 is essential for the resting α4β1-mediated firm cell adhesion, and reduced kindlin-3 expression converts the resting α4β1-mediated firm cell adhesion to rolling adhesion. Moreover, re-expression of WT kindlin-3, but not kindlin-3 W596A mutant, in kindlin-3 knockdown cells efficiently rescued the defects in cell adhesion (Fig. 3), suggesting the essential role of kindlin-3/β1 interaction in α4β1-mediated cell adhesion.
FIGURE 3.

Effect of kindlin-3 knockdown on α4β1-mediated cell adhesion in shear flow. A, rolling and firmly adherent cell numbers of K562-α4β1 control cells or transfectants on immobilized VCAM-1/Fc (5 μg/ml) substrates in 1 mm Ca2+/Mg2+ or 1 mm Mn2+ under flow condition. The adherent cell number was measured at a wall shear stress of 2 dynes/cm2. B, percentage of firmly adherent cells at a wall shear stress of 2 dynes/cm2 in 1 mm Ca2+/Mg2+ or 1 mm Mn2+. C, average rolling velocity of K562-α4β1 control cells and transfectants that adhered to VCAM-1/Fc substrates at the indicated wall shear stress in 1 mm Ca2+/Mg2+ was calculated. All experiments were performed on the surface coated with purified VCAM-1/Fc (5 μg/ml). Error bars represent ±S.D. (n = 3). ***, p < 0.001; NS, not significant (two-tailed Student's t test).
Kindlin-3 Is Required for the Stable Interaction between the Resting α4β1 and VCAM-1
To further study the effect of kindlin-3 knockdown on the strength of α4β1-mediated cell adhesion to VCAM-1, we examined resistance to detachment by increasing wall shear stress (Fig. 4). In 1 mm Ca2+/Mg2+, kindlin-3 knockdown and kindlin-3 W596A mutant-re-expressing cells detached much more rapidly from VCAM-1 than control cells (Fig. 4A), suggesting that the reduced kindlin-3/β1 interaction leads to a less stable association between the resting α4β1 and VCAM-1. In 1 mm Mn2+, all five cell lines showed comparable resistance to detachment (Fig. 4B). These data suggest that kindlin-3 binding to β1 integrin is important for stable interaction between VCAM-1 and the resting α4β1, but not Mn2+-activated, α4β1.
FIGURE 4.
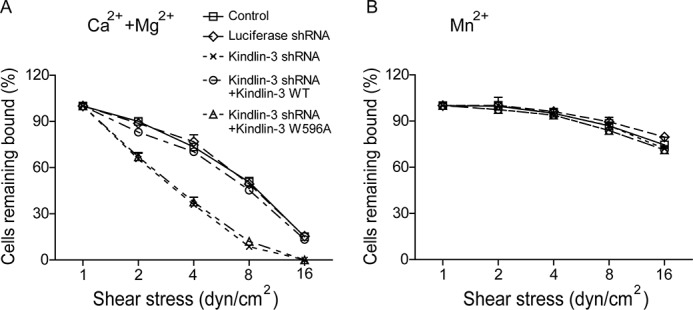
Effect of kindlin-3 knockdown on the cell resistance to detachment. A and B, resistance of K562-α4β1 control cells and transfectants to detachment at increasing wall shear stress in 1 mm Ca2+/Mg2+ (A) or 1 mm Mn2+ (B). The total number of cells remaining bound at each indicated wall shear stress was determined as a percentage of adherent cells at 1 dyne/cm2. Error bars represent ±S.D. (n = 3).
Kindlin-3 Knockdown Leads to a More Bent Conformation of α4β1
Integrin activation is accompanied by global conformational rearrangements as the headpiece of integrin folds over its legs and faces down toward the membrane in the low affinity bend conformation and extends upward in a switchblade-like opening upon activation (7, 43). We next used a FRET assay to study the effect of kindlin-3 knockdown on integrin conformation. To assess the orientation of integrin α4β1 ectodomain relative to the plasma membrane, α4β1 was labeled with Alexa Fluor 488-conjugated AIIB2 Fab fragment, which binds to the top of β1 I domain, as donor (44), and the plasma membrane was labeled with a lipophilic probe, FM4-64 FX, as acceptor (33, 39). In 1 mm Ca2+/Mg2+, kindlin-3 knockdown cells showed higher FRET efficiency than the control and luciferase shRNA-treated cells, suggesting a more bent conformation of the resting α4β1 when kindlin-3 was knocked down (Fig. 5A). Activation of integrin α4β1 by 1 mm Mn2+ significantly decreased the FRET efficiency, suggesting the extension of α4β1 ectodomain (Fig. 5A). In addition, the FRET efficiency of kindlin-3 knockdown was higher than that of controls in Mn2+, suggesting that kindlin-3 knockdown also reduces the extension of Mn2+-activated α4β1 to some degree (Fig. 5A). Re-expression of WT kindlin-3 in kindlin-3 knockdown cells fully abolished the integrin α4β1 conformational change induced by kindlin-3 knockdown in 1 mm Ca2+/Mg2+ and 1 mm Mn2+, whereas re-expression of kindlin-3 W596A mutant showed no rescue effect (Fig. 5A), suggesting that the observed defects are due to the kindlin-3 knockdown-induced deficient kindlin-3/β1 integrin binding. Thus, the binding of kindlin-3 to β1 integrin is important for maintaining a proper conformation of α4β1 in both resting and active states.
FIGURE 5.
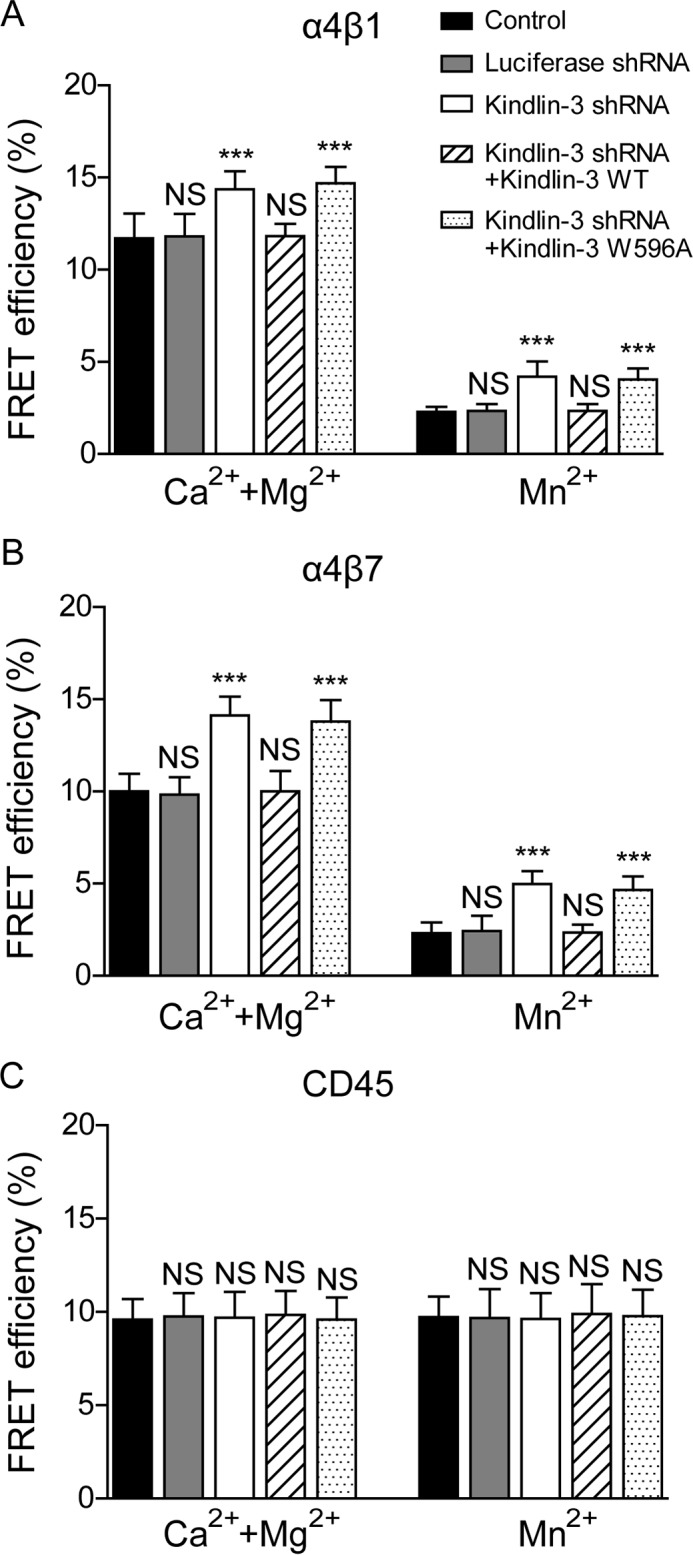
Effect of kindlin-3 knockdown on integrin conformation. A, influence of kindlin-3 expression on integrin α4β1 conformation. FRET between β1 I domain and the plasma membrane was analyzed. The FRET efficiency of K562-α4β1 control cells and transfectants in 1 mm Ca2+/Mg2+ or 1 mm Mn2+ was determined. B, influence of kindlin-3 expression on α4β7 conformation. FRET between β7 I domain and the plasma membrane was analyzed. The FRET efficiency of K562-α4β7 control cells and transfectants in 1 mm Ca2+/Mg2+ or 1 mm Mn2+ was determined. C, influence of kindlin-3 expression on CD45 conformation. FRET between CD45 and the plasma membrane was analyzed. The FRET efficiency of K562-α4β1 control cells and transfectants in 1 mm Ca2+/Mg2+ or 1 mm Mn2+ was determined. Error bars represent ±S.D. (n = 10). ***, p < 0.001; NS, not significant (two-tailed Student's t test).
To further confirm that the observed regulation is specific for integrin, we also examined the effect of kindlin-3 knockdown on the conformation of integrin α4β7 and CD45 as controls. Kindlin-3 expression level does not affect the cell surface expression of α4β7 and CD45 (18, 45–47). To examine the orientation of α4β7 ectodomain relative to the plasma membrane using the FRET system, K562 cells stably expressing human α4β7 (K562-α4β7) was labeled with Alexa Fluor 488-conjugated Act-1 Fab fragment, which binds to the top of β7 I domain (48), as donor. The FRET results showed that knockdown of kindlin-3 had similar effects on the global conformation of α4β7 as observed in α4β1 (Fig. 5B). By contrast, kindlin-3 knockdown did not change the FRET efficiency between CD45 and plasma membrane, indicating that kindlin-3 knockdown does not induce a global conformational change of CD45 (Fig. 5C).
Discussion
Kindlins serve as coactivators of integrins through binding to integrin β tails to induce integrin activation (41, 49). In addition to the major function of kindlins in integrin activation, we report that kindlin-3 has an important role in regulating the conformation and function of integrin α4β1 in its resting state. Our data show that inhibition of kindlin-3 binding to α4β1 by kindlin-3 silencing triggered a more bent conformation of the resting α4β1, leading to a transition from firm cell adhesion to rolling adhesion, higher rolling velocity, and less stable interaction between the resting α4β1 and VCAM-1.
Previous study shows that α4β1 in kindlin-3-null lymphocytes retains intrinsic rolling adhesions to VCAM-1 and exhibits partial defects in chemokine-stimulated adhesiveness to VCAM-1 (31). Moreover, it has been reported that kindlin-3-deficient lymphocytes, although deficient in optimal firm adhesions, still are able to use their residual integrin adhesiveness to enter tissues (50). Consistent with these results, our study provides additional information that inhibition of kindlin-3 binding to α4β1 converted the resting α4β1-mediated firm cell adhesion to rolling adhesion, allowing α4β1 to support robust rolling cell adhesion before activation, and only partially affected firm cell adhesion mediated by the activated integrin (Fig. 3, A and B).
Unlike most integrins that only mediate firm cell adhesion upon activation, integrin α4β1 mediates a mixture of rolling and firm cell adhesion in its resting state (3, 5). Studies have shown that rolling and firm cell adhesion are two distinct phases of adhesion with a phase transition between them, interpreted directly by integrin conformational rearrangements that can be induced by intracellular effector proteins via inside-out signaling (21, 38). A clasp formed by a salt bridge between the integrin α and β tails is crucial for maintaining integrins in the bent, inactive conformation. Forced separation of the clasp can trigger extension of ectodomains and conformational changes in the ligand-binding site, generating the activated integrin with the extended, high affinity conformation (51, 52). Intracellular proteins that interact with integrin tails, such as talin, could induce conformational activation of the integrin by disrupting the integrin clasp (53, 54). Kindlins serve as coactivators as they cooperate with talin to activate integrin (16, 55). Our data show that disassociation of kindlin-3 with the resting α4β1 triggered a more bent conformation of the resting α4β1, resulting in the transition from firm cell adhesion to rolling adhesion, implying an important role of kindlin-3 in modulating the unfolding transition of integrin α4β1, which is crucial for the phase transition between cell rolling adhesion and firm adhesion.
Integrin affinity and avidity regulation are both important for integrin-mediated cell adhesion; they are distinct processes but mutually regulated and often occur at the same time (56–58). Integrin affinity transition is associated with the conformational rearrangements of integrin molecules (7). By using an intramolecular FRET system, we found that inhibition of kindin-3 binding to β1 led to a more bent resting conformation of α4β1 as well as α4β7 (Fig. 5), suggesting the important role of kindlin-3 in triggering extended (high affinity) conformation of α4 integrins. The results are consistent with previous reports that kindlin-3 is required for the induction of the high affinity conformation of αLβ2 (31, 59). Interestingly, kindlin-3 has been shown to have little effect on the affinity of purified monomeric αIIbβ3 integrin in a cell-free system (60). Moreover, kindlin-2 increases the multivalent ligand binding to integrin αIIbβ3 by promoting the clustering of ligand-occupied αIIbβ3 in non-hematopoietic cells (60). These data suggest that kindlins may promote integrin-ligand binding by clustering αIIbβ3 rather than inducing conformational activation of monomeric integrin. It is noteworthy that the reported distinct mechanisms of kindlin-3 in regulating integrin-ligand binding are observed in different integrins, and some experiments use different kindlins. Furthermore, it has been reported that integrin β1 tails have higher binding affinity for kindlin-3 than β3 tails in a cell-free system (31, 45). Thus, it is possible that kindlin-3 regulates the ligand binding of different integrins (α4β1 and αIIbβ3) via distinct mechanisms.
Clinically, loss of kindlin-3 expression accounts for the pathogenesis of leukocyte adhesion deficiency type III that is characterized by bleeding disorders and defective recruitment of leukocytes into sites of infection (28, 29). Our finding suggests that leukocytes in these patients might have several functional deficiencies of integrin α4β1, including lack of firm cell adhesion mediated by the resting α4β1, less stable α4β1/VCAM-1 interactions, and higher rolling velocity besides the deficient activation of this integrin via inside-out signaling (26).
Taken together, kindlin-3 is crucial for maintaining a proper conformation of the resting α4β1 and its ability to mediate firm cell adhesion before activation. Defective kindlin-3 binding to the resting α4β1 leads to a transition from firm to rolling cell adhesion on VCAM-1, implying its critical role in regulating the transition between integrin-mediated rolling and firm cell adhesion.
Author Contributions
L. L., C. Lin, and J. C. designed experiments. L. L., C. Lin, Z. Y., Shu W., Y. Z., ShiHui W., J. W., and C. Liu performed experiments and analyzed data. L. L., C. Lin, and J. C. interpreted results. The manuscript was drafted by L. L. and C. Lin and edited by J. C.
This work was supported by National Basic Research Program of China Grant 2014CB541905; National Natural Science Foundation of China Grants 31525016, 31190061, and 31271487; Personalized Medicines-Molecular Signature-based Drug Discovery and Development; Strategic Priority Research Program of the Chinese Academy of Sciences Grant XDA12000000; the Chinese Academy of Sciences/State Administration of Foreign Experts Affairs International Partnership Program for Creative Research Teams; Science and Technology Commission of Shanghai Municipality Grant 11JC1414200; and the Sanofi-Aventis-Shanghai Institutes for Biological Sciences scholarship program. The authors declare that they have no conflicts of interest with the contents of this article.
- VCAM-1
- vascular cell adhesion molecule-1.
References
- 1. Hynes R. O. (2002) Integrins: bidirectional, allosteric signaling machines. Cell 110, 673–687 [DOI] [PubMed] [Google Scholar]
- 2. Berlin C., Bargatze R. F., Campbell J. J., von Andrian U. H., Szabo M. C., Hasslen S. R., Nelson R. D., Berg E. L., Erlandsen S. L., and Butcher E. C. (1995) α4 integrins mediate lymphocyte attachment and rolling under physiological flow. Cell 80, 413–422 [DOI] [PubMed] [Google Scholar]
- 3. Chen C., Mobley J. L., Dwir O., Shimron F., Grabovsky V., Lobb R. R., Shimizu Y., and Alon R. (1999) High affinity very late antigen-4 subsets expressed on T cells are mandatory for spontaneous adhesion strengthening but not for rolling on VCAM-1 in shear flow. J. Immunol. 162, 1084–1095 [PubMed] [Google Scholar]
- 4. Lim Y. C., Wakelin M. W., Henault L., Goetz D. J., Yednock T., Cabañas C., Sánchez-Madrid F., Lichtman A. H., and Luscinskas F. W. (2000) α4β1-Integrin activation is necessary for high-efficiency T-cell subset interactions with VCAM-1 under flow. Microcirculation 7, 201–214 [PubMed] [Google Scholar]
- 5. Hyduk S. J., Rullo J., Cano A. P., Xiao H., Chen M., Moser M., and Cybulsky M. I. (2011) Talin-1 and kindlin-3 regulate α4β1 integrin-mediated adhesion stabilization, but not G protein-coupled receptor-induced affinity upregulation. J. Immunol. 187, 4360–4368 [DOI] [PubMed] [Google Scholar]
- 6. Luo B. H., Carman C. V., and Springer T. A. (2007) Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 25, 619–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Takagi J., Petre B. M., Walz T., and Springer T. A. (2002) Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell 110, 599–611 [DOI] [PubMed] [Google Scholar]
- 8. Shimaoka M., and Springer T. A. (2004) Therapeutic antagonists and the conformational regulation of the β2 integrins. Curr. Top. Med. Chem. 4, 1485–1495 [DOI] [PubMed] [Google Scholar]
- 9. Arnaout M. A., Mahalingam B., and Xiong J. P. (2005) Integrin structure, allostery, and bidirectional signaling. Annu. Rev. Cell Dev. Biol. 21, 381–410 [DOI] [PubMed] [Google Scholar]
- 10. Liu S., Calderwood D. A., and Ginsberg M. H. (2000) Integrin cytoplasmic domain-binding proteins. J. Cell Sci. 113, 3563–3571 [DOI] [PubMed] [Google Scholar]
- 11. Moser M., Legate K. R., Zent R., and Fässler R. (2009) The tail of integrins, talin, and kindlins. Science 324, 895–899 [DOI] [PubMed] [Google Scholar]
- 12. Banno A., and Ginsberg M. H. (2008) Integrin activation. Biochem. Soc. Trans. 36, 229–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Calderwood D. A. (2004) Talin controls integrin activation. Biochem. Soc. Trans. 32, 434–437 [DOI] [PubMed] [Google Scholar]
- 14. Tadokoro S., Shattil S. J., Eto K., Tai V., Liddington R. C., de Pereda J. M., Ginsberg M. H., and Calderwood D. A. (2003) Talin binding to integrin β tails: a final common step in integrin activation. Science 302, 103–106 [DOI] [PubMed] [Google Scholar]
- 15. Kahner B. N., Kato H., Banno A., Ginsberg M. H., Shattil S. J., and Ye F. (2012) Kindlins, integrin activation and the regulation of talin recruitment to αIIbβ3. PLos One 7, e34056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ye F., and Petrich B. G. (2011) Kindlin: helper, co-activator, or booster of talin in integrin activation? Curr. Opin. Hematol. 18, 356–360 [DOI] [PubMed] [Google Scholar]
- 17. Bialkowska K., Ma Y. Q., Bledzka K., Sossey-Alaoui K., Izem L., Zhang X., Malinin N., Qin J., Byzova T., and Plow E. F. (2010) The integrin co-activator kindlin-3 is expressed and functional in a non-hematopoietic cell, the endothelial cell. J. Biol. Chem. 285, 18640–18649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sun H., Liu J., Zheng Y., Pan Y., Zhang K., and Chen J. (2014) Distinct chemokine signaling regulates integrin ligand specificity to dictate tissue-specific lymphocyte homing. Dev. Cell 30, 61–70 [DOI] [PubMed] [Google Scholar]
- 19. Zhang K., and Chen J. (2012) The regulation of integrin function by divalent cations. Cell Adh. Migr. 6, 20–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Leitinger B., McDowall A., Stanley P., and Hogg N. (2000) The regulation of integrin function by Ca2+. Biochim. Biophys. Acta 1498, 91–98 [DOI] [PubMed] [Google Scholar]
- 21. Chen J., Salas A., and Springer T. A. (2003) Bistable regulation of integrin adhesiveness by a bipolar metal ion cluster. Nat. Struct. Biol. 10, 995–1001 [DOI] [PubMed] [Google Scholar]
- 22. Mould A. P., Akiyama S. K., and Humphries M. J. (1995) Regulation of integrin α5β1-fibronectin interactions by divalent cations—evidence for distinct classes of binding sites for Mn2+, Mg2+, and Ca2+. J. Biol. Chem. 270, 26270–26277 [DOI] [PubMed] [Google Scholar]
- 23. Karaköse E., Schiller H. B., and Fässler R. (2010) The kindlins at a glance. J. Cell Sci. 123, 2353–2356 [DOI] [PubMed] [Google Scholar]
- 24. Malinin N. L., Plow E. F., and Byzova T. V. (2010) Kindlins in FERM adhesion. Blood 115, 4011–4017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Moretti F., Schmidt S., Ruppert R., Fassler R., and Moser M. (2010) A critical kindlin-3 level is required for efficient integrin activation in hematopoietic cells. Eur. J. Cell Biol. 89, 51 [Google Scholar]
- 26. Malinin N. L., Zhang L., Choi J., Ciocea A., Razorenova O., Ma Y. Q., Podrez E. A., Tosi M., Lennon D. P., Caplan A. I., Shurin S. B., Plow E. F., and Byzova T. V. (2009) A point mutation in KINDLIN3 ablates activation of three integrin subfamilies in humans. Nat. Med. 15, 313–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Moser M., Nieswandt B., Ussar S., Pozgajova M., and Fässler R. (2008) Kindlin-3 is essential for integrin activation and platelet aggregation. Nat. Med. 14, 325–330 [DOI] [PubMed] [Google Scholar]
- 28. McDowall A., Svensson L., Stanley P., Patzak I., Chakravarty P., Howarth K., Sabnis H., Briones M., and Hogg N. (2010) Two mutations in the KINDLIN3 gene of a new leukocyte adhesion deficiency III patient reveal distinct effects on leukocyte function in vitro. Blood 115, 4834–4842 [DOI] [PubMed] [Google Scholar]
- 29. Meller J., Malinin N. L., Panigrahi S., Kerr B. A., Patil A., Ma Y., Venkateswaran L., Rogozin I. B., Mohandas N., Ehlayel M. S., Podrez E. A., Chinen J., and Byzova T. V. (2012) Novel aspects of kindlin-3 function in humans based on a new case of leukocyte adhesion deficiency III. J. Thromb. Haemost. 10, 1397–1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Svensson L., Howarth K., McDowall A., Patzak I., Evans R., Ussar S., Moser M., Metin A., Fried M., Tomlinson I., and Hogg N. (2009) Leukocyte adhesion deficiency-III is caused by mutations in KINDLIN3 affecting integrin activation. Nat. Med. 15, 306–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Manevich-Mendelson E., Feigelson S. W., Pasvolsky R., Aker M., Grabovsky V., Shulman Z., Kilic S. S., Rosenthal-Allieri M. A., Ben-Dor S., Mory A., Bernard A., Moser M., Etzioni A., and Alon R. (2009) Loss of kindlin-3 in LAD-III eliminates LFA-1 but not VLA-4 adhesiveness developed under shear flow conditions. Blood 114, 2344–2353 [DOI] [PubMed] [Google Scholar]
- 32. Kassner P. D., and Hemler M. E. (1993) Interchangeable α-chain cytoplasmic domains play a positive role in control of cell adhesion mediated by Vla-4, a β1 integrin. J. Exp. Med. 178, 649–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yue J., Pan Y., Sun L., Zhang K., Liu J., Lu L., and Chen J. (2013) The unique disulfide bond-stabilized W1 β4-β1 loop in the α4β-propeller domain regulates integrin α4β7 affinity and signaling. J. Biol. Chem. 288, 14228–14237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Askari J. A., Tynan C. J., Webb S. E., Martin-Fernandez M. L., Ballestrem C., and Humphries M. J. (2010) Focal adhesions are sites of integrin extension. J. Cell Biol. 188, 891–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rose D. M., Cardarelli P. M., Cobb R. R., and Ginsberg M. H. (2000) Soluble VCAM-1 binding to α4 integrins is cell-type specific and activation dependent and is disrupted during apoptosis in T cells. Blood 95, 602–609 [PubMed] [Google Scholar]
- 36. Liao Z., Kato H., Pandey M., Cantor J. M., Ablooglu A. J., Ginsberg M. H., and Shattil S. J. (2015) Interaction of kindlin-2 with integrin β3 promotes outside-in signaling responses by the αVβ3 vitronectin receptor. Blood 125, 1995–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen J., Yang W., Kim M., Carman C. V., and Springer T. A. (2006) Regulation of outside-in signaling and affinity by the β2 I domain of integrin αLβ2. Proc. Natl. Acad. Sci. U.S.A. 103, 13062–13067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Salas A., Shimaoka M., Chen S., Carman C. V., and Springer T. (2002) Transition from rolling to firm adhesion is regulated by the conformation of the I domain of the integrin lymphocyte function-associated antigen-1. J. Biol. Chem. 277, 50255–50262 [DOI] [PubMed] [Google Scholar]
- 39. Pan Y., Zhang K., Qi J., Yue J., Springer T. A., and Chen J. (2010) Cation-π interaction regulates ligand-binding affinity and signaling of integrin α4β7. Proc. Natl. Acad. Sci. U.S.A. 107, 21388–21393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ussar S., Wang H. V., Linder S., Fässler R., and Moser M. (2006) The kindlins: subcellular localization and expression during murine development. Exp. Cell Res. 312, 3142–3151 [DOI] [PubMed] [Google Scholar]
- 41. Harburger D. S., Bouaouina M., and Calderwood D. A. (2009) Kindlin-1 and-2 directly bind the C-terminal region of β integrin cytoplasmic tails and exert integrin-specific activation effects. J. Biol. Chem. 284, 11485–11497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. de Château M., Chen S., Salas A., and Springer T. A. (2001) Kinetic and mechanical basis of rolling through an integrin and novel Ca2+-dependent rolling and Mg2+-dependent firm adhesion modalities for the α4β7-MAdCAM-1 interaction. Biochemistry 40, 13972–13979 [DOI] [PubMed] [Google Scholar]
- 43. Chen X., Xie C., Nishida N., Li Z., Walz T., and Springer T. A. (2010) Requirement of open headpiece conformation for activation of leukocyte integrin αXβ2. Proc. Natl. Acad. Sci. U.S.A. 107, 14727–14732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Takada Y., and Puzon W. (1993) Identification of a regulatory region of integrin-β1 subunit using activating and inhibiting antibodies. J. Biol. Chem. 268, 17597–17601 [PubMed] [Google Scholar]
- 45. Moser M., Bauer M., Schmid S., Ruppert R., Schmidt S., Sixt M., Wang H. V., Sperandio M., and Fässler R. (2009) Kindlin-3 is required for β2 integrin-mediated leukocyte adhesion to endothelial cells. Nat. Med. 15, 300–305 [DOI] [PubMed] [Google Scholar]
- 46. Dixit N., Kim M. H., Rossaint J., Yamayoshi I., Zarbock A., and Simon S. I. (2012) Leukocyte function antigen-1, kindlin-3, and calcium flux orchestrate neutrophil recruitment during inflammation. J. Immunol. 189, 5954–5964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Moretti F. A., Moser M., Lyck R., Abadier M., Ruppert R., Engelhardt B., and Fässler R. (2013) Kindlin-3 regulates integrin activation and adhesion reinforcement of effector T cells. Proc. Natl. Acad. Sci. U.S.A. 110, 17005–17010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tidswell M., Pachynski R., Wu S. W., Qiu S. Q., Dunham E., Cochran N., Briskin M. J., Kilshaw P. J., Lazarovits A. I., Andrew D. P., Butcher E. C., Yednock T. A., and Erle D. J. (1997) Structure-function analysis of the integrin β7 subunit—identification of domains involved in adhesion to MAdCAM-1. J. Immunol. 159, 1497–1505 [PubMed] [Google Scholar]
- 49. Ma Y. Q., Qin J., Wu C., and Plow E. F. (2008) Kindlin-2 (Mig-2): a co-activator of β3 integrins. J. Cell Biol. 181, 439–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cohen S. J., Gurevich I., Feigelson S. W., Petrovich E., Moser M., Shakhar G., Fassler R., and Alon R. (2013) The integrin coactivator kindlin-3 is not required for lymphocyte diapedesis. Blood 122, 2609–2617 [DOI] [PubMed] [Google Scholar]
- 51. O'Toole T. E., Katagiri Y., Faull R. J., Peter K., Tamura R., Quaranta V., Loftus J. C., Shattil S. J., and Ginsberg M. H. (1994) Integrin cytoplasmic domains mediate inside-out signal transduction. J. Cell Biol. 124, 1047–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vinogradova O., Velyvis A., Velyviene A., Hu B., Haas T., Plow E., and Qin J. (2002) A structural mechanism of integrin αIIbβ3 “inside-out” activation as regulated by its cytoplasmic face. Cell 110, 587–597 [DOI] [PubMed] [Google Scholar]
- 53. Kinashi T. (2005) Intracellular signalling controlling integrin activation in lymphocytes. Nat. Rev. Immunol. 5, 546–559 [DOI] [PubMed] [Google Scholar]
- 54. Wegener K. L., Partridge A. W., Han J., Pickford A. R., Liddington R. C., Ginsberg M. H., and Campbell I. D. (2007) Structural basis of integrin activation by talin. Cell 128, 171–182 [DOI] [PubMed] [Google Scholar]
- 55. Theodosiou M., Widmaier M., Böttcher R. T., Rognoni E., Veelders M., Bharadwaj M., Lambacher A., Austen K., Müller D. J., Zent R., and Fässler R. (2016) Kindlin-2 cooperates with talin to activate integrins and induces cell spreading by directly binding paxillin. Elife 5, e10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hato T., Pampori N., and Shattil S. J. (1998) Complementary roles for receptor clustering and conformational change in the adhesive and signaling functions of integrin αIIbβ3. J. Cell Biol. 141, 1685–1695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Carman C. V., and Springer T. A. (2003) Integrin avidity regulation: are changes in affinity and conformation underemphasized? Curr. Opin. Cell Biol. 15, 547–556 [DOI] [PubMed] [Google Scholar]
- 58. Chigaev A., Zwartz G., Graves S. W., Dwyer D. C., Tsuji H., Foutz T. D., Edwards B. S., Prossnitz E. R., Larson R. S., and Sklar L. A. (2003) α4β1 integrin affinity changes govern cell adhesion. J. Biol. Chem. 278, 38174–38182 [DOI] [PubMed] [Google Scholar]
- 59. Lefort C. T., Rossaint J., Moser M., Petrich B. G., Zarbock A., Monkley S. J., Critchley D. R., Ginsberg M. H., Fässler R., and Ley K. (2012) Distinct roles for talin-1 and kindlin-3 in LFA-1 extension and affinity regulation. Blood 119, 4275–4282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ye F., Petrich B. G., Anekal P., Lefort C. T., Kasirer-Friede A., Shattil S. J., Ruppert R., Moser M., Fässler R., and Ginsberg M. H. (2013) The mechanism of kindlin-mediated activation of integrin αIIbβ3. Curr. Biol. 23, 2288–2295 [DOI] [PMC free article] [PubMed] [Google Scholar]