

Abstract
Background
Current recommendations for lipoprotein(a) (Lp[a]) focus on the control of other risk factors, including lowering low‐density lipoprotein cholesterol (LDL‐C), with little evidence to support this approach. Identifying interactions between Lp(a) and other risk factors could identify individuals at increased risk for Lp(a)‐mediated disease.
Methods and Results
We used a case‐only study design and included 939 participants (median age=49 years, interquartile range 46–53, women=33.1%) from the GENdEr and Sex determInantS of cardiovascular disease: from bench to beyond‐Premature Acute Coronary Syndrome (GENESIS‐PRAXY) study, a multicenter prospective cohort study of premature acute coronary syndrome. There was a higher prevalence of elevated Lp(a) levels (>50 mg/dL; 80th percentile) in PRAXY participants as compared to the general population (31% versus 20%; P<0.001). Lp(a) was strongly associated with LDL‐C (adjusted β 0.17; P<0.001). Individuals with high Lp(a) were more likely to have LDL‐C >2.5 mmol/L, indicating a synergistic interaction (adjusted odds ratio 1.51; 95% CI 1.08–2.09; P=0.015). The interaction with high Lp(a) was stronger at increasing LDL‐C levels (LDL‐C >3.5, adjusted odds ratio 1.87; LDL‐C >4.5, adjusted odds ratio 2.72). In a polytomous logistic model comparing mutually exclusive LDL‐C categories, the interaction with high Lp(a) became attenuated at LDL‐C ≤3.5 mmol/L (odds ratio 1.16; 95% CI 0.80–1.68, P=0.447). Other risk factors were not associated with high Lp(a).
Conclusions
In young acute coronary syndrome patients, high Lp(a) is more prevalent than in the general population and is strongly associated with high LDL‐C, suggesting that Lp(a) confers greater risk for acute coronary syndrome when LDL‐C is elevated. Individuals with high Lp(a) and LDL‐C >3.5 mmol/L may warrant aggressive LDL‐C lowering.
Keywords: acute coronary syndrome, atherosclerosis, lipids and lipoproteins, lipoprotein(a), prevention, risk factor
Subject Categories: Lipids and Cholesterol, Risk Factors, Coronary Artery Disease, Epidemiology
Introduction
Elevated lipoprotein(a) (Lp[a]) represents one of the most common genetic dyslipidemias worldwide, affecting 1 in 5 individuals.1 Lp(a) has recently been confirmed as a causal factor for cardiovascular disease including myocardial infarction (MI)2 and aortic stenosis.3 To date, the appropriate management of high Lp(a) is not known as there are limited therapeutic options to directly lower Lp(a).4 Current recommended strategies focus on the control of other risk factors, including lowering low‐density lipoprotein cholesterol (LDL‐C), with little evidence to support this approach.5 Identifying interactions between Lp(a) and other risk factors could point to patient groups at increased risk for Lp(a)‐mediated disease, as well as preventative strategies to mitigate the risk conferred by high Lp(a) (eg, LDL‐C lowering and smoking cessation). However, whether Lp(a) interacts with LDL‐C or other cardiovascular risk factors in the development of premature acute coronary syndrome (ACS) has not been well established.
Accordingly, we sought to evaluate whether high Lp(a) interacts with LDL‐C levels and other risk factors in premature ACS using a case‐only design. The case‐only study has been used to identify several gene–environment interactions and provides valid estimates when the genetic exposure and environmental exposure are not associated in the general population.6, 7 Since Lp(a) is under strong genetic regulation with modest environmental influence from other cardiovascular risk factors,6 and has been shown to be independent of most cardiovascular risk factors, including LDL‐C, in the general population,8, 9, 10, 11 the case‐only study design provides a valid approach to identify interactions between cardiovascular risk factors and Lp(a).
Methods
Study Population
Patients included in the analysis were from the Gender and Sex determinants of cardiovascular disease: From bench to beyond—Premature Acute Coronary Syndrome (GENESIS‐PRAXY) study. GENESIS‐PRAXY is a multicenter prospective cohort study of premature ACS patients from Canada, the United States, and Switzerland who are 18 to 55 years old. Details of this study have been previously published elsewhere.12 Of the 1123 patients enrolled in GENESIS‐PRAXY, 184 were excluded for missing Lp(a) levels, leaving a final sample of 939 patients included in this analysis. In Canada, the McGill University Health Center acted as the central ethics review board for all centers. All other centers received approval from their respective ethics review boards. Informed consent was obtained from all of the participating subjects.
ACS in the Study Population
The GENESIS‐PRAXY study population included individuals who developed symptoms consistent with acute cardiac ischemia within the first 24 hours of hospital admission. These individuals were considered to have an ACS, which included unstable or intermediate coronary syndromes and/or acute MI. In addition to symptoms, these patients were required to fulfill at least 1 of the following criteria: (1) New electrocardiographic (ECG) changes in ≥2 contiguous leads including ST elevations by ≥1 mm, ST‐segment depressions by ≥1 mm, dynamic T‐wave inversions of ≥1 mm, pseudo‐normalization of negative T‐waves, significant Q‐waves defined by being at least one third the height of the corresponding R wave or ≥0.04 seconds in duration, R‐wave >S‐wave in lead V1 suggestive of posterior MI, left bundle branch block; and/or (2) Increased cardiac enzymes above the hospital's normal range: creatine kinase‐MB >2 times the upper limit and/or creatine phosphokinase >2 times upper limit, elevated troponin I and/or elevated troponin T.
Data Collection
Patients fulfilling the above criteria were identified within the first 48 hours of hospital admission. Standardized chart reviews in addition to self‐administered questionnaires were used to obtain demographic data and information on risk factors. We defined hypertension, hyperlipidemia, and diabetes based on self‐report of these conditions, and/or use of antihypertensive, lipid‐lowering, or hypoglycemic medications, respectively, at admission. If patients reported cigarette smoking >1 cigarette per day within 30 days prior to admission, they were defined as current smokers. Standard sphygmomanometers were used to measure blood pressure during the first 48 hours of the admission. Family history of coronary artery disease was defined as having a first‐degree relative with coronary artery disease at any age. The 10‐year general cardiovascular disease Framingham Risk Score was calculated using previously reported methods.13 Fasting plasma samples obtained within the first 48 hours of admission were used to measure LDL‐C levels via standard methods. Lp(a) plasma concentration (in mg/dL) was measured using rate nephelometry technique on an Immage system analyzer (Beckman‐Coulter Inc., Fullerton, CA) using an anti‐lipoprotein(a) rabbit polyclonal antibody, independent of the Apo(a) isoform size, in frozen plasma samples (Immage; Beckman‐Coulter).
Case‐Only Study Design
A case‐only study design is a modern epidemiological method used to identify genetic by environment (G×E) interactions for a disease of interest without the use of controls. Under conditions of no association between the genetic and environmental exposure in the general population, a case‐only study estimates the enrichment of the presence of both a genetic and environmental exposure in cases.6, 7 Simply put, since Lp(a) is predominantly genetically mediated,8 and Lp(a) levels are not associated with environmental factors in the general population, including cardiovascular risk factors such as LDL‐C, as previously reported,8, 9, 10, 11 any observed association between Lp(a) and these risk factors among cases of premature ACS is indicative of a departure from a multiplicative interaction. Previous work has shown that the odds ratio obtained from the case‐only studies is the same as the odds ratio from case–control studies with unmatched controls.14 Furthermore, this type of design reduces variation and can result in greater precision with smaller standard errors.7 Finally, if a positive association is detected between the factors that influence disease risk (eg, LDL and Lp[a]), it suggests a synergistic interaction.15, 16
Statistical Analysis
We present continuous data as means with SDs and discrete data as percentages. Bivariate analyses were performed using t tests for continuous variables and Fisher exact tests for discrete data. To evaluate whether high Lp(a) was enriched in our study sample of premature ACS, we compared the prevalence in our study sample to the population prevalence using the χ2 test for continuous variables. The population prevalence of high Lp(a) was based on published estimates from the Copenhagen General Population Study.2
To evaluate association between risk factors (age, sex, smoking status, family history, body mass index, hypertension, diabetes, LDL‐C levels) and continuous log‐normalized Lp(a) levels, we used linear regression models. In these models, the effect size reported is in terms of transformed Lp(a) levels. Because Lp(a) pathogenicity increases significantly at high levels, we also used logistic regression models to evaluate the association between high Lp(a) >50 mg/dL (80th population percentile) and each risk factor. To further explore the strength of the association between high Lp(a) and LDL‐C, we performed sensitivity analyses by increasing LDL‐C thresholds. All models were adjusted for age, sex, smoking status, hypertension, and diabetes. To identify a clinical LDL‐C threshold at which the interaction with Lp(a) was attenuated, we performed a polytomous logistic model across mutually exclusive LDL‐C groups. A polytomous logistic model was used as there were more than 2 clinically relevant LDL‐C groups as possible outcomes. In secondary analyses, to ensure that the associations were not an artifact of the LDL‐C measurement, we repeated all analyses using corrected LDL‐C levels using the Dahlen's formula, which corrects the LDL‐C measurements for the presence of cholesterol contained in Lp(a).17 We also performed additional adjustments of all models for statin, angiotensin‐converting enzyme inhibitor, or angiotensin II receptor blocker use. All statistical analyses were performed using R version 3.1.3 software.18 Statistical significance was considered at a 2‐sided α<0.05.
Results
Baseline Characteristics
Table 1 summarizes study sample characteristics stratified by Lp(a) level with a cut‐off value of 50 mg/dL. Median age of study participants was 49 years (interquartile range 46–53) and 33.1% were women. Most participants were considered to be at moderate risk for cardiovascular disease based on the Framingham 10‐year cardiovascular disease risk score of 18%, with no statistically significant difference between the 2 groups.
Table 1.
Baseline Characteristics of GENESIS‐PRAXY Study Subjects
| All Patients (N=939) | Lp(a) ≤50 mg/dL (N=647) | Lp(a) >50 mg/dL (N=292) | P Value | |
|---|---|---|---|---|
| Median age [IQR] | 49 [46–53] | 49 [45–53] | 50 [46–53] | 0.862 |
| Female | 311 (33.1) | 205 (31.7) | 106 (36.3) | 0.178 |
| Framingham 10‐year risk | 18.0 (7.4) | 17.8 (7.3) | 18.3 (7.7) | 0.438 |
| Current smokers | 404 (43.5) | 280 (43.9) | 124 (42.8) | 0.775 |
| Family history | 458 (48.8) | 312 (48.2) | 146 (50.0) | 0.622 |
| Body mass index | 29.7 (6.4) | 29.7 (6.2) | 29.5 (6.8) | 0.739 |
| Hypertension | 445 (47.5) | 314 (48.6) | 131 (45.1) | 0.358 |
| Hyperlipidemia | 511 (54.4) | 336 (51.9) | 175 (59.9) | 0.024 |
| Diabetes | 147 (15.7) | 110 (17.0) | 37 (12.7) | 0.099 |
| Statin at admission | 200 (35.7) | 126 (32.4) | 74 (43.0) | 0.017 |
| ACEI/ARB | 195 (34.8) | 133 (34.2) | 62 (36.0) | 0.700 |
Continuous variables are expressed as mean and categorical variables as percentage.
P‐value calculated by t test for continuous variables and Fisher exact test for categorical variables.
ACEI indicates angiotensin‐converting‐enzyme inhibitor; ARB, angiotensin II receptor blockers; GENESIS‐PRAXY, GENdEr and Sex determInantS of cardiovascular disease: from bench to beyond‐Premature Acute Coronary Syndrome; IQR, interquartile range; Lp(a), lipoprotein(a).
High Lp(a) Is Enriched in Early‐Onset ACS and Is Associated With LDL‐C
Table 2 shows that PRAXY study participants had a higher prevalence of high Lp(a) compared to the general population (20% versus 31%; P<0.001). As compared to patients with lower Lp(a), patients with high Lp(a) levels (>50 mg/dL) had a higher prevalence of hyperlipidemia (P=0.024), but there were no significant differences in smoking status, family history of cardiovascular disease, body mass index, hypertension, or diabetes (P>0.050 for all). In linear regression analysis, only LDL‐C was significantly associated with elevated Lp(a) (β=0.16; P<0.001), even after adjusting for age, sex, smoking status, hypertension, diabetes, and body mass index (β=0.17; P<0.001) (Table 3).
Table 2.
Prevalence of High Lp(a) in GENESIS‐PRAXY Versus the General Population
| Lp(a) ≤50 mg/dL N (%) | Lp(a) >50 mg/dL N (%) | OR (95% CI) P Value | |
|---|---|---|---|
| CGPS (N=5543)a | 4434 (80.0) | 1109 (20.0) |
1.81 (1.55, 2.10) <0.001 |
| GENESIS‐PRAXY (N=939) | 647 (69.0) | 292 (31.0) |
GENESIS‐PRAXY indicates GENdEr and Sex determInantS of cardiovascular disease: from bench to beyond‐Premature Acute Coronary Syndrome; Lp(a), lipoprotein(a); OR, odds ratio.
P‐value calculated by χ2 test for continuous variables.
In the Copenhagen General Population Study (CGPS) (N=5543), 20% of the population had elevated Lp(a).2
Table 3.
Associations Between Continuous Lp(a) and Traditional Cardiovascular Risk Factors in Premature ACS
| βb | 95% CI | P Value | Adj. βa | 95% CI | P Value | |
|---|---|---|---|---|---|---|
| Age, per y | −0.0032 | (−0.017, 0.011) | 0.654 | −0.0018 | (−0.016, 0.012) | 0.806 |
| Women | 0.098 | (−0.071, 0.270) | 0.256 | 0.11 | (−0.068, 0.280) | 0.233 |
| Smoking | −0.11 | (−0.270, 0.054) | 0.190 | −0.12 | (−0.280, 0.045) | 0.157 |
| Hypertension | −0.11 | (−0.270, 0.048) | 0.190 | −0.11 | (−0.280, 0.056) | 0.193 |
| Diabetes | −0.13 | (−0.350, 0.087) | 0.238 | −0.13 | (−0.360, 0.100) | 0.272 |
| FHx | 0.034 | (−0.160, 0.220) | 0.731 | 0.045 | (−0.150, 0.240) | 0.650 |
| BMI | −0.0047 | (−0.017, 0.008) | 0.461 | −0.0023 | (−0.016, 0.011) | 0.735 |
| LDL‐C | 0.16 | (0.090, 0.240) | <0.001 | 0.17 | (0.089, 0.240) | <0.001 |
ACS indicates acute coronary syndrome; BMI, body mass index; FHx, family history; LDL‐C, low‐density lipoprotein cholesterol; Lp(a), lipoprotein(a).
Adjusted for age, sex, smoking status, hypertension, diabetes, and BMI.
βs are in terms of ln (Lp[a]).
High Lp(a) Interaction With LDL‐C
In logistic regression analysis, elevated Lp(a) >50 mg/dL was associated with LDL‐C >2.5 mmol/L in both unadjusted (odds ratio 1.53, 95% CI 1.11–2.11; P=0.009) and adjusted models (odds ratio 1.51; 95% CI 1.08–2.09; P=0.015). There were no significant interactions by sex (P>0.050). Given that Lp(a) levels are genetically mediated and not associated with LDL‐C in the general population, the observed odds ratio >1 among cases is indicative of a synergistic interaction.
Increasing Strength of the Lp(a) and LDL‐C Interaction at Higher LDL‐C Thresholds
In sensitivity analyses, we analyzed the interaction between elevated Lp(a) with increasing LDL‐C levels. We demonstrate that the magnitude of the interaction between elevated Lp(a) increases with increasing LDL‐C thresholds. The adjusted odds ratio increased from 1.51 (95% CI 1.08–2.09, P=0.015) at LDL‐C >2.5 mmol/L, to 2.72 (95% CI 1.67–4.42; P<0.001) at an LDL‐C >4.5 mmol/L.
Interaction of High Lp(a) and LDL‐C Becomes Attenuated at LDL‐C ≤3.5 mmol/L
To identify a possible threshold for the interaction of high Lp(a) and LDL‐C, we used a polytomous logistic regression analysis comparing mutually exclusive groups based on LDL‐C levels. As compared to LDL‐C <2.5 mmol/L, the adjusted Lp(a) interaction odds ratios across LDL‐C groups were 1.16 (95% CI 0.80–1.68, P=0.447), 1.66 (95% CI 1.11–2.51, P=0.015), and 3.31 (95% CI 1.93–5.67, P<0.001) for LDL‐C 2.5 to 3.5 mmol/L, 3.5 to 4.5 mmol/L, and >4.5 mmol/L, respectively. We observed a marked attenuation of the interaction of high Lp(a) and LDL‐C at the threshold value of ≤3.5 mmol/L (Figure).
Figure 1.
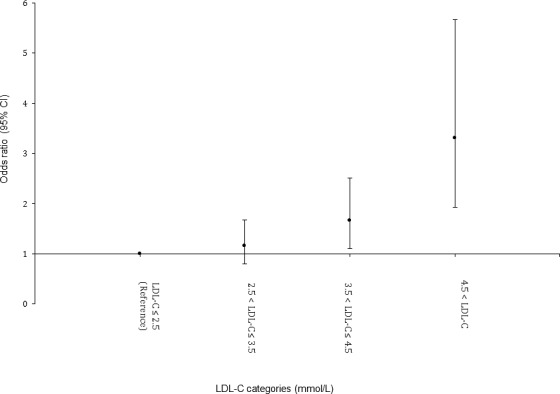
Interaction between lipoprotein(a) >50 mg/dL and mutually exclusive LDL‐C categories in patients with premature ACS. ACS indicates acute coronary syndrome; LDL‐C, low‐density lipoprotein cholesterol.
Sensitivity Analyses Using Corrected LDL‐C and Other Secondary Analyses
Using Dahlen's formula and correction of LDL‐C levels for high Lp(a), the interaction between Lp(a) and LDL‐C remained significant; however, in our threshold analyses, the threshold for attenuation of the Lp(a) interaction with LDL‐C decreased, to a corrected LDL‐C value of >2.5 mmol/L (Table 4). In secondary analyses, all of the above associations between Lp(a) and LDL‐C remained significant even after adjustment for statin and angiotensin‐converting enzyme inhibitor/angiotensin II receptor blocker use (results not shown).
Table 4.
Polytomous Logistic Regression Analysis of the Interaction Between High Lp(a) and Mutually Exclusive Increasing Corrected LDL‐C Levels, Using Dahlen's Equation, in Premature ACS
| OR for Lp(a) >30 mg/dLa | 95% CI | P Value | OR for Lp(a) >50 mg/dLa | 95% CI | P Value | |
|---|---|---|---|---|---|---|
| cLDL‐C ≤2.5 | Reference | |||||
| 2.5<cLDL‐C≤3.5 | 1.40 | (0.97, 2.00) | 0.07 | 1.60 | (1.08, 2.38) | 0.020 |
| 3.5<cLDL‐C≤4.5 | 1.51 | (0.95, 2.40) | 0.08 | 1.54 | (0.93, 2.55) | 0.096 |
| 4.5 <cLDL‐C | 3.17 | (1.49, 6.76) | 0.002 | 4.04 | (1.96, 8.34) | >0.001 |
ACS indicates acute coronary syndrome; BMI, body mass index; LDL‐C, low‐density lipoprotein cholesterol; Lp(a), lipoprotein(a); OR, odds ratio.
cLDL‐C is corrected LDL‐C using Dahlen's equation (LDL=TC−HDL−0.2TG−0.3Lp[a]) (references 8).
Adjusted for age, sex, smoking status, hypertension, diabetes, and BMI.
Discussion
In this study of 939 premature ACS cases, we demonstrate a higher prevalence of high Lp(a) level, compared to the general population, confirming that elevated Lp(a) is an important risk factor for premature ACS. We also demonstrate that Lp(a) appears to be strongly associated with LDL‐C, but not with other cardiovascular risk factors, in young ACS cases, highlighting the potential importance of LDL‐C in patients with elevated Lp(a). Given that previous studies have confirmed that Lp(a) and LDL‐C are not associated in the general population,10, 11 our finding that Lp(a) and LDL‐C are strongly associated in young ACS cases is indicative of an interaction between these lipids in the development of premature ACS. Our results suggest that Lp(a) excess may promote initiation and early development of vascular plaque that may be accelerated by the presence of other pro‐atherogenic lipids, such as LDL‐C, leading to the synergistic interaction observed in our analysis. We note that among premature ACS patients, individuals with high Lp(a) were >1.5 times more likely to have LDL‐C values of >2.5 mmol/L, as compared with those with lower Lp(a) levels. Moreover, the interaction between elevated Lp(a) and LDL‐C levels became increasingly stronger at higher LDL‐C levels. Young ACS cases with high Lp(a) had 80% higher odds of having LDL‐C levels above 3.5 mmol/L and had almost 300% greater odds for having LDL‐C levels above 4.5 mmol/L, as compared to those with lower Lp(a). Most importantly, we demonstrate that below a LDL‐C threshold of <3.5 mmol/L, the interaction with Lp(a) became markedly attenuated (and statistically nonsignificant), suggesting that LDL‐C below this threshold may reduce the risk of premature ACS in those with high Lp(a). Given the absence of approved specific Lp(a)‐lowering medications at the current time, our finding further highlights the importance of appropriate LDL management in patients with elevated Lp(a) and lends support to current recommendations for more aggressive LDL‐C lowering in individuals with high Lp(a).
Several prior studies have explored possible interactions between Lp(a) and other risk factors, including LDL‐C, in primary prevention, but many of these studies have been performed in small cohorts with several limitations.19, 20, 21, 22, 23, 24, 25 The Bruneck study prospectively evaluated 500 participants free of carotid atherosclerosis and demonstrated that Lp(a) was a predictor of accelerated progression of carotid atherosclerosis, only among participants with LDL‐C levels >3.3 mmol/L. However, this interaction could not be further evaluated with clinical cardiovascular events due to a low event rate (n=64).20 In the PROCAM study, with data from 788 participants with 44 cardiovascular events, Lp(a) was found to be predictive of cardiovascular events, especially at higher LDL‐C levels (>4.1 mmol/L), but formal testing for interaction was not performed.21 The PRIME study reported data from 9133 individuals followed for 5 years (n=288 cardiovascular events) and demonstrated the presence of an interaction between elevated Lp(a) ≥33 mg/dL, and LDL‐C >4.3 mmol/L,22 but did not identify a specific LDL‐C threshold for this interaction, likely due to the relatively small number of available cases. Similarly, in a small nested case–control study with 195 cases and controls from the Physician's Health Study, elevated Lp(a) (>95th percentile) was associated with the development of angina only among individuals with LDL‐C >4.15 mmol/L,23 but did not report other cardiovascular events (including MI). The most robust data to date regarding the interaction of Lp(a) and LDL‐C originate from the Women's Health Study (n=27 791 women with 899 incident cardiovascular events) in which a strong interaction between elevated Lp(a) levels (>44 mg/dL) and high LDL‐C levels (>3.1 mmol/L) was observed in women.24 Finally, although a recent observational study has suggested that patients with ACS and high Lp(a) have recurrent cardiovascular events despite low LDL‐C,25 a recent large meta‐analysis of statin trials in secondary prevention suggested that Lp(a) was only predictive of recurrent events in individuals with high LDL‐C >3.4 mmol/L.26 Although these studies have all demonstrated some evidence for interaction between elevated Lp(a) and LDL‐C levels, these analyses have been limited by small sample sizes, the lack of formal interaction tests, the inclusion of only women or the use of surrogate end points (ie, angina or vascular imaging). Our study therefore extends the evidence regarding this interaction in a sizeable contemporary sample of much younger ACS patients (≤55 years) that includes both men and women, using a unique case‐only approach, and adds to the existing evidence that elevated Lp(a) is most strongly associated with ACS under conditions of elevated LDL‐C. We also used a more clinically relevant Lp(a) value of 50 mg/dL for high Lp(a), which is considered to be the level where Lp(a) is most strongly associated with cardiovascular risk.27 Finally, the large number of ACS cases allowed us to identify a LDL‐C threshold at which the interaction was attenuated, which was not previously possible and is of clinical relevance.
Several studies have also used the modified Friedewald equation, also known as Dahlen's formula, to calculate corrected LDL‐C levels10, 11 to account for the increased cholesterol content of Lp(a) particles. In sensitivity analyses, using Dahlen's corrected LDL‐C levels, the interaction between elevated Lp(a) and corrected LDL‐C remained significant but at a lower corrected LDL‐C threshold of <2.5 mmol/L.17 Nonetheless, given that Dahlen's modified LDL‐C formula is not frequently used clinically, an uncorrected LDL‐C threshold >3.5 mmol/L is likely more relevant in clinical practice.
High Lp(a) has been determined as the strongest independent genetic risk factor of CV disease,27, 28 and a causal role for circulating Lp(a) in cardiovascular events has been demonstrated by Mendelian randomization.2 Lp(a) has also been shown to be a potential causal factor for valve calcification and aortic stenosis in a large genome‐wide association study,3 and in several subsequent reports.29, 30 Despite these important associations, Lp(a) screening remains controversial in general practice largely due to the limited therapeutic options for lowering Lp(a). Nonetheless, the European Atherosclerotic Society consensus panel as well as the National Lipid Association recommends measuring Lp(a) in patients with early‐onset ACS, a family history of early ACS, or who are at intermediate risk (ie, 10–20% 10‐year risk) for cardiovascular events.1 This approach is supported by recent data suggesting that Lp(a) may reclassify intermediate risk individuals to high risk,31 especially when Lp(a) levels are very high (>95% percentile).5, 32 Our results suggest that in addition to these groups, Lp(a) screening should also be recommended for individuals with LDL‐C >3.5 mmol/L, as the presence of both high LDL‐C and high Lp(a) may accelerate progression of vascular disease and markedly increase the risk of cardiovascular events. This may have particular importance in young patients with LDL‐C >3.5 mmol/L who are frequently at low 10‐year predicted cardiovascular risk and would therefore not be otherwise eligible for statin therapy. Our results suggest that statin therapy among those with high Lp(a) and concomitant LDL‐C >3.5 mmol/L could lead to meaningful reductions in premature cardiovascular events. Such an approach is supported by a post‐hoc analysis of the Familial Atherosclerosis Treatment Study (FATS) randomized trial in which the reductions in coronary stenosis progression and clinical events from lipid‐lowering treatment were greatest among individuals with high Lp(a) (>90th percentile) who had the greatest LDL‐C reductions from treatment.33
Future trials targeting individuals with high Lp(a) using statins (to lower LDL‐C), niacin, proprotein convertase subtilisin kexin type 9 (PCSK9) inhibitors,34, 35 or specific Lp(a)‐lowering antisense therapies36 are clearly warranted to confirm the benefits of lipid‐lowering in individuals with high Lp(a), and should pay special attention to the subgroup with concomitant elevations of LDL‐C, who may be at highest cardiovascular risk. It is noteworthy that PCSK9 inhibitors have shown promising results with respect to significantly lowering both Lp(a) and LDL‐C and may serve as the future preferred treatment strategy for patients with familial hypercholesterolemia with combined elevations of Lp(a) and LDL‐C.34, 35, 37 Anacetrapib, the only cholesteryl ester transfer protein inhibitor currently under investigation, has also been suggested to effectively reduce both LDL‐C and Lp(a) levels.38 In contrast, Lp(a)‐lowering agents such as tibolone,39 as well as recent antisense therapies could potentially be used as the primary approach to treat individuals with isolated high Lp(a) levels.36 A randomized trial with Lp(a)‐lowering antisense agents could also provide the final confirmatory test of causality for the role of Lp(a) in coronary artery disease and aortic valve disease.40
Our study has several strengths including a sizeable sample of young ACS patients typically encountered in clinical practice and the use of a case‐only design, which provides a robust approach for identifying relevant risk factor interactions. Several limitations deserve mention. First, although the case‐only study maximizes power and provides robust interaction estimates, this depends on a key assumption that the interacting factors observed in cases are independent in the general population. Although we could not directly confirm this assumption, several studies have shown that Lp(a) and LDL‐C levels do not associate in the general population.10, 11 Furthermore, our estimates for this interaction are consistent with the evidence from smaller case–control and prospective studies, suggesting that our results, based on the case‐only approach, are valid. Second, our study was cross‐sectional and may not fully consider the lifelong impact of elevations of both Lp(a) and LDL‐C on ACS risk. However, because Lp(a) levels are primarily genetically mediated, Lp(a) has been shown to remain constant over time.2 Third, use of the case‐only approach precludes reporting the direct association of Lp(a) or LDL‐C with MI; however, both of these lipid markers have been shown to be strongly associated with MI and cardiovascular events in large meta‐analyses and have recently been shown to be causal using Mendelian randomization. Finally, we did not correct LDL‐C levels for lipid‐lowering treatment, and this could lead to some misclassification in LDL‐C levels; however, <40% of PRAXY participants were on lipid‐lowering therapies prior to their index event and any misclassification would tend to bias our results to the null; therefore, we may underestimate the true magnitude of this interaction.
Conclusions
In young ACS patients, high Lp(a) is more prevalent than in the general population and is strongly associated with high LDL‐C levels. Our results demonstrate that Lp(a) confers greater risk for ACS when LDL‐C is elevated. In individuals with high Lp(a) and concomitant elevations in LDL‐C >3.5 mmol/L, LDL‐C lowering may be warranted to reduce the risk of premature ACS. Larger prospective studies and randomized trials in individuals with high Lp(a) are needed to confirm these findings.
Author Contributions
Afshar participated in the design of the study, statistical analysis, and draft of the manuscript. Pilote developed GENESIS‐PRAXY, obtained funding, and conducted data collection. Dufresne performed the statistical analysis and was involved in revision of the manuscript. Engert participated in the design of the study and revisions of the manuscript. Thanassoulis conceived the study, was involved in its design, coordination, statistical analysis, and helped to draft the manuscript. All authors have read and approved the final manuscript.
Sources of Funding
This work was supported by Canadian Institute of Health Research (CIHR) grant MOP‐119380 to Dr Thanassoulis. The GENESIS‐PRAXY study was funded by the CIHR and the Heart and Stroke Foundations of Québec, Nova Scotia, Alberta, Ontario, Yukon, and British Columbia, Canada. Dr Thanassoulis is supported by a FRQS Chercheur Boursier Clinicien Salary Award. Dr Pilote holds a James McGill Chair in Medicine. Preventive and Genomic Cardiology at the MUHC is supported by the MUHC Foundation and the Doggone Foundation.
Disclosures
Dr Thanassoulis has received speaker's bureau honoraria from Servier Canada and has participated in an advisory board for ISIS Pharmaceuticals. The other authors have no conflicts to disclose.
Acknowledgments
We would like to thank all of the GENESIS‐PRAXY investigators and patients.
(J Am Heart Assoc. 2016;5:e003012 doi: 10.1161/JAHA.115.003012)
References
- 1. Nordestgaard BG, Chapman MJ, Ray K, Boren J, Andreotti F, Watts GF, Ginsberg H, Amarenco P, Catapano A, Descamps OS, Fisher E, Kovanen PT, Kuivenhoven JA, Lesnik P, Masana L, Reiner Z, Taskinen MR, Tokgozoglu L, Tybjaerg‐Hansen A; European Atherosclerosis Society Consensus Panel . Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31:2844–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kamstrup PR, Tybjaerg‐Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301:2331–2339. [DOI] [PubMed] [Google Scholar]
- 3. Thanassoulis G, Campbell CY, Owens DS, Smith JG, Smith AV, Peloso GM, Kerr KF, Pechlivanis S, Budoff MJ, Harris TB, Malhotra R, O'Brien KD, Kamstrup PR, Nordestgaard BG, Tybjaerg‐Hansen A, Allison MA, Aspelund T, Criqui MH, Heckbert SR, Hwang SJ, Liu Y, Sjogren M, Van der Pals J, Kalsch H, Muhleisen TW, Nothen MM, Cupples LA, Caslake M, Di Angelantonio E, Danesh J, Rotter JL, Sigurdsson S, Wong Q, Erbel R, Kathiresan S, Melander O, Gudnason V, O'Donnell CJ, Post WS; CHARGE Extracoronary Calcium Working Group . Genetic associations with valvular calcification and aortic stenosis. N Engl J Med. 2013;368:503–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brown WV, Ballantyne CM, Jones PH, Marcovina S. Management of Lp(a). J Clin Lipidol. 2010;4:240–247. [DOI] [PubMed] [Google Scholar]
- 5. Davidson MH, Ballantyne CM, Jacobson TA, Bittner VA, Braun LT, Brown AS, Brown WV, Cromwell WC, Goldberg RB, McKenney JM, Remaley AT, Sniderman AD, Toth PP, Tsimikas S, Ziajka PE, Maki KC, Dicklin MR. Clinical utility of inflammatory markers and advanced lipoprotein testing: advice from an expert panel of lipid specialists. J Clin Lipidol. 2011;5:338–367. [DOI] [PubMed] [Google Scholar]
- 6. Khoury MJ, Flanders WD. Non‐traditional epidemiologic approaches in the analysis of gene‐environment interaction: case‐control studies with no controls. Am J Epidemiol. 1996;144:207–213. [DOI] [PubMed] [Google Scholar]
- 7. Yang Q, Khoury MJ, Flanders WD. Sample size requirements in case‐only designs to detect gene‐environment interaction. Am J Epidemiol. 1997;146:713–720. [DOI] [PubMed] [Google Scholar]
- 8. Boerwinkle E, Leffert CC, Lin J, Lackner C, Chiesa G, Hobbs HH. Apolipoprotein(a) gene accounts for greater than 90% of the variation in plasma lipoprotein(a) concentrations. J Clin Invest. 1992;90:52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Danesh J, Collins R, Peto R. Lipoprotein(a) and coronary heart disease: meta‐analysis of prospective studies. Circulation. 2000;102:1082–1085. [DOI] [PubMed] [Google Scholar]
- 10. Li KM, Wilcken DEL, Dudman NPB. Effect of serum lipoprotein(a) on estimation of low‐density lipoprotein cholesterol by the Friedewald formula. Clin Chem. 1994;40:571–573. [PubMed] [Google Scholar]
- 11. Saeedi R, Li M, Allard M, Frohlich J. Marked effects of extreme levels of lipoprotein(a) on estimation of low‐density lipoprotein cholesterol. Clin Biochem. 2014;47:1098–1099. [DOI] [PubMed] [Google Scholar]
- 12. Pilote L, Karp I. GENESIS‐PRAXY (GENdEr and Sex determInantS of cardiovascular disease: from bench to beyond‐Premature Acute Coronary SYndrome). Am Heart J. 2012;163:741–746. [DOI] [PubMed] [Google Scholar]
- 13. D'Agostino RB Sr, Vasan RS, Pencina MJ, Wolf PA, Cobain M, Massaro JM, Kannel WB. General cardiovascular risk profile for use in primary care: the Framingham Heart Study. Circulation. 2008;117:743–753. [DOI] [PubMed] [Google Scholar]
- 14. Piegorsch WW, Weinberg CR, Taylor JA. Non‐hierarchical logistic models and case‐only designs for assessing susceptibility in population‐based case‐control studies. Stat Med. 1994;13:153–162. [DOI] [PubMed] [Google Scholar]
- 15. Ottman R. Gene‐environment interaction: definitions and study designs. Prev Med. 1996;25:764–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pierce BL, Ahsan H. Case‐only genome‐wide interaction study of disease risk, prognosis and treatment. Genet Epidemiol. 2010;34:7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dahlen GH. Incidence of Lp(a) among populations In: Scanuu AM, ed. Lipoprotein(a). New York, NY: Academic Press; 1990:151–173. [Google Scholar]
- 18. R Core Team . R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2015. Available at: http://www.R-project.org/. [Google Scholar]
- 19. Hopkins PN, Hunt SC, Schreiner PJ, Eckfeldt JH, Borecki IB, Ellison CR, Williams RR, Siegmund KD. Lipoprotein(a) interactions with lipid and non‐lipid risk factors in patients with early onset coronary artery disease: results from the NHLBI Family Heart Study. Atherosclerosis. 1998;141:333–345. [DOI] [PubMed] [Google Scholar]
- 20. Kronenberg F, Kronenberg MF, Kiechl S, Trenkwalder E, Santer P, Oberhollenzer F, Egger G, Utermann G, Willeit J. Role of lipoprotein(a) and apolipoprotein(a) phenotype in atherogenesis: prospective results from the Bruneck study. Circulation. 1999;100:1154–1160. [DOI] [PubMed] [Google Scholar]
- 21. Von Eckardstein A, Schulte H, Cullen P, Assmann G. Lipoprotein(a) further increases the risk of coronary events in men with high global cardiovascular risk. J Am Coll Cardiol. 2001;37:434–439. [DOI] [PubMed] [Google Scholar]
- 22. Luc G, Bard JM, Arveiler D, Ferrieres J, Evans A, Amouyel P, Fruchart JC, Ducimetiere P; PRIME Study Group . Lipoprotein(a) as a predictor of coronary heart disease: the PRIME Study. Atherosclerosis. 2002;163:377–384. [DOI] [PubMed] [Google Scholar]
- 23. Rifai N, Ma J, Sacks FM, Ridker PM, Hernandez WJ, Stampfer MJ, Marcovina SM. Apolipoprotein(a) size and lipoprotein(a) concentration and future risk of angina pectoris with evidence of severe coronary atherosclerosis in men: the Physicians’ Health Study. Clin Chem. 2004;50:1364–1371. [DOI] [PubMed] [Google Scholar]
- 24. Suk Danik J, Rifai N, Buring JE, Ridker PM. Lipoprotein(a), measured with an assay independent of apolipoprotein(a) isoform size, and risk of future cardiovascular events among initially healthy women. JAMA. 2006;296:1363–1370. [DOI] [PubMed] [Google Scholar]
- 25. Konishi H, Miyauchi K, Kasai T, Tsuboi S, Ogita M, Naito R, Sai E, Fukushima Y, Katoh Y, Okai I, Tamura H, Okazaki S, Daida H. Impact of lipoprotein(A) as residual risk on long‐term outcomes in patients after percutaneous coronary intervention. Am J Cardiol. 2015;115:157–160. [DOI] [PubMed] [Google Scholar]
- 26. O'Donoghue ML, Morrow DA, Tsimikas S, Sloan S, Ren AF, Hoffman EB, Desai NR, Solomon SD, Domanski M, Arai K, Chiuve SE, Cannon CP, Sacks FM, Sabatine MS. Lipoprotein(a) for risk assessment in patients with established coronary artery disease. J Am Coll Cardiol. 2014;63:520–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Emerging Risk Factors Collaboration , Erqou S, Kaptoge S, Perry PL, Di Angelantonio E, Thompson A, White IR, Marcovina SM, Collins R, Thompson SG, Danesh J. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302:412–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. CARDIoGRAMplusC4D Consortium , Deloukas P, Kanoni S, Willenborg C, Farral M, Assimes TL, Thompson JR, Ingelsson E, Saleheen D, Erdmann J, Goldstein BA, Stirrups K, Konig IR, Cazier JB, Johansson A, Hall AS, Lee JY, Willer CJ, Chambers JC, Esko T, Folkersen L, Goel A, Grundberg E, Havulinna AS, Ho WK, Hopewell JC, Eriksson N, Kleber ME, Kristiansson K, Lundmark P, Lyytikainen LP, Rafelt S, Shungin D, Strawbridge RJ, Thorleifsson G, Tikkanen E, Van Zuydam N, Voight BF, Waite LL, Zhang W, Ziegler A, Absher D, Altshuler D, Balmforth AJ, Barroso I, Braund PS, Burgdorf C, Claudi‐Boehm S, Cox D, Dimitriou M, Do R; DIAGRAM Consortium; CARDIOGENICS Consortium , Doney AS, El Mokhtari N, Eriksson P, Fischer K, Fontanillas P, Franco‐Cereceda A, Gigante B, Groop L, Gustafsson S, Hager J, Hallmans G, Han BG, Hunt SE, Kang HM, Illig T, Kessler T, Knowles JW, Kolovou G, Kuusisto J, Langenberg C, Langford C, Leander K, Lokki ML, Lundmark A, McCarthy MI, Meisinger C, Melander O, Mihailov E, Maouche S, Morris AD, Muller‐Nurasyid M; MuTHER Consortium , Nikus K, Peden JF, Rayner NW, Rasheed A, Rosinger S, Rubin D, Rumpf MP, Schafer A, Sivananthan M, Song C, Stewart AF, Tan ST, Thorgeirsson G, Van der Schoot CE, Wagner PJ; Wellcome Trust Case Control Consortium , Wells GA, Wild PS, Yang TP, Amouyel P, Arveiler D, Basart H, Boehnke M, Boerwinkle E, Brambilla P, Cambien F, Cupples AL, de Faire U, Dehghan A, Diemert P, Epstein SE, Evans A, Ferrario MM, Ferrieres J, Gauguier D, Go AS, Goodall AH, Gudnason V, Hazen SL, Holm H, Iribarren C, Jang Y, Kahonen M, Kee F, Kim HS, Klopp N, Koenig W, Kratzer W, Kuulasmaa K, Laakso M, Laaksonen R, Lee JY, Lind L, Ouwehand WH, Parish S, Park JE, Pedersen NL, Peteres A, Quertermous T, Rader DJ, Salomaa V, Schadt E, Shah SH, Sinisalo J, Stark K, Stefansson K, Tregouet DA, Virtamo J, Wallentin L, Wareham N, Zimmermann ME, Nieminen MS, Hangstenberg C, Sandhu MS, Pastinen T, Syvanen AC, Hovingh GK, Dedoussis G, Franks PW, Lehtimaki T, Metspalu A, Zalloua PA, Siegbahn A, Schreiber S, Ripatti S, Blankenberg SS, Perola M, Clarke R, Boehm BO, O'Donnell C, Reilly MP, Marz W, Collins R, Kathiresan S, Hamsten A, Kooner JS, Thorsteinsdottir U, Danesh J, Palmer CN, Roberts R, Watkins H, Schunkert H, Samani NJ.Large‐scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45:25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kamstrup PR, Tybjaerg‐Hansen A, Nordestgaard BG. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J Am Coll Cardiol. 2014;63:470–477. [DOI] [PubMed] [Google Scholar]
- 30. Arsenault BJ, Boekholdt SM, Dube MP, Rheaume E, Wareham NJ, Khaw KT, Sandhu MS, Tardif JC. Lipoprotein(a) levels, genotype, and incident aortic valve stenosis: a prospective Mendelian randomization study and replication in a case‐control cohort. Circ Cardiovasc Genet. 2014;7:304–310. [DOI] [PubMed] [Google Scholar]
- 31. Willeit P, Kiechl S, Kronenberg F, Witztum JL, Santer P, Mayr M, Xu Q, Mayr A, Willeit J, Tsimikas S. Discrimination and net reclassification of cardiovascular risk with lipoprotein(a): prospective 15‐year outcomes in the Bruneck Study. J Am Coll Cardiol. 2014;64:851–860. [DOI] [PubMed] [Google Scholar]
- 32. Kamstrup PR, Tybjaerg‐Hansen A, Nordestgaard BG. Extreme lipoprotein(a) levels and improved cardiovascular risk prediction. J Am Coll Cardiol. 2013;61:1146–1156. [DOI] [PubMed] [Google Scholar]
- 33. Meher VM, Brown BG, Marcovina SM, Hillger LA, Zhao XQ, Albers JJ. Effects of lowering elevated LDL cholesterol on the cardiovascular risk of lipoprotein(a). JAMA. 1995;274:1771–1774. [PubMed] [Google Scholar]
- 34. Sabatine MS, Giugliano RP, Wiviott SD, Raal FJ, Blom DJ, Robinson J, Ballantyne CM, Somaratne R, Legg J, Wasserman SM, Scott R, Koren MJ, Stein EA; Open‐Label Study of Long‐Term Evaluation against LDL‐Cholesterol (OSLER) Investigators . Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1500–1509. [DOI] [PubMed] [Google Scholar]
- 35. Robinson JG, Farnier M, Krempf M, Bergeron J, Luc G, Averna M, Stroes ES, Langslet G, Raal FJ, El Shahawy M, Koren MJ, Lepor NE, Lorenzato C, Pordy R, Chaudhari U, Kastelein JJ; ODYSSEY LONG TERM Investigators . Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1489–1499. [DOI] [PubMed] [Google Scholar]
- 36. Tsimikas S, Viney NJ, Hughes SG, Singleton W, Graham MJ, Baker BF, Burkey JL, Yang Q, Marcovina SM, Geary RS, Crooke RM, Witztum JL. Antisense therapy targeting apolipoprotein(a): a randomised, double‐blind, placebo‐controlled phase 1 study. Lancet. 2015;386:1472–1483. [DOI] [PubMed] [Google Scholar]
- 37. Dragan S, Serban MC, Banach M. Proprotein convertase subtilisin/kexin 9 inhibitors: an emerging lipid‐lowering therapy? J Cardiovasc Pharmacol Ther. 2015;20:157–168. [DOI] [PubMed] [Google Scholar]
- 38. Cannon CP, Shah S, Dansky HM, Davidson M, Brinton EA, Gotto AM, Stepanavage M, Liu SX, Gibbons P, Ashraf TB, Zafarino J, Mitchel Y, Barter P; Determining the Efficacy and Tolerability Investigators . Safety of anacetrapib in patients with or at high risk for coronary heart disease. N Engl J Med. 2010;363:2406–2415. [DOI] [PubMed] [Google Scholar]
- 39. Kotani K, Sahebkar A, Serban C, Andrica F, Toth PP, Jones SR, Kostner K, Blaha MJ, Martin S, Rysz J, Glasser S, Ray KK, Watts GF, Mikhailidis DP, Banach M; Lipid and Blood Pressure Meta‐analysis Collaboration (LBPMC) Group . Tibolone decreases lipoprotein(a) levels in postmenopausal women: a systematic review and meta‐analysis of 12 studies with 1009 patients. Atherosclerosis. 2015;242:87–96. [DOI] [PubMed] [Google Scholar]
- 40. Hung MY, Tsimikas S. What is the ultimate test that lowering lipoprotein(a) is beneficial for cardiovascular disease and aortic stenosis? Curr Opin Lipidol. 2014;25:423–430. [DOI] [PubMed] [Google Scholar]