

Abstract
A few years ago therapeutic options in advanced melanoma were very limited and the prognosis was somber. Although recent progresses are far from providing a cure for advanced melanoma, yet these have kindled new hopes and searching for a cure does not seem unreasonable. Seven new medicines have been authorized in various regions of the world in the recent past in the therapy of advanced melanoma, over half of them acting by mechanisms involving the immune system of the host. The anti-CTLA-4 (cytotoxic T lymphocyte associated protein-4) ipilimumab has been followed by anti-PD1 (programmed death1) inhibitors, more effective and safer. Very recently, the first oncolytic immunotherapy, talimogene laherparepvec (T-VEC) has been authorized for placing on the market and a variety of combinations of the new therapies are currently being evaluated or considered. Besides, a plethora of other molecules and approaches, especially monoclonal antibodies, are in the preliminary phases of clinical investigation and are likely to bring new benefits for the treatment of this potentially fatal form of cancer.
Keywords: Approved drugs, immune checkpoints, immune-therapy, melanoma, oncolytic immunotherapy
Melanoma and its complex relationship with the host immune system
Melanoma is the most lethal form of skin cancer (rarer forms located elsewhere than skin are also possible), a tumour occurring by the malignant transformation of melanocytes1. The number of new cases has been increasing in recent decades2,3 and an association with higher socio-economic status has been described3. Detected early, melanoma may be cured by surgical excision, but the advanced (metastatic) disease is commonly incurable, with 5-year overall survival (OS) under 10 per cent and a median survival under one year4. Until recently, the therapeutic options in stage IV melanoma were very limited, mostly centered on dacarbazine used as monotherapy or in various combinations. Although initially dacarbazine was reported to induce response rates of up to 25 per cent, larger studies conducted later have indicated lower response rates (12% or less), median progression free survival (PFS) under six months and OS of less than 2 per cent at six years5. Interleukin-2 (IL-2), never tested in a properly designed pivotal clinical trial, at no time approved in Europe for melanoma treatment, was authorized by FDA (Food and Drug Administration) 18 years ago in the treatment of advanced melanoma5. Interleukin-2 had modest efficacy, at best an objective response rate (ORR) of 16 per cent in a retrospective analysis of clinical studies (which included no phase III trials), although some respondents had longer duration of response but the treatment was accompanied by severe, although mostly reversible adverse effects5.
The immune system is physiologically equipped to detect cells with abnormal proliferation and thus destroy them during early neoplasia development6. Hence, immune surveillance is the complex pathway responsible for the surveillance and eradication of transformed self, but the theory of immunoediting, developed more than 10 years ago7, has gained, in the meantime, valuable experimental and clinical proofs. Immunity is involved in neoplastic cellular transformation but it can also preclude or restrict tumour growth and mould the immunogenicity of tumours. Although these functions seem both pro- and anti-tumoural, specific immune actions get involved in different stages. This is a dynamic progression; the immune system safeguards the host against tumour cell development but it also frames the features of emerging tumours. The process has three stages - Elimination, Equilibrium and Escape8.
In the Escape phase the defense cells fail to control the tumour and lead to a clinically visible tumour. Tumour cells lose their specific antigens, express anti-apoptotic molecule Bcl-2 and immunosuppression inducing molecules [indoleamine 2,3-dioxygenase (IDO), tryptophan 2,3-dioxygenase (TDO), programmed death-ligand 1 (PD-L1), galectin-1/3/9, CD39, CD73, adenosine receptors]. Tumour cells become active in secreting vascular endothelial growth factor (VEGF), transforming growth factor beta (TGF-ß), interleukin-6 (IL-6), macrophage colony-stimulating factor (M-CSF) thus enhancing the angiogenesis. Myeloid-derived suppressor cells (MDSCs), M2 macrophages and dendritic cells (DCs) are converted to pro-tumoural action, express arginase, inducible nitric oxide synthase (iNOS) and IDO and secrete immunosuppressive regulatory molecules such as IL-10 and TGF-ß. The effector activity of cytotoxic lymphocytes is inhibited. MDSCs and DCs generate regulatory T cells (Treg) with high immunosuppressive action. T lymphocytes express inhibitory receptor proteins such as PD-1, CTLA-4, Tim-3 and lymphocyte activation gene (LAG-3) suppressing anti-tumour immune response. Immune suppressant cytokines and other signaling proteins like IL-10, TGF-ß, VEGF, IDO, PD-L1 sustain a pro-tumoural milieu9 (Figure).
Figure.
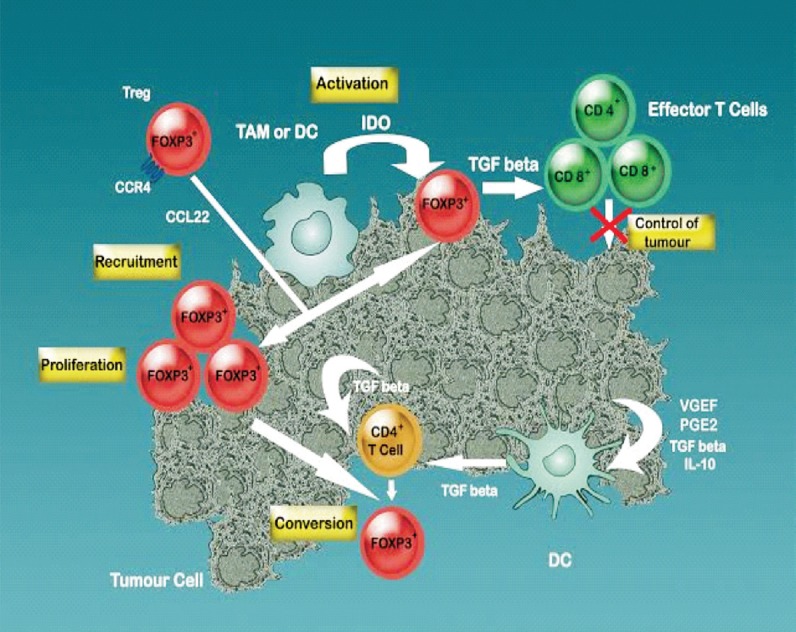
Immune-suppression mechanisms - therapy targets. Macrophages secrete indoleamine 2,3-dioxygenase (IDO) that induce an inhibition of T cell proliferation due to tryptophan depletion (activation). Moreover, IDO recruits regulatory T cells (FOXP3+) at the tumoural site. Recruiting more TGF beta-secreting Tregs the suppression induced on the effector couple CD4-CD8 increases and therefore, the control of tumour development decreases. Tumoural cells by themselves secrete TGFbeta, IL-10, VEGF, PGE2 that induce DCs to secrete more TGFbeta contributing to the conversion of CD4+ T cells to Tregs phenotype enhancing the cellular immune suppression (conversion). Skin-homing T cells CC-chemokine receptor 4 (CCR4) binds to the CCL22 (macrophage-derived chemokine) of the tumour associated macrophages (TAM) and are recruited to the tumoural site (recruitment). On the whole, an immune suppressive microenvironment is created by the concerted action of several elements, action that induces Tregs proliferation, further hindering CD4+-CD8+ cooperation and therefore, abolishing the effector activity anti-tumoural cytotoxic cells.
Source: Ref. 6; Reproduced with permission.
In cutaneous melanoma immune therapy, the three stages have been well studied and the panel of immune therapies includes immune checkpoints inhibitors, monoclonal antibodies, anti-tumour vaccines, adoptive cell transfer therapies, and a variety of their associations10.
Melanoma is notorious for its complex relationship with the immune system, a relationship known for over 25 years. The first convincing proof of the participation of the immunological mechanisms in the response of the host organism against the autologous melanoma was reported in 1987 about the presence of a large number of tumour-infiltrating lymphocytes (TILs) in skin melanoma of a small number of patients, as well as their cytolytic activity against malignant melanocytes when activated with IL-211,12. Numerous therapeutic options have been explored along time in an attempt to improve the immune response of the host against the melanoma cells, but they often resulted in unimpressive successes and even in impressive failures. Recently new hopes have been kindled, as seven new medicines are being or are authorized in various regions of the world in the treatment of late stage melanoma. At least four of these (ipilimumab, nivolumab, pembrolizumab and talimogene laherparepvec) act directly on the immune system, whereas the others interfere at least indirectly with it. The first three belong to the so-called class of “immune checkpoint inhibitors”13. Talimogene laherparepvec, an oncolytic virus expressing granulocyte macrophage (GM)-CSF has just been approved in October 2015 both in European Union and United States for the treatment of melanoma14,15.
Immune checkpoints and co-stimulatory molecules
Immune checkpoints are molecules (receptors and their corresponding ligands) playing a central part in preserving peripheral immune tolerance, exerting a suppressing role on the immune system and thus preventing the occurrence of autoimmune disturbances. Negation of the negation, their blockade or inhibition is, therefore, equivalent to a boost for the immune system, a boost whose efficacy in controlling tumours has revolutionized cancer therapy in recent years, not only for melanoma, but also for other forms of cancer16. Data accumulated over time indicate that tumour cells exploit immune checkpoints as one of their major mechanisms of getting resistant to the immune system, especially to evade the action of T cells that should normally target tumour antigens17, and this may explain the efficacy of these products acting through an indirect mechanism (not directly on the tumour cells, but through an action oriented towards the immune system cells).
Over six immune checkpoints have been described. TIM-3, belonging to the TIM family (transmembrane immunoglobulin and mucin domain), regulates primarily the Th1 function, its overexpression being accompanied by poor prognosis in different forms of cancer18. BTLA (B- and T-lymphocyte attenuator, CD272) is a receptor of the CD28 family, that binds to HVEM (herpes virus entry mediator a TNFR family protein), co-inhibiting T-cells together with CD160, an immunoglobulin-like molecule19. VISTA (V-domain immunoglobulin suppressor of T-cell activation) is a programmed death-ligand 1 (PD-L1) -like ligand with a single IgV domain, expressed mostly on myeloid cells and able to suppress strongly T cells, being intensely expressed within the tumour microenvironment20. KIR (killer immunoglobulin receptor) is expressed on natural killer (NK) cells, involved in the inhibitory regulation of their cytotoxic functions and apparently also co-opted by tumours to bypass the defense system of the host21. LAG-3, a type I transmembrane receptor inhibits T lymphocyte proliferation, activity and homeostasis18. The two most widely known immune checkpoints exploited for therapeutic purposes in melanoma are cytotoxic T-lymphocyte associated protein-4 (CTLA-4) and PD. CTLA-4 (CD152), the first one to be targeted for therapeutic purposes, is a transmembrane glycoprotein which interacts with its specific ligands CD80 (B7-1) and CD86 (B7-2) and downregulates the activity of T-lymphocytes through several ways, including competition for CD28 ligand binding, tryptophan depletion, production of regulatory cytokines or removal of ligands by a transendocytosis process22. The CTLA-4 negatively regulates T-lymphocyte activation, PD-1 (programmed death 1, an immune receptor of the same CTLA-4 family) signaling through a PD-L1 ligand regulates mainly the effector function of T, B, and NK cells within tissues and tumours23. CTLA-4 is considered to intervene in the early immune response, while PD-1 is thought to intervene in the later immune response; the former acts largely in lymph nodes, while the latter occurs predominantly in peripheral tissues24.
On the other side, at least four activating co-stimulatory molecules, all belonging to the tumour necrosis factor receptor superfamily (TNFRS), are in the focus of anticancer research groups for their potential of improving the therapy over the current options. CD40, a first member of TNFRS, interacts with its specific ligand, CD40L, providing thus a co-stimulatory signal to antigen-presenting cells (APC), resulting in the end in enhanced cytotoxic responses of T cell against tumour cells25. OX40 is a second member of the TNFRS with co-stimulatory effects on T lymphocytes, inducing stimulation of CD4+ and CD8+ T-cells and prolonging their survival, triggering inhibition of the T-regulatory cells subset, and seemingly inducing anti-tumour effects, although the intricacy of the immune signaling pathways related to its activation limits our current understanding of the effects of this ligand-receptor pair26. CD137 (also known as 4-1BB and ILA - because it was initially described as a gene induced by lymphocyte activation) is a third component of the TNFRS, participating to the co-stimulation of T and NK cells, its activation being associated with improved anti-tumour responses in a melanoma non-clinical model27. GITR (glucocorticoid-induced TNFR-related protein) also belongs to the TNFRS and is expressed on activated T cells, NK cells, B cells, as well as on T regulatory cells; it has not yet been clarified to what extent its activation and consecutive anti-tumour effects are the result of its signaling on typical T lymphocytes or on the CD4+CD25+and similar subpopulations (Tregs)28,29.
In the following two sections we discuss the medicines (monoclonal antibodies) interfering with the CTLA-4 and PD-1 checkpoints that managed to go through the ‘Caudine Forks’ of the full clinical development and regulatory scrutiny, being approved for melanoma treatment.
CTLA-4 inhibitors
Generation of an anti-tumour immune response has a complexity that starts with the recognition of tumour antigen with the help of HLA proteins by T cells. The process is strengthened by further cross-talks between T cells and antigen presenting cells (APC). One of the molecules that adds co-stimulatory events is CD28, an activating checkpoint expressed on T lymphocytes, that interacts with its specific ligands CD80 and CD86 on APC. This co-stimulatory binding leads to the stimulation of the T lymphocytes. CTLA-4 is also present on activated T lymphocytes. T regulatory cells play a critical role in the preservation of immune self-tolerance and equilibrium30 and these cells harbour a series of immune checkpoints where CTLA-4 is one of the earliest molecular checkpoints that controls T cells response to antigen. CTLA-4 in physiological conditions prevents autoimmune reactions and influences self-tolerance31.
One of the first reports showing the clear therapeutical effect of an anti-CTLA-4 antibody in melanoma was presented in 200432. Ribas et al32 have shown that this approach can “break peripheral immunologic tolerance” and further develop an efficient anti-tumoural response. Since then, several positive clinical trials have led to ipilimumab being approved for the first time by FDA in 2011 (followed by other regions, including European Union) to treat late-stage melanoma and then in October 2015 its use extended in the adjuvant setting in the subpopulation with advanced melanoma, to diminish the recurrence hazard33. Tremelimumab targets the same receptor and was also initially developed for the the same indication, but after a failed phase III trial it was put on hold at least temporarily for this malignancy34; it was later shown that this failure might have been related to the access of a number of patients from the control arm to ipilimumab35 (leading to misclassification bias) and currently there seems to be a renewed interest in its potential use in melanoma36.
In a phase 3 study, ipilimumab was compared with a glycoprotein (gp100) peptide vaccine and a combination of the two therapies in a subpopulation with late stage melanoma (randomization 1:1:3). Median OS in the combination group was 10 months, in the ipilimumab monotherapy group 10.1 months, whereas the gp100 vaccine group had only 6.4 months. OS did not differ significantly between the two ipilimumab groups37. In the EORTC 18071 study, a multi-centric, double blind, phase III trial in treatment-naïve subjects with advanced disease (475 on ipilimumab, 476 on placebo), on which the extension of indication to this subpopulation was based, the median recurrence-free survival was 26.1 months (95% CI 19.3-39.3) for the ipilimumab group and 17.1 months (95% CI 13.4-21.6) for the control group38. However, the improved efficacy of ipilimumab came at a safety price, about 85 per cent of patients experienced adverse events of any grade, and although the majority were of minor or reasonable gravity, in a small proportion of patients these were severe and life threatening. Careful monitoring and possibly early use of corticosteroids might be necessary39.
Anti-PD1 inhibitors
PD1 is an immune checkpoint inhibitor expressed on activated T, B and NK lymphocytes, as well as on peripheral myeloid cells, acting through its two ligands to induce T cell tolerance40. Its ligand PD-L1 is expressed on a large variety of human tumours including melanoma, being exploited by these to evade the host immune system, for instance by suppressing cytotoxic T lymphocytes41. Unlike PD-L1, PD-L2 seems to be expressed in a restricted manner, only on APC cells (dendritic cells, B cells, etc.)23,40. PD-1 expression or that of its ligand was correlated with poor outcome in several malignancies42, and thus, it was to be expected that inhibition of PD-1 signaling, e.g. by use of monoclonal antibodies should result in improved outcome. Two such antibodies, nivolumab and pembrolizumab confirmed this hypothesis. Nivolumab has been the earliest anti-PD-1 antibody to be authorized in the European Union, pembrolizumab being the second; in the US, the reverse was true (nivolumab was the second, pembrolizumab the first)43,44,45,46.
Nivolumab, a fully human antibody blocking the interaction between PD-1 and its ligands, disrupts the T-cell toleration induced by the checkpoint inhibitor. After a promising phase I study in advanced melanoma on pre-treated, nivolumab was tested in a phase III, open label trial against chemotherapy selected by investigator (investigator's choice chemotherapy, ICC - dacarbazine 1000 mg/m2 Q3W, or carboplatin AUC 6 + paclitaxel 175 mg/m2 Q3W), in subjects with metastatic melanoma refractory to ipilimumab and to a BRAF inhibitor where relevant. In this open-label study the objective response rate (ORR) for nivolumab was 31.7% (95% CI: 23.5, 40.8), whereas for the ICC group it was only about one third (10.6%; 95% CI: 3.5, 23.1). Nivolumab was also better than ICC in terms of reduction of 50 per cent or more in target lesion burden (82 versus 60%), of median response time (2.1 versus 3.5 months) and in terms of median response duration (not yet attained versus 3.5 months)47,48. The pivotal study for market approval was a randomized, double-blind trial comparing nivolumab (3 mg/kg Q2W, plus a dacarbazine-matched placebo Q3W) with dacarbazine (1000 mg/m2 Q3W, plus a nivolumab-matched placebo Q2W) in treatment-naïve patients with no BRAF mutations. The one year OS was 72.9 per cent (95% CI 65.5 to 78.9) for nivolumab, whereas for dacarbazine it was almost half, 42.1 per cent (95% CI 33.0 to 50.9); hazard ratio (HR) for death was 0.42 per cent (99.79% CI, 0.25 to 0.73; P<0.001). In terms of PFS (progression free survival) and ORR (objective response rate), nivolumab was also sizably superior to dacarbazine and the benefit was observed irrespective of the PD-L1 status subgroup49.
Pembrolizumab (lambrolizumab, MK-3475) is only humanized (such antibodies have in most part human sequences, except for the complementarity-determining regions). It also targets PD-1 and has obtained approval for use in the therapy of late stage melanoma. It showed positive outcomes with respect to both efficacy and safety in the phase I trials50, doses of 2 and 10 mg/kg having comparable safety profiles51, which led to the use of the latter, higher dose in the pivotal study. This was a randomized, controlled trial with three treatment arms (randomization 1:1:1)52: the first two arms were allocated to pembrolizumab (10 mg/kg and every three weeks, respectively) and the third to ipilimumab (3 mg/kg every three weeks). The results were very convincing with regard to both OS and PFS, pembrolizumab being shown to have an efficacy far superior to ipilimumab, but with less adverse reactions. Six-months PFS proportions were 47.3 and 46.4 per cent in the two anti-PD1 arms, versus only 26.5 per cent in the control group (HR with respect to disease progression 0.58, P<0.001 for each treatment arm versus control). Twelve-months OS rates were 74.1 and 68.4 per cent for the two pembrolizumab regimens, versus 58.2 per cent for ipilimumab. The frequency of responses was about three times higher for pembrolizumab (33.7 and 32.9%) than for ipilimumab (11.9%; P<0.001) for each comparison, but adverse events of grade 3-5 were only about half as frequent in the pembrolizumab arms (13.3 and 10.1 versus 19.9%)52. A small, randomized study in subjects non-responding to ipilimumab and, if relevant, to a BRAF or a mitogen-activated protein kinase kinase (MEK) inhibitor or both, compared pembrolizumab (2 and 10 mg/kg) with ICC paclitaxel plus carboplatin, paclitaxel, carboplatin, dacarbazine, or oral temozolomide)53. Pembrolizumab showed better PFS (38% for 10 mg/kg versus 16% ICC; HR 0.50, P<0.0001 for 10 mg/kg) and a better safety profile (grade 3-4 adverse events about half as frequent in those treated with the monoclonal antibody as in the ICC arm, 14 against 26%)53. Data obtained in patients with other forms of cancer (colorectal, renal) indicate that the mismatch repair status54, the interferon-γ signature 10 gene55 or baseline differential expression ≥ 1.3-fold of a relatively large number of genes (311 genes)56 may be able to predict treatment response to pembrolizumab, a hypothesis which should be tested in melanoma and also of interest for nivolumab.
Oncolytic immunotherapy in melanoma (talimogene laherparepvec)
Talimogene laherparepvec (T-VEC) is the earliest oncolytic immune therapy to be approved for cancer treatment, a medicinal product using a herpes simplex virus type 1 modified to reproduce in a selective manner inside tumours and to biosynthesize GM-CSF which in turn will induce a systemic anti-tumour immune response57. The official prescribing information or assessment reports have not yet been published, but the press releases by the European and American regulatory authorities indicate that the approval is based on the phase III trial recently published57,58, which showed that talimogene laherparepvec induced a lasting response in 16.3 per cent of the patients (95% CI, 12.1-20.5) versus 2.1 per cent only (95% CI, 0-4.5) for GM-CSF (OR 8.9, P<0.001). The ORR also favoured talimogene laherparepvec, but the difference in median OS, although favouring the oncolytic therapy marginally failed to reach the conventional significance threshold of 0.05 (23.3 months versus 18.9 months, HR=0.79, 95CI for HR 0.62 to 1.00; P=0.051)57. In Europe the target population is represented by “adults with unresectable melanoma that is regionally or distantly metastatic (Stage IIIB, IIIC and IVM1a) with no bone, brain, lung or other visceral disease”. In this target population the analyses indicated a 25 per cent DRR for the test product versus 1 per cent in the GM-CSF control58.
Combination therapy
With a variety of therapeutic approaches available now in melanoma, the idea of combining medicines with different mechanisms of activity is very appealing. Therefore, various combinations have already been assessed, at least tentatively, in clinical trials. Although in this mini-review we have focused especially on the drugs that managed to break through the regulatory wall into clinical practice, there is currently much excitement about combination therapy in melanoma, with several studies published or underway evaluating molecules not yet approved in combination with some already authorized for use in melanoma, based on the same concept of complementary mechanisms of action. Of the combinations involving at least one mechanism with immunological mechanism are T-VEC (talimogene laherparepvec) + ipilimumab, durvalamab (MEDI4736, another PD-1 inhibitor) + dabrafenib + trametinib59, ipilimumab + sargramostim (with improved OS, but not PFS in a relatively large clinical study)60, ipilimumab + bevacizumab61.
The idea of simultaneously blocking CTLA-4 and PD-1 was supported by non-clinical evidence and confirmed by phase III clinical studies. Nivolumab plus ipilimumab is superior to ipilimumab monotherapy in advanced melanoma, as evidenced in an early trial62, followed by a phase III one, the benefit being reached for patients irrespective of their BRAF-mutation status63. In treatment naïve patients with metastatic melanoma, nivolumab alone had better outcomes than ipilimumab, but their combination, especially in subjects with PD-L1-negative malignancies, has enhanced efficacy compared to each agent administered as monotherapy64. Although various therapeutic vaccines tested so far have not yet fulfilled their promise, a recent small trial explored the efficacy and safety of nivolumab co-administered with a multi-peptide vaccine and reported apparent good results65, but given its non-controlled character, the results are difficult to be placed in context. One of the first phase I studies with nivolumab also assessed it as monotherapy or co-administered with a peptide vaccine66.
Data accumulated so far indicate that increased toxicity may be a limiting factor of combinations and although with the arrival of newcomers in the field, with novel mechanisms of action, the temptation to use more complex combinations will increase, but safety considerations may impose limits on the number of entities to be combined or will narrow their use. The combination of vemurafenib and ipilimumab, for instance, despite good theoretical reasons supporting it, is currently not recommended because of the increased risk of hepatotoxicity, as evidenced in an early phase trial of the association67.
Conclusions and perspectives
A large body of investigations carried out within important research centres and organizations has led to considerable research advancements in the field, including therapies with new, immunological mechanisms of activity, whose efficacy has been proven in the treatment of late stage of melanoma. The inhibitors of the BRAF/MEK/ERK pathway, anti-CTLA-4 and anti-PD1/PD1-L monoclonal antibodies and the first oncolytic virus have all opened the gates of regulatory approval and are now used in the routine clinical setting68. But melanoma as well as other malignancies have not yet been defeated; thus, the exploration of new pathways and new approaches for old(er) targets goes on. For most checkpoint inhibitors and co-stimulatory molecules discussed in this paper, monoclonal antibodies targeting them have already been developed, although not all have been clinically tested in melanoma. IMP-321, a LAG-3Ig fusion protein has been evaluated in an early development trial69 and an anti-LAG-3 antibody (BMS-986016) is also tested together with nivolumab70 against melanoma. Lirilumab has been developed against KIR (killer cell immunoglobulin like receptor)71, but it has not yet been tested against melanoma. Preliminary data indicate a potential benefit for therapies targeting TIM-372 BLTA/CD-16073 or VISTA20, especially in dual blockade combinations. CP-870,893, a human monoclonal antibody74 and dacetuzumab (SGN-40)75 are potent CD40 agonists; MEDI6469 is an agonist murine monoclonal antibody (mAb) for the human OX40 receptor tested in phase I in various solid tumours76; an early development trial for urelumab (an agonistic anti-hCD137 antibody)77 was withdrawn before the start of enrollment and TRX518, a monoclonal antibody targeting GITR, is currently evaluated in an early development trial in several advanced malignancies including melanoma78. We have left out the whole field of therapeutic vaccines and adaptive cell transfer, which have also to be regarded as forms of immune therapy of considerable interest in melanoma. Due to the well-known and complex relationship of melanoma with the immune system, other forms of therapy with immunological mechanism will continue to be explored against this form of skin cancer.
Acknowledgment
The first author (RA) received honorarium from pharmaceutical companies (UCB, Angelini Pharmaceuticals, Zentiva, Stada Teva, Laropharm and Sodimed). The second author (MN) was partially supported by Research Grant PN-II-PT-PCCA-2013-4-1407 (MELTAG).
References
- 1.McKibbin T. Melanoma: understanding relevant molecular pathways as well as available and emerging therapies. Am J Manag Care. 2015;21:s224–33. [PubMed] [Google Scholar]
- 2.Chen ST, Geller AC, Tsao H. Update on the epidemiology of melanoma. Curr Dermatol Rep. 2013;2:24–34. doi: 10.1007/s13671-012-0035-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson-Obaseki SE, Labajian V, Corsten MJ, McDonald JT. Incidence of cutaneous malignant melanoma by socioeconomic status in Canada: 1992-2006. J Otolaryngol Head Neck Surg. 2015;44:53. doi: 10.1186/s40463-015-0107-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jang S, Atkins MB. Treatment of BRAF-mutant melanoma: The role of vemurafenib and other therapies. Clin Pharmacol Ther. 2013;95:24–31. doi: 10.1038/clpt.2013.197. [DOI] [PubMed] [Google Scholar]
- 5.Gogas H, Polyzos A, Kirkwood J. Immunotherapy for advanced melanoma: Fulfilling the promise. Cancer Treat Rev. 2013;39:879–85. doi: 10.1016/j.ctrv.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 6.Neagu M. The immune system - a hidden treasure for biomarker discovery in cutaneous melanoma. Adv Clin Chem. 2012;58:89–140. doi: 10.1016/b978-0-12-394383-5.00011-4. [DOI] [PubMed] [Google Scholar]
- 7.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–8. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 8.Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases - elimination, equilibrium and escape. Curr Opin Immunol. 2014;27:16–25. doi: 10.1016/j.coi.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21:137–48. doi: 10.1016/j.immuni.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 10.Raval RR, Sharabi AB, Walker AJ, Drake CG, Sharma P. Tumor immunology and cancer immunotherapy: summary of the 2013 SITC primer. J Immunother Cancer. 2014;2:14. doi: 10.1186/2051-1426-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karachaliou N, Pilotto S, Teixidó C, Viteri S, González-Cao M, Riso A, et al. Melanoma: oncogenic drivers and the immune system. Ann Transl Med. 2015;3:265. doi: 10.3978/j.issn.2305-5839.2015.08.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muul LM, Spiess PJ, Director EP, Rosenberg SA. Identification of specific cytolytic immune responses against autologous tumor in humans bearing malignant melanoma. J Immunol Baltim Md. 1987;138:989–95. [PubMed] [Google Scholar]
- 13.Márquez-Rodas I, Cerezuela P, Soria A, Berrocal A, Riso A, González-Cao M, et al. Immune checkpoint inhibitors: therapeutic advances in melanoma. Ann Transl Med. 2015;3:267. doi: 10.3978/j.issn.2305-5839.2015.10.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.European Medicine Agency. Imlygic 2015. [accessed on February 29, 2016]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002771/smops/Positive/human_smop_000894jsp&mid=WC0b01ac058001d127 .
- 15.Food and Drug Administration. FDA approves first-ofits-kind product for the treatment of melanoma. 2015. [accessed on February 29, 2016]. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm469571.htm .
- 16.Haanen JBAG, Robert C. Immune checkpoint inhibitors. Prog Tumor Res. 2015;42:55–66. doi: 10.1159/000437178. [DOI] [PubMed] [Google Scholar]
- 17.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li X, Hu W, Zheng X, Zhang C, Du P, Zheng Z, et al. Emerging immune checkpoints for cancer therapy. Acta Oncol Stockh Swed. 2015;54:1706–13. doi: 10.3109/0284186X.2015.1071918. [DOI] [PubMed] [Google Scholar]
- 19.Paulos CM, June CH. Putting the brakes on BTLA in T cell-mediated cancer immunotherapy. J Clin Invest. 2010;120:76–80. doi: 10.1172/JCI41811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lines JL, Pantazi E, Mak J, Sempere LF, Wang L, O’Connell S, et al. VISTA is an immune checkpoint molecule for human T cells. Cancer Res. 2014;74:1924–32. doi: 10.1158/0008-5472.CAN-13-1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Creelan BC. Update on immune checkpoint inhibitors in lung cancer. Cancer Control J Moffitt Cancer Cent. 2014;21:80–9. doi: 10.1177/107327481402100112. [DOI] [PubMed] [Google Scholar]
- 22.Walker LSK, Sansom DM. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat Rev Immunol. 2011;11:852–63. doi: 10.1038/nri3108. [DOI] [PubMed] [Google Scholar]
- 23.Honeychurch J, Cheadle EJ, Dovedi SJ, Illidge TM. Immuno-regulatory antibodies for the treatment of cancer. Expert Opin Biol Ther. 2015;15:787–801. doi: 10.1517/14712598.2015.1036737. [DOI] [PubMed] [Google Scholar]
- 24.Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: Similarities, differences, and implications of their inhibition. Am J Clin Oncol. 2016;39:98–106. doi: 10.1097/COC.0000000000000239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y, Yuan J, Yang Q, Cao W, Zhou X, Xie Y, et al. Immunoliposome co-delivery of bufalin and anti-CD40 antibody adjuvant induces synergetic therapeutic efficacy against melanoma. Int J Nanomedicine. 2014;9:5683–700. doi: 10.2147/IJN.S73651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aspeslagh S, Postel-Vinay S, Rusakiewicz S, Soria J-C, Zitvogel L, Marabelle A. Rationale for anti-OX40 cancer immunotherapy. Eur J Cancer Oxf Engl. 1990;52:50–66. doi: 10.1016/j.ejca.2015.08.021. [DOI] [PubMed] [Google Scholar]
- 27.Weigelin B, Bolaños E, Teijeira A, Martinez-Forero I, Labiano S, Azpilikueta A, et al. Focusing and sustaining the antitumor CTL effector killer response by agonist anti-CD137 mAb. Proc Natl Acad Sci USA. 2015;112:7551–6. doi: 10.1073/pnas.1506357112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schaer DA, Budhu S, Liu C, Bryson C, Malandro N, Cohen A, et al. GITR pathway activation abrogates tumor immune suppression through loss of regulatory T-cell lineage stability. Cancer Immunol Res. 2013;1:320–31. doi: 10.1158/2326-6066.CIR-13-0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu L, Xu X, Zhang B, Zhang R, Ji H, Wang X. Combined PD-1 blockade and GITR triggering induce a potent antitumor immunity in murine cancer models and synergizes with chemotherapeutic drugs. J Transl Med. 2014;12:36. doi: 10.1186/1479-5876-12-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271–5. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 31.Sansom DM. Moving CTLA-4 from the trash to recycling. Science. 2015;349:377–8. doi: 10.1126/science.aac7888. [DOI] [PubMed] [Google Scholar]
- 32.Ribas A. Antitumor activity in melanoma and anti-self responses in a phase I trial with the anti-cytotoxic T lymphocyte-associated antigen 4 monoclonal antibody CP-675, 206. J Clin Oncol. 2005;23:8968–77. doi: 10.1200/JCO.2005.01.109. [DOI] [PubMed] [Google Scholar]
- 33.Food and Drug Administration. FDA approves Yervoy to reduce the risk of melanoma returning after surgery 2015. [accessed on February 29, 2016]. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm469944.htm .
- 34.Piérard GE, Aubin F, Humbert P. Ipilimumab, a promising immunotherapy with increased overall survival in metastatic melanoma? Dermatol Res Pract 2012. 2012:182157. doi: 10.1155/2012/182157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ribas A, Hauschild A, Kefford R. Reply to K.S. Wilson et al. J Clin Oncol. 2013;31:2836–7. doi: 10.1200/JCO.2013.50.2120. [DOI] [PubMed] [Google Scholar]
- 36.Marabelle A, Eggermont A. How should we use anti-CTLA-4 antibodies? Eur J Cancer. 2015;51:2686–8. doi: 10.1016/j.ejca.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 37.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eggermont AMM, Chiarion-Sileni V, Grob J-J, Dummer R, Wolchok JD, Schmidt H, et al. Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2015;16:522–30. doi: 10.1016/S1470-2045(15)70122-1. [DOI] [PubMed] [Google Scholar]
- 39.Camacho LH. CTLA-4 blockade with ipilimumab: biology, safety, efficacy, and future considerations. Cancer Med. 2015;4:661–72. doi: 10.1002/cam4.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol. 2013;14:1212–8. doi: 10.1038/ni.2762. [DOI] [PubMed] [Google Scholar]
- 41.Tarhini AA, Zahoor H, Yearley JH, Gibson C, Rahman Z, Dubner R, et al. Tumor associated PD-L1 expression pattern in microscopically tumor positive sentinel lymph nodes in patients with melanoma. J Transl Med. 2015;13:319. doi: 10.1186/s12967-015-0678-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Improta G, Leone I, Donia M, Gieri S, Pelosi G, Fraggetta F. New developments in the management of advanced melanoma - role of pembrolizumab. OncoTargets Ther. 2015;8:2535–43. doi: 10.2147/OTT.S72823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.European Medicines Agency. Opdivo. Authorization details. [accessed on March 10, 2016]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/003985/human_med_ 001876jsp&mid=WC0b01ac058001d124 .
- 44.European Medicines Agency. Keytruda. Authorization details. [accessed on February 10, 2016]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/003820/human_med_001886jsp&mid=WC0b01ac058001d124 .
- 45.US Food and Drug Administration. FDA approves Keytruda for advanced melanoma. [accessed on February 10, 2016]. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm412802.htm .
- 46.US Food and Drug Administration. Nivolumab. [accessed on February 10, 2016]. Available from: http://www.fda.gov/Drugs/InformationOnDrugs/Approved Drugs/ucm427807.htm .
- 47.Weber JS, Minor DR, D’Angelo SP, Hodi FS, Gutzmer R, Neyns B, et al. LBA3_PRA phase 3 randomized, open-label study of nivolumab (Anti-PD-1; BMS-936558; ONO-4538) versus investigator's choice chemotherapy (ICC) in patients with advanced melanoma after prior anti-CTLA-4 therapy. [accessed on February 10, 2016];Ann Oncol. 2014 25(Suppl 4) Available from: http://annonc.oxfordjournals.org/content/25/suppl_4/mdu438.34 . [Google Scholar]
- 48.Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375–84. doi: 10.1016/S1470-2045(15)70076-8. [DOI] [PubMed] [Google Scholar]
- 49.Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320–30. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 50.Hamid O, Robert C, Daud A, Hodi FS, Hwu W-J, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134–44. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet Lond Engl. 2014;384:1109–17. doi: 10.1016/S0140-6736(14)60958-2. [DOI] [PubMed] [Google Scholar]
- 52.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2521–32. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 53.Ribas A, Puzanov I, Dummer R, Schadendorf D, Hamid O, Robert C, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16:908–18. doi: 10.1016/S1470-2045(15)00083-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in Tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–20. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Choueiri T, Fishman M, Escudier B, McDermott D, Kluger H, Stadler W, et al. Immunomodulatory activity of nivolumab in metastatic renal cell carcinoma (mRCC): Association of biomarkers with clinical outcomes. J Clin Oncol. 2015;33 doi: 10.1158/1078-0432.CCR-15-2839. suppl; abstr 2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ribas A, Robert C, Hodi FS, Wolchok JD, Joshua AM, Hwu W-J, et al. Association of response to programmed death receptor 1 (PD-1) blockade with pembrolizumab (MK-3475) with an interferon-inflammatory immune gene signature. J Clin Oncol. 2015 suppl; abstr 3001. [Google Scholar]
- 57.Andtbacka RHI, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33:2780–8. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 58.European Medicine Agency. First oncolytic immunotherapy medicine recommended for approval. [accessed on February 27, 2016]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2015/10/news_detail_002421.jsp&mid=WC0b01ac058004d5c1 .
- 59.Ascierto PA, Marincola FM, Atkins MB. What's new in melanoma? Combination! J Transl Med. 2015;13:213. doi: 10.1186/s12967-015-0582-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hodi FS, Lee S, McDermott DF, Rao UN, Butterfield LH, Tarhini AA, et al. Ipilimumab plus sargramostim vs ipilimumab alone for treatment of metastatic melanoma: a randomized clinical trial. JAMA. 2014;312:1744–53. doi: 10.1001/jama.2014.13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hodi FS, Lawrence D, Lezcano C, Wu X, Zhou J, Sasada T, et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol Res. 2014;2:632–42. doi: 10.1158/2326-6066.CIR-14-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–33. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372:2006–17. doi: 10.1056/NEJMoa1414428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gibney GT, Kudchadkar RR, DeConti RC, Thebeau MS, Czupryn MP, Tetteh L, et al. Safety, correlative markers, and clinical results of adjuvant nivolumab in combination with vaccine in resected high-risk metastatic melanoma. Clin Cancer Res. 2015;21:712–20. doi: 10.1158/1078-0432.CCR-14-2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weber JS, Kudchadkar RR, Yu B, Gallenstein D, Horak CE, Inzunza HD, et al. Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -Naive melanoma. J Clin Oncol. 2013;31:4311–8. doi: 10.1200/JCO.2013.51.4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2013;368:1365–6. doi: 10.1056/NEJMc1302338. [DOI] [PubMed] [Google Scholar]
- 68.Khattak M, Fisher R, Turajlic S, Larkin J. Targeted therapy and immunotherapy in advanced melanoma: an evolving paradigm. Ther Adv Med Oncol. 2013;5:105–18. doi: 10.1177/1758834012466280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Romano E, Michielin O, Voelter V, Laurent J, Bichat H, Stravodimou A, et al. MART-1 peptide vaccination plus IMP321 (LAG-3Ig fusion protein) in patients receiving autologous PBMCs after lymphodepletion: results of a Phase I trial. J Transl Med. 2014;12:97. doi: 10.1186/1479-5876-12-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jackson SR, Yuan J, Teague RM. Targeting CD8 + T-cell tolerance for cancer immunotherapy. Immunotherapy. 2014;6:833–52. doi: 10.2217/imt.14.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kohrt HE, Thielens A, Marabelle A, Sagiv-Barfi I, Sola C, Chanuc F, et al. Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells as monotherapy and in combination with anti-CD20 antibodies. Blood. 2014;123:678–86. doi: 10.1182/blood-2013-08-519199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MWL, Smyth MJ. Anti-TIM3 antibody promotes T cell IFN-γ-mediated antitumor immunity and suppresses established tumors. Cancer Res. 2011;71:3540–51. doi: 10.1158/0008-5472.CAN-11-0096. [DOI] [PubMed] [Google Scholar]
- 73.Ascierto PA, Atkins M, Bifulco C, Botti G, Cochran A, Davies M, et al. Future perspectives in melanoma research: meeting report from the “Melanoma Bridge”: Napoli, December 3rd-6th 2014. J Transl Med. 2015;13:374. doi: 10.1186/s12967-015-0736-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bajor DL, Xu X, Torigian DA, Mick R, Garcia LR, Richman LP, et al. Immune activation and a 9-year ongoing complete remission following CD40 antibody therapy and metastasectomy in a patient with metastatic melanoma. Cancer Immunol Res. 2014;2:1051–8. doi: 10.1158/2326-6066.CIR-14-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.de Vos S, Forero-Torres A, Ansell SM, Kahl B, Cheson BD, Bartlett NL, et al. A phase II study of dacetuzumab (SGN-40) in patients with relapsed diffuse large B-cell lymphoma (DLBCL) and correlative analyses of patient-specific factors. J Hematol Oncol J Hematol Oncol. 2014;7:44. doi: 10.1186/1756-8722-7-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Powderly JD, Gutierrez M, Wang D, Chae YK, Mahadevan D, Braiteh FS, et al. A phase 1b/2, open-label study to evaluate the safety and tolerability of MEDI6469 in combination with immune therapeutic agents or therapeutic mAbs in patients with selected advanced solid tumors or aggressive B-cell lymphomas. J Clin Oncol. 2015;33 suppl; abstr TPS3091. [Google Scholar]
- 77.Sanmamed MF, Rodriguez I, Schalper KA, Oñate C, Azpilikueta A, Rodriguez-Ruiz ME, et al. Nivolumab and urelumab enhance antitumor activity of human T lymphocytes engrafted in Rag2-/-IL2Rγnull immunodeficient mice. Cancer Res. 2015;75:3466–78. doi: 10.1158/0008-5472.CAN-14-3510. [DOI] [PubMed] [Google Scholar]
- 78.Trial of TRX518 (Anti-GITR mAb) in Stage III or IV malignant melanoma or other solid tumors (TRX518-001) [accessed on February 29, 2016]. Available from: http://clinicaltrials.gov/ct2/show/NCT01239134 .