

Abstract
Background
Two‐pore K+ channels have emerged as potential targets to selectively regulate cardiac cell membrane excitability; however, lack of specific inhibitors and relevant animal models has impeded the effort to understand the role of 2‐pore K+ channels in the heart and their potential as a therapeutic target. The objective of this study was to determine the role of mechanosensitive 2‐pore K+ channel family member TREK‐1 in control of cardiac excitability.
Methods and Results
Cardiac‐specific TREK‐1–deficient mice (αMHC‐Kcnk f/f) were generated and found to have a prevalent sinoatrial phenotype characterized by bradycardia with frequent episodes of sinus pause following stress. Action potential measurements from isolated αMHC‐Kcnk2 f/f sinoatrial node cells demonstrated decreased background K+ current and abnormal sinoatrial cell membrane excitability. To identify novel pathways for regulating TREK‐1 activity and sinoatrial node excitability, mice expressing a truncated allele of the TREK‐1–associated cytoskeletal protein βIV‐spectrin (qv 4J mice) were analyzed and found to display defects in cell electrophysiology as well as loss of normal TREK‐1 membrane localization. Finally, the βIV‐spectrin/TREK‐1 complex was found to be downregulated in the right atrium from a canine model of sinoatrial node dysfunction and in human cardiac disease.
Conclusions
These findings identify a TREK‐1–dependent pathway essential for normal sinoatrial node cell excitability that serves as a potential target for selectively regulating sinoatrial node cell function.
Keywords: automaticity, K channel, sinoatrial node, spectrin, TREK‐1
Subject Categories: Arrhythmias, Pacemaker
Introduction
The importance of background (“leak”) K+ channels for excitable cell function has been known for the better part of a century.1 Despite the central role of these channels in maintaining normal membrane excitability in a wide variety of cells, including cardiac myocytes,2 our knowledge of their specific identity and physiological functions lags behind that of other K+ channel families (eg, voltage‐dependent and inward rectifier K+ channels), in part because of the lack of specific pharmacological agents and animal models. The mammalian 2‐pore K+ channel (K2P) superfamily is rapidly emerging as a likely source of background K+ channels in heart. The almost 20 K2P channels identified to date form as hetero‐ or homodimers of subunits with 4 transmembrane and 2 pore domains and have been divided into subfamilies (TWIK, TREK, TASK, TALK, THIK, and TRESK) based on functional characteristics.3, 4, 5, 6 Among the most interesting of these channels is TREK‐1, distinguished by its sensitivity to mechanical membrane deformation, polyunsaturated fatty acids (eg, arachidonic acid), temperature, volatile anesthetics, and intracellular pH. TREK‐1 is highly expressed in the nervous system, where it regulates depression phenotypes and nociception. Expression of TREK‐1 has also been identified in heart with proposed roles in regulation of myocyte membrane excitability,7, 8, 9, 10, 11, 12, 13, 14 although its role in specific cardiac phenotypes remains unclear.
We developed a novel cardiac‐specific TREK‐1–deficient mouse model (αMHC‐Kcnk2 f/f) that we used to investigate the role of TREK‐1 in heart. We found that TREK‐1 is highly expressed in the sinoatrial node (SAN) and regulates cardiac pacemaking. At the cellular level, TREK‐1 influenced the diastolic depolarization rate, spontaneous firing, and repolarization of the SAN action potential (AP). We also provided evidence that normal TREK‐1 membrane localization and activity in SAN cells depend on association with the actin‐associated cytoskeletal protein βIV‐spectrin. Finally, we reported downregulation of the βIV‐spectrin/TREK‐1 complex in the right atrium from a preclinical model of sinus node dysfunction and in human cardiac disease, suggesting that loss of this novel complex contributes to sinus node disease. Based on these observations, we hypothesize that the spectrin/TREK‐1 complex is an important pathway for regulating automaticity in response to environmental cues (eg, mechanical load, temperature, pH), whereas loss of spectrin/TREK‐1 may contribute to SAN disease. We propose that TREK‐1, with its SAN enrichment and unique gating properties, represents a potential target for selectively regulating SAN excitability.
Methods
Molecular Biology
The cDNA for βIV‐spectrin and TREK‐1 (C‐terminal cytoplasmic region 305–422) fusion proteins was cloned from a human heart cDNA library, as described previously.8 Constructs for in vitro translation and fusion protein expression were generated by engineering cDNAs in frame into pcDNA3.1+ (Invitrogen) and pGEX6P1 (GE Healthcare).
Animals
TREK‐1 conditional knockout mice were generated by the Ohio State University Genetically Engineered Mouse Modeling Core using standard embryonic stem cell homologous recombination technology. Briefly, the targeting vector “conditional ready” (PG00255_Z_1_C03) was obtained from the International Knockout Mouse Consortium (project number 100110). After sequencing, the vector was linearized by AsiI restriction digest and electroporated into S1B6a, a mixed genetic background 129Sv/C57BL/6 mouse embryonic stem cell line. G418‐resistant clones were generated, as described previously,15 and screened by southern blotting with probes specific to regions outside the 5′ of the Trek1 locus. Correctly targeted embryonic stem cell clones were used to generate chimeras that transmitted the mutated allele to the germ line.15 For experimental purposes, mice were crossed first to a Flpe ubiquitous line16 to remove the LacZ‐Neo cassette flanked by frt sites and then backcrossed onto the C57Bl/6 strain background. Experimental mice were generated by crossing these animals with mice expressing Cre under the cardiac promoter α‐myosin heavy chain (αMHC‐Cre), resulting in cardiac‐specific deletion of TREK‐1 (αMHC‐Kcnk2 f/f). The qv 4J mice (which express a mutant βIV‐spectrin allele with a premature stop codon in spectrin repeat 10)8, 17 were obtained from Jackson Laboratory (Bar Harbor, ME). All experiments were performed in male mice aged 2 months (30 wild‐type [WT], 30 αMHC‐Kcnk2 f/f, and 12 qv 4J animals were used for studies). Animals were euthanized using CO2 and cervical dislocation, followed by collection of tissue or cell isolation. Studies were conducted in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health, following protocols that were reviewed and approved by the institutional animal care and use committee at the Ohio State University. Right atrial samples from 3 dogs with heart failure and sinus node dysfunction and 3 control dogs were generous gifts from Dr Peng‐Sheng Chen.18
Human Tissue
Deidentified coded human hearts were obtained from the Lifeline of Ohio organ procurement organization and the Division of Cardiac Surgery at the Ohio State University Wexner Medical Center. Immunostaining of tissue sections was performed on 1 nondiseased and 1 diseased heart, as described.19 The study was approved by the Ohio State University institutional review board, and participants gave informed consent.
Biochemistry
Equal quantities of auricular, ventricular, and SAN lysates (determined by standard bicinchoninic acid protocols) were analyzed by SDS–PAGE and immunoblot. Equal protein loading was verified through Coomassie and Ponceau stains of gels and blots. In addition, small differences in protein loading were corrected using normalization to levels of actin or GAPDH. Adult heart immunoprecipitations were performed, as described previously.8 The following antibodies were used for immunoblotting, immunoprecipitation, or immunostaining: TREK‐1 (C‐terminal: sc50412, N‐terminal: sc11554; Santa Cruz Biotechnology), βIV‐spectrin (N‐terminal, HPA043370, lot A96519; Sigma‐Aldrich), Kir2.1 (Alomone), Kv2.1 (NeuroMab), Kv4.2 (Alomone), Kv4.3, TASK‐1 (APC024; Alomone), HCN4 (32675, lot GR153269‐7; Abcam), N‐cadherin (333900, lot 1397763A; Invitrogen), Connexin43 (MAB3067, lot 23070602; Invitrogen), and GAPDH (10R‐G109A; Fitzgerald).
Imaging
SAN cardiomyocytes were isolated from adult mouse right atrial explants, as described.20, 21, 22 Cells were blocked in phosphate‐buffered saline containing 0.15% Triton X‐100, 3% normal goat serum (Sigma‐Aldrich), and 1% BSA (Sigma‐Aldrich) and incubated in primary antibody for 2 hours at room temperature. Cells were washed and then incubated in secondary antibody (Alexa 488, 568) for 2 hours at room temperature and mounted using Vectashield with DAPI (Vector) and No. 1 coverslips. Images were collected on a Zeiss 780 confocal microscope (Objective W Plan Apochromat 40×/1.0 DIC, pinhole equals 1.0 Airy Disc) using the Carl Zeiss Imaging software.
Electrophysiology
Current recordings from isolated SAN cardiomyocytes were measured using a conventional whole‐cell patch‐clamp technique with an Axon 200B patch‐clamp amplifier controlled by a personal computer using a Digidata 1320A acquisition board driven by pClamp 8.0 software (Axon Instruments). TREK‐1–like background (leak) K+ current was measured using a slow (6 mV/s) voltage ramp from −90 mV to +30 mV, as described.11 Briefly, bath solution contained (in mmol/L) 150 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 8 glucose, and 10 HEPES (pH 7.4). Pipette solution contained (in mmol/L) 120 K‐aspartate, 20 KCl, 10 EGTA, 1.0 MgCl2, 2.0 Na2ATP, 0.1 mmol/L Na4GTP, and 20 mmol/L HEPES. Moreover, 1 μmol/L glibenclamide and 100 μmol/L CdCl2 were added to block ATP‐sensitive K+ channels and voltage‐gated Ca2+ channels. Pacemaker current (funny current [I f]) was recorded, as described.23 All current recording experiments were conducted at room temperature (21–23°C). Spontaneous APs were recorded using the perforated (amphotericin B) patch‐clamp technique at room temperature in Tyrode solution (bath). The pipette contained (in mmol/L) 130 potassium aspartate, 10 NaCl, 10 HEPES, 0.04 CaCl2, 2.0 MgATP, 7.0 phosphocreatine, 0.1 NaGTP, and 240 μg/mL amphotericin B, with the pH adjusted to 7.2 with KOH. Recording pipettes, fabricated from borosilicate glass, had resistance of 2 to 4 MΩ when filled with recording solution. All solutions were adjusted to 275 to 295 mOsm.
Echocardiography
Echocardiography was performed on male mice aged 2 months to assess baseline cardiac function using the Vevo 2100 (Visualsonics), as described.24 The MS‐400 transducer was used in the short‐axis M‐mode to assess heart function and contractile features.
Mathematical Model
Ion channel kinetics underlying the SAN AP were simulated using a model of the mammalian central SAN cell25 modified to include formulations for TREK‐1 and Cav1.3 currents (Data S1). Parameter sensitivity analysis was performed using regression to determine the relationship between model parameters and AP properties, as described.26 Briefly, random scale factors for Cav1.2‐ and Cav1.3‐dependent L‐type Ca2+ current (I CaL,α1C and I CaL,α1D, respectively), T‐type Ca2+ current (I CaT), hyperpolarization‐activated funny current (I H), rapid delayed rectifier K+ current (I Kr), transient outward K+ current (I to), Na+/Ca2+ exchanger (I NaCa), Na+/K+ ATPase (I NaK), and TREK‐1 (I TREK1) were selected from a log‐normal distribution with a median value of 1 (SD 0.2) to create 2000 unique model parameter sets. For each set of parameter values, model outputs (AP firing frequency, diastolic depolarization rate, AP amplitude, maximum diastolic potential [MDP], AP duration [APD] at 50% repolarization, AP upstroke velocity [dVm/dtmax], Ca2+AP upstroke velocity transient amplitude, Ca2+ transient width at 50% amplitude, and minimum diastolic intracellular Ca2+ concentration [Ca2+]i) were determined following 20 000 ms of spontaneous activity. Model inputs and right‐skewed outputs (AP firing frequency and APD at 50% repolarization) were log‐transformed. Model inputs and outputs were then mean centered and normalized by standard deviations to create an input matrix X and an output matrix Y. The regression coefficient matrix, B, was obtained using standard multivariable linear regression [B=(X TˣX)−1ˣX TˣY]. Computer code was written in C++ and compiled using Intel Composer XE 2011 for Linux. Computer simulations were performed on a Dell PowerEdge R515 server (dual 6 core, 32 GB RAM running CentOS 6.2), as described.21, 22, 27, 28, 29, 30 Regression was performed using MATLAB R2014b (MathWorks) on a MacBook Pro with a 2.5‐GHz Intel Core i7 processor (Apple Inc).
Statistics
SigmaPlot 12.0 (Systat Software, Inc) was used for statistical analysis. Data distributions were tested for normality using the Shapiro–Wilk test. The Student t test was used as a parametric test for single comparisons of nonpaired data. One‐way ANOVA was used for multiple comparisons with the Fisher least significant difference test for post hoc testing (data presented as mean±SEM). If the data distribution failed normality tests, the Wilcoxon‐Mann–Whitney U test was used to determine P values for single comparisons, or a Kruskal–Wallis 1‐way ANOVA on ranks was applied with a Dunn multiple‐comparisons test for significant P values (data presented as median with 25th and 75th percentiles [box] and 10th and 90th percentiles [whiskers]). The null hypothesis was rejected for P<0.05.
Results
Cardiac‐Specific Deletion of TREK‐1 Produces SAN Dysfunction
Although TREK‐1 channel function in brain and vasculature has been investigated in a global TREK‐1 knockout mouse,31 the model has not been used to study the specific role of TREK‐1 in heart in part because of phenotypes related to loss of channel function in other cell types (eg, neurons, endothelial, immune cells).31, 32, 33, 34 Consequently, a heart‐specific TREK‐1–deficient mouse (αMHC‐Kcnk2 f/f) was generated to eliminate off‐target effects (Figure 1A and 1B). Immunoblots verified cardiac‐specific loss of TREK‐1 from heart but not from other tissue (brain, pancreas) (Figure 1C). Based on established mechanosensitivity of TREK‐1, echocardiography was initially performed to determine whether loss of TREK‐1 altered cardiac function at baseline. Interestingly, cardiac function of adult αMHC‐Kcnk2 f/f was similar to WT, without significant changes in ejection fraction, ventricular wall thickness, or ventricular chamber diameter, indicating that overt cardiac remodeling does not occur at baseline in the absence of TREK‐1 (Table).
Figure 1.
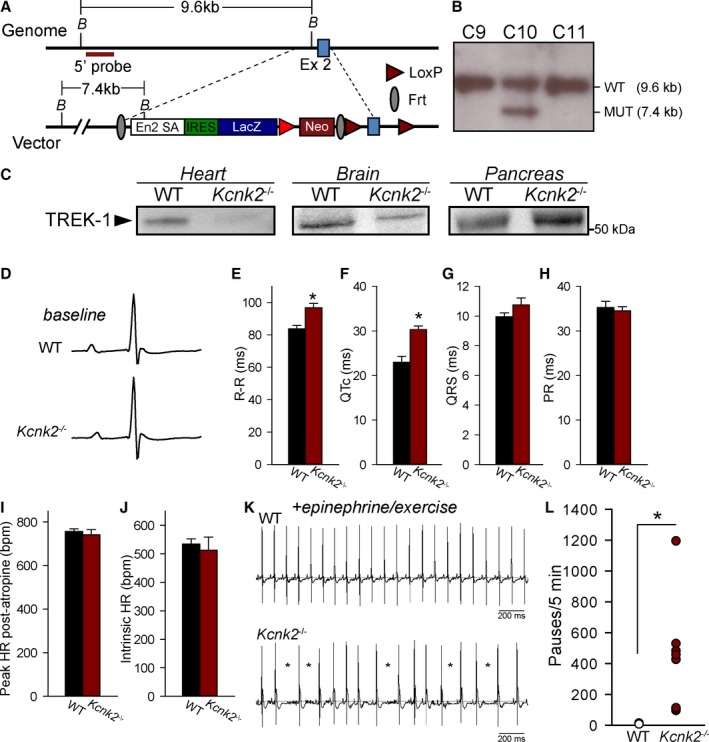
Generation of a heart‐specific TREK‐1–deficient mouse. A, TREK‐1 conditional knockout targeting strategy. The targeting vector includes a 5′ homology arm of about 5.9 kb and a 3′ homology arm of about 4.9 kb. Exon 2 of the gene Kcnk2 is flanked by 2 loxP sites. The mutated locus contains an En2‐IRES‐LAcZ cassette to survey Kcnk2 gene expression. Exon 2 can be removed by Cre recombination in a tissue‐specific manner. The targeting vector also includes a DTa cassette for in vitro negative selection of embryonic stem cell clones with random integrations. B, Representative southern blot screening of S1B6a embryonic stem colonies were positively targeted. DNA was digested with Bam HI (B in A) and hybridized with a 5′ probe. Clone C10 shows the targeted mutated band of 7.4 kb and was used to generate the Kcnk2 floxed mouse line. The presence of the distal loxP on the 3′ of exon 2 was confirmed by PCR. C, Immunoblots for TREK‐1 in heart, brain, and pancreas lysates from WT and αMHC‐Kcnk2 f/f mice (Kcnk2 −/−). TREK‐1 was first immunoprecipitated using an antibody against c‐terminal epitope (H‐75; Santa Cruz Biotechnology), and then immunoblot was performed using antibody against N‐terminal epitope (N‐20; Santa Cruz Biotechnology). D, Representative signal‐averaged telemetry recordings from awake unanesthetized WT and Kcnk2 −/− animals at baseline. E through H, Summary ECG data in WT and Kcnk2 −/− animals at rest (n=5 for WT, n=8 for Kcnk2 −/−, n represents the number of animals from which ECGs were recorded; *P<0.05 vs WT). I, Peak heart rate following atropine injection (1 mg/kg) and (J) intrinsic heart rate following atropine and propranolol injection (3 mg/kg) in WT and αMHC‐Kcnk2 f/f mice (Kcnk2 −/−). P not significant, n=4 per genotype (n represents number of animals from which recordings were made). K, Representative telemetry recordings during continuous recording showing frequent episodes of sinus pause (marked by asterisks) in Kcnk2 −/− but not WT following treadmill exercise and epinephrine injection (2 mg/kg). L, Summary data on number of sinus pause events (defined as R‐R interval 50% greater than average R‐R interval) in WT and Kcnk2 −/− animals over a 5‐minute interval following epinephrine challenge. HR indicates heart rate; kb, kilobase; MUT, mutation; WT, wild type.
Table 1.
Baseline Echocardiographic Features in WT and Cardiac‐Specific TREK‐1–Deficient Mice
| WT (n=4) | αMHC‐Kcnk2 f/f (n=7) | |
|---|---|---|
| LVID,d, mm | 4.02±0.17 | 3.95±0.11 |
| EF, % | 57.1±1.5 | 53.5±1.7 |
| FS, % | 29.6±0.9 | 27.2±1.0 |
| LVAW,d, mm | 0.59±0.01 | 0.59±0.01 |
| LVPW,d, mm | 0.61±0.01 | 0.61±0.01 |
Data presented as mean value±SEM. P not significant vs WT. EF indicates ejection fraction; FS, fractional shortening; LVAW,d, LV anterior wall thickness in diastole; LVID,d, left ventricular inner chamber diameter in diastole; LVPW,d, LV posterior wall thickness in diastole; WT, wild type.
Potential effects of TREK‐1 deficiency on cardiac electrophysiology were screened in vivo using telemetry (all data recorded during the day). Surprisingly, electrocardiograms from conscious αMHC‐Kcnk2 f/f mice showed significant increases in R‐R and corrected QT (QTc) intervals (measured, as described24) at rest (Figure 1D through 1H). No significant difference was observed in QRS or PR intervals of αMHC‐Kcnk2 f/f and WT animals. Intrinsic heart rate was also assessed (atropine 1 mg/kg followed by propranolol 3 mg/kg)35 and was found not to be different between WT and αMHC‐Kcnk2 f/f mice (Figure 1I and 1J). Following baseline recordings, animals then underwent a stress protocol consisting of treadmill exercise followed by epinephrine injections (2 mg/kg). Despite the slight but significant QTc prolongation in αMHC‐Kcnk2 f/f animals, no ventricular arrhythmias were observed, even following stress. Instead, αMHC‐Kcnk2 f/f animals demonstrated a pronounced increase in susceptibility to stress‐induced sinus pause (without premature atrial contractions or atrioventricular block) compared with WT (Figure 1K and 1L). Together, these data indicated that TREK‐1 is essential for normal SAN automaticity, with loss of TREK‐1 promoting stress‐induced SAN dysfunction.
TREK‐1 Regulates SAN Cell Membrane Excitability
TREK‐1 expression has been reported throughout all regions of the heart3, 8, 10, 36; however, few data are available on relative levels of expressed protein in SAN, atrium, and ventricle. TREK‐1 protein expression was assessed in different mouse heart regions to determine whether differential expression could contribute to the predominant SAN phenotypes observed in αMHC‐Kcnk2 f/f mice. Consistent with the proposed role in regulating SAN automaticity, TREK‐1 was detectable by immunoblot in SAN lysates and in fact was higher in the SAN and atrium than in the ventricle (Figure 2A and 2B). Significant TREK‐1 immunoreactive signal was also observed in the SAN region (HCN4‐positive, connexin 43‐negative) of right atrial tissue sections from mouse heart (Figure 2C). This pattern of expression was also observed in rabbit, in which the relative expression of TREK‐1 in SAN compared with other heart regions was even higher than in mouse (Figure 2D and 2E). These data revealed enrichment of TREK‐1 in SAN, consistent with the observed phenotype.
Figure 2.
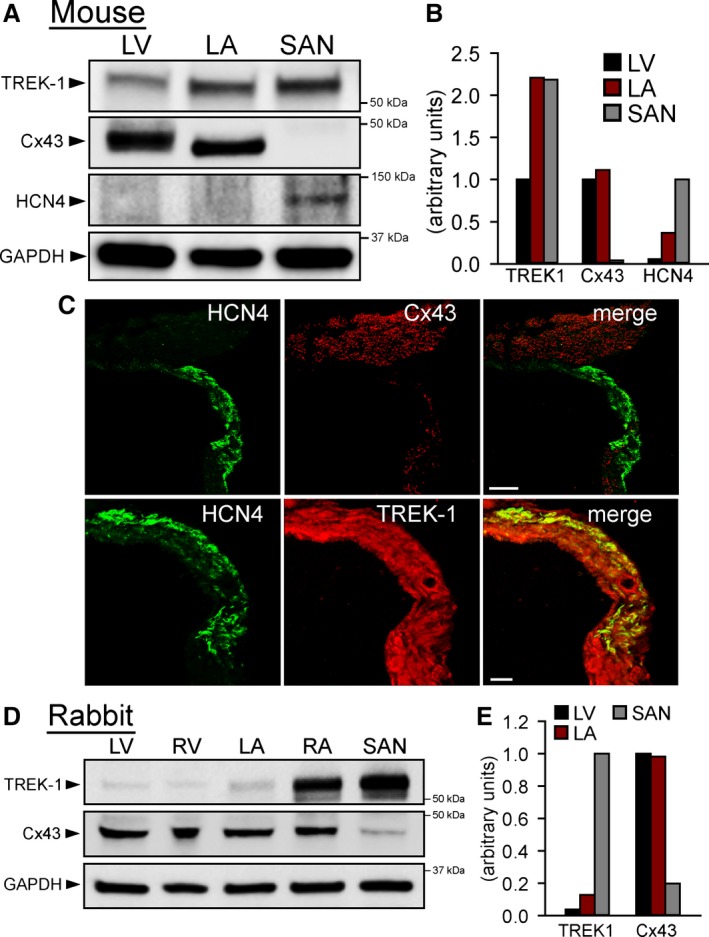
TREK‐1 is highly expressed in mouse SAN. A, Immunoblots and (B) densitometric measurments (normalized to GAPDH loading control) showing expression of TREK‐1, Cx43, and HCN4 in detergent‐soluble lysates from mouse LV, LA, and SAN (LA and SAN lysates are from 3 pooled samples from 3 preparations, thus no error bars are provided for densitometric measurements). TREK‐1 immunoblot was performed using C‐terminal antibody. C, Confocal images of mouse RA tissue section (14 μm thick) immunostained for Cx43 and HCN4 to show intact SAN region (HCN4‐positive, Cx43‐negative area). Scale bar=100 μm. Higher magnification images (scale bar=50 μm) show sections from same region immunostained for HCN4 and TREK‐1. D, Immunoblots and (E) densitometric measurments (normalized to GAPDH) showing expression of TREK‐1 and Cx43 in detergent‐soluble lysates from rabbit LV and RV, LA and RA, and SAN (LA, RA, and SAN are from 3 pooled samples from 3 preparations). Cx43 indicates connexin 43; LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle; SAN, sinoatrial node.
With robust expression of TREK‐1 in mammalian SAN having been established, spontaneous APs were measured from isolated adult αMHC‐Kcnk2 f/f and SAN myocytes to determine whether defects in excitability at the level of the single SAN cell underlie SAN dysfunction observed in TREK‐1–deficient animals. The αMHC‐Kcnk2 f/f SAN APs showed a significant increase in spontaneous firing rate, diastolic depolarization rate, and APD at 50% repolarization (Figure 3). Surprisingly, TREK‐1 deficiency had no significant effect on AP amplitude or APD at 90% repolarization, with a significant but small effect on MDP.
Figure 3.
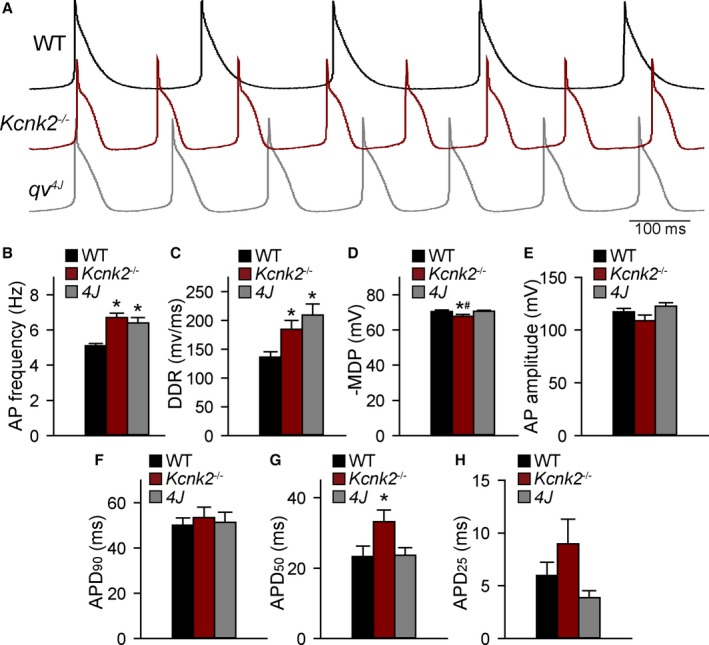
Loss of TREK‐1 expression alters SAN cell excitability. A, Spontaneous action potentials from isolated WT, αMHC‐Kcnk2 f/f (Kcnk2 −/−), and qv 4J (lack spectrin/TREK‐1 interaction) SAN cells. B through H, Summary data showing AP firing frequency; DDR; MDP; AP amplitude; and APD at 90%, 50%, and 25% repolarization in WT, Kcnk2 −/−, and qv 4J SAN cells. (n=9 from 6 preparations for WT and qv 4J and n=7 from 5 preparations for Kcnk2 −/−, *P<0.05 vs WT). AP indicates action potential; APD, action potential duration; DDR, diastolic depolarization rate; MDP, maximum diastolic potential; SAN, sinoatrial node; WT, wild type.
To determine the mechanism for altered SAN cell membrane excitability in TREK‐1–deficient SAN cells, background TREK‐1–like K+ current was measured in WT and αMHC‐Kcnk2 f/f SAN myocytes. Consistent with loss of TREK‐1, background K+ current was significantly decreased in αMHC‐Kcnk2 f/f myocytes compared with WT (Figure 4A and 4B). In support of a specific role for loss of TREK‐1 in observed AP and current differences, no difference was measured in I f between αMHC‐Kcnk2 f/f and WT SAN myocytes compared with WT (Figure 5). Furthermore, the levels of other ion channels (HCN4, Kv2.1, Kv4.2, Kv4.3, and the K2P family member TASK‐1) were similar between WT and αMHC‐Kcnk2 f/f SAN (Figure 5). Finally, although a TREK‐1 immunoreactive signal was not detectable in isolated SAN myocytes from αMHC‐Kcnk2 f/f hearts, localization of TASK‐1 was normal (Figure 5). Together these data indicate that cardiac‐specific deletion of TREK‐1 reduces background K+ current without altering levels or localization of other channels important for SAN cell membrane excitability.
Figure 4.
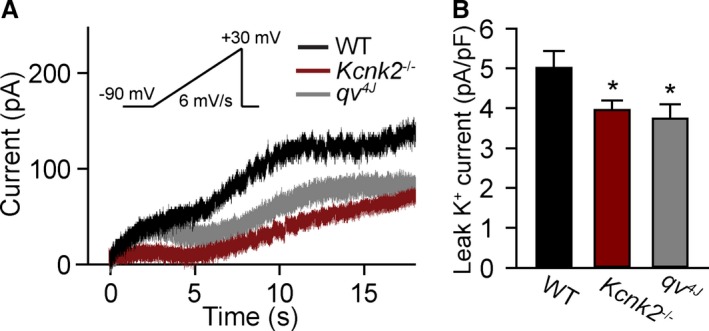
Loss of TREK‐1 expression reduces background K+ current. A and B, Representative current traces of background TREK‐1–like K+ current and summary data of the current amplitude at +30 mV (end of ramp protocol) in WT, Kcnk2 −/−, and qv 4J sinoatrial node cells (n=6 from 4 preparations for WT, n=8 from 5 preparations for Kcnk2 −/− and qv 4J, *P<0.05 vs WT). Voltage ramp protocol is shown in inset. WT indicates wild type.
Figure 5.
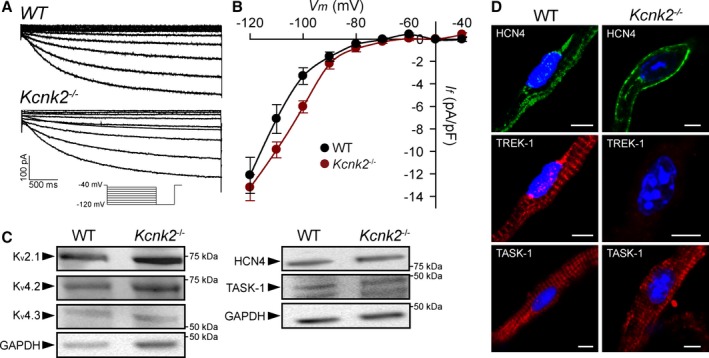
TREK‐1 deficiency does not affect activity or expression of other channels important for SAN function. A and B, Representative current traces of pacemaker current (I f) and summary current‐voltage data in WT and Kcnk2 −/− SAN cells (n=5 from 4 preparations for WT, n=5 from 3 preparations for Kcnk2 −/−, P=NS). Voltage ramp protocol is shown in inset. C, Immunoblots showing expression of voltage‐gated K+ channels important for SAN cell repolarization (Kv2.1, Kv4.2, Kv4.3), HCN4, and K2P family member TASK‐1 in detergent‐soluble lysates from WT and αMHC ‐Kcnk2 f/f (Kcnk2 −/−) SAN (3 pooled samples from 3 preparations). D, Isolated WT and Kcnk2 −/− adult SAN myocytes immunostained for HCN4, TREK‐1, and TASK‐1. Scale bar=5 μm. SAN indicates sinoatrial node; WT, wild type.
βIV‐Spectrin Regulates TREK‐1 Membrane Localization and Cell Membrane Excitability in SAN
Previous work from our group has identified the cytoskeletal protein βIV‐spectrin as an interacting partner for TREK‐1, using a qv 4J mouse expressing truncated βIV‐spectrin.8 With the goal of identifying new pathways for regulating TREK‐1 activity and atrial automaticity, the expression and function of the TREK‐1–interacting partner βIV‐spectrin in SAN was next explored. βIV‐spectrin was detectable in WT mouse atrium and SAN by both immunoblot and immunostaining and was not affected by TREK‐1 deficiency (Figure 6A, 6B, and 6D). In contrast, SAN myocytes from qv 4J mice lacking spectrin/TREK‐1 interaction8 demonstrated abnormal membrane localization of TREK‐1 (Figure 6C). Based on previous findings that qv 4J animals show increased susceptibility to stress‐induced sinus pause,8 similar to αMHC‐Kcnk2 f/f animals, spontaneous APs were measured in isolated qv 4J SAN cells (Figure 3A). Interestingly, diastolic depolarization rate and spontaneous firing were faster in qv 4J SAN cells compared with WT, similar to the observed difference in αMHC‐Kcnk2 f/f myocytes (Figure 3B and 3C). Also similar to αMHC‐Kcnk2 f/f, AP amplitude and APD at 90% repolarization were not affected in qv 4J SAN myocytes (Figure 3E and 3F). In contrast, αMHC‐Kcnk2 f/f and qv 4J SAN APs showed differences with respect to MDP and APD at 50% repolarization, (Figure 3D and 3G), suggesting that, aside from TREK‐1, there may be defects in other currents that contribute to the qv 4J phenotype. To test decreased TREK‐1 current due to loss of TREK‐1 membrane localization as a mechanism for abnormal qv 4J SAN myocyte excitability, background K+ current was measured in qv 4J SAN myocytes. Consistent with loss of TREK‐1, background K+ current was significantly decreased in qv 4J myocytes compared with WT, similar to levels measured in αMHC‐Kcnk2 f/f (Figure 4). Together these data identify a new spectrin‐based pathway for regulating TREK‐1 membrane localization and cell membrane excitability in SAN myocytes.
Figure 6.
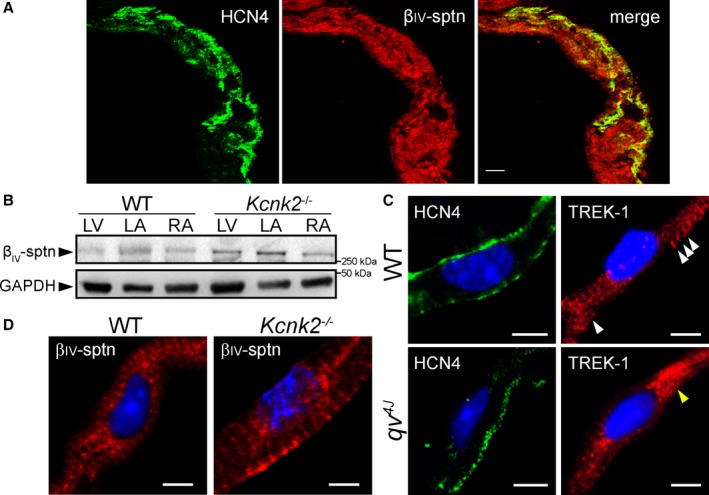
βIV‐spectrin is expressed in sinoatrial node and regulates TREK‐1 membrane localization. A, Confocal image of mouse RA tissue section (14 μm thick) immunostained for βIV‐spectrin as well as HCN4 to show intact SAN region. Scale bar=50 μm. B, Immunoblots showing expression of βIV‐spectrin in detergent‐soluble lysates from WT and aMHC‐Kcnk2 f/f (Kcnk2 −/−) LV, LA, and RA. GAPDH is shown as loading control. C, Isolated permeabilized WT and qv 4J SAN cells immunostained for HCN4 and TREK‐1. TREK‐1 is localized to membrane and sarcomeric structures (white arrows) in WT SAN cells. In contrast, SAN cells from qv 4J animals lacking spectrin/TREK‐1 interaction show increased perinuclear TREK‐1 localization (yellow arrow). Scale bar=5 μm. D, Isolated permeabilized WT and Kcnk2 −/− SAN cells immunostained for βIV‐spectrin. Scale bar=5 μm. βIV‐sptn indicates βIV‐spectrin; LA indicates left atrium, LV, left ventricle; RA, right atrium; SAN, sinoatrial node; WT, wild type.
As a first step in determining a potential role for the spectrin/TREK‐1 complex in SAN disease, βIV‐spectrin levels were analyzed in right atrial samples from dogs with pacing‐induced heart failure and SAN dysfunction. Interestingly, a dramatic reduction in βIV‐spectrin was found in right atria of dogs with SAN dysfunction compared with control (Figure 7). βIV‐spectrin expression was also examined in human SAN, and a prominent immunoreactive signal was found in tissue sections containing the SAN from a nondiseased human heart (Figure S1). Interestingly, βIV‐spectrin immunoreactive signal was apparent but weaker in the SAN region from a patient with documented coronary artery disease.
Figure 7.
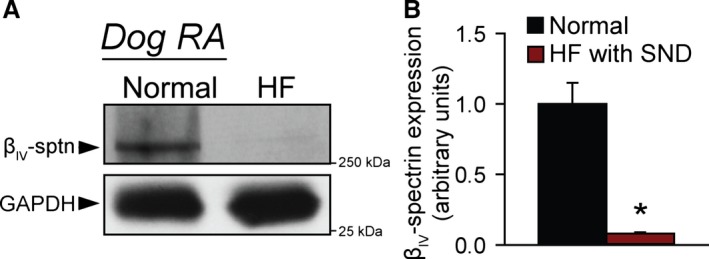
βIV‐spectrin is altered in canine model of SND. A, Representative immunoblots and (B) densitometry on βIV‐spectrin levels in detergent‐soluble lysates from RA of normal control dogs or dogs with pacing induced HF and SND (n=3 from 3 preparations, *P<0.01). βIV‐sptn indicates βIV‐spectrin; HF, heart failure; RA, right atrium; SND, sinus node dysfunction.
To further define a potential link between spectrin/TREK‐1 dysfunction and SAN disease, we performed computer simulations using a mathematical model of the mammalian SAN cell AP,25 modified to incorporate a representation of TREK‐1 current based on experimental data (Figure 8A). Elimination of TREK‐1 from our model resulted in a faster diastolic depolarization rate and firing of spontaneous APs, with much smaller effects on maximum diastolic potential and AP amplitude compared with the control model, consistent with differences observed between qv 4J or αMHC‐Kcnk2 f/f and WT animals (Figure 8B through 8F). Analysis of TREK‐1 current (Figure 8C) during the spontaneous WT AP (Figure 8B) provides insight into why loss of TREK‐1 has a relatively small effect on MDP. At the WT MDP of −57.6 mV, TREK‐1 is about 9 times smaller than the major repolarizing current I Kr (0.0761 μA/μF versus 0.6549 μA/μF); however, TREK‐1 increases during the spontaneous diastolic depolarization phase, whereas I Kr decreases (due to rapid inactivation) such that at −40 mV (close to takeoff potential), the 2 currents are much closer in amplitude (0.1628 μA/μF versus 0.4644 μA/μF). To more fully understand the role of TREK‐1 in the SAN, we performed parametric sensitivity analysis using regression26 on the modified SAN AP model (Figure 8G through 8J and Table S1). Regression coefficients express the relative impact of changing model parameters on AP properties and show that changing the conductance of TREK‐1 produced changes to AP firing, diastolic depolarization rate, AP amplitude, and MDP that closely matched expected changes measured in qv 4J or αMHC‐Kcnk2 f/f SAN myocytes. Together, these data support the hypothesis that TREK‐1 regulates SAN cell excitability (in particular, diastolic depolarization) and automaticity and that defects in TREK‐1 activity promote abnormal SAN cell membrane excitability.
Figure 8.
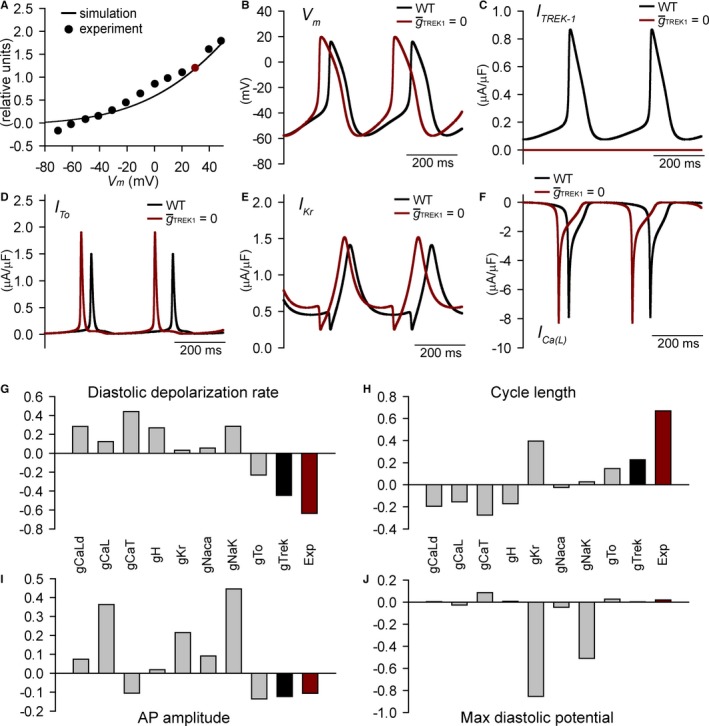
Mathematical model of spectrin/TREK‐1 dysfunction and abnormal sinoatrial node cell excitability. A, Simulated and experimentally measured37 TREK‐1 current‐voltage relationship. Current was normalized to 1.2 μA/μF at +30 mV in agreement with difference in background K+ current measured between WT and Kcnk2 −/− sinoatrial node cells. B through F, Simulated spontaneous APs, TREK‐1 current, transient outward K+ current (I to), rapid delayed rectifier K+ current (IK r), and L‐type Ca2+ current (IC a(L)) in the control model (black lines) or with maximal TREK‐1 conductance () set to zero (red lines). G through J, Regression coefficients indicating how changes in model parameters affect AP properties. Also shown are predicted regression coefficients for perturbation (5‐SD decrease in single parameter) that would produce experimentally measured changes in qv 4J AP properties (red bars). AP indicates action potential; gCaL, L‐type Ca2+ channel Cav1.2; gCaLd, L‐type Ca2+ channel Cav1.3; gCaT, T‐type Ca2+ channel; gH, funny current; Exp, experimental; gKr, Rapid delayed rectifier K+ current; gNaca, Na+/Ca2+ exchanger velocity; gNaK, Na+/K+ ATPase transport rate; gTo, transient outward K+ current; max, maximum; WT, wild type.
Discussion
The first K2P channel was identified in yeast 20 years ago38, 39, 40 and sparked a remarkable wave of discovery such that today we know of 15 channels encoded by distinct genes in humans alone. In fact, the K2P superfamily is as diverse as the much better characterized inward rectifier K+ channel family, and yet we have only begun to understand the importance of these channels for normal excitable cell function. The prevailing paradigm is that K2P channels form background leak K+ conductances that help set the resting membrane potential in excitable cells. Our findings paint a more complex picture of how K2P family member TREK‐1 contributes to cell automaticity and cardiac pacemaking. Specifically, using a novel cardiac‐specific TREK‐1–deficient mouse, we demonstrated that TREK‐1 controls automaticity, in part, by regulating the diastolic depolarization phase of the SAN cell AP. Importantly, our findings align with an identified role for ORK1, a TREK‐1 orthologue, in control of pacemaking in Drosophila.41 Based on this combined work and the fact that TREK‐1 is sensitive to a wide variety of environmental factors, it is interesting to consider the possibility that TREK‐1 has evolved to allow for rapid control of automaticity in response to stress cues.
Although relatively little is known about the function of TREK‐1 and other K2P channels in heart, even less is known about molecular pathways that are important for its membrane targeting or regulation. Previous studies have described an interaction between Popeye domain‐containing proteins (Popdc1 and Popdc2) and TREK‐1 that is important for its regulation by protein kinase A, with implications for pacemaking in the mouse.7 Similarly, even earlier work identified an association between TREK‐1 and A kinase anchor protein 150 that controls response of channel to Gs‐coupled receptor activation.42 Our new findings together with our previous work8 indicate that βIV‐spectrin associates with TREK‐1 and is essential for its proper membrane targeting in myocytes throughout the heart. The precise nature of this association remains to be determined, but it is interesting to speculate that spectrin may target a distinct subpopulation of TREK‐1 with unique localization and regulatory properties, similar to its involvement in Ca2+/calmodulin‐dependent protein kinase II–dependent regulation of the cardiac voltage‐gated Na+ channel Nav1.5.24, 29, 43
SAN dysfunction is a constellation of disorders related to compromised cardiac pacemaking function, with clinical signs of bradycardia, sinus pause or arrest, and even supraventricular arrhythmias.44 SAN dysfunction is common in older patients (≈1 in 600 patients aged >65 years) but may be observed at any age due to a variety of factors, including inherited disease; drugs; myocardial ischemia; and endocrine, thermoregulatory, or metabolic imbalance. Importantly, SAN failure is associated with increased mortality in heart failure patients, in which bradyarrhythmias account for >40% of sudden deaths.45 Underlying causes of SAN dysfunction likely involve changes in tissue structure (eg, fibrosis), ion channel defects (eg, mutations in Scn5a or HCN genes), and neurohumoral dysregulation, although the precise molecular and cellular pathways leading to aberrant pacemaking remain unknown.46, 47 Moreover, despite the prevalence of SAN dysfunction, available therapies are limited by their efficacy or risk of procedural complication. Our findings suggest (1) that downregulation of TREK‐1–associated βIV‐spectrin occurs in specific cardiac disease states to promote SAN dysfunction and (2) that loss of TREK‐1 function promotes abnormal SAN cell excitability and pacemaking defects. Interestingly, our new data align with recent studies that have shown dramatic reduction in TREK‐1 in the right atrium during persistent atrial fibrillation in the pig, suggesting a broader role for TREK‐1 dysfunction in atrial arrhythmogenesis.36 It is interesting to note that αMHC‐Kcnk2 f/f animals show mild bradycardia at rest despite an increase in SAN cell membrane excitability (increased AP firing rate). TREK‐1 deficiency, however, did not affect intrinsic heart rate, suggesting that compensatory changes (eg, altered sympathetic or parasympathetic tone) in vivo are responsible for basal bradycardia observed in absence of TREK‐1. It will be important for future studies to more fully understand the relationship between changes at the single‐cell and whole‐animal levels and whether additional SAN phenotypes emerge with aging or chronic stress. Based on our new findings, a logical next step would also be to determine the underlying mechanism for altered βIV‐spectrin/TREK‐1 expression in disease and whether the remodeling process may be prevented for therapeutic benefit.
Sources of Funding
This work was supported by National Institutes of Health (NIH) (grant numbers HL114893 to Hund, HL084583, HL083422, HL114383 to Mohler, HL079031, HL070250, HL096652, HL113001 to Anderson, HL115580 to Fedorov, HL129766 to Onal); James S. McDonnell Foundation (to Hund); Saving Tiny Hearts Society (to Mohler); American Heart Association (to Mohler).
Disclosures
None.
Supporting information
Data S1. Supplemental methods.
Table S1. Regression Coefficients Indicating How Changes in Model Parameters Affect Action Potential and Ca2+ Transient Properties
Figure S1. Masson's trichrome staining (left) or immunostaining for βIV‐spectrin (green) and α‐actinin (red; right) of paraffin‐embedded SAN sections from a nondiseased human heart (identification number 921821) and from a patient with coronary artery disease (identification number 168021). CT indicates crista terminalis; IAS, intraatrial septum; RAFW, right atrial free wall; SAN, sinoatrial node; SVC, superior vena cava.
(J Am Heart Assoc. 2016;5:e002865 doi: 10.1161/JAHA.115.002865)
References
- 1. Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952;117:500–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Backx PH, Marban E. Background potassium current active during the plateau of the action potential in guinea pig ventricular myocytes. Circ Res. 1993;72:890–900. [DOI] [PubMed] [Google Scholar]
- 3. Goonetilleke L, Quayle J. TREK‐1 K+ channels in the cardiovascular system: their significance and potential as a therapeutic target. Cardiovasc Ther. 2012;30:e23–e29. [DOI] [PubMed] [Google Scholar]
- 4. Gurney A, Manoury B. Two‐pore potassium channels in the cardiovascular system. Eur Biophys J. 2009;38:305–318. [DOI] [PubMed] [Google Scholar]
- 5. Honore E. The neuronal background K2P channels: focus on TREK1. Nat Rev Neurosci. 2007;8:251–261. [DOI] [PubMed] [Google Scholar]
- 6. Feliciangeli S, Chatelain FC, Bichet D, Lesage F. The family of K2P channels: salient structural and functional properties. J Physiol. 2015;593:2587–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Froese A, Breher SS, Waldeyer C, Schindler RF, Nikolaev VO, Rinne S, Wischmeyer E, Schlueter J, Becher J, Simrick S, Vauti F, Kuhtz J, Meister P, Kreissl S, Torlopp A, Liebig SK, Laakmann S, Muller TD, Neumann J, Stieber J, Ludwig A, Maier SK, Decher N, Arnold HH, Kirchhof P, Fabritz L, Brand T. Popeye domain containing proteins are essential for stress‐mediated modulation of cardiac pacemaking in mice. J Clin Invest. 2012;122:1119–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hund TJ, Snyder JS, Wu X, Glynn P, Koval OM, Onal B, Leymaster ND, Unudurthi SD, Curran J, Camardo C, Wright PJ, Binkley PF, Anderson ME, Mohler PJ. betaIV‐Spectrin regulates TREK‐1 membrane targeting in the heart. Cardiovasc Res. 2014;102:166–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tan JH, Liu W, Saint DA. Trek‐like potassium channels in rat cardiac ventricular myocytes are activated by intracellular ATP. J Membr Biol. 2002;185:201–207. [DOI] [PubMed] [Google Scholar]
- 10. Terrenoire C, Lauritzen I, Lesage F, Romey G, Lazdunski M. A TREK‐1‐like potassium channel in atrial cells inhibited by beta‐adrenergic stimulation and activated by volatile anesthetics. Circ Res. 2001;89:336–342. [DOI] [PubMed] [Google Scholar]
- 11. Xian Tao L, Dyachenko V, Zuzarte M, Putzke C, Preisig‐Muller R, Isenberg G, Daut J. The stretch‐activated potassium channel TREK‐1 in rat cardiac ventricular muscle. Cardiovasc Res. 2006;69:86–97. [DOI] [PubMed] [Google Scholar]
- 12. Bond RC, Choisy SC, Bryant SM, Hancox JC, James AF. Inhibition of a TREK‐like K+ channel current by noradrenaline requires both beta1‐ and beta2‐adrenoceptors in rat atrial myocytes. Cardiovasc Res. 2014;104:206–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bodnar M, Schlichthorl G, Daut J. The potassium current carried by TREK‐1 channels in rat cardiac ventricular muscle. Pflugers Arch. 2015;467:1069–1079. [DOI] [PubMed] [Google Scholar]
- 14. Wang W, Zhang M, Li P, Yuan H, Feng N, Peng Y, Wang L, Wang X. An increased TREK‐1‐like potassium current in ventricular myocytes during rat cardiac hypertrophy. J Cardiovasc Pharmacol. 2013;61:302–310. [DOI] [PubMed] [Google Scholar]
- 15. Piovan C, Amari F, Lovat F, Chen Q, Coppola V. Generation of mouse lines conditionally over‐expressing microRNA using the Rosa26‐Lox‐Stop‐Lox system. Methods Mol Biol. 2014;1194:203–224. [DOI] [PubMed] [Google Scholar]
- 16. Rodriguez CI, Buchholz F, Galloway J, Sequerra R, Kasper J, Ayala R, Stewart AF, Dymecki SM. High‐efficiency deleter mice show that FLPe is an alternative to Cre‐loxP. Nat Genet. 2000;25:139–140. [DOI] [PubMed] [Google Scholar]
- 17. Parkinson NJ, Olsson CL, Hallows JL, McKee‐Johnson J, Keogh BP, Noben‐Trauth K, Kujawa SG, Tempel BL. Mutant beta‐spectrin 4 causes auditory and motor neuropathies in quivering mice. Nat Genet. 2001;29:61–65. [DOI] [PubMed] [Google Scholar]
- 18. Ogawa M, Zhou S, Tan AY, Song J, Gholmieh G, Fishbein MC, Luo H, Siegel RJ, Karagueuzian HS, Chen LS, Lin SF, Chen PS. Left stellate ganglion and vagal nerve activity and cardiac arrhythmias in ambulatory dogs with pacing‐induced congestive heart failure. J Am Coll Cardiol. 2007;50:335–343. [DOI] [PubMed] [Google Scholar]
- 19. Li N, Csepe TA, Hansen BJ, Dobrzynski H, Higgins RS, Kilic A, Mohler PJ, Janssen PM, Rosen MR, Biesiadecki BJ, Fedorov VV. Molecular mapping of sinoatrial node HCN channel expression in the human heart. Circ Arrhythm Electrophysiol. 2015;8:1219–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Le Scouarnec S, Bhasin N, Vieyres C, Hund TJ, Cunha SR, Koval O, Marionneau C, Chen B, Wu Y, Demolombe S, Song LS, Le Marec H, Probst V, Schott JJ, Anderson ME, Mohler PJ. Dysfunction in ankyrin‐B‐dependent ion channel and transporter targeting causes human sinus node disease. Proc Natl Acad Sci USA. 2008;105:15617–15622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Swaminathan PD, Purohit A, Soni S, Voigt N, Singh MV, Glukhov AV, Gao Z, He JB, Luczak ED, Joiner ML, Kutschke W, Yang J, Donahue JK, Weiss RM, Grumbach IM, Ogawa M, Chen PS, Efimov IR, Dobrev D, Mohler PJ, Hund TJ, Anderson ME. Oxidized CaMKII causes sinus node dysfunction in mice. J Clin Invest. 2011;121:3277–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Luo M, Guan X, Di L, Kutschke W, Gao Z, Yang J, Luczak ED, Glynn P, Swaminathan PD, Weiss RM, Yang B, Rokita AG, Sossalla S, Maier LS, Efimov IR, Hund TJ, Anderson ME. Diabetes increases mortality after myocardial infarction by oxidizing CaMKII. J Clin Invest. 2013;123:1262–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gao Z, Rasmussen TP, Li Y, Kutschke W, Koval OM, Wu Y, Hall DD, Joiner ML, Wu XQ, Swaminathan PD, Purohit A, Zimmerman K, Weiss RM, Philipson KD, Song LS, Hund TJ, Anderson ME. Genetic inhibition of Na+‐Ca2+ exchanger current disables fight or flight sinoatrial node activity without affecting resting heart rate. Circ Res. 2013;112:309–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Glynn P, Musa H, Wu X, Unudurthi SD, Little S, Qian L, Wright PJ, Radwanski PB, Gyorke S, Mohler PJ, Hund TJ. Voltage‐gated sodium channel phosphorylation at Ser571 regulates late current, arrhythmia, and cardiac function in vivo. Circulation. 2015;132:567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kurata Y, Matsuda H, Hisatome I, Shibamoto T. Regional difference in dynamical property of sinoatrial node pacemaking: role of Na+ channel current. Biophys J. 2008;95:951–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sobie EA. Parameter sensitivity analysis in electrophysiological models using multivariable regression. Biophys J. 2009;96:1264–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wolf RM, Glynn P, Hashemi S, Zarei K, Mitchell CC, Anderson ME, Mohler PJ, Hund TJ. Atrial fibrillation and sinus node dysfunction in human ankyrin‐B syndrome: a computational analysis. Am J Physiol Heart Circ Physiol. 2013;304:H1253–H1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wolf RM, Mitchell CC, Christensen MD, Mohler PJ, Hund TJ. Defining new insight into atypical arrhythmia: a computational model of ankyrin‐B‐syndrome. Am J Physiol Heart Circ Physiol. 2010;299:H1505–H1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Koval OM, Snyder JS, Wolf RM, Pavlovicz RE, Glynn P, Curran J, Leymaster ND, Dun W, Wright PJ, Cardona N, Qian L, Mitchell CC, Boyden PA, Binkley PF, Li C, Anderson ME, Mohler PJ, Hund TJ. Ca2+/calmodulin‐dependent protein kinase II‐based regulation of voltage‐gated Na+ channel in cardiac disease. Circulation. 2012;126:2084–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Glynn P, Onal B, Hund TJ. Cycle length restitution in sinoatrial node cells: a theory for understanding spontaneous action potential dynamics. PLoS One. 2014;9:e89049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Heurteaux C, Guy N, Laigle C, Blondeau N, Duprat F, Mazzuca M, Lang‐Lazdunski L, Widmann C, Zanzouri M, Romey G, Lazdunski M. TREK‐1, a K+ channel involved in neuroprotection and general anesthesia. EMBO J. 2004;23:2684–2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Heurteaux C, Lucas G, Guy N, El Yacoubi M, Thummler S, Peng XD, Noble F, Blondeau N, Widmann C, Borsotto M, Gobbi G, Vaugeois JM, Debonnel G, Lazdunski M. Deletion of the background potassium channel TREK‐1 results in a depression‐resistant phenotype. Nat Neurosci. 2006;9:1134–1141. [DOI] [PubMed] [Google Scholar]
- 33. Garry A, Fromy B, Blondeau N, Henrion D, Brau F, Gounon P, Guy N, Heurteaux C, Lazdunski M, Saumet JL. Altered acetylcholine, bradykinin and cutaneous pressure‐induced vasodilation in mice lacking the TREK1 potassium channel: the endothelial link. EMBO Rep. 2007;8:354–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Blondeau N, Petrault O, Manta S, Giordanengo V, Gounon P, Bordet R, Lazdunski M, Heurteaux C. Polyunsaturated fatty acids are cerebral vasodilators via the TREK‐1 potassium channel. Circ Res. 2007;101:176–184. [DOI] [PubMed] [Google Scholar]
- 35. Little SC, Curran J, Makara MA, Kline CF, Ho HT, Xu Z, Wu X, Polina I, Musa H, Meadows AM, Carnes CA, Biesiadecki BJ, Davis JP, Weisleder N, Gyorke S, Wehrens XH, Hund TJ, Mohler PJ. Protein phosphatase 2A regulatory subunit B56alpha limits phosphatase activity in the heart. Sci Signal. 2015;8:ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schmidt C, Wiedmann F, Tristram F, Anand P, Wenzel W, Lugenbiel P, Schweizer PA, Katus HA, Thomas D. Cardiac expression and atrial fibrillation‐associated remodeling of K2p2.1 (TREK‐1) K+ channels in a porcine model. Life Sci. 2014;97:107–115. [DOI] [PubMed] [Google Scholar]
- 37. Aimond F, Rauzier JM, Bony C, Vassort G. Simultaneous activation of p38 MAPK and p42/44 MAPK by ATP stimulates the K+ current ITREK in cardiomyocytes. J Biol Chem. 2000;275:39110–39116. [DOI] [PubMed] [Google Scholar]
- 38. Ketchum KA, Joiner WJ, Sellers AJ, Kaczmarek LK, Goldstein SA. A new family of outwardly rectifying potassium channel proteins with two pore domains in tandem. Nature. 1995;376:690–695. [DOI] [PubMed] [Google Scholar]
- 39. Reid JD, Lukas W, Shafaatian R, Bertl A, Scheurmann‐Kettner C, Guy HR, North RA. The S. cerevisiae outwardly‐rectifying potassium channel (DUK1) identifies a new family of channels with duplicated pore domains. Receptors Channels. 1996;4:51–62. [PubMed] [Google Scholar]
- 40. Zhou XL, Vaillant B, Loukin SH, Kung C, Saimi Y. YKC1 encodes the depolarization‐activated K+ channel in the plasma membrane of yeast. FEBS Lett. 1995;373:170–176. [DOI] [PubMed] [Google Scholar]
- 41. Lalevee N, Monier B, Senatore S, Perrin L, Semeriva M. Control of cardiac rhythm by ORK1, a Drosophila two‐pore domain potassium channel. Curr Biol. 2006;16:1502–1508. [DOI] [PubMed] [Google Scholar]
- 42. Sandoz G, Thummler S, Duprat F, Feliciangeli S, Vinh J, Escoubas P, Guy N, Lazdunski M, Lesage F. AKAP150, a switch to convert mechano‐, pH‐ and arachidonic acid‐sensitive TREK K(+) channels into open leak channels. EMBO J. 2006;25:5864–5872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hund TJ, Koval OM, Li J, Wright PJ, Qian L, Snyder JS, Gudmundsson H, Kline CF, Davidson NP, Cardona N, Rasband MN, Anderson ME, Mohler PJ. A betaIV spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J Clin Invest. 2010;120:3508–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Monfredi O, Boyett MR. Sick sinus syndrome and atrial fibrillation in older persons—a view from the sinoatrial nodal myocyte. J Mol Cell Cardiol. 2015;83:88–100. [DOI] [PubMed] [Google Scholar]
- 45. Sanders P, Kistler PM, Morton JB, Spence SJ, Kalman JM. Remodeling of sinus node function in patients with congestive heart failure: reduction in sinus node reserve. Circulation. 2004;110:897–903. [DOI] [PubMed] [Google Scholar]
- 46. Lou Q, Hansen BJ, Fedorenko O, Csepe TA, Kalyanasundaram A, Li N, Hage LT, Glukhov AV, Billman GE, Weiss R, Mohler PJ, Gyorke S, Biesiadecki BJ, Carnes CA, Fedorov VV. Upregulation of adenosine A1 receptors facilitates sinoatrial node dysfunction in chronic canine heart failure by exacerbating nodal conduction abnormalities revealed by novel dual‐sided intramural optical mapping. Circulation. 2014;130:315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Unudurthi SD, Wolf RM, Hund TJ. Role of sinoatrial node architecture in maintaining a balanced source‐sink relationship and synchronous cardiac pacemaking. Front Physiol. 2014;5:446. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplemental methods.
Table S1. Regression Coefficients Indicating How Changes in Model Parameters Affect Action Potential and Ca2+ Transient Properties
Figure S1. Masson's trichrome staining (left) or immunostaining for βIV‐spectrin (green) and α‐actinin (red; right) of paraffin‐embedded SAN sections from a nondiseased human heart (identification number 921821) and from a patient with coronary artery disease (identification number 168021). CT indicates crista terminalis; IAS, intraatrial septum; RAFW, right atrial free wall; SAN, sinoatrial node; SVC, superior vena cava.