

Abstract
The G-protein-coupled chemokine receptor CXCR4 generates signals that lead to cell migration, cell proliferation, and other survival mechanisms that result in the metastatic spread of primary tumor cells to distal organs. Numerous studies have demonstrated that CXCR4 can form homodimers or can heterodimerize with other G-protein-coupled receptors to form receptor complexes that can amplify or decrease the signaling capacity of each individual receptor. Using biophysical and biochemical approaches, we found that CXCR4 can form an induced heterodimer with cannabinoid receptor 2 (CB2) in human breast and prostate cancer cells. Simultaneous, agonist-dependent activation of CXCR4 and CB2 resulted in reduced CXCR4-mediated expression of phosphorylated ERK1/2 and ultimately reduced cancer cell functions such as calcium mobilization and cellular chemotaxis. Given that treatment with cannabinoids has been shown to reduce invasiveness of cancer cells as well as CXCR4-mediated migration of immune cells, it is plausible that CXCR4 signaling can be silenced through a physical heterodimeric association with CB2, thereby inhibiting subsequent functions of CXCR4. Taken together, the data illustrate a mechanism by which the cannabinoid system can negatively modulate CXCR4 receptor function and perhaps tumor progression.
Keywords: CXC chemokine receptor type 4 (CXCR-4), cannabinoid receptor, dimerization, G protein-coupled receptor (GPCR), metastasis
Introduction
G-protein-coupled receptors (GPCRs)2 constitute the largest family of transmembrane receptors (1, 2), and their activation by an appropriate agonist triggers signaling through G-protein α (Gα) and/or βγ subunits (3, 4), leading to context-dependent outcomes. GPCRs have been reported to form homodimers, homomultimers, or heterodimers with related or unrelated GPCRs (5). The resultant heterodimers often generate pharmacological outcomes that are distinct from those of GPCR homodimers. Hence, GPCR heterodimers have become attractive targets for new drug development.
The G-protein-coupled chemokine receptor CXCR4 is expressed on the surface of endothelial and epithelial cells of many tissues (6, 7), and upon activation by its agonist, stromal cell-derived 1α (SDF1α), CXCR4 generates signals resulting in processes that favor tissue remodeling such as hematopoiesis, angiogenesis, normal tissue maintenance and development, cell migration, and cell proliferation (8–19). These functions make CXCR4 a key participant in cancer development, progression, and metastasis (20–24). Clinically, expression of CXCR4 protein in tumors is used to predict tumor aggressiveness, survival probability, and metastasis-associated mortality (17, 20, 21, 25–28). Therefore, developing agents that can inhibit the action of CXCR4 in early and advanced stages of cancer may be effective in preventing and managing metastasis (26).
Cannabinoid receptors 1 (CB1) and 2 (CB2) (29, 30) are systems comprising receptors, their agonists (exocannabinoids and endocannabinoids), and enzymes for their metabolism (29, 30). CB1 is highly expressed in the brain (31), whereas CB2 is expressed in a variety of other tissues (32–36). The most notable cannabinoid is tetrahydrocannabinol, the primary psychoactive compound derived from marijuana. Humans naturally produce endogenous cannabinoids that activate the same receptors as tetrahydrocannabinol, resulting in pharmacological, behavioral, and immunological effects (37–39). However, cannabinoids have received considerable attention in cancer due to their abilities to induce apoptosis and cell growth arrest (40–42), diminution of angiogenesis (43, 44), and inhibition of cellular movement among diverse tissue types (45–50).
Chemokine receptors form heterodimers with related and non-related GPCRs, resulting in inhibition or amplification of signaling normally individually mediated by these receptors (51–56). Mellado et al. (57) demonstrated that, in cells simultaneously stimulated with respective agonists for chemokine receptors CCR2 and CCR5, this treatment induced their dimerization, resulting in altered signaling through the G-protein Gαq/11 instead of the traditional Gαi (57). As a result, the receptors resisted internalization and were desensitized, a functional contrast to the activity of their individual receptor homodimers (57). Pello et al. (58) identified a dimer comprising CXCR4 and the δ-opioid receptor in immune cells whereby each homodimeric receptor complex mediated signaling when stimulated with their respective agonists SDF1α and [d-Pen2,d-Pen5]enkephalin. However, as evidenced by fluorescence resonance energy transfer (FRET) analysis, simultaneous treatment with both agonists promoted the formation of CXCR4/δ-opioid receptor heterodimers that, although unable to signal, still retained the ability to bind their respective agonists (58). As another example, simultaneous treatment with respective CXCR4 and CB2 agonists caused a reduction in CB2-induced analgesia (relief of pain), suggesting a functional desensitization of CB2 (59), although a mechanism was not analyzed. Moreover, CB2 agonists inhibited CXCR4-tropic HIV infection by altering CD4+ T cell activation (60).
In the present study, we hypothesized that CXCR4-mediated functions can be abrogated by simultaneous, agonist-induced heterodimerization of CXCR4 with CB2, leading to an overall attenuation of cell metastasis and ultimately progression of tumors. To this end, we analyzed a physical heterodimeric association between CXCR4 and CB2, cellular migration, and subsequent biological functions upon simultaneous stimulation by their respective ligands. Our results complement other reported model systems in which the formation of a heterodimerized GPCR receptor complex attenuated downstream functions that would otherwise result from individually signaling receptors.
Materials and Methods
Cell Lines, Ligands, and Antibodies
Human metastatic mammary adenocarcinoma cell line MDA-MB-231, human metastatic prostate adenocarcinoma cell line PC3, and human embryonic kidney cell line 293T (HEK 293T) were purchased from American Type Culture Collection (ATCC). All cell lines were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 1% non-essential amino acids, and 1% antibiotic/antimycotic. Cells were cultured in a humidified incubator (5% CO2) at 37 °C. Human SDF1α ligand (CXCR4 agonist; 100 ng/ml) was from PeproTech, Inc. Human AM1241 ligand (CB2 agonist; 1 μm) was from Cayman Chemicals. AMD3100 (CXCR4 antagonist; 1 μg/ml) was from Sigma-Aldrich. AM630 (CB2 antagonist; 1 μm) and JWH-015 (CB2 agonist; 1 μm) were from Cayman Chemicals. Agonists and antagonists were diluted in RPMI 1640 medium unless otherwise indicated. For agonist studies, cells were serum-starved in RPMI 1640 medium only (0% serum, 0% non-essential amino acids, and 0% antibiotic/antimycotic) for 24 h in 5% CO2 at 37 °C prior to treating with ligands. Samples marked as “control” or “untreated” received fresh starvation medium supplemented with corresponding vehicles (e.g. 0.1% bovine serum albumin (BSA) in PBS, dimethyl sulfoxide, etc.) for each agonist or antagonist at the time of experiment.
Anti-CXCR4 monoclonal antibody was from R&D Systems (MAB172), and anti-CXCR4 polyclonal antibody was from Santa Cruz Biotechnology (sc-9046); mouse anti-CXCR4 monoclonal antibody (Santa Cruz Biotechnology; sc-53534) was used for the Duolink® assay only. Anti-CB2 was originally from Chemicon (now EMD Millipore; 216407-200UG) and Cayman (101550). All secondary antibodies were from Jackson ImmunoResearch Laboratories. Anti-phospho-ERK1/2 (4370), anti-phospho-AKT (4060), and anti-total AKT (4685) antibodies were from Cell Signaling Technology. Total anti-ERK1/2 was from Invitrogen BIOSOURCE (44-654G), and anti-EP-2 antibody was from Santa Cruz Biotechnology (sc-20675). Human melanocortin-5 receptor antibody was from R&D Systems (MAB8205).
The calcium assay was from Enzo Life Sciences, and the Duolink proximity ligation assay was from Sigma-Aldrich. CHAPS buffer was from Amresco, radioimmune precipitation assay (RIPA) buffer was from Boston Bioproducts, and cell lysis buffer was from Cell Signaling Technology.
FACScan Analysis of CXCR4 Expression
One million MDA-MB-231 cells were serum-starved for 24 h followed by treatment with 100 ng/ml SDF1α, 1 μm AM1241, or SDF1α and AM1241 simultaneously for 10 min. Cells were detached by incubation with 1× citric saline at 37 °C, washed once in 1× PBS, and then centrifuged at 200 × g for 5 min. Thereafter cells were fixed in 4% cold paraformaldehyde for 30 min, and flocculent cells were washed in 0.1 m Tris, glycine, pH 7.3, followed by 1× PBS, then centrifuged at 300 × g for 5 min at 4 °C, and blocked in 1% BSA in PBS on ice for 60 min. Cells were incubated with human anti-CXCR4 (1:100; R&D Systems) at 4 °C overnight in blocking buffer followed by a wash in 1× PBS and centrifugation at 300 × g for 5 min at 4 °C. Cells were incubated with FITC-conjugated secondary antibody (1:500) for 45 min at 4 °C and washed twice in 1× PBS, and cell aggregates were removed using a mesh filter and stored in 0.1% BSA in 1× PBS until analyzed. Analysis was done on a FACScan (BD Biosciences) using BD CellQuest software. All experiments were performed at least thrice, and the mean and S.E. were calculated. Significant differences were analyzed by a t test (***, p < 0.05; **, p < 0.01; *, p < 0.001).
Immunoprecipitation and Western Blotting Analysis
Fig. 3A
FIGURE 3.

Simultaneous SDF1α/AM1241 treatment induced physical association between CXCR4 and CB2. A, PC3 cells were treated for 15 min with SDF1α, AM1241, or SDF1α/AM1241 simultaneously. Cells were lysed and then incubated with anti-CXCR4 antibody for 1 h at 4 °C prior to adding Protein A/G PLUS-agarose beads and further incubating overnight at 4 °C. Immunoprecipitates were separated by 10% SDS-PAGE and then immunoblotted for CB2 protein expression. CB2 Eluent represents immunoprecipitated proteins attached to Protein A/G beads; CB2 Supernatant represents protein lysate after immunoprecipitation and the first centrifugal spin; CXCR4 Input represents lysate prior to immunoprecipitation. B, MDA-MB-231 cells were harvested as described above. Immunoprecipitates were separated by 10% SDS-PAGE and then immunoblotted for CB2 or EP-2 protein expression. C, PC3 cells were harvested as described above, and immunoprecipitates were separated by 10% SDS-PAGE and then immunoblotted for CB2 or melanocortin-5 (MC5). Each experiment was performed at least twice. IP, immunoprecipitation; WB, Western blotting.
One million five hundred cells were serum-starved for 24 h prior to treatment with 100 ng/ml SDF1α, 1 μm AM1241, or SDF1α and AM1241 simultaneously for 10 min. Cells were solubilized in 0.01% (w/v) CHAPS buffer for 30 min at room temperature, and then radioimmune precipitation assay buffer was added to each sample at a 1:1 dilution at a final volume of 500 μl per sample as described (61). Samples were passed through a 25-gauge needle 15 times, and then equal amounts of protein were incubated with an anti-CXCR4 antibody (1:100; Santa Cruz Biotechnology) for 1 h at 4 °C followed by the addition of Protein A/G PLUS-agarose beads (1:50; Santa Cruz Biotechnology) to samples for an overnight incubation at 4 °C. Alternatively, 30 μg of whole cell lysate per sample were loaded and separated by SDS-PAGE to demonstrate that equal amounts of CXCR4 were immunoprecipitated from samples; this served as input. After immunoprecipitation, cell lysates were separated from the protein-bead complexes by centrifugation (1000 × g for 5 min at 4 °C) and saved as supernatant; supernatant was used to demonstrate that CB2 was immunoprecipitated from samples. The beads were washed thrice with 1× Tris-buffered saline (TBS), and the resulting proteins were eluted from the beads by boiling in 1× Laemmli buffer and then separated by 10% SDS-PAGE. The proteins eluted from the beads were denoted as eluent. Protein-bound PVDF membranes were blocked in 5% nonfat dried milk (milk) in Tris-buffered saline and Tween 20 (TBST) and immunoblotted in 3% milk in TBST with an anti-CB2 antibody (1:500; Cayman Chemicals). Primary antibodies were detected with corresponding HRP-conjugated secondary antibodies (1:1000) and enhanced chemiluminescence (SuperSignal West Pico Chemiluminescent Substrate, Pierce/Thermo Scientific).
Fig. 3, B and C
One million cells per sample were serum-starved for 24 h prior to treatment with 100 ng/ml SDF1α, 1 μm AM1241, or SDF1α and AM1241 simultaneously for 10 min. Cells were lysed and sonicated in cell lysis buffer, and then the lysate was incubated on ice and isolated by centrifugation at maximal speed for 20 min at 4 °C. Equal amounts of protein per sample were precleared with rabbit IgG TrueBlot beads (1:50; Santa Cruz Biotechnology) for 2 h at 4 °C and centrifuged at maximal speed, and the cleared lysate was immunoprecipitated with anti-CXCR4 antibody (1:100; Santa Cruz Biotechnology) overnight at 4 °C followed by an incubation with Protein A/G beads for 2 h at 4 °C. Immunocomplexes were washed in 1× PBS, boiled in 1× Laemmli buffer, and separated by 10% SDS-PAGE. Protein-bound PVDF membranes were blocked in 5% milk in TBST and subsequently incubated with anti-CB2 (1:1000), anti-EP-2 (1:1000), or anti-melanocortin-5 (1:1000) in 3% BSA in TBST overnight at 4 °C. Bound antibodies were detected by HRP-conjugated secondary antibody and enhanced chemiluminescence (SuperSignal West Pico Chemiluminescent Substrate, Pierce/Thermo Scientific).
Western Blotting Analysis
Two hundred fifty thousand cells per sample were serum-starved for 24 h prior treatment with 100 ng/ml SDF1α, 1 μm AM1241, or SDF1α and AM1241 simultaneously at various time points. Cells were lysed and sonicated in 1× cell lysis buffer prior to incubation on ice for 30 min. Lysates were centrifuged at maximal speed for 20 min at 4 °C, and then equal amounts of protein per sample were separated by SDS-PAGE and transferred to PVDF membrane. Protein-bound membranes were blocked in 5% milk in TBST and subsequently incubated with primary antibodies (1:1000) overnight at 4 °C in 1–3% BSA in TBST or 5% milk in TBST. Primary antibodies were detected by HRP-conjugated secondary antibodies and enhanced chemiluminescence (SuperSignal West Pico Chemiluminescent Substrate).
Cell Migration
Fig. 7C
FIGURE 7.


Interaction between CXCR4 and CB2 inhibited CXCR4-mediated functions. Serum-starved PC3 (A) or MDA-MB-231 (B) cells, both preincubated with calcium dye for 1 h, were treated with SDF1α, AM1241, AMD3100, or SDF1α/AM1241 simultaneously at 37 °C for 15 min (A) or 30 s (B). Calcium flux was determined at an excitation of 490 nm and emission of 525 nm on a microplate reader. C, serum-starved MDA-MB-231 cells were treated with SDF1α, AM1241, or SDF1α/AM1241 simultaneously for a 1 h prior to detaching with 1× citric saline. Transwell plates were prepared for migration by placing RPMI 1640 medium alone, SDF1α, AM1241, or SDF1α/AM1241 simultaneously in the lower (bottom) chamber. Equal cell numbers (∼ 2 × 104) were seeded in the upper well of 24-well transmigration chambers to allow migration/chemotaxis toward ligand solutions in the bottom chamber at 37 °C for 4 h. Chambers were cleaned, stained, and counted on a light microscope. D–G, serum-starved MDA-MB-231 cells were seeded in the upper chamber of Transwell inserts, and various combinations of RPMI 1640 medium alone, SDF1α (D), AM1241 (D, E, and G), CXCL1 (100 ng/ml) (E), or JWH-015 (1 μm) and AM630 (1 μm) (F) were added to the lower (bottom) chambers. Cells were allowed to migrate toward ligand combinations at 37 °C prior to harvesting as described above. The migration index was calculated as the x-fold change in migration observed over untreated treated cells or cells that migrated to SDF1α alone. Statistical analysis is described under “Materials and Methods.” ***, p < 0.05; **, p < 0.01; *, p < 0.001. Scale bar, 50 μm. Each experiment above was performed at least thrice. Error bars represent S.E. A graphical representation of the restoration of CXCR4-mediated migration in the presence of AMD630 is shown in the bottom panel.
Two hundred fifty thousand cells were serum-starved for 24 h prior to treatment with 100 ng/ml SDF1α, 1 μm AM1241, or SDF1α/AM1241 simultaneously. Cells were detached with 1× citric saline at 37 °C, washed in 1× PBS, centrifuged at 300 × g, and counted. Twenty thousand cells per experimental sample were diluted in 100 μl of RPMI 1640 medium and seeded into the upper well of 24-well transmigration chambers (8.0-μm pore; Transwell, Costar) to allow for chemotaxis. Plates were incubated for 4 h in 5% CO2 at 37 °C, then the inner-upper chambers were cleaned with a cotton swab, and cells attached to the outer-upper chamber were stained with the Hema 3 system (Fisher Scientific). Alternatively, ligands diluted in 400 μl of RPMI 1640 medium were added to the lower (bottom) chambers as indicated, and 2.5 × 105 cells diluted in 100 μl of RPMI 1640 medium were added to each upper chamber. Plates were incubated for 4 h in 5% CO2 at 37 °C, then the inner-upper chambers were cleaned with a cotton swab, and cells attached to the outer-upper chamber were stained with the Hema 3 system. Five representative fields of each insert were counted on a light microscope, and the migration index was calculated and graphed as the x-fold change in migration observed over control cells. Experiments were performed at least thrice, and the mean and S.E. were calculated. Significant differences were analyzed by a t test or one-way analysis of variance (***, p < 0.05; **, p < 0.01; *, p < 0.001).
Fig. 7, D–F
After a 24-h serum starvation, 4 × 104 cells were seeded in the upper chamber of Transwell inserts, and either SDF1α (100 ng/ml), AM1241 (1 μm), chemokine (CXC motif) ligand 1 (CXCL1) (100 ng/ml), JWH-015 (1 μm), or any combination of the preceding diluted in RPMI 1640 medium was added to the lower chambers. Cells were allowed to chemotax for 5 h in 5% CO2 at 37 °C. After incubation, non-migrated cells were removed, and remaining cells were then fixed and stained as described immediately above.
Fig. 7G
After a 24-h serum starvation, cells were pretreated with AM630 (1 μm) for 30 min prior to harvesting cells as described immediately above. The scale bar is 50 μm in Fig. 7, D–G.
Live Cell Binding Assay
Two hundred fifty thousand MDA-MB-231 (CXCR4-positive) or 293T (CXCR4-negative) cells were plated in 96-well puncher plates in complete medium (six rows down and eight rows across) and grown to a density of 75%. To assign absolute affinity of each ligand for CXCR4 receptor, a competitive displacement assay was used as described by Misra et al. (62) using cold ligand AM1241 as a test compound along with [99mTc-MAS3]SDF1α and [99mTc-MAS3]AM1241 as radiotracers on the surfaces of cell lines. To avoid internalization of the radioligand due to constitutive endocytosis (63), live cell binding was performed at 4 °C. Cells were washed twice with ice-cold 1× PBS, pH 7.4, and incubated for 20 min at 4 °C with 0.5 μCi of radiotracer in the presence or absence of the test compound. Cells were then washed thrice with 1× PBS, and the well contents were transferred directly to 12 × 75-mm plastic tubes placed in γ counter racks. Transfer was accomplished using a modified 96-well puncher and disposable punch tips. Well contents were counted on a model 1470 Wallac Wizard 10-detector γ counter.
SDF1α siRNA Transfection
Cells were transfected with 100 nm SDF1α-targeted siRNA (Santa Cruz Biotechnology or Bioneer (Fig. 5C)) and grown to 80% confluence in 6-well plates. siRNA and Lipofectamine 2000 were premixed in Opti-MEM Transfection Reagent and applied to cells for incubation. After 24 h, the sample media were removed and replaced with serum-free RPMI 1640 medium for an additional 24 h prior to treatment with SDF1α and/or AM1241. Cells were then harvested for the respective assays.
FIGURE 5.

Diminution of endogenous SDF1α did not inhibit endogenous CXCR4/CB2 heterodimerization. MDA-MB-231 (A) and PC3 (E) cells were transfected with siRNA (Santa Cruz Biotechnology; 100 nm) targeting SDF1α prior to harvesting for Duolink. Transfected siRNA reduced endogenous SDF1α production in MDA-MB-231 (D) and PC3 (G) cells as well as secretion into the medium in MDA-MB-231 cells (C) (Bioneer; 100 nm). α-Tubulin was used as a loading control. Homodimerization of MDA-MB-231 (B) and PC3 (F) cells was analyzed to ensure that the siRNA did not alter receptor function. Magnification, 40×; scale bars, 20 μm. Each experiment was performed at least twice.
SDF1α Secretion Assay
Secretion of SDF1α into the medium samples was determined as described (64). After a 24-h transfection as described above, the medium was removed and replaced with serum-free RPMI 1640 medium for an additional 24 h. RPMI 1640 medium was collected and vortexed, and 100 μl of each sample plus 1× Laemmli buffer was prepared for separation by 10% SDS-PAGE. Immunoblots were processed using standard Western blotting procedures.
Calcium (Ca2+) Mobilization Assay
Detection of calcium mobilization was conducted according to the manufacturer's instructions (Enzo Biosciences). Briefly, 6 × 104 cells per sample were plated in 96-well plates and serum-starved cells for 24 h in phenol-free RPMI 1640 medium. At hour 23 of the serum starvation, 100 μl of Fluo-Forte calcium dye (Enzo Biosciences) was added to each well; alternatively, AMD3100 (1 μg/ml diluted in 100 μl of dye) was added to designated samples. Plates were wrapped in foil and incubated for 45 min at 37 °C and then for 15 min at room temperature. Twenty microliters of SDF1α (100 ng/ml), AM1241 (1 μm), or SDF1α and AM1241 simultaneously was added to the dye solution in designated wells on ice, bringing the final volume of each well to 120 μl and a final ratio of 1:6. Plates were again incubated at 37 °C for 15 min (PC3) or 30 s (MDA-MB-231) and immediately returned to ice to stop the reaction. Calcium flux was monitored immediately on a microplate reader at an excitation of 490 nm and emission of 525 nm. Mobilization was performed at least thrice, and the mean and S.E. were calculated. Significant differences were analyzed by a t test or one-way analysis of variance (***, p < 0.05; **, p < 0.01; *, p < 0.001).
Duolink Proximity Ligation Assay
Physical association of CXCR4 and CB2 was analyzed according to the manufacturer's instructions (Sigma-Aldrich). Briefly, 3 × 105 cells were plated on tissue culture treated coverslips and serum-starved for 24 h prior to fixing in 2% paraformaldehyde or 100% methanol. Cell lines were washed in 0.1 m Tris, glycine, pH 7.3, and washed in 1× PBS, and then cells were blocked in kit-provided buffer for 30 min at 37 °C. Solutions were tapped off (not aspirated), blocked in kit-provided buffer, and then incubated with the following primary antibodies that were diluted in kit-provided diluent, mouse anti-CXCR4 monoclonal (1:100; Santa Cruz Biotechnology) and rabbit anti-CB2 monoclonal antibodies (1:100; Cayman Chemicals), overnight at 37 °C. Solutions were tapped off and washed with Wash Buffer A prior to adding PLUS anti-rabbit and MINUS anti-mouse proximity ligation assay (PLA) probes (1:5 dilution in kit diluent). Coverslips were incubated for 1 h at 37 °C, solutions were tapped off and washed as described above, and then kit-provided ligation buffer was added to each sample for further incubation for 30 min at 37 °C. Solutions were tapped off and washed as described above, and then amplification buffer was added to each slide for a final incubation at 37 °C for 100 min. Slides were tapped off and washed in Wash Buffer B and then mounted in the provided DAPI-prestained mounting medium. Images were viewed and captured via a Zeiss LSM 700 confocal microscope using a 20× objective (Zeiss Plan-Apochromat, 0.8 numerical aperture) at room temperature. CXCR4/CB2 heterodimers were detected via positive ligation of PLA probes on a rhodamine filter (excitation, 571 nm; emission, 590 nm). Data were acquired and analyzed using Zen (Blue Edition) software. Scale bars are 20 or 50 μm.
Results
Simultaneous Treatment with SDF1α and AM1241 Antagonized Internalization of CXCR4
Exposure to SDF1α causes CXCR4 to be rapidly internalized to attenuate signaling and recycled back to the plasma membrane for later ligand binding (65, 66). Therefore, a reduction in detectable CXCR4 surface expression correlates with receptor activation (67). Using MDA-MB-231 human breast cancer cells, we observed a reduction in the surface expression of CXCR4 upon treatment with SDF1α, the agonist for CXCR4 (Fig. 1). AM1241 did not trigger surface reduction of CXCR4 as it is a CB2 agonist; however, simultaneous (combined) treatment of MDA-MB-231 cells with SDF1α and AM1241 failed to reduce surface expression of CXCR4 compared with treatment with SDF1α alone (Fig. 1).
FIGURE 1.
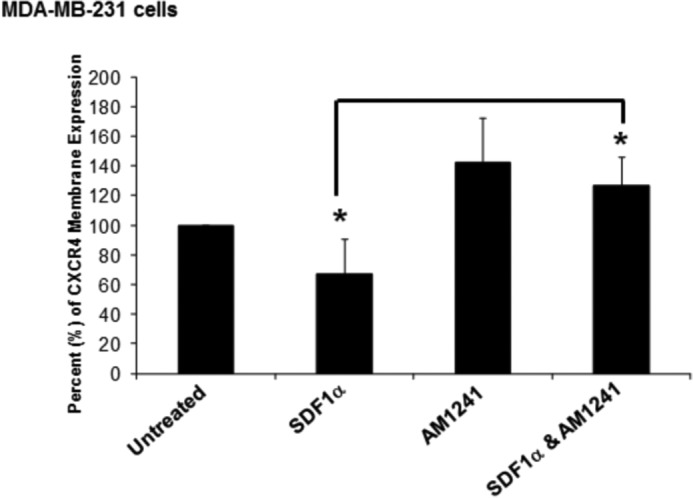
Simultaneous SDF1α/AM1241 treatment inhibited internalization of CXCR4. One million MDA-MB-231 cells were serum-starved for 24 h prior to treatment with SDF1α, AM1241, or SDF1α/AM1241 simultaneously for 10 min. Cells were detached by incubation with 1× citric saline at 37 °C, washed in 1× PBS, centrifuged, then resuspended, and fixed in 4% cold paraformaldehyde. Cells were blocked in 1% BSA in PBS on ice for 60 min prior to incubation with a CXCR4-specific antibody at 4 °C overnight in blocking buffer. The next day, cells were washed, centrifuged at maximal speed, and then incubated with a FITC-conjugated secondary antibody for 30 min at 4 °C. After washes in 1× PBS, cells were stored in 0.1% BSA in PBS until analyzed by FACS. Statistical analysis is described under “Materials and Methods.” *, p < 0.001. Experiments were performed at least thrice. Error bars represent S.E.
AM1241 Did Not Compete for CXCR4 Binding
There is growing evidence that GPCRs form heterodimers with other GPCRs; however, one concern has been that abrogated receptor function, as a result GPCR heterodimerization, might be an artifact of misguided ligands (68, 69). For this reason, we evaluated whether AM1241 competes with SDF1α for binding to CXCR4 during simultaneous stimulation with both agonists (62). To quantify the dissociation of SDF1α from CXCR4, which inversely correlates with ligand-receptor affinity on the surface of living MBA-MD-231 cells, a competition assay was used as described by Misra et al. (62). 293T human embryonic kidney cells were used as a negative control as they do not express CXCR4 (62, 70). SDF1α and AM1241 were radiolabeled as [99mTc-MAS3]SDF1α and [99mTc-MAS3]AM1241, respectively. Treatment of MDA-MB-231 cells with radiolabeled SDF1α exhibited a low dissociation constant (KD) for CXCR4 at KD = 2.4 + 0.2 nm (mean ± S.D.), inversely indicating a high affinity and preferred binding of SDF1α to CXCR4 (Fig. 2A); the measured affinity was consistent with previously published values for SDF1α binding to CXCR4 (62). The dissociation constant of AM1241 for CXCR4 was much higher at KD = 16.3 + 1.4 nm (mean ± S.D.), inversely correlating to a low affinity of AM1241 to CXCR4 (Fig. 2A). For both SDF1α and AM1241, the binding sites per cell (Bmax) were 1.21 × 104, demonstrating that the total density (concentration) of CXCR4 receptors available to ligands for binding was equivalent (Fig. 2A). To determine whether SDF1α competes with AM1241 for binding to CXCR4, we measured the dissociation constant of [99mTc-MAS3]SDF1α, simultaneously with unlabeled AM1241, for CXCR4 binding. AM1241 did not compete for binding to CXCR4 as the dissociation constant of [99mTc-MAS3]SDF1α remained low at KD = 3.0 + 1.02 nm (Bmax = 1.36 × 104), aligning with the affinity values of cells treated with SDF1α alone. These results rule out the notion that the purported abrogation of CXCR4-mediated functions may be due to competitive, or misguided, agonist binding of AM1241 to CXCR4.
FIGURE 2.

SDF1α and AM1241 did not compete for binding to CXCR4. MDA-MB-231 (A and B) or 293T cells (A) were plated in 96-well puncher plates and grown to a density of 75% prior to the experiment. To assign absolute affinity of each ligand for CXCR4 receptor, a competitive displacement assay was used as discussed by Misra et al. (62) using cold ligand AM1241 as a test compound along with [99mTc-MAS3]SDF1α and [99mTc-MAS3]AM1241 as radiotracers on the surface of MDA-MB-231 (CXCR4-positive) and 293T (CXCR4-negative) cell lines. Cells were washed twice with ice-cold 1× PBS, pH 7.4, and incubated for 20 min at 4 °C with 0.5 μCi of radiotracer in the presence or absence of the test compound. Cells were then washed thrice with 1× PBS, and the well contents were transferred directly to plastic tubes in γ counter racks. Well contents were counted on a Wallac Wizard 1470 γ counter. Experiments were performed at least thrice. Error bars represent S.E.
CXCR4 and CB2 Dimerized in Vitro
Despite the reported effects of cannabinoids on CXCR4-mediated cell migration (50, 71), a physical association between CXCR4 and CB2 and the subsequent biological effects have yet to be demonstrated. To establish that CXCR4 and CB2 associated in vitro, we first examined receptor co-immunoprecipitation. PC3 human prostate cancer cells were treated individually with either SDF1α, AM1241, or the ligands in combination prior to immunoprecipitating CXCR4 from whole cell lysate and immunoblotting with an anti-CB2 antibody. Results revealed that CXCR4 and CB2 displayed a basal level interaction in untreated control cells (Fig. 3A), an observation that has been described between CXCR4 and GPCRs in the following other model systems: (i) δ-opioid receptor (58) and (ii) CCR5 chemokine receptor (72, 73). However, the association between CXCR4 and CB2 increased upon simultaneous stimulation with both SDF1α and AM1241 (Fig. 3A). The SDF1α/AM1241-mediated induced association between CXCR4 and CB2 was specific as we were unable to immunoprecipitate CXCR4 in complex with non-related GPCRs EP-2 (Fig. 3B) in MDA-MB-231 cells and melanocortin-5 (Fig. 3C) in PC3 cells.
Next, we examined the association between CXCR4 and CB2 by performing a PLA (Duolink). This direct method required both CXCR4 and CB2 receptors to be in close proximity to allow the two separate antibody-PLA probes to ligate, implying a physical interaction (Figs. 4 and 5) (74, 75). A fluorescence signal was observed upon combined SDF1α/AM1241 treatment (Fig. 4, A and B), indicating that CXCR4 and CB2 receptors physically dimerized solely in response to simultaneous ligand binding. It is important to note that PC3 cells (Fig. 4A) were fixed in methanol, which permeabilized cells, and MDA-MB-231 cells (Fig. 4B) were fixed in paraformaldehyde, which was not permeant. Following SDF1α/AM1241 simultaneous treatment, a CXCR4-CB2 complex was detected ubiquitously throughout PC3 cells, suggesting that the two receptors maintained their association upon internalization (76). Similar to untreated cells (negative control), significant positive signal (punctate red fluorescence), indicative of physical interaction, was not detected in SDF1α-treated cells.
FIGURE 4.


Endogenous CXCR4 and CB2 formed heterodimers in cancer cells. Heterodimerization of CXCR4 and CB2 was recognized by incubating cells with primary antibodies raised in two different species and secondary antibodies linked to different DNA oligomers, one designated PLUS and one designated MINUS. If the two receptors were close enough, the two different antibody-DNA probes were able to ligate and hybridize. Following an amplification process, the presence of fluorescently tagged nucleotides allowed detection of a punctate fluorescence signal by confocal microscopy. PC3 (A) and MDA-MB-231 (B) cells were each plated on coverslips; serum-starved for 24 h; and then treated with SDF1α, AM1241, or SDF1α/AM1241 simultaneously for 1 min prior to fixing with methanol (PC3) or 2% paraformaldehyde (MDA-MB-231). Cells were prepared as described above (Duolink). A CXCR4/CB2 heterodimer was detected via positive ligation of PLA probes on a rhodamine filter (excitation, 571 nm; emission, 590 nm), and nuclei were stained with DAPI. Images were reviewed and captured using a Zeiss LSM 700 confocal microscope. Scale bars, 50 μm. B, merged images of MDA-MD-231 cells only; colocalization and heterodimerization in merged images are indicated by punctate foci and white arrows. Designated PC3 (C) or MDA-MBA-231 (D) cells were pretreated with AMD3100 (1 μg/ml) prior to treatment with SDF1α and/or AM1241. Cells were later harvested for Duolink as described above. Magnification, 20×; scale bars, 50 μm. Each experiment was performed at least twice.
Surprisingly, we also detected basal heterodimerization in AM124-treated PC3 (Fig. 4, A and C) and MDA-MB-231 cells (Fig. 4, B and D) and in our immunoprecipitation of the CXCR4-CB2 receptor complex (Fig. 3A). Because, aggressive cancer cells can produce and secrete endogenous SDF1α, we antagonized endogenous SDF1α binding to CXCR4 with the chemical inhibitor AMD3100 (77, 78), which diminished red fluorescence in AM1241-treated cells (Fig. 4, C and D). We also reduced endogenous SDF1α production and secretion into the medium via siRNA (Fig. 5, C, D, and G) and observed that red fluorescence was also greatly reduced in AM1241-treated MDA-MB-231 (Fig. 5A) and PC3 cells (Fig. 5E), which did not affect heterodimerization upon simultaneous exogenous agonist treatment (Fig. 5, A and E) or homodimerization of CXCR4 receptors (Fig. 5, B and F).
AM1241 Modulated SDF1α-induced Phosphorylation of ERK1/2
A common and pharmacologically essential attribute of GPCR heterodimers is the ability to amplify or antagonize downstream signaling that would otherwise result from activation of single constituent receptors (50, 58, 68, 71, 78). Therefore, we investigated whether changes occurred in the expression levels of phosphorylated kinases ERK1/2 and AKT when cancer cells were simultaneously stimulated with SDF1α and AM1241. A time point (15 min) was chosen at which an obvious diminution of phosphorylated kinases would be demonstrated (71), and we observed that treatment with SDF1α induced expression of phosphorylated ERK1/2 as compared with untreated control in MDA-MB-231 and PC3 cells (Fig. 6, A and B). However, simultaneous SDF1α/AM1241 treatment diminished expression of phosphorylated ERK1/2. We previously demonstrated in prostate cancer cells that treatment with SDF1α resulted in phosphorylation of ERK1/2 in a biphasic manner, whereas no changes in AKT phosphorylation status were observed (77, 78). Likewise, we observed that combined agonist treatment with SDF1α/AM1241 did not significantly reduce the phosphorylation status of AKT compared with cells treated with SDF1α alone in MDA-MB-231 cells (Fig. 6A).
FIGURE 6.

CXCR4/CB2 heterodimer modulated ERK1/2 phosphorylation. A, serum-starved MDA-MB-231 cells were treated with SDF1α, AM1241, or SDF1α/AM1241 simultaneously for 15 min. Whole protein lysates were isolated prior to separation by 10% SDS-PAGE and transferred to PVDF membranes. Protein-bound membranes were processed for Western blotting analysis as described under “Materials and Methods.” B, PC3 cells were treated with ligands for 15 min prior to harvesting as described above. α-Tubulin was used as a loading control. Each experiment was performed at least twice.
Simultaneous Treatment of SDF1α and AM1241 Blocks CXCR4-mediated Functions
In evaluating whether simultaneous treatment with SDF1α/AM1241 affected Ca2+ mobilization (79, 80), we observed that Ca2+ mobilization in PC3 (Fig. 7A) and MDA-MB-231 cells (Fig. 7B) was attenuated by simultaneous SDF1α/AM1241 treatment compared with SDF1α stimulation alone. The inability of CXCR4, as a heterodimer with CB2, to mobilize Ca2+ was comparable with treatment with AMD3100 where the antagonist inhibited CXCR4-mediated mobilization of Ca2+ (Fig. 7B).
The hallmark of CXCR4 antagonism is reducing the ability of tumor cells to migrate or chemotax. Therefore, we tested the ability of cancer cells to migrate after being pretreated with SDF1α and/or AM1241 toward culture medium only in the bottom chamber or toward combinations of SDF1α and/or AM124 in the bottom chamber. Pretreatment of MDA-MB-231 cells with SDF1α triggered cell migration through Transwell inserts toward culture medium only, an effect that was significantly reduced in cells pretreated with SDF1α and AM1241 simultaneously (Fig. 7C). Pretreating cells with SDF1α and then allowing them to migrate toward AM1241 in the bottom chamber demonstrated very little movement among the cells (Fig. 7C). Likewise, pretreating cells with AM1241 and then allowing them to migrate toward SDF1α in the bottom chamber triggered insignificant cellular movement (Fig. 7C). Combining SDF1α and AM1241 in the bottom chamber did not trigger significant chemotaxis through Transwell inserts (Fig. 7C), suggesting that agonists acting simultaneously on CXCR4 and CB2 receptors modulate cancer cell migration.
First, to determine whether cancer cells retained the capacity to migrate to chemokines other than SDF1α, supporting the argument that abrogation of CXCR4 function(s) is specific to the interaction between CXCR4 and CB2, MDA-MB-231 cells were first harvested for Transwell migration toward either CXCL1, AM1241 alone, or both in combination (Fig. 7E). Breast cancer cells migrated toward CXCL1; however, CXCL1-induced migration was reduced by the combined presence of CXCL1 and AM1241 in the bottom chamber. Second, we used an alternate CB2 agonist, JWH-015, instead of AM1241 to show that this phenomenon was not exclusive to the ligand but rather due to an agonist-induced dimerization of the receptors. We observed that the combination of agonists SDF1α and JWH-015 in the lower chamber reduced migration of breast cancer cells (Fig. 7F). Lastly, breast cancer cells were pretreated with a CB2 antagonist (AM630) prior to harvesting cells for Transwell migration toward SDF1α or AM1241 either alone or in combination. Pretreatment with a CB2 inhibitor prevented the binding of AM1241 to CB2 (Fig. 7, bottom panel), and presumably heterodimerization of CXCR4 and CB2. Therefore, AM630 restored the ability of breast cancer cells to migrate toward SDF1α even in the presence of simultaneous SDF1α and AM1241 treatment (Fig. 7G).
Discussion
In the present study, we explored the effects of agonist-induced GPCR heterodimerization on cancer cell function. In contrast to a report by McKallip et al. (81) where human breast cancer cell lines MCF-7 and MDA-MB-231 expressed low to undetectable levels of cannabinoid receptors CB1 and CB2, we and others found that human breast and prostate cancer cells express functional CB2 receptors (46, 59, 82, 83).
Here, we observed that the combination of CXCR4 and CB2 agonists specifically reduced CXCR4-mediated migration and that the CB2 antagonist AM630 reversed this effect. This study also confirmed that CXCR4 and CB2 form a physical heterodimer and that the dimer could be induced by both exogenous and endogenous agonists. Comparable data on GPCR heterodimerization have been demonstrated in other model systems. Ghosh et al. (50) observed that CB2 modulated the CXCR4-induced transendothelial migration of T cells and therefore altered multiple immune and inflammatory responses. Pello et al. (58) reported that simultaneous treatment with SDF1α and [d-Pen2,d-Pen5]enkephalin blocked CXCR4-mediated adhesion of leukocytes.
Until recently, studies on CB2 in cancer cells have focused on antitumor activity (81, 82) and proliferation (58). Now, synthetic cannabinoids are reported to inhibit the migration of cancer cells through CB2 signaling (83). Nonetheless, these reports have not described direct biochemical mechanisms by which CB2 or cannabinoids inhibit migration. Herein, we diminished endogenous SDF1α, which disrupted the heterodimer in AM1241-treated cells. This observation was significant because it suggests and possibly explains how tumors that overexpress endogenous SDF1α respond to exogenous cannabinoid treatment. Therefore, we posit that metastatic regulation via CXCR4 may be a result of GPCR heterodimerization with CB2 and subsequent heterodimer-induced antagonism of signaling and or/function.
Homodimerization of CXCR4 receptors in malignant cells is important in directing migration of cancer cells, and receptor desensitization is an important regulatory mechanism for CXCR4 signaling (66, 84). We also know that receptors involved in pain sensation are emerging as desensitizers of chemokine responses (58, 85). For instance, preincubation with the μ-opioid receptor agonist [d-Ala2,N-Me-Phe4,Gly5-ol]enkephalin (DAMGO) abrogated CCL5-induced chemotaxis in Chinese hamster ovary cells co-expressing μ-opioid receptor and CCR5 (86). Conversely, Pello et al. (58) excluded desensitization in their model of immune cells expressing a CXCR4/δ-opioid receptor heterodimer for reasons such as (i) cells incubated with the opioid ligand [d-Pen2,d-Pen5]enkephalin alone did not internalize CXCR4 and (ii) cells incubated with [d-Pen2,d-Pen5]enkephalin alone showed no alteration in CXCL12-mediated adhesion (50, 87). We agree with Pello et al. (58) in that our observations were not based on AM1241-induced desensitization of CXCR4 especially because AM1241 did not compete with SDF1α for binding to CXCR4 and that treatment with AM1241 did not trigger internalization of CXCR4, indicating that CXCR4 is not a target of AM1241.
Our studies and others (58, 86, 88) strongly support that simultaneous stimulation with GPCR agonists could result in a dynamic receptor complex that will disrupt downstream signaling. The decrease observed in phosphorylated ERK1/2 protein expression in both MDA-MB-231 and PC3 cells indicates that GPCR receptor heterodimerization induces distinct and specific signaling events (57). In the context of this study, the induced dimer diminished subsequent expression of phosphorylated ERK1/2 that would have otherwise come from individually activated CXCR4. In our experience working with SDF1α (77, 78) and as reported by Nasser et al. (71), ERK1/2 is more responsive to the SDF1α/CXCR4 signaling axis than AKT, especially in the context of cell migration. Robust CB2-mediated activation of ERK1/2 is cell line-specific, agonist-specific, and/or coupled with other signaling pathways (89, 90). Moreover, activation of MAPK/ERK1/2 signaling is more directly downstream of CXCR4 receptor signaling than activation of AKT signaling (91). It is possible that cancer cells may choose either ERK1/2 or AKT signaling with regard to desired biological function. In our experience and work with cellular migration and movement, CXCR4 signals through ERK1/2.
GPCR heterodimerization could naturally control signals triggered individually by CXCR4 without the side effects of antagonists, especially in situations where the heterodimerizing receptors are expressed on the same tumor cell. Moreover, stabilization of the CXCR4/CB2 receptor heterodimer could represent a natural mechanism that alerts the organism to tumor formation by reducing the signaling potential of CXCR4. Whereas a single antagonist would inhibit CXCR4 function in a wide spectrum of cell types, including immune cells, a combination of CXCR4 and CB2 agonists intended to promote inhibition via heterodimerization could exert this inhibitory effect more selectively on tumor cells with high expression of both receptors. Although one could argue that such an approach might induce permanent or long term effects, a counterargument is that this outcome is unlikely because agonist-dependent dimerization of GPCRs is rapid and reversible, and such dimers can be disassembled by endocytosis (92).
Author Contributions
C. V. H. contributed to Figs. 1 and 2, designed the study, and wrote the manuscript. C. J. C. performed and analyzed experiments in Figs. 3–6 and contributed to the preparation of these figures. K. A. S. performed and analyzed experiments in Fig. 4 and contributed to the preparation of the figure. K. J. J. performed and analyzed experiments in Fig. 6 and contributed to the preparation of the figure. A. S. D. performed experiments in Fig. 6. M. A. C. performed and analyzed experiments in Figs. 1 and 5 and contributed to the preparation of the figures and editing of the manuscript. B. J. S. performed experiments in Fig. 3 and contributed to the preparation of the figure. A. I. M. contributed to the analysis of Figs. 4 and 5.
Acknowledgments
We thank Dr. Hava K. Avraham of the Center of Drug Discovery at Northeastern University and Drs. John Frangioni and Preeti Misra of the Center for Molecular Imaging, Beth Israel Deaconess Medical Center and Harvard Medical School for technical expertise and advice.
This work was supported by National Institutes of Health Grants 1R01GM106020, 3R01GM106020, and R25GM060414 from the NIGMS and Grant 5G12MD007590 from the Research Centers in Minority Institutions Program. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- GPCR
- G-protein-coupled receptor
- AKT
- protein kinase B
- CB1
- cannabinoid receptor 1
- CB2
- cannabinoid receptor 2
- CCR2
- CC chemokine receptor type 2
- CCR5
- CC chemokine receptor type 5
- CXCR4
- chemokine (CXC motif) receptor 4
- SDF1α
- stromal cell-derived factor 1α
- MAS3
- S-acetylmercaptoacetyltriserine
- PLA
- proximity ligation assay
- CXCL1
- chemokine (CXC motif) ligand 1
- Pen
- penicillamine.
References
- 1. Albizu L., Cottet M., Kralikova M., Stoev S., Seyer R., Brabet I., Roux T., Bazin H., Bourrier E., Lamarque L., Breton C., Rives M. L., Newman A., Javitch J., Trinquet E., et al. (2010) Time-resolved FRET between GPCR ligands reveals oligomers in native tissues. Nat. Chem. Biol. 6, 587–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Satake H., and Sakai T. (2008) Recent advances and perceptions in studies of heterodimerization between G protein-coupled receptors. Protein Pept. Lett. 15, 300–308 [DOI] [PubMed] [Google Scholar]
- 3. Pierce K. L., Premont R. T., and Lefkowitz R. J. (2002) Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 3, 639–650 [DOI] [PubMed] [Google Scholar]
- 4. Gilman A. G. (1987) G proteins: transducers of receptor-generated signals. Annu. Rev. Biochem. 56, 615–649 [DOI] [PubMed] [Google Scholar]
- 5. Franco R., Casadó V., Cortés A., Ferrada C., Mallol J., Woods A., Lluis C., Canela E. I., and Ferré S. (2007) Basic concepts in G-protein-coupled receptor homo- and heterodimerization. ScientificWorldJournal 7, 48–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gupta S. K., Lysko P. G., Pillarisetti K., Ohlstein E., and Stadel J. M. (1998) Chemokine receptors in human endothelial cells. Functional expression of CXCR4 and its transcriptional regulation by inflammatory cytokines. J. Biol. Chem. 273, 4282–4287 [DOI] [PubMed] [Google Scholar]
- 7. Zlotnik A., and Yoshie O. (2000) Chemokines: a new classification system and their role in immunity. Immunity 12, 121–127 [DOI] [PubMed] [Google Scholar]
- 8. Rollins B. J. (1997) Chemokines. Blood 90, 909–928 [PubMed] [Google Scholar]
- 9. Premack B. A., and Schall T. J. (1996) Chemokine receptors: gateways to inflammation and infection. Nat. Med. 2, 1174–1178 [DOI] [PubMed] [Google Scholar]
- 10. Bokoch G. M. (1995) Chemoattractant signaling and leukocyte activation. Blood 86, 1649–1660 [PubMed] [Google Scholar]
- 11. Gerard C., and Gerard N. P. (1994) The pro-inflammatory seven-transmembrane segment receptors of the leukocyte. Curr. Opin. Immunol. 6, 140–145 [DOI] [PubMed] [Google Scholar]
- 12. Taub D. D., and Oppenheim J. J. (1994) Chemokines, inflammation and the immune system. Ther. Immunol. 1, 229–246 [PubMed] [Google Scholar]
- 13. Luster A. D. (1998) Chemokines—chemotactic cytokines that mediate inflammation. N. Engl. J. Med. 338, 436–445 [DOI] [PubMed] [Google Scholar]
- 14. Shirozu M., Nakano T., Inazawa J., Tashiro K., Tada H., Shinohara T., and Honjo T. (1995) Structure and chromosomal localization of the human stromal cell-derived factor 1 (SDF1) gene. Genomics 28, 495–500 [DOI] [PubMed] [Google Scholar]
- 15. Hamada T., Tashiro K., Tada H., Inazawa J., Shirozu M., Shibahara K., Nakamura T., Martina N., Nakano T., and Honjo T. (1996) Isolation and characterization of a novel secretory protein, stromal cell-derived factor-2 (SDF-2) using the signal sequence trap method. Gene 176, 211–214 [DOI] [PubMed] [Google Scholar]
- 16. Ohtani Y., Minami M., Kawaguchi N., Nishiyori A., Yamamoto J., Takami S., and Satoh M. (1998) Expression of stromal cell-derived factor-1 and CXCR4 chemokine receptor mRNAs in cultured rat glial and neuronal cells. Neurosci. Lett. 249, 163–166 [DOI] [PubMed] [Google Scholar]
- 17. Wang J., Wang J., Sun Y., Song W., Nor J. E., Wang C. Y., and Taichman R. S. (2005) Diverse signaling pathways through the SDF-1/CXCR4 chemokine axis in prostate cancer cell lines leads to altered patterns of cytokine secretion and angiogenesis. Cell. Signal. 17, 1578–1592 [DOI] [PubMed] [Google Scholar]
- 18. Karnoub A. E., and Weinberg R. A. (2006) Chemokine networks and breast cancer metastasis. Breast Dis. 26, 75–85 [DOI] [PubMed] [Google Scholar]
- 19. Chinni S. R., Yamamoto H., Dong Z., Sabbota A., Bonfil R. D., and Cher M. L. (2008) CXCL12/CXCR4 transactivates HER2 in lipid rafts of prostate cancer cells and promotes growth of metastatic deposits in bone. Mol. Cancer Res. 6, 446–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Müller A., Homey B., Soto H., Ge N., Catron D., Buchanan M. E., McClanahan T., Murphy E., Yuan W., Wagner S. N., Barrera J. L., Mohar A., Verástegui E., and Zlotnik A. (2001) Involvement of chemokine receptors in breast cancer metastasis. Nature 410, 50–56 [DOI] [PubMed] [Google Scholar]
- 21. Taichman R. S., Cooper C., Keller E. T., Pienta K. J., Taichman N. S., and McCauley L. K. (2002) Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 62, 1832–1837 [PubMed] [Google Scholar]
- 22. Singh S., Singh U. P., Grizzle W. E., and Lillard J. W. Jr. (2004) CXCL12-CXCR4 interactions modulate prostate cancer cell migration, metalloproteinase expression and invasion. Lab. Invest. 84, 1666–1676 [DOI] [PubMed] [Google Scholar]
- 23. Chinni S. R., Sivalogan S., Dong Z., Filho J. C., Deng X., Bonfil R. D., and Cher M. L. (2006) CXCL12/CXCR4 signaling activates Akt-1 and MMP-9 expression in prostate cancer cells: the role of bone microenvironment-associated CXCL12. Prostate 66, 32–48 [DOI] [PubMed] [Google Scholar]
- 24. Engl T., Relja B., Marian D., Blumenberg C., Müller I., Beecken W. D., Jones J., Ringel E. M., Bereiter-Hahn J., Jonas D., and Blaheta R. A. (2006) CXCR4 chemokine receptor mediates prostate tumor cell adhesion through alpha5 and beta3 integrins. Neoplasia 8, 290–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee B. C., Lee T. H., Avraham S., and Avraham H. K. (2004) Involvement of the chemokine receptor CXCR4 and its ligand stromal cell-derived factor 1α in breast cancer cell migration through human brain microvascular endothelial cells. Mol. Cancer Res. 2, 327–338 [PubMed] [Google Scholar]
- 26. Kato M., Kitayama J., Kazama S., and Nagawa H. (2003) Expression pattern of CXC chemokine receptor-4 is correlated with lymph node metastasis in human invasive ductal carcinoma. Breast Cancer Res. 5, R144–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang J., Loberg R., and Taichman R. S. (2006) The pivotal role of CXCL12 (SDF-1)/CXCR4 axis in bone metastasis. Cancer Metastasis Rev. 25, 573–587 [DOI] [PubMed] [Google Scholar]
- 28. Varambally S., Yu J., Laxman B., Rhodes D. R., Mehra R., Tomlins S. A., Shah R. B., Chandran U., Monzon F. A., Becich M. J., Wei J. T., Pienta K. J., Ghosh D., Rubin M. A., and Chinnaiyan A. M. (2005) Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell 8, 393–406 [DOI] [PubMed] [Google Scholar]
- 29. Howlett A. C., Barth F., Bonner T. I., Cabral G., Casellas P., Devane W. A., Felder C. C., Herkenham M., Mackie K., Martin B. R., Mechoulam R., and Pertwee R. G. (2002) International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol. Rev. 54, 161–202 [DOI] [PubMed] [Google Scholar]
- 30. Pertwee R. G. (1995) Cannabinoids, Springer, London [Google Scholar]
- 31. Ameri A. (1999) The effects of cannabinoids on the brain. Prog. Neurobiol. 58, 315–348 [DOI] [PubMed] [Google Scholar]
- 32. Croci T., Manara L., Aureggi G., Guagnini F., Rinaldi-Carmona M., Maffrand J. P., Le Fur G., Mukenge S., and Ferla G. (1998) In vitro functional evidence of neuronal cannabinoid CB1 receptors in human ileum. Br. J. Pharmacol. 125, 1393–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pertwee R. G. (1997) Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol. Ther. 74, 129–180 [DOI] [PubMed] [Google Scholar]
- 34. Pertwee R. G. (2001) Cannabinoid receptors and pain. Prog. Neurobiol. 63, 569–611 [DOI] [PubMed] [Google Scholar]
- 35. Szabo B., Nordheim U., and Niederhoffer N. (2001) Effects of cannabinoids on sympathetic and parasympathetic neuroeffector transmission in the rabbit heart. J. Pharmacol. Exp. Ther. 297, 819–826 [PubMed] [Google Scholar]
- 36. Wagner J. A., Járai Z., Bátkai S., and Kunos G. (2001) Hemodynamic effects of cannabinoids: coronary and cerebral vasodilation mediated by cannabinoid CB1 receptors. Eur. J. Pharmacol. 423, 203–210 [DOI] [PubMed] [Google Scholar]
- 37. Svízenská I., Dubový P., and Sulcová A. (2008) Cannabinoid receptors 1 and 2 (CB1 and CB2), their distribution, ligands and functional involvement in nervous system structures—a short review. Pharmacol. Biochem. Behav. 90, 501–511 [DOI] [PubMed] [Google Scholar]
- 38. Howlett A. C. (1984) Inhibition of neuroblastoma adenylate cyclase by cannabinoid and nantradol compounds. Life Sci. 35, 1803–1810 [DOI] [PubMed] [Google Scholar]
- 39. Howlett A. C. (1985) Cannabinoid inhibition of adenylate cyclase. Biochemistry of the response in neuroblastoma cell membranes. Mol. Pharmacol. 27, 429–436 [PubMed] [Google Scholar]
- 40. Ruiz L., Miguel A., and Díaz-Laviada I. (1999) Δ9-Tetrahydrocannabinol induces apoptosis in human prostate PC-3 cells via a receptor-independent mechanism. FEBS Lett. 458, 400–404 [DOI] [PubMed] [Google Scholar]
- 41. Guzmán M., Sánchez C., and Galve-Roperh I. (2001) Control of the cell survival/death decision by cannabinoids. J. Mol. Med. 78, 613–625 [DOI] [PubMed] [Google Scholar]
- 42. Melck D., De Petrocellis L., Orlando P., Bisogno T., Laezza C., Bifulco M., and Di Marzo V. (2000) Suppression of nerve growth factor Trk receptors and prolactin receptors by endocannabinoids leads to inhibition of human breast and prostate cancer cell proliferation. Endocrinology 141, 118–126 [DOI] [PubMed] [Google Scholar]
- 43. Sarfaraz S., Adhami V. M., Syed D. N., Afaq F., and Mukhtar H. (2008) Cannabinoids for cancer treatment: progress and promise. Cancer Res. 68, 339–342 [DOI] [PubMed] [Google Scholar]
- 44. Pisanti S., Malfitano A. M., Grimaldi C., Santoro A., Gazzerro P., Laezza C., and Bifulco M. (2009) Use of cannabinoid receptor agonists in cancer therapy as palliative and curative agents. Best Pract. Res. Clin. Endocrinol. Metab. 23, 117–131 [DOI] [PubMed] [Google Scholar]
- 45. Qamri Z., Preet A., Nasser M. W., Bass C. E., Leone G., Barsky S. H., and Ganju R. K. (2009) Synthetic cannabinoid receptor agonists inhibit tumor growth and metastasis of breast cancer. Mol. Cancer Ther. 8, 3117–3129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Blázquez C., Casanova M. L., Planas A., Gómez Del Pulgar T., Villanueva C., Fernández-Aceñero M. J., Aragonés J., Huffman J. W., Jorcano J. L., and Guzmán M. (2003) Inhibition of tumor angiogenesis by cannabinoids. FASEB J. 17, 529–531 [DOI] [PubMed] [Google Scholar]
- 47. Jordà M. A., Verbakel S. E., Valk P. J., Vankan-Berkhoudt Y. V., Maccarrone M., Finazzi-Agrò A., Löwenberg B., and Delwel R. (2002) Hematopoietic cells expressing the peripheral cannabinoid receptor migrate in response to the endocannabinoid 2-arachidonoylglycerol. Blood 99, 2786–2793 [DOI] [PubMed] [Google Scholar]
- 48. Joseph J., Niggemann B., Zaenker K. S., and Entschladen F. (2004) Anandamide is an endogenous inhibitor for the migration of tumor cells and T lymphocytes. Cancer Immunol. Immunother. 53, 723–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pereira J. P., An J., Xu Y., Huang Y., and Cyster J. G. (2009) Cannabinoid receptor 2 mediates the retention of immature B cells in bone marrow sinusoids. Nat. Immunol. 10, 403–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ghosh S., Preet A., Groopman J. E., and Ganju R. K. (2006) Cannabinoid receptor CB2 modulates the CXCL12/CXCR4-mediated chemotaxis of T lymphocytes. Mol. Immunol. 43, 2169–2179 [DOI] [PubMed] [Google Scholar]
- 51. Muñoz L. M., Lucas P., Holgado B. L., Barroso R., Vega B., Rodríguez-Frade J. M., and Mellado M. (2011) Receptor oligomerization: a pivotal mechanism for regulating chemokine function. Pharmacol. Ther. 131, 351–358 [DOI] [PubMed] [Google Scholar]
- 52. Springael J. Y., Urizar E., and Parmentier M. (2005) Dimerization of chemokine receptors and its functional consequences. Cytokine Growth Factor Rev. 16, 611–623 [DOI] [PubMed] [Google Scholar]
- 53. Springael J. Y., Le Minh P. N., Urizar E., Costagliola S., Vassart G., and Parmentier M. (2006) Allosteric modulation of binding properties between units of chemokine receptor homo- and hetero-oligomers. Mol. Pharmacol. 69, 1652–1661 [DOI] [PubMed] [Google Scholar]
- 54. Sohy D., Parmentier M., and Springael J. Y. (2007) Allosteric transinhibition by specific antagonists in CCR2/CXCR4 heterodimers. J. Biol. Chem. 282, 30062–30069 [DOI] [PubMed] [Google Scholar]
- 55. Isik N., Hereld D., and Jin T. (2008) Fluorescence resonance energy transfer imaging reveals that chemokine-binding modulates heterodimers of CXCR4 and CCR5 receptors. PLoS One 3, e3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rodríguez-Frade J., Muñoz L. M., and Mellado M. (2009) Chapter 5. Multiple approaches to the study of chemokine receptor homo- and heterodimerization. Methods Enzymol. 461, 105–122 [DOI] [PubMed] [Google Scholar]
- 57. Mellado M., Rodríguez-Frade J. M., Vila-Coro A. J., Fernández S., Martín de Ana A., Jones D. R., Torán J. L., and Martínez-A C. (2001) Chemokine receptor homo- or heterodimerization activates distinct signaling pathways. EMBO J. 20, 2497–2507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pello O. M., Martínez-Muñoz L., Parrillas V., Serrano A., Rodríguez-Frade J. M., Toro M. J., Lucas P., Monterrubio M., Martínez-A C., and Mellado M. (2008) Ligand stabilization of CXCR4/δ-opioid receptor heterodimers reveals a mechanism for immune response regulation. Eur. J. Immunol. 38, 537–549 [DOI] [PubMed] [Google Scholar]
- 59. Benamar K., Geller E. B., and Adler M. W. (2008) First in vivo evidence for a functional interaction between chemokine and cannabinoid systems in the brain. J. Pharmacol. Exp. Ther. 325, 641–645 [DOI] [PubMed] [Google Scholar]
- 60. Costantino C. M., Gupta A., Yewdall A. W., Dale B. M., Devi L. A., and Chen B. K. (2012) Cannabinoid receptor 2-mediated attenuation of CXCR4-tropic HIV infection in primary CD4+ T cells. PLoS One 7, e33961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dias J., Ferrão F. M., Axelband F., Carmona A. K., Lara L. S., and Vieyra A. (2014) ANG-(3–4) inhibits renal Na+-ATPase in hypertensive rats through a mechanism that involves dissociation of ANG II receptors, heterodimers, and PKA. Am. J. Physiol. Renal Physiol. 306, F855–F863 [DOI] [PubMed] [Google Scholar]
- 62. Misra P., Lebeche D., Ly H., Schwarzkopf M., Diaz G., Hajjar R. J., Schecter A. D., and Frangioni J. V. (2008) Quantitation of CXCR4 expression in myocardial infarction using 99mTc-labeled SDF-1α. J. Nucl. Med. 49, 963–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Humblet V., Lapidus R., Williams L. R., Tsukamoto T., Rojas C., Majer P., Hin B., Ohnishi S., De Grand A. M., Zaheer A., Renze J. T., Nakayama A., Slusher B. S., and Frangioni J. V. (2005) High-affinity near-infrared fluorescent small-molecule contrast agents for in vivo imaging of prostate-specific membrane antigen. Mol. Imaging 4, 448–462 [DOI] [PubMed] [Google Scholar]
- 64. Geminder H., Sagi-Assif O., Goldberg L., Meshel T., Rechavi G., Witz I. P., and Ben-Baruch A. (2001) A possible role for CXCR4 and its ligand, the CXC chemokine stromal cell-derived factor-1, in the development of bone marrow metastases in neuroblastoma. J. Immunol. 167, 4747–4757 [DOI] [PubMed] [Google Scholar]
- 65. Kukreja P., Abdel-Mageed A. B., Mondal D., Liu K., and Agrawal K. C. (2005) Up-regulation of CXCR4 expression in PC-3 cells by stromal-derived factor-1α (CXCL12) increases endothelial adhesion and transendothelial migration: role of MEK/ERK signaling pathway-dependent NF-κB activation. Cancer Res. 65, 9891–9898 [DOI] [PubMed] [Google Scholar]
- 66. Haribabu B., Richardson R. M., Fisher I., Sozzani S., Peiper S. C., Horuk R., Ali H., and Snyderman R. (1997) Regulation of human chemokine receptors CXCR4. Role of phosphorylation in desensitization and internalization. J. Biol. Chem. 272, 28726–28731 [DOI] [PubMed] [Google Scholar]
- 67. Ali H., Haribabu B., Richardson R. M., and Snyderman R. (1997) Mechanisms of inflammation and leukocyte activation. Med. Clin. North Am. 81, 1–28 [DOI] [PubMed] [Google Scholar]
- 68. Prinster S. C., Hague C., and Hall R. A. (2005) Heterodimerization of g protein-coupled receptors: specificity and functional significance. Pharmacol. Rev. 57, 289–298 [DOI] [PubMed] [Google Scholar]
- 69. Christopoulos A., and Kenakin T. (2002) G protein-coupled receptor allosterism and complexing. Pharmacol. Rev. 54, 323–374 [DOI] [PubMed] [Google Scholar]
- 70. Deng H. K., Unutmaz D., KewalRamani V. N., and Littman D. R. (1997) Expression cloning of new receptors used by simian and human immunodeficiency viruses. Nature 388, 296–300 [DOI] [PubMed] [Google Scholar]
- 71. Nasser M. W., Qamri Z., Deol Y. S., Smith D., Shilo K., Zou X., and Ganju R. K. (2011) Crosstalk between chemokine receptor CXCR4 and cannabinoid receptor CB2 in modulating breast cancer growth and invasion. PLoS One 6, e23901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Steele A. D., Szabo I., Bednar F., and Rogers T. J. (2002) Interactions between opioid and chemokine receptors: heterologous desensitization. Cytokine Growth Factor Rev. 13, 209–222 [DOI] [PubMed] [Google Scholar]
- 73. Wang J., Alvarez R., Roderiquez G., Guan E., and Norcross M. A. (2004) Constitutive association of cell surface CCR5 and CXCR4 in the presence of CD4. J. Cell. Biochem. 93, 753–760 [DOI] [PubMed] [Google Scholar]
- 74. Söderberg O., Leuchowius K. J., Gullberg M., Jarvius M., Weibrecht I., Larsson L. G., and Landegren U. (2008) Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods 45, 227–232 [DOI] [PubMed] [Google Scholar]
- 75. Callén L., Moreno E., Barroso-Chinea P., Moreno-Delgado D., Cortés A., Mallol J., Casadó V., Lanciego J. L., Franco R., Lluis C., Canela E. I., and McCormick P. J. (2012) Cannabinoid receptors CB1 and CB2 form functional heteromers in brain. J. Biol. Chem. 287, 20851–20865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Terrillon S., and Bouvier M. (2004) Roles of G-protein-coupled receptor dimerization. EMBO Rep. 5, 30–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Chetram M. A., Odero-Marah V., and Hinton C. V. (2011) Loss of PTEN permits CXCR4-mediated tumorigenesis through ERK1/2 in prostate cancer cells. Mol. Cancer Res. 9, 90–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Chetram M. A., Don-Salu-Hewage A. S., and Hinton C. V. (2011) ROS enhances CXCR4-mediated functions through inactivation of PTEN in prostate cancer cells. Biochem. Biophys. Res. Commun. 410, 195–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Don-Salu-Hewage A. S., Chan S. Y., McAndrews K. M., Chetram M. A., Dawson M. R., Bethea D. A., and Hinton C. V. (2013) Cysteine (C)-x-C receptor 4 undergoes transportin 1-dependent nuclear localization and remains functional at the nucleus of metastatic prostate cancer cells. PLoS One 8, e57194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Agle K. A., Vongsa R. A., and Dwinell M. B. (2010) Calcium mobilization triggered by the chemokine CXCL12 regulates migration in wounded intestinal epithelial monolayers. J. Biol. Chem. 285, 16066–16075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. McKallip R. J., Nagarkatti M., and Nagarkatti P. S. (2005) Δ9-Tetrahydrocannabinol enhances breast cancer growth and metastasis by suppression of the antitumor immune response. J. Immunol. 174, 3281–3289 [DOI] [PubMed] [Google Scholar]
- 82. Ligresti A., Moriello A. S., Starowicz K., Matias I., Pisanti S., De Petrocellis L., Laezza C., Portella G., Bifulco M., and Di Marzo V. (2006) Antitumor activity of plant cannabinoids with emphasis on the effect of cannabidiol on human breast carcinoma. J Pharmacol. Exp. Ther. 318, 1375–1387 [DOI] [PubMed] [Google Scholar]
- 83. Prasad A., Qamri Z., Wu J., and Ganju R. K. (2007) Slit-2/Robo-1 modulates the CXCL12/CXCR4-induced chemotaxis of T cells. J. Leukoc. Biol. 82, 465–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wang J., He L., Combs C. A., Roderiquez G., and Norcross M. A. (2006) Dimerization of CXCR4 in living malignant cells: control of cell migration by a synthetic peptide that reduces homologous CXCR4 interactions. Mol. Cancer Ther. 5, 2474–2483 [DOI] [PubMed] [Google Scholar]
- 85. Hereld D., and Jin T. (2008) Slamming the DOR on chemokine receptor signaling: heterodimerization silences ligand-occupied CXCR4 and δ-opioid receptors. Eur. J. Immunol. 38, 334–337 [DOI] [PubMed] [Google Scholar]
- 86. Chen C., Li J., Bot G., Szabo I., Rogers T. J., and Liu-Chen L. Y. (2004) Heterodimerization and cross-desensitization between the μ-opioid receptor and the chemokine CCR5 receptor. Eur. J. Pharmacol. 483, 175–186 [DOI] [PubMed] [Google Scholar]
- 87. Kohout T. A., Nicholas S. L., Perry S. J., Reinhart G., Junger S., and Struthers R. S. (2004) Differential desensitization, receptor phosphorylation, β-arrestin recruitment, and ERK1/2 activation by the two endogenous ligands for the CC chemokine receptor 7. J. Biol. Chem. 279, 23214–23222 [DOI] [PubMed] [Google Scholar]
- 88. Gomes I., Jordan B. A., Gupta A., Trapaidze N., Nagy V., and Devi L. A. (2000) Heterodimerization of μ and δ opioid receptors: a role in opiate synergy. J. Neurosci. 20, RC110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Reggio P. H. (2009) The Cannabinoid Receptors, pp. 160–162, Humana, New York [Google Scholar]
- 90. Correa F., Mestre L., Docagne F., and Guaza C. (2005) Activation of cannabinoid CB2 receptor negatively regulates IL-12p40 production in murine macrophages: role of IL-10 and ERK1/2 kinase signaling. Br. J. Pharmacol. 145, 441–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Duda D. G., Kozin S. V., Kirkpatrick N. D., Xu L., Fukumura D., and Jain R. K. (2011) CXCL12 (SDF1α)-CXCR4/CXCR7 pathway inhibition: an emerging sensitizer for anticancer therapies? Clin. Cancer Res. 17, 2074–2080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. McCarty N. A., and Haack K. K. (2011) Functional consequences of GPCR heterodimerization: GPCRs as allosteric modulators. Pharmaceuticals 4, 509–523 [Google Scholar]