

Abstract
Background
Foam cells are central to two major pathogenic processes in atherogenesis: cholesterol buildup in arteries and inflammation. The main underlying cause of cholesterol deposition in arteries is hypercholesterolemia. This study aimed to assess, in vivo, whether elevated plasma cholesterol also alters the inflammatory balance of foam cells.
Methods and Results
Apolipoprotein E–deficient mice were fed regular mouse chow through the study or were switched to a Western‐type diet (WD) 2 or 14 weeks before death. Consecutive sections of the aortic sinus were used for lesion quantification or to isolate RNA from foam cells by laser‐capture microdissection (LCM) for microarray and quantitative polymerase chain reaction analyses. WD feeding for 2 or 14 weeks significantly increased plasma cholesterol, but the size of atherosclerotic lesions increased only in the 14‐week WD group. Expression of more genes was affected in foam cells of mice under prolonged hypercholesterolemia than in mice fed WD for 2 weeks. However, most transcripts coding for inflammatory mediators remained unchanged in both WD groups. Among the main players in inflammatory or immune responses, chemokine (C‐X‐C motif) ligand 13 was induced in foam cells of mice under WD for 2 weeks. The interferon‐inducible GTPases, guanylate‐binding proteins (GBP)3 and GBP6, were induced in the 14‐week WD group, and other GBP family members were moderately increased.
Conclusions
Our results indicate that acceleration of atherosclerosis by hypercholesterolemia is not linked to global changes in the inflammatory balance of foam cells. However, induction of GBPs uncovers a novel family of immune modulators with a potential role in atherogenesis.
Keywords: atherosclerosis, cholesterol, gene expression, inflammation, macrophages
Subject Categories: Atherosclerosis, Gene Expression & Regulation
Introduction
Lipid‐laden macrophages, or foam cells, are chief cellular components of atherosclerotic lesions through all stages of development.1 Circulating monocytes are recruited to the arterial wall in response to inflammatory stimuli induced, among other factors, by modified low‐density lipoprotein (mLDL) particles deposited in the subendothelial space. Monocytes differentiate into macrophages that take up mLDL in an unfettered fashion and become heavily loaded with lipoprotein‐derived cholesterol, while they also orchestrate the development of a local inflammatory process.2, 3, 4 Given that, presumably, the amount of mLDL deposited in arteries is related to the concentration of circulating LDL, it might be inferred that, by promoting monocyte immigration, hypercholesterolemia directly contributes to vascular inflammation.3 However, it could also be possible that exposure to different lipoprotein concentrations alters the inflammatory balance of foam cells. The main LDL modifications related to atherosclerosis development involve lipid peroxidation.5 Transcriptional response of macrophages to exposure to oxidized (ox) LDL has been tested in cultured macrophages of different sources, with diverse results that, depending on the study, suggested predominantly proinflammatory or anti‐inflammatory effects.6, 7, 8, 9 In some cases, the outcome was significantly affected by changes in experimental variables, such as the cell type, form of LDL modification and presentation to cells, or time of exposure to the lipoproteins. For example, Brand et al. found that short‐term exposure of THP‐1 macrophages to oxLDL induced activation of nuclear factor kappa B (NF‐κB), whereas long‐term exposure to oxLDL not only did not activate NF‐κB, but actually prevented NF‐κB activation by lipopolysaccharide10; Hammad et al. observed different effects on inflammatory gene expression depending on whether U937 monocytic cells were treated with oxLDL or with oxLDL immune complexes11; and Shiffman et al. identified gene clusters in THP‐1 macrophages with different temporal patterns of expression in response to treatment with oxLDL. An added challenge that might contribute to the variability of studies on macrophages is the remarkable plasticity of these cells, which allows them to change their phenotype depending on the surrounding environment.12 Thus, given that the complex atherosclerotic milieu is difficult to reproduce in vitro, cell‐culture approaches to characterize macrophages may not represent the functional relevance of the variables being studied as well as studies performed on macrophages resident within actual atherosclerotic lesions.
The apolipoprotein E‐deficient (apoE−/−) mouse is a widely used mouse model of atherosclerosis.13, 14, 15 From a molecular point of view, gene expression patterns in mouse aortas that defined different stages of atherosclerosis development also correlated with severity of human coronary lesions.16 Like in humans, atherosclerosis development in apoE−/− mice is driven by hypercholesterolemia, and plasma cholesterol levels and the extent of lesion development are directly related to the cholesterol content in their diet.17 Thus, to study the effect of hypercholesterolemia on the transcriptional response of lesional foam cells, here we have fed apoE−/− mice regular chow through the study or have switched their diet to a Western‐type diet (WD) for a short (2 weeks) or for a longer (14 weeks) time period. We have assessed the effects of the WD on atherosclerosis development in cross‐sections of the aortic sinus, and we have used sections consecutive to the used for lesion quantification to selectively isolate RNA by laser‐capture microdissection (LCM) from lesional macrophages to perform a broad analysis of gene expression, with an emphasis on the expression of genes that regulate inflammatory and immune responses.
Methods
Experimental Design
At 8 weeks of age, 15 female apoE−/− mice in C57BL6/J background were divided into 3 groups with similar cholesterol levels. One group was maintained on regular mouse chow (2020X Teklad Global Soy Protein‐Free Extruded Rodent Diet; Harlan Laboratories, Indianapolis, IN) until death at 22 weeks. This is a cholesterol‐free diet that provides 3.5 kcal/g, including 16% of calories from fat. From 8 weeks of age to the end of the study, the diet of a second group of 5 mice was switched from regular chow to a WD (TD.88137; Harlan Laboratories). This diet provides 4.5 kcal/g and contains 20% (wt/wt) milk fat (42% of total calories) and 0.15% (wt/wt) cholesterol. The third group of mice remained on regular chow until the age of 20 weeks, when mice were fed WD for the remaining 2 weeks of the study. The study design is summarized in Figure 1A. Cholesterol measurements and lipoprotein fractionation by fast‐performance liquid chromatography were performed as we previously described.18 Plasma oxLDL levels were determined by ELISA (USCN Life Science Inc., Wuhan, China), following the manufacturer's instructions.19, 20 Additional mice were used to obtain aortic arches for RNA isolation from whole artery, isolate thioglycollate‐elicited peritoneal macrophages, and assess the rate of increase of plasma cholesterol upon introduction of the WD. All animal experiments were conducted following protocols approved by the institutional animal care and use committees at Albany Medical College (Albany, NY) and Baylor College of Medicine (Houston, TX).
Figure 1.
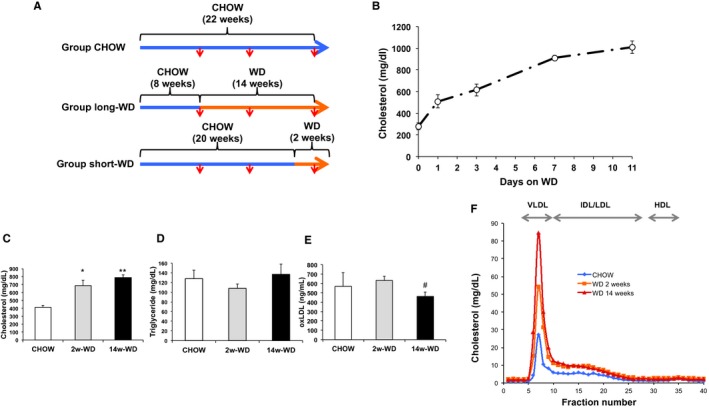
A, Study design. Blue lines represent time periods in which mice were fed regular chow; periods of WD feeding are represented in orange. Red arrows indicate times of blood sampling. B, Time course of plasma cholesterol elevation after introduction of WD. Mice were not fasted for these plasma measurements (n=5). C, D, and E, Fasting plasma cholesterol (C), triglyceride (D), and oxLDL (E) levels at time of death in mice used for gene expression studies (n=5). F, Cholesterol distribution in different lipoprotein fractions in pooled plasmas obtained at week 22 under overnight fasting. *P<0.005 and **P<0.001 with respect to CHOW; # P<0.05 with respect to 2‐week WD. HDL indicates high‐density lipoprotein; IDL, intermediate‐density lipoprotein; LDL, low‐density lipoprotein; oxLDL, oxidized low‐density lipoprotein; VLDL, very low‐density lipoprotein; WD, Western‐type diet.
Lesion Analysis
After death, mouse hearts were perfused with sterile PBS, bisected, and the upper half immediately embedded in OCT (Sakura, Torrance, CA) and stored at −80°C. Approximately 30 consecutive 7‐μm cryosections of each aortic sinus were sequentially mounted on 3 slides. The first and third slides, which were used for macrophage isolation by LCM, were immediately fixed, cell nuclei were stained with toluene blue, and sections were dehydrated with the HistoGene LCM Frozen Section Staining Kit (Applied Biosystems, Foster City, CA). The second slide was stained for macrophages with anti‐Lamp2/Mac3 antibody (Santa Cruz Biotechnology, Santa Cruz, CA) and used as a template to identify macrophage‐rich areas within the lesions. Images were acquired with a Zeiss AxioObserver.D1 microscope using an AxioCam MRc camera (Carl Zeiss, Jena, Germany), and analyzed with AxioVision software (Zeiss). Lesion area was measured by outlining the perimeter of the area between the vessel lumen and media layer of the arteries. The areas within lesions that stained positive for macrophages were determined using ImageJ software (National Institutes of Health [NIH], Bethesda, MD), as we previously described.21
LCM and RNA Amplification
LCM of macrophages and RNA processing were performed as we previously described.22 Briefly, ≈2000 laser shots (power, ≈65 mV; pulse, ≈2500 μs) were performed with a Veritas Microdissection System (Applied Biosystems), and cells were collected on CapSure HS LCM Caps (Applied Biosystems). RNA was extracted with the PicoPure RNA Isolation Kit (Applied Biosystems). mRNA was submitted to 2 rounds of amplification with the RiboAmp HS RNA Amplification Kit (Applied Biosystems), each of which consisted of synthesis of double‐stranded cDNA using oligo(dT) primers tagged with the T7 promoter sequence, followed by in vitro transcription (IVT) with T7 polymerase.22 A260/A280 ratios and size distribution analysis of amplified cRNAs were assessed with an Agilent 2100 bioanalyzer (Agilent Technologies Inc., Santa Clara, CA).
Gene Expression Analysis
Fifteen micrograms of each cRNA were biotin‐labeled with the TURBO Labeling Biotin kit (Applied Biosystems), fragmented, and hybridized to Mouse Genome 430 2.0 Arrays (Affymetrix, San Diego, CA), as we previously described.22 Data were filtered with dChip software to include probes with ≥50% presence call in the arrays and with expression levels of ≥25 in ≥50% samples. The filtered data were transferred to MeV software,23 log2 transformed, and differential gene expression in the 3 experimental groups was assessed by ANOVA. Welch's t test (assuming unequal variances) was used for pair‐wise comparisons. Differences were considered significant when P≤0.01. To visualize the patterns of gene expression between data sets, volcano plots were generated by plotting significance versus fold change in the Y and X axes, respectively. Pathway Express was used to identify the most relevant pathways affected by the dietary manipulations.24 This software calculates P values based on the number of genes differentially expressed in each pathway in relationship to the number of genes expected to change by chance. It also produces a gamma P value that, in addition to classical statistics, takes into consideration parameters such as the fold change and the topology of genes within each given pathway.24 Thus, the gamma P value is influenced by biologically meaningful factors that are not usually captured by classic statistics.
Primers for quantitative real‐time polymerase chain reaction (qPCR) were designed in the 3′‐terminus region of mRNA, including the 3′ untranslated region (Table). Relative gene expression was determined from threshold cycle values normalized to cyclophilin A, as we previously described.22 For gene expression analyses in cultured primary macrophages, thioglycollate‐elicited peritoneal macrophages were cultured in DMEM/0.2% BSA containing oxLDL (1, 50 or 100 μg/mL; Alfa Aesar, Ward Hill, Ma) for 4 or 24 hours. RNA was purified and digested with DNase using the Absolutely RNA miniprep kit (Agilent Technologies). Reverse transcription of 500 ng of total RNA was performed with SuperScript III (Invitrogen, Carlsbad, CA). The qPCR protocol was the same used for analysis of samples isolated by LCM.
Table 1.
Primer Sequences
| Name | Sequence |
|---|---|
| Cyclophilin A | Forward 5′‐TGGTCTTTGGGAAGGTGAAA ‐3′ |
| Reverse 5′‐CACAGTCGGAAATGGTGATCT‐3′ | |
| CD68 | Forward 5′‐ CCTCACCCTGGTGCTCAT ‐3′ |
| Reverse 5′‐ ATTGATTCCCCACCCCTATT‐3′ | |
| CD14 | Forward 5′‐ GTGGCCTTGTCAGGAACTCT ‐3′ |
| Reverse 5′‐ ATCAGGGGTCAAGTTTGCTG ‐3′ | |
| SR‐A1 | Forward 5′‐ GCCCTGTTCAGAAGCATCA ‐3′ |
| Reverse 5′‐ CTTGATCACGAGCACAGCAT ‐3′ | |
| ABCA1 | Forward 5′‐TGGATCTATTTTTGCACTGGA‐3′ |
| Reverse 5′‐CAGCAGGACTGTCACAGCTTTA‐3′ | |
| α‐actin | Forward 5′‐ TCAACAGAGGAAGGTCCACTT ‐3′ |
| Reverse 5′‐ ACTTGCCAAATTTTAAATACACG ‐3′ | |
| SM22 | Forward 5′‐CAGAGGAAGCAGGCTATGGA‐3′ |
| Reverse 5′‐AACCCAAACAAGTCCACCAC‐3′ | |
| MYH11 | Forward 5′‐CACAGGAAACTTCGCAGTGA‐3′ |
| Reverse 5′‐TTCTGTTTTCCCTGACATGGT ‐3′ | |
| VE‐cadherin | Forward 5′‐ AGGAAGGGGCATACAGACAC ‐3′ |
| Reverse 5′‐ CCTGCAGAAAGGCCTTGTTG ‐3′ | |
| IL‐6 | Forward 5′‐TGACAATATGAATGTTGGGACA‐3′ |
| Reverse 5′‐TTCCAAGAAACCATCTGGCTA‐3′ | |
| CCL2 | Forward 5′‐TCCCTCTCTGTGAATCCAGA‐3′ |
| Reverse 5′‐ ACCTTGGAATCTCAAACACAAAG‐3′ | |
| CXCL13 | Forward 5′‐ CCTGGGGGAAACAGTCCTAC ‐3′ |
| Reverse 5′‐ GCCTGGACCTTTAAGCTGAG ‐3′ | |
| TNF‐α | Forward 5′‐GTCCTGGAGGACCCAGTGT‐3′ |
| Reverse 5′‐GGGAGCAGAGGTTCAGTGAT‐3′ | |
| IL‐18 | Forward 5′‐ GTTAGGTGGGGAGGGTTTGT‐3′ |
| Reverse 5′‐ GCAGCCTCGGGTATTCTGT ‐3′ | |
| GBP3 | Forward 5′‐ ACATGGCCAAATGAAGACACA‐3′ |
| Reverse 5′‐ TGAAAACCCACTTGTGCGTT‐3′ | |
| GBP6 | Forward 5′‐ TCATCTTGGTGGTTGGAACA‐3′ |
| Reverse 5′‐TCATGAGAAACAATGTCACAAGG‐3′ | |
| GBP8 | Forward 5′‐ TGAGGGTATTTCATCACAGCA ‐3′ |
| Reverse 5′‐ TTGCCAATCTAACTCAGGGATG‐3′ | |
| GBP4 | Forward 5′‐CCTTGTGATATTGTTTCCACGGT‐3′ |
| Reverse 5′‐AATTGGGAAGGTTGCAGGTG‐3′ | |
| GBP2 | Forward 5′‐ TGTTCCTCTTTCCTACAGATAGC ‐3′ |
| Reverse 5′‐ AAGTCCTCAGAGAAATGAAAGGG ‐3′ |
ABCA1 indicates ATP binding cassette A1; CCL2 indicates chemokine (C‐C motif) ligand 2; CXCL, chemokine (C‐X‐C motif) ligand 13; GBP, guanylate‐binding protein; IL, interleukin; MYH11, myosin heavy chain 11; SM22, smooth muscle protein 22; SR‐A1, scavenger receptor A1; TNFα, tumor necrosis factor alpha.
Statistical Analysis
Statistical analysis of nonmicroarray data was carried out using parametric methods when the data followed a normal distribution and the samples had equal variances. Otherwise, nonparametric tests were used for the analysis. When parametric tests were used, multiple comparisons were analyzed by ANOVA, and post‐hoc pair‐wise comparisons were performed using the Holm–Sidak test. When nonparametric tests were used, multiple comparisons were performed with the Kruskal–Wallis test, followed by pair‐wise comparisons with the Mann–Whitney U test. Statistical analysis involving comparisons between two groups were performed using a 2‐tailed Student t test (parametric) or the Mann–Whitney U test (nonparametric). Differences were considered significant when P<0.05. Values are presented as mean±SEM.
Results
Effects of WD on Plasma Lipids and Atherosclerosis Development
To determine the adequate length of the study in which foam cells were exposed to hypercholesterolemia for a short time period, we performed a preliminary study to assess the rate of increase of plasma cholesterol in apoE−/− mice upon introduction of the WD. As seen in Figure 1B, plasma cholesterol was markedly higher at 24 hours of WD feeding, but levels continued to increase gradually to reach concentrations of ≈1000 mg/dL (under fed conditions) at day 11. Thus, in order to expose foam cells to plasma cholesterol levels similar to the achieved under prolonged WD, but for a period of time not long enough to affect atherosclerosis development, mice were fed WD for 2 weeks. Fasting plasma cholesterol levels measured at the time of death in mice used for gene expression studies were still moderately (≈10%) higher in mice fed WD for 14 weeks than in mice that were fed WD for only 2 weeks, although these differences did not reach statistical significance. However, in both WD groups, cholesterol levels were significantly (≈2‐fold) higher than in mice fed regular mouse chow (Figure 1C and 1F). As expected in apoE−/− mice, plasma triglycerides did not increase in response to WD (Figure 1D). Although oxidative modifications are believed to take place mainly on apoB‐containing lipoproteins retained at the arterial wall, oxLDL is also present in the circulation.25, 26 Plasma oxLDL were very similar between mice fed chow through the study and mice fed WD for the last 2 weeks, whereas oxLDL levels were slightly reduced in mice fed WD for 14 weeks (Figure 1E). This observation is consistent with recent studies that showed that plasma oxLDL decreases concomitantly with increase in lesion size.26 Indeed, the prolonged hypercholesterolemia in the 14‐week WD group resulted in a very substantial (>3‐fold) increase in atherosclerosis development at the aortic sinus: 696±32×103 μm2 in mice fed WD for 14 weeks versus 221±13×103 μm2 in mice fed regular chow through the study. However, the size of the lesions in the 2‐week WD group was similar to that of mice fed regular chow: 263±37 μm2 (Figure 2A and 2B). Next, we performed immunohistochemical analyses to quantify the areas of macrophages within lesions. The relative macrophage content was similar in mice fed chow through the study and in mice fed WD for 2 weeks (≈35–40%), but it was lower (≈25%) in mice fed WD for 14 weeks (Figure 2C). However, the total area of macrophages, calculated by multiplying the percentage of macrophages by the total lesion area, was higher in mice fed WD for 14 weeks (Figure 2D). Thus, WD feeding for either 2 or 14 weeks significantly increased plasma cholesterol levels. However, lesions in mice fed WD for 2 week were similar in size and contained a similar amount of macrophages than those of mice fed chow through the study, whereas the longer WD feeding protocol markedly increased the progression of atherosclerotic lesions.
Figure 2.
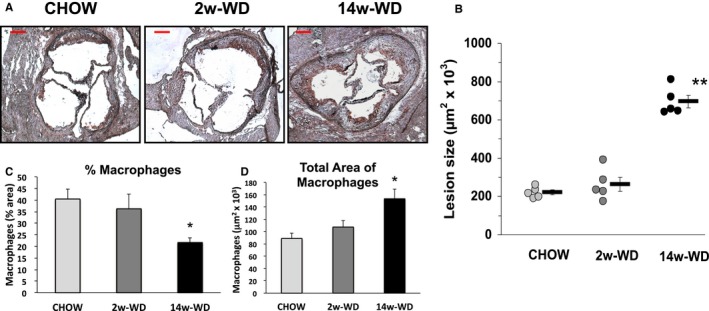
A, Representative images of sections of the aortic sinus in the 3 experimental groups stained with the macrophage marker, Mac‐3 (Brown), and counterstained with hematoxylin. Bar=200 μm. B, Area of atherosclerosis involvement was determined by quantifying the area between the media layer and the vessel lumen. Dots represent individual values. Bars represent average±SEM. C, Percentage of lesion area occupied by macrophages and (D) absolute of macrophages at the aortic sinus. *P<0.05 and **P<0.001 with respect to CHOW. WD indicates Western‐type diet.
Gene Expression Analyses of Lesional Foam Cells
RNA from lesional foam cells was isolated by LCM and amplified by IVT to obtain enough material for a broad gene expression analysis.22 First, to control the quality of the isolation and amplification processes, we assessed whether the gene expression patterns in the cRNA amplified from cells captured by LCM were the expected in macrophage/foam cell populations. Using qPCR, we compared expression of several macrophage markers between these cRNA samples and cRNA amplified from lysates of aortic arches (whole artery) of apoE−/− mice. As seen in Figure 3A through 3D, levels of transcripts that are typically elevated in macrophage populations, including CD68, CD14, scavenger receptor A1 (SR‐A1), and ATP binding cassette A1 (ABCA1), were enriched in LCM‐cRNAs. Conversely, levels of transcripts coding for the smooth muscle cell (SMC) markers, α‐actin, smooth muscle myosin heavy chain 11 (MYH11), and smooth muscle protein 22 (SM22), were significantly reduced in macrophages isolated by LCM (Figure 3E through 3G). Likewise, mRNA coding for the endothelial cell marker VE‐cadherin, was also reduced in LCM samples (Figure 3H). Importantly, both the enrichment in macrophage markers and the decrease in SMC markers and VE‐cadherin were similar in LCM‐cRNAs in the 3 experimental groups.
Figure 3.
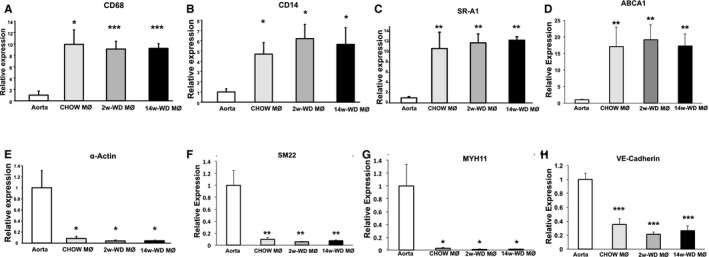
qPCR analysis comparing the expression of genes between RNA isolated from macrophage/foam cell populations (MΦ) by LCM and RNA isolated from aortic arches (aorta). A through D, Are genes highly expressed by foam cells, including CD68 (A), CD14 (B), SR‐A1 (C) and ABCA1 (D). E through H, Are markers of other cell types, including the smooth‐muscle cell markers α‐actin (E), SM22 (F) and MYH11 (G), and the endothelial cell marker VE‐cadherin (H). n=5. *P<0.05, **P<0.01 and ***P<0.005 with respect to aorta. LCM indicates laser‐capture microdissection; qPCR, quantitative polymerase chain reaction; WD, Western‐type diet.
For broad gene expression profiling of foam cells, amplified cRNAs were biotinylated and hybridized to Affymetrix DNA chips. The microarray data have been deposited at the GEO (Gene Expression Omnibus) repository and can be accessed through the accession number GSE70619. Data was filtered to remove probes with low presence of call and/or low intensity across the board of arrays, and 15 433 targets satisfied the filtering criteria. Among them, ANOVA analysis at a P value of 0.01 followed by pair‐wise comparisons against the “CHOW” group identified 52 targets significantly affected in the 2‐week WD group and 366 targets in the 14‐week WD group. Volcano plots summarizing these changes are shown in Figure 4A and 4B, and the genes affected by the dietary manipulations are listed in Table S1 (2‐week WD vs CHOW) and Table S2 (14‐week WD vs CHOW). To determine the main biological processes affected in response to hypercholesterolemia, the data were analyzed with Pathway Express. Two pathways, “antigen processing and presentation” and “ubiquitin‐mediated proteolysis,” were commonly affected in both WD groups (Figure 4C). Changes in genes in the antigen processing and presentation pathway could be related to the increased uptake of modified lipoproteins by foam cells of mice under WD. The reasons for the over‐representation of genes in the ubiquitin‐mediated proteolysis pathway are not clear, though it was reported that protein ubiquitination was increased in atherosclerotic plaques, and ubiquitin‐mediated proteolysis was the most over‐represented pathway in plaques of diabetic patients.27, 28 Whereas these two pathways were the only ones affected in the 2‐week WD group, the higher number of genes differently expressed in the 14‐week WD group was reflected in 11 additional pathways significantly affected in this group (Figure 4C). Given that lesions were more developed in response to prolonged WD, but the plasma lipid profiles were similar in both WD groups, it is likely that changes in the lesional microenvironment contributed to the more‐robust changes in gene expression observed in the 14‐week WD group. Indeed, the modest changes in gene expression observed in mice fed WD for only 2 weeks suggest that hypercholesterolemia is not a major determinant of transcriptional response of foam cells. However, whether the changes in gene expression were directly related to the plasma cholesterol concentrations, or were the result of other changes in the lesional milieu, neither WD feeding protocol affected the expression of the vast majority of genes in inflammatory and immune response categories. The most noteworthy exceptions were chemokine (C‐X‐C motif) ligand 13 (CXCL13), which was upregulated in the 2‐week WD group by ≈4.4‐fold, and two members of the p65 guanylate‐binding proteins (GBP) family of interferon‐inducible GTPases, GBP3 and GBP6, which were induced in the 14‐week WD group by ≈3.2‐ and ≈5.2‐fold, respectively.
Figure 4.
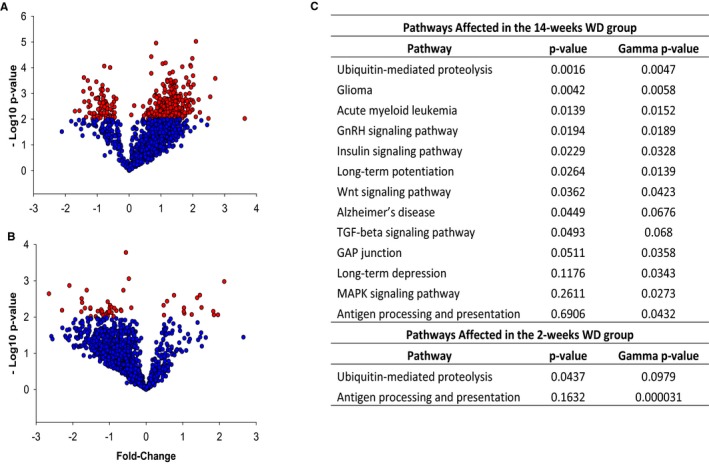
A and B, Volcano plots showing the fold change and level of significance of the genes affected in the 14‐week WD group (A) and in the 2‐week WD group (B). Red dots represent genes statistically significant at P<0.01. C, Pathways affected in foam cells isolated from lesions of mice fed WD for 2 and 14 weeks were determined using Pathway Express. The P value is calculated by a classical analysis based on the set of genes differentially expressed in each pathway related to the number of genes expected to be found by chance. The gamma P value is calculated by an impact analysis that, in addition to classical statistics, includes important biological factors such as fold change and topology of genes within each given pathway. GnRH indicates gonadotropin‐releasing hormone; MAPK indicates mitogen‐activated protein kinase; TGF, transforming growth factor; WD, Western‐type diet.
Analysis of Inflammatory Mediators
The inflammatory nature of atherosclerosis has been supported by numerous studies, including analysis of gene expression in both clinical samples and animal models of atherosclerosis.2, 4 Thus, to assess whether genes involved in inflammation could have been affected, but to an extent that did not meet the criteria of this analysis, we performed a similar analysis at a significance level of P<0.05 instead of P<0.01. As expected, the less‐conservative analysis yielded a larger number of genes significantly affected by both dietary manipulations (78 targets significantly changed when comparing 2‐week WD vs CHOW and 571 targets when comparing 14‐week WD vs CHOW). However, the absence of immune and inflammatory mediators was still evident, and, accordingly, pathways related to the inflammatory and immune responses were not significantly affected (data not shown). Next, we focused the analysis on expression of transcripts coding for major cytokines and chemokines that had been related to progression of atherosclerosis or development of more‐severe plaque phenotypes.16, 28, 29, 30, 31 As seen in Figure 5A, in both WD groups, the levels of most transcripts coding for interleukins or for CC or CXC cytokines were very close or within ≈1‐fold change of the levels observed in the CHOW group. There were no obvious trends toward higher or lower ratios in any of the 2 WD groups that suggested an overall proinflammatory or anti‐inflammatory effect. Among these players in inflammation, only CXCL13 (in the 2‐week WD group) was significantly elevated. This result was confirmed by qPCR analysis (Figure 5B). Interleukin‐6 (IL‐6) was increased by ≈5‐fold in the 14‐week WD group, although the signal intensities for IL‐6 were very variable and the differences did not reach statistical significance. Changes in IL‐6 mRNA levels were not supported by qPCR analysis (Figure 5C). Other inflammatory mediators that displayed modest and nonstatistically significant increases in the microarray analyses, namely, chemokine (C‐C motif) ligand 2 (CCL2) in the 2‐week WD group or IL‐18 in the 14‐week WD group, also remained similar among groups by qPCR analysis (Figure 5D and 5E). Interestingly, whereas CXCL13 was induced by short‐term hypercholesterolemia, it was not elevated in foam cells from chow‐fed mice when compared to whole aortic arches. In contrast, levels of transcripts coding for IL‐6, CCL2, and IL‐18 were significantly higher in all 3 foam cell populations.
Figure 5.
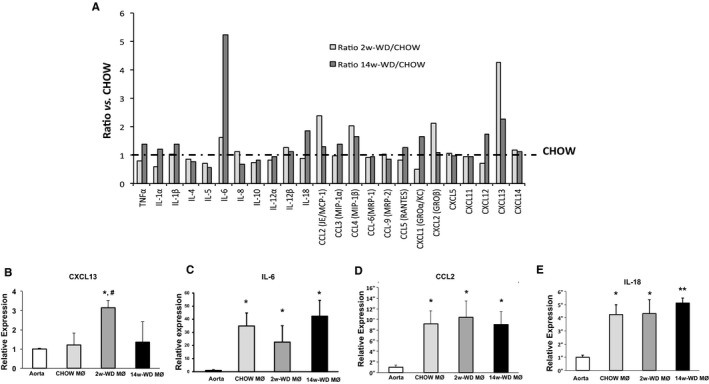
A, Average signal intensity for some of the main macrophage cytokines and chemokines in the microarray analysis. Bars represent the ratios (fold increase) with respect to LCM macrophages in the CHOW group (dashed line). B through E, qPCR analysis of mRNA expression of CXCL13, IL‐6, CCL2, and IL‐18 in aortas and LCM macrophages of the 3 experimental groups. *P<0.05 and **P<0.001 with respect to RNA isolated from aortic arches (Aorta); # P<0.05 with respect to CHOW macrophages (MØ). CCL indicates chemokine (C‐C motif) ligand; CXCL, chemokine (C‐X‐C motif) ligand; IL, interleukin; LCM, laser‐capture microdissection; qPCR, quantitative polymerase chain reaction; TNFα, tumor necrosis factor alpha; WD, Western‐type diet.
Induction of GBPs by Hypercholesterolemia
A remarkable observation in the microarray analyses was the induction of two p65‐GBPs (GBP3 and GBP6) in foam cells of mice fed WD for 14 weeks. Several studies have shown that different GBPs are concomitantly induced.32 Thus, we asked whether in the 14‐week WD group there would also be some degree of induction of other GBP family members. Interestingly, as seen in Figure 6A, levels of 5 of the 6 GBPs included in the microarray was higher in foam cells isolated from mice fed WD for 14 weeks than in mice fed regular chow. Intensity of GBP2, GBP7, and GBP8 was ≈2‐fold higher, although these differences were not statistically significant. In general, GBPs were not elevated in mice fed WD for only 2 weeks, although GBP6 was relatively higher than in control CHOW‐fed mice both in the microarray and qPCR analyses (Figure 6A and 6C). Results of qPCR analyses were consistent with the microarray data (Figure 6B through 6F). Next, we asked whether the changes observed in vivo would also be observed in vitro in peritoneal macrophages treated with oxLDL. It was reported that oxLDL in human endarterectomy specimens was nearly 70 times higher than plasma oxLDL in the same patients.33 Thus, given that measurements of oxLDL concentrations within mouse atherosclerotic lesions are extremely challenging, we performed a dose‐response study that included a low dose of oxLDL close to the circulating levels (1 μg/mL) and 2 higher doses of 50 and 100 μg/mL. Mouse peritoneal macrophages were exposed to these concentrations of oxLDL for 4 and 24 hours. None of the treatments altered the levels of the CXCL13 mRNA. However, as seen in Figure 7A, we observed significantly increased GBP3 and GBP6 mRNA in peritoneal macrophages cultured with 100 μg/mL of oxLDL. Overall, GBP6 seems to be the most responsive GBP family member both in vivo and in vitro. Next, we assessed whether expression of other GBP family members was also induced in response to oxLDL, and found that GBP2 and GBP7 were also increased upon treatment with oxLDL (100 μg/mL). Thus, whereas CXCL13 was not induced by oxLDL in vitro, several GBP family members were induced, suggesting that oxLDL might be one of the factors involved in the induction of GBPs in response to hypercholesterolemia.
Figure 6.
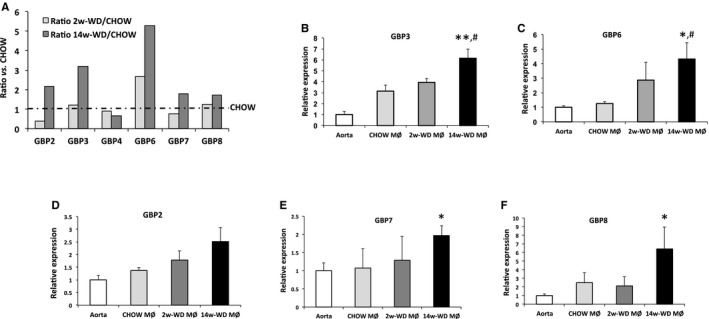
A, Average signal intensity for GBPs present in the microarray. Bars represent the ratios (fold increase) with respect to LCM macrophages in the CHOW group (dashed line). B through F, qPCR analysis of expression of GBP3, GBP6, GBP2, GBP7, and GBP8 in aortas and LCM macrophages of the 3 experimental groups. *P<0.05 and **P<0.001 with respect to RNA isolated from aortic arches (Aorta); # P<0.05 with respect to CHOW macrophages (MØ). GBP indicates guanylate‐binding protein; LCM, laser‐capture microdissection; qPCR, quantitative polymerase chain reaction; WD, Western‐type diet.
Figure 7.
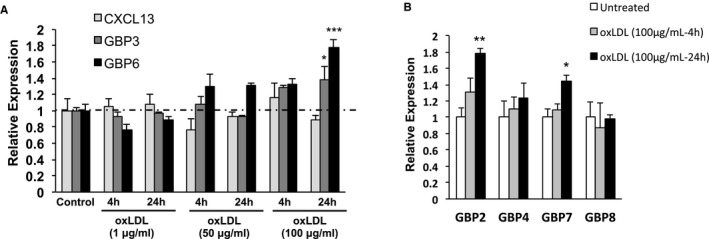
A, qPCR analysis of expression of CXCL13, GBP3, and GBP6 in mouse peritoneal macrophages that remained untreated or were stimulated with oxLDL (1, 50, or 100 μg/mL) for 4 or 24 hours. B, Expression of other GBP family members in response to oxLDL (100 μg/mL for 4 or 24 hours; n=3). *P<0.05, **P<0.01 and ***P<0.001 with respect to untreated control. CXCL, chemokine (C‐X‐C motif) ligand; GBP, guanylate‐binding protein; oxLDL, oxidized low‐density lipoprotein; qPCR, quantitative polymerase chain reaction.
Discussion
Hypercholesterolemia is a leading risk factor for development of atherosclerosis, an inflammatory disease of the arterial wall.2 There is compelling evidence supporting the notion that hypercholesterolemia enhances vascular inflammation, including gene expression profiles of aortic sinuses isolated from apoE−/− mice that showed increased expression of inflammatory mediators after the introduction of a Western diet.30, 34 Upregulation of proinflammatory genes observed in these studies could be the logical consequence of the increased influx of inflammatory cells to the arterial wall during progression of the disease, but could also be related to other factors, such as the activation of lesional cells, in response to hypercholesterolemia. In this line, there is evidence that exposure to modified lipoproteins might regulate the inflammatory response of foam cells. For example, oxLDL was shown to activate NF‐κB by binding to Toll‐like receptors (TLRs) such as TLR2 and TLR4.35, 36 Accumulation of cholesterol crystals in the cytoplasm of macrophages was also proposed to stimulate a proinflammatory cascade.37 Alternatively, lipoprotein‐derived oxysterols are natural liver X receptor ligands, which can counter‐regulate induction of inflammatory gene expression by NF‐κB by recruiting corepressors to the promoters of inflammatory genes.37, 38, 39 Thus, we asked whether increases in plasma cholesterol that significantly accelerate atherosclerosis development would actually affect the inflammatory balance of lesional foam cells. Answering this question using complex heterogeneous samples, such as fragments of atheromatous plaques or diseased arteries, may be quite challenging, mainly because of the variable number of inflammatory cells that can be found in these specimens. To circumvent this challenge, here we have used LCM to specifically isolate and assess the inflammatory status of macrophages resident within atherosclerotic lesions. We fed apoE−/− mice for a period of time that was not long enough to affect the size of atherosclerotic lesions (2 weeks) or for a longer period that significantly enhanced the development of atherosclerosis (14 weeks). Whereas WD feeding resulted in a ≈2‐fold elevation in plasma cholesterol, neither dietary manipulation affected expression of the vast majority of genes coding for inflammatory mediators. Thus, a first and foremost conclusion of this study is that accelerated development of atherosclerosis in response to hypercholesterolemia was not linked to major changes in the inflammatory balance of foam cells, as it would be expected if the activity of central regulators of inflammation, such as NF‐κB, were affected.
Although we did not observe global changes in inflammation, we observed certain changes that might be relevant to the pathogenesis of the disease. Expression of the chemokine CXCL13, was elevated only in the short‐term WD group. CXCL13 is a homeostatic chemokine that has been primarily linked to lymphocyte trafficking, but has also been shown to influence other key processes such as activation of T cells and macrophages.40 CXCL13 is produced by macrophages and is expressed in human atherosclerotic lesions.40, 41 However, its role in atherogenesis remains poorly characterized, and it has actually been proposed to play a role both in plaque stabilization and plaque destabilization.40, 41, 42 It is noteworthy that the in vivo changes in CXCL13 expression were not recapitulated in peritoneal macrophages cultured with various doses of oxLDL. This may simply stress the importance of performing gene expression analyses on macrophages within actual atheroma, but could also indicate that CXCL13 expression is induced by other factors involved in the pathogenesis of atherosclerosis.
In the long‐term WD group, we observed a significant induction of 2 GBPs, GBP3 and GBP6, as well as a more‐moderate elevation of other GBP family members. The GBPs were among the first interferon (IFN)‐inducible genes identified, and, like other interferon target genes, the function of GBPs has been primarily associated to protection against viral and bacterial infections.43, 44 Although their mechanism of action is still under investigation, p65‐GBPs have been shown to localize to vacuoles containing pathogens and play a role in transport of autophagic machinery, antimicrobial peptides, and NADPH oxidase (NOX) enzymes for assembly on phagosomal membranes.44, 45 Interestingly, both the phagocytic clearance of apoptotic cells, known as efferocytosis, and the production of reactive oxygen species by NOX enzymes are processes associated with the development of atherosclerosis.46, 47 Furthermore, similar to what happens during bacterial phagocytosis, efferocytosis was shown to induce an oxidative burst in macrophages in a NOX‐dependent fashion.48, 49 Importantly, in follow‐up studies using cultured macrophages, we found that several GBPs were induced in vitro by oxLDL, which indicates that oxLDL may be one of the factors responsible for the induction observed in vivo. However, the reason for the specific upregulation of GBPs among the various players in inflammation and immunity is not clear. A possible explanation is that the genes coding for GBPs may be more sensitive to modest changes in inflammation that may take place in more‐advanced lesions. Indeed, GBPs are known to be very strongly induced by IFN and other inflammatory stimuli, a fact that even facilitated the characterization of signaling pathways such as the Janus kinase/signal transducer and activator of transcription and the IFN‐γ and IFN‐α/β pathways.32 However, to our knowledge, this is the first report linking this family of IFN‐induced GTPases with the pathogenesis of atherosclerosis. Thus, additional studies will be necessary to determine whether GBPs play a significant role in regulation of atherogenesis, whether it is through regulation of macrophage function during efferocytosis or by other mechanisms.
In conclusion, this study challenges the notion that acceleration of atherogenesis by hypercholesterolemia is linked to a global impact on the inflammatory balance of foam cells. Significant changes among inflammatory and immune mediators included induction of CXCL13 in response to short‐term increases in plasma cholesterol, and induction of GBPs in foam cells resident within the more‐advanced lesions that formed in response to prolonged hypercholesterolemia. Further research will be necessary to elucidate the role of these players in the development of atherosclerosis.
Sources of Funding
This work was supported by NIH grants HL104251 (to Paul) and R01DK097160 (to Yechoor). Goo was partly supported by an American Heart Association Scientist Development Grant (14SDG19690016).
Disclosures
None.
Supporting information
Table S1. Genes Differentially Expressed in Foam Cells of Mice Fed WD for 2 Weeks With Respect to Mice Fed Regular Chow Through the Entire Study
Table S2. Genes Differentially Expressed in Foam Cells of Mice Fed WD for 14 Weeks With Respect to Mice Fed Regular Chow Through the Entire Study
Acknowledgments
We thank the members of the Genomic and RNA Profiling Core at Baylor College of Medicine for their help in sample processing and discussion.
(J Am Heart Assoc. 2016;5:e002663 doi: 10.1161/JAHA.115.002663)
References
- 1. Glass CK, Witztum JL. Atherosclerosis: the road ahead. Cell. 2001;104:503–516. [DOI] [PubMed] [Google Scholar]
- 2. Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med. 1999;340:115–126. [DOI] [PubMed] [Google Scholar]
- 3. Steinberg D. Atherogenesis in perspective: hypercholesterolemia and inflammation as partners in crime. Nat Med. 2002;8:1211–1217. [DOI] [PubMed] [Google Scholar]
- 4. Libby P. Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:2045–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Steinberg D. The LDL modification hypothesis of atherogenesis: an update. J Lipid Res. 2009;50:S376–S381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tuomisto TT, Riekkinen MS, Viita H, Levonen AL, Yla‐Herttuala S. Analysis of gene and protein expression during monocyte‐macrophage differentiation and cholesterol loading–cDNA and protein array study. Atherosclerosis. 2005;180:283–291. [DOI] [PubMed] [Google Scholar]
- 7. Eligini S, Colli S, Basso F, Sironi L, Tremoli E. Oxidized low density lipoprotein suppresses expression of inducible cyclooxygenase in human macrophages. Arterioscler Thromb Vasc Biol. 1999;19:1719–1725. [DOI] [PubMed] [Google Scholar]
- 8. Conway JP, Kinter M. Proteomic and transcriptomic analyses of macrophages with an increased resistance to oxidized low density lipoprotein (oxLDL)‐induced cytotoxicity generated by chronic exposure to oxLDL. Mol Cell Proteomics. 2005;4:1522–1540. [DOI] [PubMed] [Google Scholar]
- 9. Spann NJ, Garmire LX, McDonald JG, Myers DS, Milne SB, Shibata N, Reichart D, Fox JN, Shaked I, Heudobler D, Raetz CR, Wang EW, Kelly SL, Sullards MC, Murphy RC, Merrill AH Jr, Brown HA, Dennis EA, Li AC, Ley K, Tsimikas S, Fahy E, Subramaniam S, Quehenberger O, Russell DW, Glass CK. Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell. 2012;151:138–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brand K, Eisele T, Kreusel U, Page M, Page S, Haas M, Gerling A, Kaltschmidt C, Neumann F‐J, Mackman N, Baeuerle PA, Walli AK, Neumeier D. Dysregulation of monocytic nuclear factor‐κB by oxidized low‐density lipoprotein. Arterioscler Thromb Vasc Biol. 1997;17:1901–1909. [DOI] [PubMed] [Google Scholar]
- 11. Hammad SM, Twal WO, Barth JL, Smith KJ, Saad AF, Virella G, Argraves WS, Lopes‐Virella MF. Oxidized LDL immune complexes and oxidized LDL differentially affect the expression of genes involved with inflammation and survival in human U937 monocytic cells. Atherosclerosis. 2009;202:394–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Verschoor C, Puchta A, Bowdish DM. The Macrophage. Methods Mol Biol. 2012;844:139–156. [DOI] [PubMed] [Google Scholar]
- 13. Reddick RL, Zhang SH, Maeda N. Atherosclerosis in mice lacking apo E. Evaluation of lesional development and progression. Arterioscler Thromb. 1994;14:141–147. [DOI] [PubMed] [Google Scholar]
- 14. Zhang SH, Reddick RL, Piedrahita JA, Maeda N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science. 1992;258:468–471. [DOI] [PubMed] [Google Scholar]
- 15. Meir KS, Leitersdorf E. Atherosclerosis in the apolipoprotein E‐deficient mouse: a decade of progress. Arterioscler Thromb Vasc Biol. 2004;24:1006–1014. [DOI] [PubMed] [Google Scholar]
- 16. Tabibiazar R, Wagner RA, Ashley EA, King JY, Ferrara R, Spin JM, Sanan DA, Narasimhan B, Tibshirani R, Tsao PS, Efron B, Quertermous T. Signature patterns of gene expression in mouse atherosclerosis and their correlation to human coronary disease. Physiol Genomics. 2005;22:213–226. [DOI] [PubMed] [Google Scholar]
- 17. Getz GS, Reardon CA. Diet and murine atherosclerosis. Arterioscler Thromb Vasc Biol. 2006;26:242–249. [DOI] [PubMed] [Google Scholar]
- 18. Paul A, Chang BH, Li L, Yechoor VK, Chan L. Deficiency of adipose differentiation‐related protein impairs foam cell formation and protects against atherosclerosis. Circ Res. 2008;102:1492–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Douglas G, Bendall JK, Crabtree MJ, Tatham AL, Carter EE, Hale AB, Channon KM. Endothelial‐specific Nox2 overexpression increases vascular superoxide and macrophage recruitment in ApoE(−/−) mice. Cardiovasc Res. 2012;94:20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Harmon EY, Fronhofer V, Keller RS, Feustel PJ, Zhu X, Xu H, Avram D, Jones DM, Nagarajan S, Lennartz MR. Anti‐inflammatory immune skewing is atheroprotective: Apoe(−/−)FcγRIIb(−/−) mice develop fibrous carotid plaques. J Am Heart Assoc. 2014;3:e001232 doi: 10.1161/JAHA.114.001232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Son S‐H, Goo Y‐H, Choi M, Saha PK, Oka K, Chan LCB, Paul A. Enhanced atheroprotection and lesion remodelling by targeting the foam cell and increasing plasma cholesterol acceptors. Cardiovasc Res. 2016;109:294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Paul A, Yechoor V, Raja R, Li L, Chan L. Microarray gene profiling of laser‐captured cells: a new tool to study atherosclerosis in mice. Atherosclerosis. 2008;200:257–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J. TM4: a free, open‐source system for microarray data management and analysis. Biotechniques. 2003;34:374–378. [DOI] [PubMed] [Google Scholar]
- 24. Draghici S, Khatri P, Tarca AL, Amin K, Done A, Voichita C, Georgescu C, Romero R. A systems biology approach for pathway level analysis. Genome Res. 2007;17:1537–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tabas I, Williams KJ, Borén J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116:1832–1844. [DOI] [PubMed] [Google Scholar]
- 26. Itabe H, Obama T, Kato R. The dynamics of oxidized LDL during atherogenesis. J Lipids. 2011;2011:418313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang Z, Guo D, Yang B, Wang J, Wang R, Wang X, Zhang Q. Integrated analysis of microarray data of atherosclerotic plaques: modulation of the ubiquitin‐proteasome system. PLoS One. 2014;9:e110288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. King JY, Ferrara R, Tabibiazar R, Spin JM, Chen MM, Kuchinsky A, Vailaya A, Kincaid R, Tsalenko A, Deng DX, Connolly A, Zhang P, Yang E, Watt C, Yakhini Z, Ben‐Dor A, Adler A, Bruhn L, Tsao P, Quertermous T, Ashley EA. Pathway analysis of coronary atherosclerosis. Physiol Genomics. 2005;23:103–118. [DOI] [PubMed] [Google Scholar]
- 29. Satterthwaite G, Francis SE, Suvarna K, Blakemore S, Ward C, Wallace D, Braddock M, Crossman D. Differential gene expression in coronary arteries from patients presenting with ischemic heart disease: further evidence for the inflammatory basis of atherosclerosis. Am Heart J. 2005;150:488–499. [DOI] [PubMed] [Google Scholar]
- 30. Lutgens E, Faber B, Schapira K, Evelo CT, van Haaften R, Heeneman S, Cleutjens KB, Bijnens AP, Beckers L, Porter JG, Mackay CR, Rennert P, Bailly V, Jarpe M, Dolinski B, Koteliansky V, de Fougerolles T, Daemen MJ. Gene profiling in atherosclerosis reveals a key role for small inducible cytokines: validation using a novel monocyte chemoattractant protein monoclonal antibody. Circulation. 2005;111:3443–3452. [DOI] [PubMed] [Google Scholar]
- 31. Cagnin S, Biscuola M, Patuzzo C, Trabetti E, Pasquali A, Laveder P, Faggian G, Iafrancesco M, Mazzucco A, Pignatti PF, Lanfranchi G. Reconstruction and functional analysis of altered molecular pathways in human atherosclerotic arteries. BMC Genom. 2009;10:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martens S, Howard J. The interferon‐inducible GTPases. Annu Rev Cell Dev Biol. 2006;22:559–589. [DOI] [PubMed] [Google Scholar]
- 33. Nishi K, Itabe H, Uno M, Kitazato KT, Horiguchi H, Shinno K, Nagahiro S. Oxidized LDL in carotid plaques and plasma associates with plaque instability. Arterioscler Thromb Vasc Biol. 2002;22:1649–1654. [DOI] [PubMed] [Google Scholar]
- 34. Castro C, Campistol JM, Barettino D, Andres V. Transcriptional profiling of early onset diet‐induced atherosclerosis in apolipoprotein E‐deficient mice. Front Biosci. 2005;10:1932–1945. [DOI] [PubMed] [Google Scholar]
- 35. Miller YI, Viriyakosol S, Binder CJ, Feramisco JR, Kirkland TN, Witztum JL. Minimally modified LDL binds to CD14, induces macrophage spreading via TLR4/MD‐2, and inhibits phagocytosis of apoptotic cells. J Biol Chem. 2003;278:1561–1568. [DOI] [PubMed] [Google Scholar]
- 36. Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12:204–212. [DOI] [PubMed] [Google Scholar]
- 37. Im SS, Osborne TF. Liver X receptors in atherosclerosis and inflammation. Circ Res. 2011;108:996–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat Med. 2003;9:213–219. [DOI] [PubMed] [Google Scholar]
- 39. Shibata N, Glass CK. Regulation of macrophage function in inflammation and atherosclerosis. J Lipid Res. 2009;50:S277–S281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Smedbakken LM, Halvorsen B, Daissormont I, Ranheim T, Michelsen AE, Skjelland M, Sagen EL, Folkersen L, Krohg‐Sorensen K, Russell D, Holm S, Ueland T, Fevang B, Hedin U, Yndestad A, Gullestad L, Hansson GK, Biessen EA, Aukrust P. Increased levels of the homeostatic chemokine CXCL13 in human atherosclerosis—potential role in plaque stabilization. Atherosclerosis. 2012;224:266–273. [DOI] [PubMed] [Google Scholar]
- 41. Carlsen HS, Baekkevold ES, Morton HC, Haraldsen G, Brandtzaeg P. Monocyte‐like and mature macrophages produce CXCL13 (B cell–attracting chemokine 1) in inflammatory lesions with lymphoid neogenesis. Blood. 2004;104:3021–3027. [DOI] [PubMed] [Google Scholar]
- 42. van Dijk RA, Duinisveld AJ, Schaapherder AF, Mulder‐Stapel A, Hamming JF, Kuiper J, de Boer OJ, van der Wal AC, Kolodgie FD, Virmani R, Lindeman JH. A change in inflammatory footprint precedes plaque instability: a systematic evaluation of cellular aspects of the adaptive immune response in human atherosclerosis. J Am Heart Assoc. 2015;4:e001403 doi: 10.1161/JAHA.114.001403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Carter CC, Gorbacheva VY, Vestal DJ. Inhibition of VSV and EMCV replication by the interferon‐induced GTPase, mGBP‐2: differential requirement for wild‐type GTP binding domain. Arch Virol. 2005;150:1213–1220. [DOI] [PubMed] [Google Scholar]
- 44. Kim B‐H, Shenoy AR, Kumar P, Das R, Tiwari S, MacMicking JD. A family of IFN‐γ–inducible 65‐kD GTPases protects against bacterial infection. Science. 2011;332:717–721. [DOI] [PubMed] [Google Scholar]
- 45. Dupont CD, Hunter CA. Guanylate‐binding proteins: niche recruiters for antimicrobial effectors. Immunity. 2012;37:191–193. [DOI] [PubMed] [Google Scholar]
- 46. Thorp E, Subramanian M, Tabas I. The role of macrophages and dendritic cells in the clearance of apoptotic cells in advanced atherosclerosis. Eur J Immunol. 2011;41:2515–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lassègue B, San Martín A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res. 2012;110:1364–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lee H‐N, Surh Y‐J. Resolvin D1‐mediated NOX2 inactivation rescues macrophages undertaking efferocytosis from oxidative stress‐induced apoptosis. Biochem Pharmacol. 2013;86:759–769. [DOI] [PubMed] [Google Scholar]
- 49. Yvan‐Charvet L, Pagler TA, Seimon TA, Thorp E, Welch CL, Witztum JL, Tabas I, Tall AR. ABCA1 and ABCG1 protect against oxidative stress‐induced macrophage apoptosis during efferocytosis. Circ Res. 2010;106:1861–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Genes Differentially Expressed in Foam Cells of Mice Fed WD for 2 Weeks With Respect to Mice Fed Regular Chow Through the Entire Study
Table S2. Genes Differentially Expressed in Foam Cells of Mice Fed WD for 14 Weeks With Respect to Mice Fed Regular Chow Through the Entire Study