

Abstract
Background
Microvascular leakage of plasma proteins is a hallmark of inflammation that leads to tissue dysfunction. There are no current therapeutic strategies to reduce microvascular permeability. The purpose of this study was to identify the role of Rnd3, an atypical Rho family GTPase, in the control of endothelial barrier integrity. The potential therapeutic benefit of Rnd3 protein delivery to ameliorate microvascular leakage was also investigated.
Methods and Results
Using immunofluorescence microscopy, Rnd3 was observed primarily in cytoplasmic areas around the nuclei of human umbilical vein endothelial cells. Permeability to fluorescein isothiocyanate–albumin and transendothelial electrical resistance of human umbilical vein endothelial cell monolayers served as indices of barrier function, and RhoA, Rac1, and Cdc42 activities were determined using G‐LISA assays. Overexpression of Rnd3 significantly reduced the magnitude of thrombin‐induced barrier dysfunction, and abolished thrombin‐induced Rac1 inactivation. Depleting Rnd3 expression with siRNA significantly extended the time course of thrombin‐induced barrier dysfunction and Rac1 inactivation. Time‐lapse microscopy of human umbilical vein endothelial cells expressing GFP‐actin showed that co‐expression of mCherry‐Rnd3 attenuated thrombin‐induced reductions in local lamellipodia that accompany endothelial barrier dysfunction. Lastly, a novel Rnd3 protein delivery method reduced microvascular leakage in a rat model of hemorrhagic shock and resuscitation, assessed by both intravital microscopic observation of extravasation of fluorescein isothiocyanate–albumin from the mesenteric microcirculation, and direct determination of solute permeability in intact isolated venules.
Conclusions
The data suggest that Rnd3 can shift the balance of RhoA and Rac1 signaling in endothelial cells. In addition, our findings suggest the therapeutic, anti‐inflammatory potential of delivering Rnd3 to promote endothelial barrier recovery during inflammatory challenge.
Keywords: actin cytoskeleton, inflammation, lamellipodia, microcirculation, microvascular permeability
Subject Categories: Vascular Disease, Cell Signalling/Signal Transduction
Introduction
Elevated microvascular permeability is a significant problem that accompanies uncontrolled inflammation, contributing to tissue injury and dysfunction. Much work over the past several decades has improved our understanding of the endothelium, the main permselective barrier at leakage sites, in inflammation. However, a successful strategy to ameliorate microvascular hyperpermeability in a clinical setting remains elusive.1
Previous work has highlighted the active role of endothelial cells in regulating microvascular permeability with attention paid to their subcellular structures, including adherens junctions, focal adhesions, and the cytoskeleton.2 Activation of endothelial cells by inflammatory cells or soluble inflammatory mediators triggers several signaling cascades that lead to cytoskeletal rearrangement and disruption of intercellular adherens junctions, enhancing paracellular flux of fluids and solutes.3, 4, 5, 6 Several experiments have shown that the hyperpermeability response to inflammatory stimuli is often transient,3, 6, 7, 8 although the precise mechanisms involved in its termination are not well defined.
We previously reported that the small GTPase RhoA and its downstream effector Rho‐associated kinase (ROCK) mediate neutrophil‐induced hyperpermeability.5, 6 Others have also reported that RhoA/ROCK can increase hydraulic conductivity in single‐perfused microvessels.9, 10 ROCK can phosphorylate the myosin light chain phosphatase targeting subunit (MYPT‐1), leading to inactivation of myosin light chain phosphatase, and accumulation of myosin light chain phosphorylated on Thr‐18 and Ser‐19 by myosin light chain kinase.11, 12 Several reports have suggested that inflammatory mediators promote excess phosphorylated myosin light chain, which may interact with actin to open intercellular junctions, either through contractile or retractile mechanisms.3, 13, 14, 15, 16, 17, 18 Despite these advances, a caveat to using inhibition of RhoA/ROCK to reduce microvascular hyperpermeability is the fact that this pathway is also important for maintaining endothelial barrier function under noninflammatory conditions,17, 19, 20 as RhoA activation near cell borders promotes endothelial barrier integrity.21 Thus, inhibition of RhoA may lead to different outcomes depending on specific subcellular sites of action. Additionally, other Rho family small GTPases, particularly Rac1 and Cdc42, have been shown to be important for maintaining microvascular barrier function and may work by either stabilizing cortical actin or promoting local membrane protrusions to promote junctional stability.17, 22, 23, 24 In addition, the expression levels and activities of various Rho family GTPases are also known to influence one other.25 Thus, altering the balance of activities among different Rho family GTPases may also represent a way to improve microvascular barrier function.
One group of Rho family GTPases that has not received much attention is the Rnd subfamily. Unlike most GTPases, which cycle between GTP‐ and GDP‐bound states and thus act like molecular switches in signaling pathways, Rnd proteins are deficient in GTPase activity, and are probably regulated in large part at the transcriptional level.26 Rnd3 is the most commonly expressed member of this subfamily and is ubiquitously expressed.27, 28, 29 In addition to transcriptional regulation, Rnd3 is also phosphorylated by ROCK1 and PKCα, promoting its stabilization and translocation between the membrane and cytosolic fractions.30, 31 Rnd3 overexpression can inhibit the RhoA‐ROCK signaling pathway by suppressing RhoA activity through a p190RhoGAP‐dependent pathway, as well as by binding to and inhibiting downstream ROCK I signaling.32, 33 This Rnd3‐mediated inhibition of RhoA has also been shown to enhance tight junction barrier function in mammary epithelial cells.34, 35 In endothelial cells, Rnd3 overexpression was reported to initially reduce actin stress fibers, but after 24 hours result in elevated actin stress fiber formation and changes in junctional structure due to upregulation of RhoB.36 However, it has not been previously reported whether modulation of Rnd3 expression causes changes in endothelial barrier function.
In the current study, we investigated the potential role of Rnd3 in the control of endothelial permeability. We first confirmed the expression of Rnd3 in microvessels and vascular endothelial cells. Next, using thrombin‐induced endothelial barrier dysfunction, which we previously reported involves activation of RhoA and inactivation of Rac1, we then tested the hypothesis that Rnd3 can promote resolution of thrombin‐induced endothelial barrier dysfunction through its influence on Rho family GTPase activities and cytoskeletal mechanisms. For these studies we used Rnd3 overexpression and depletion in cultured endothelial cell monolayers. We then tested the ability of delivering Rnd3 protein into the microcirculation as a strategy to reduce microvascular hyperpermeability caused by injury, using an established rat hemorrhagic shock and resuscitation (HSR) model.
Methods
Cell Culture
Human umbilical vein endothelial cells (HUVEC) were obtained from Lonza (Basel, Switzerland) and grown in Clonetics endothelial growth medium‐2 (EGM‐2, Lonza). Prior to experiments, the medium was changed to endothelial basal medium as indicated.
Generation of Rnd3 Fusion Proteins
Open Biosystems clone #MHS1010‐58455 (RND3 cDNA) was purified from DH10B Escherichia coli and selection was confirmed by HindIII restriction digest and agarose gel electrophoresis. The gene was amplified using Phusion high‐fidelity DNA polymerase (New England Biolabs, Ipswich, MA) for subcloning into pCMV‐MAT‐FLAG‐1 (Sigma, St. Louis, MO) using the oligonucleotides: 5′‐AAGGCAGATATCATGAAGGAGAGAAGAGCCAGC‐3′ (RND3 EcoRV forward primer) and 5′‐AATGGGTACCTCACATCACAGTGCAGC‐3′ (RND3 TGA KpnI reverse primer). The vector and amplicon were EcoRV/KpnI digested, purified, and ligated overnight with T4 DNA ligase. The ligation mix was used to transform competent Escherichia coli DH5a, and clones were selected on LB plates with 100 μg/mL ampicillin. This yielded human MAT‐FLAG‐RND3 (in frame), controlled by a CMV promoter. The Rnd3 cDNA was also subcloned into the N‐terminal mCherry destination vector using the Gateway system (Invitrogen, Grand Island, NY). All constructs were verified by sequencing, and the plasmids were purified for transfection with the EndoFree MaxiPrep kit (Qiagen, Valencia, CA).
Plasmid Transfection
Cells were transfected with plasmids encoding MAT‐FLAG‐Rnd3, mCherry‐Rnd3, GFP‐Actin, GFP, or with no vector (mock transfection) using the Nucleofector™ II System (Lonza) according to the manufacturer's instructions. Briefly, HUVEC grown to 80% to 90% confluence in EGM‐2 were trypsinized, and washed with PBS. The number of cells was counted, the suspension was centrifuged at 100g for 10 minutes, and the pellet was resuspended in HUVEC Nucleofector solution (Lonza) at 5×106 cells/mL. Plasmid DNA (2 μg) was added to 100 μL of the cell suspension, and the mixture was transferred into a cuvette for nucleofection. Immediately after nucleofection, 500 μL of prewarmed EGM‐2MV was added to the cuvette, and after a 15‐minute incubation at 37°C, the cells were seeded into either 35‐mm culture dishes, electrode arrays, or Transwell plates for study. Medium was changed 4 to 6 hours posttransfection. Cells were used for study at 1 to 2 days posttransfection. Expression of fluorescent proteins was confirmed using a microscope with appropriate epifluorescent illumination. Expression of MAT‐FLAG‐Rnd3 was confirmed by Western blotting and immunofluorescence labeling.
Rnd3 and RhoA siRNA
Specific siRNAs for inhibiting Rnd3 (siGENOME SMARTpool M‐007794‐02) or RhoA (M‐003860‐03) expression were obtained from Dharmacon (Chicago, IL). Cells were transfected siRNA (15–150 pmol) with the Nucleofector II system as described above. Nontargeting siRNA (siCONTROL) served as control. Cells were used 2 days posttransfection. Pools of siRNA were chosen over individual sequences in order to maximize loss of target function while minimizing off‐target effects.37, 38
Western Blotting
HUVEC were washed with ice‐cold PBS and protein lysates were obtained by lysis in ice‐cold RIPA buffer (Upstate) containing 1X HALT protease inhibitor cocktail (Pierce, Rockford, IL). Protein concentrations were determined with the Bradford assay, and equal amounts of protein were loaded for SDS‐PAGE. Proteins were transferred to nitrocellulose for Western blotting with the following primary antibodies overnight at 4°C: mouse anti‐RhoE/Rnd3, clone 4 (Merck Millipore 05‐723, 1:1000 dilution, Billerica, MA), rabbit anti‐Rho (Merck Millipore 06‐770, 1:1000), or rabbit anti‐tubulin (Santa Cruz Biotechnology, H‐235/sc‐9104, Santa Cruz, CA). Anti‐rabbit and anti‐mouse secondary (HRP conjugated) antibodies were from Jackson Immunoresearch (West Grove, PA). To identify the FLAG probe, we used mouse anti‐FLAG MS‐peroxidase conjugate (Sigma A8592, 1:500) at room temperature for 1 hour. Bands were visualized by chemiluminescence.
Electrical Cell‐Substrate Impedance Sensing
Transendothelial electrical resistance (TER) was determined as an index of endothelial barrier function as previously described6, 20 with an electrical cell‐substrate impedance sensing 1600R system (Applied Biophysics, Troy, NY). Briefly, cells were cultured (105 cells/cm2) on 8W1E small gold electrode arrays (Applied Biophysics). Medium served as the electrolyte and barrier function was dynamically measured by determining the impedance of a cell‐covered electrode. A 1‐V, 4000‐Hz AC signal was supplied through a 1‐MΩ resistor to approximate a constant‐current source. The in‐phase voltage (proportional to resistance) and the out‐of‐phase voltage (proportional to capacitive resistance) were measured. Endothelial monolayers with baseline resistance values below 5000 Ω were excluded from the study.
Endothelial Monolayer Permeability Assay
HUVEC were grown 4 to 5 days on gelatin‐coated Costar Transwell membranes (VWR, Houston, TX) as previously described.4, 5 Endothelial basal medium containing fluorescein isothiocyanate–albumin was added to the upper (luminal) chamber. Thrombin (1 U/mL) or vehicle control (PBS) was also added to the upper chamber. Samples were collected from both the upper and lower (abluminal) chambers at various time points for fluorometry analysis. Albumin concentrations were determined using a standard curve, and the apparent permeability coefficient for albumin (P s albumin) was calculated as: P s albumin=[A]/t×1/A×V/[L]; where [A] is the abluminal albumin concentration, t is time in seconds, A is the area of the membrane in cm2, V is the volume of the abluminal chamber, and [L] is the luminal albumin concentration.
RhoA, Rac1, and Cdc42G‐LISA Activation Assays
GTP‐bound RhoA, Rac1, and Cdc42 were used as indicators of RhoA, Rac1, and Cdc42 activation, respectively. These were determined as previously described17 using the RhoA and Rac1G‐LISA™ activation assay luminescence kits and Cdc42G‐LISA™ activation assay colorimetric kit (Cytoskeleton, Inc., Denver, CO) according to the manufacturer's instructions. HUVEC were transfected with plasmid or siRNA (or appropriate controls) and cultured for 2 days on 60‐mm plates. Cells were serum starved overnight prior to the start of the experiment. Thrombin (1 U/mL) was added for 5, 30, 60, or 120 minutes. The cells were lysed and 1 set of lysate aliquots was snap frozen until the start of the assay. Another set of aliquots was used to determine protein concentrations. Protein concentrations were then equilibrated in the first set of aliquots and then for each sample, lysate (25 μL) and binding buffer (25 μL) were added to wells in a RhoA‐GTP, Rac1‐GTP, or Cdc42‐GTP affinity plate for 30 minutes. Bound RhoA‐GTP, Rac1‐GTP, or Cdc42‐GTP was detected with specific primary antibodies, and HRP‐conjugated secondary antibody. For the RhoA and Rac1 assays, chemiluminescence agent was added to the wells and luminescence was measured in a SpectraMax2 plate reader (Molecular Devices, Sunnyvale, CA). For the Cdc42 assay, a colorimetric reagent was added and the plates were read at 490 nm in a Tecan Infinite 200 plate reader (Tecan Group, Ltd., Männedorf, Switzerland). This technique was chosen to quantify RhoA‐, Rac1‐, and Cdc42‐GTP rather than the Western blot method because (1) this technique produces quantitative data whereas Western blots are only semiquantitative, and (2) the ELISA can be completed in ≈3 hours versus over 2 days for the more traditional pulldown assay and Western blot, enabling us to perform a more detailed study of the time course of RhoA, Rac1, and Cdc42 activation.
Time Lapse Imaging of Endothelial Cells
Experiments were performed as previously described17, 18, 39 using a Nikon Eclipse TE2000U inverted microscope system (Nikon Instruments, Melville, NY). Briefly, HUVEC expressing GFP‐actin, with or without co‐expression of mCherryRnd3, were perfused with warm (37°C) albumin–physiological salt solution (APSS; NaCl, 120 mmol/L; KCl, 4.7 mmol/L; CaCl2·2H2O, 2 mmol/L; MgSO4·7H2O, 1.2 mmol/L; NaH2PO4, 1.2 mmol/L; Na pyruvate, 2 mmol/L; glucose, 5 mmol/L; EDTA, 0.02 mmol/L; MOPS, 3 mmol/L; purified BSA 1 g/100 mL; pH 7.4). Fluorescent images (S492 and S572 excitation filters) were acquired every 15 seconds. Time‐lapse image sets were then analyzed by counting the number of local lamellipodia that appear over time, using ImageJ.
Cell‐Permeable (CP)‐Rnd3 Mixture Preparation
Purified Rnd3 protein was obtained from Origene (Rockville, MD) and mixed with TransITLT‐1 polyamine transfection reagent40, 41 in APSS. The mixture was applied directly to the mesentery (topically) or in the isolated microvessel bath at final concentrations of 2 μg protein/mL and 10 μL TransIT‐LT1/mL. As a control, a mixture with only 10 μL TransIT‐LT1/mL in APSS was used. We have previously used this method to successfully deliver purified proteins into isolated venules and lymphatic vessels, producing functional changes.6, 40, 41, 42, 43
Animal Procedures
All animal procedures were approved by the University of South Florida and LSUHSC‐NO Institutional Animal Care and Use committees and were performed in strict accordance with the U.S. Animal Welfare Act, U.S. Public Health Service Policy on the Humane Care and Use of Laboratory Animals, and the NIH Guide for the Care and Use of Laboratory Animals. All surgery was performed after the rats were anesthetized, and all efforts were made to minimize pain. Male Sprague Dawley rats were obtained from Charles River Laboratories (Wilmington, MA) and provided a standard diet (Purina Rat Chow, Ralston Purina, St. Louis, MO) and water ad libitum. The rats were housed in a 12 hours light/12 hours dark cycle for a 1‐week acclimation period prior to surgery.
Collection of Rat Mesentery for Immunofluorescence Labeling
The rats (200–250 g) were anesthetized with ketamine/xylazine (40/5 mg/kg, i.m.). After a rat was anesthetized, a midline laparotomy was performed. A section of small intestine (ileum) and mesentery was excised and placed into cold PBS. Each rat was then immediately euthanized by an overdose of sodium pentobarbital (120 mg/kg, i.v.) followed by cervical dislocation. The luminal contents of the intestine were removed by compression in order to avoid enzyme digestion of the tissue in subsequent steps. The mesentery was dissected away from the intestine, washed several times with cold PBS, and mounted with pins on a silicone surface for fixation and immunolabeling.
Immunofluorescence Microscopy
HUVEC grown on gelatin‐coated coverslips or mesentery specimens were fixed with freshly prepared 4% paraformaldehyde and permeabilized with 0.2% Triton X‐100. Specimens were then blocked in 2% donkey serum (Sigma). For labeling of recombinant MAT‐FLAG‐Rnd3, the fixed cells were incubated with a monoclonal anti‐FLAG M2 antibody (Sigma #F1804, 1:1000 dilution) for 2 hours at 37°C. Native Rnd3 in HUVEC was labeled overnight at 4°C with mouse anti‐RhoE/Rnd3, clone 4 (Merck‐Millipore #05‐723, 1:100). Native Rnd3 and VE‐cadherin in rat mesentery were labeled overnight at 4°C with rabbit anti‐Rnd3 (ProteinTech #12534‐1‐AP, 1:500) and goat anti‐VE‐cadherin (Santa Cruz #sc‐6458), respectively. Secondary antibodies (Jackson Immunoresearch), namely, Cy2‐conjugated donkey anti‐mouse IgG (#715‐225‐150, 1:100), Cy2‐conjugated donkey anti‐goat IgG (#705‐225‐003, 1:100), or Texas Red–conjugated donkey anti‐rabbit IgG (#711‐075‐152, 1:100), were applied for 1.5 hours at room temperature. The mesenteric samples were transferred to slides, and whole mounted with Vectashield (Vector Laboratories, Burlingame, CA). Coverslips with HUVEC were mounted in a similar fashion. Fluorescent microscopy was performed with either a Nikon Eclipse TE2000‐U inverted fluorescent microscope equipped with a Photometrics CoolSnap HQ digital camera and Metamorph 6.1 software for acquisition and analysis (Nikon Instruments, Lewisville, TX), or a Zeiss Axiovert 200M fluorescent microscope equipped with an Axiocam camera and Axiovision software (Carl Zeiss, Thornwood, NY).
Conscious Rat Model of HSR
A previously established model of hemorrhagic shock in conscious, unrestrained rats44, 45 was used to produce microvascular hyperpermeability in the gut. At least 48 hours prior to the day of the experiment, rats were anesthetized with ketamine and xylazine (90 and 9 mg/kg, respectively), and sterile catheters were implanted in the left common carotid artery and right external jugular vein. The catheters were flushed with 0.9% sterile sodium chloride USP (Baxter, Deerfield, IL), thermally sealed, and routed subcutaneously with a trocar to the dorsal nape of the neck. The catheters were then secured to the closed incision with a suture, coiled, and wrapped in masking tape to prevent damage. The carotid catheters were used to monitor blood pressure and for removal of blood. The jugular catheters were used for administration of fluorescent tracers for intravital microscopy. After surgery, the rats were allowed 2 to 3 days of recovery prior to experiments.
On the days of the experiments, the rats were placed into smaller cages positioned over a heating pad. The arterial catheter was attached to a pressure transducer connected to a PowerLab 4/35 Quad Bridge amplifier and computer running LabChart Pro software (ADInstruments, Colorado Springs, CO). Baseline arterial pressure was measured for at least 1 hour. Next, blood was withdrawn from conscious, unrestrained rats via the arterial catheter to produce a fixed‐pressure hemorrhage to ≈40 mm Hg mean arterial pressure. After this was maintained for 60 minutes, fluid resuscitation was provided using Lactate‐Ringer's solution USP, delivered through the intravenous catheter initially as a bolus equal to 40% of the total blood volume removed to produce the hemorrhage, plus an additional 2× the total blood volume removed as an intravenous infusion over 1 hour. Blood pressure measurements continued during all parts of the protocol except during the time when blood was removed from the arterial catheter. Time‐matched, sham control rats did not have blood removed during the 1‐hour hemorrhage period.
Assessment of Extravasation by Intravital Microscopy
Immediately following the hemorrhage/resuscitation protocol, rats were anesthetized and prepared for intravital microscopy as previously described.46 Briefly, the ventral fur was shaved and skin cleaned, and a midline laparotomy was performed in order to exteriorize the small intestine and associated mesentery over an optical stage. The mesentery was superfused with APSS at 37°C. Body temperature was monitored using a rectal thermometer connected to a thermostat‐controlled heating pad to keep the rat warm. Blood pressure was monitored via the arterial catheter. Fluorescein isothiocyanate–albumin, dissolved in Lactate Ringer's solution, which served as a tracer for extravasation of plasma proteins, was administered via the jugular vein catheter as a bolus (0.1 mg/kg) followed by continuous infusion (0.15 mg/kg per minute) to generate a steady‐state blood concentration. After a 30‐minute period to allow for stabilization, epifluorescent images of the mesenteric microcirculation were obtained at 10‐minute intervals, initially just prior to addition of the CP‐Rnd3 mixture and for an additional 1 hour. Extravasation was quantified by measuring the integrated optical intensity of the interstitial spaces adjacent to postcapillary venules as previously described.46, 47, 48 Arteriole diameters were also measured within the same images to test for potential changes in blood flow to the tissue.1
Isolated, Perfused Venule Permeability Model
Isolation of mesenteric venules was performed as previously described.17 Briefly, after the hemorrhagic shock/resuscitation protocol, rats were anesthetized, a midline laparotomy was performed, and the small intestine and associated mesentery were excised and pinned into a cold dissection chamber. The rats were euthanized by an overdose of ketamine/xylazine. A mesenteric venule (40–80‐μm diameter, 0.5–1.0‐mm length) was carefully dissected, excised, and mounted onto 2 resistance‐matched glass micropipettes, preserving the natural length of the vessel, in a custom isolated vessel chamber containing warm (37°C) APSS. The 2 pipettes were connected to inflow and outflow reservoirs containing APSS. A third, smaller micropipette, inserted into the inflow pipette, served as a source for Alexa Fluor‐488 albumin in APSS, which served as a tracer. Venular permeability was determined by measuring transvascular flux and transmural concentration of the Alexa Fluor‐488 tracer. P s albumin was calculated as P s albumin=(1/ΔIf)(dIf/dt)0(r/2), where ΔIf is the initial step increase in fluorescent intensity when switching to the APSS containing Alexa Fluor 488‐albumin, (dIf/dt)0 is the initial rate of the more gradual increase in fluorescence intensity due to flux of the tracer across the vessel wall into the surrounding bath, and r is the radius of the venule.49 For all experiments the net perfusion pressure was held at 10 cm H2O and the flow velocity was 7 mm/s.
Data Analysis
Data are presented as means±SE. For some experiments, data were normalized to baseline prior to calculating the means and SE. For time course experiments involving repeated measures, intergroup differences were tested with repeated‐measures ANOVA followed by Bonferroni t‐tests. For other studies, differences were evaluated using 1‐way ANOVA followed by Tukey's multiple comparisons test. Significance was accepted at P<0.05.
Results
Rnd3 Expression in Endothelial Cells
Rnd3 was detected by immunofluorescence microscopy in both the rat mesenteric microcirculation (Figure 1A) and within individual endothelial cells (Figure 1B and 1C). The subcellular localization of Rnd3 was typically concentrated within a perinuclear region resembling tubule‐like structures surrounding the nucleus (Figure 1B). Confocal images with additional immunolabeling of TGN46 showed that some of the Rnd3 is localized within the trans‐Golgi network (Figure 1C).
Figure 1.
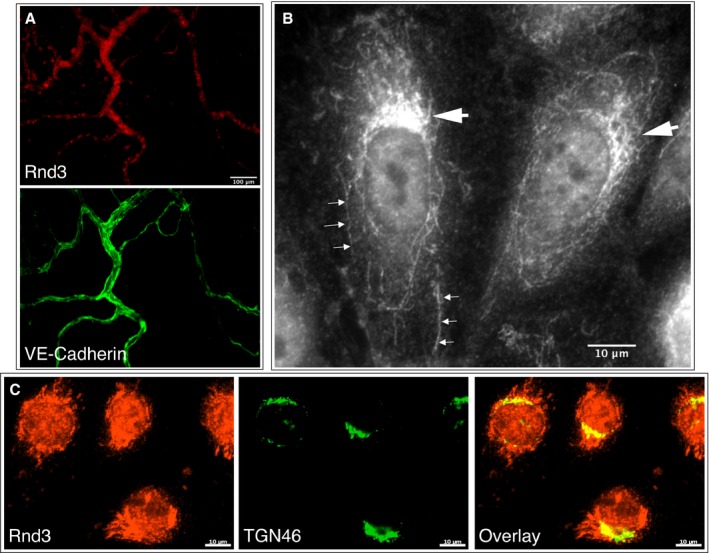
Rnd3 in the microcirculation and in cultured endothelial cells. A, Widefield images of a rat mesenteric window labeled with anti‐Rnd3 (top) and anti‐VE‐cadherin antibodies (bottom), showing a relatively high degree of Rnd3 labeling in microvessels. B, Widefield image of HUVEC labeled with anti‐Rnd3 antibodies, revealing strong patches of Rnd3 labeling adjacent to the nuclei (large arrows), and tubule‐like labeling reaching into the cytoplasm (small arrows). C, Single confocal slice images of Rnd3 labeling (left), with counter‐labeling for the trans‐Golgi network with TGN46 (middle), in HUVEC. The overlay image shows that a portion of the intense areas of Rnd3 labeling near the nuclei are within the trans‐Golgi network. Images are representative of at least 3 separate labeling experiments. These labeling patterns were not observed in control experiments with no primary antibody (not shown). HUVEC indicates human umbilical vein endothelial cells.
Inhibition of Rnd3 Expression Extends Thrombin‐Induced Endothelial Barrier Dysfunction
We first aimed to determine the extent to which reducing endogenous Rnd3 expression may affect endothelial barrier function. Transfection of HUVEC with Rnd3 siRNA reduced Rnd3 protein in a concentration‐dependent manner, without affecting RhoA or β‐tubulin, which served as loading controls (Figure 2A). In HUVEC monolayers transfected with Rnd3 siRNA or siCONTROL nontargeting RNA, there were no differences in mean baseline TER (Table 1). There was also no difference in the initial reduction in TER caused by the barrier‐disrupting agent thrombin (Figure 2B). However, there was a significant difference in the return to baseline between the 2 groups. Rnd3 siRNA treatment caused a prolonged decrease in TER that did not recover as quickly as with siCONTROL RNA‐treated cells (Figure 2B). This same trend was also observed when we determined changes in the apparent permeability to albumin (P s albumin) for these 2 groups. There was no difference in the initial increase in P s albumin caused by thrombin; however, P s albumin remained elevated much longer in the Rnd3 siRNA‐treated cells than in the siCONTROL RNA‐treated cells (Figure 2C).
Figure 2.
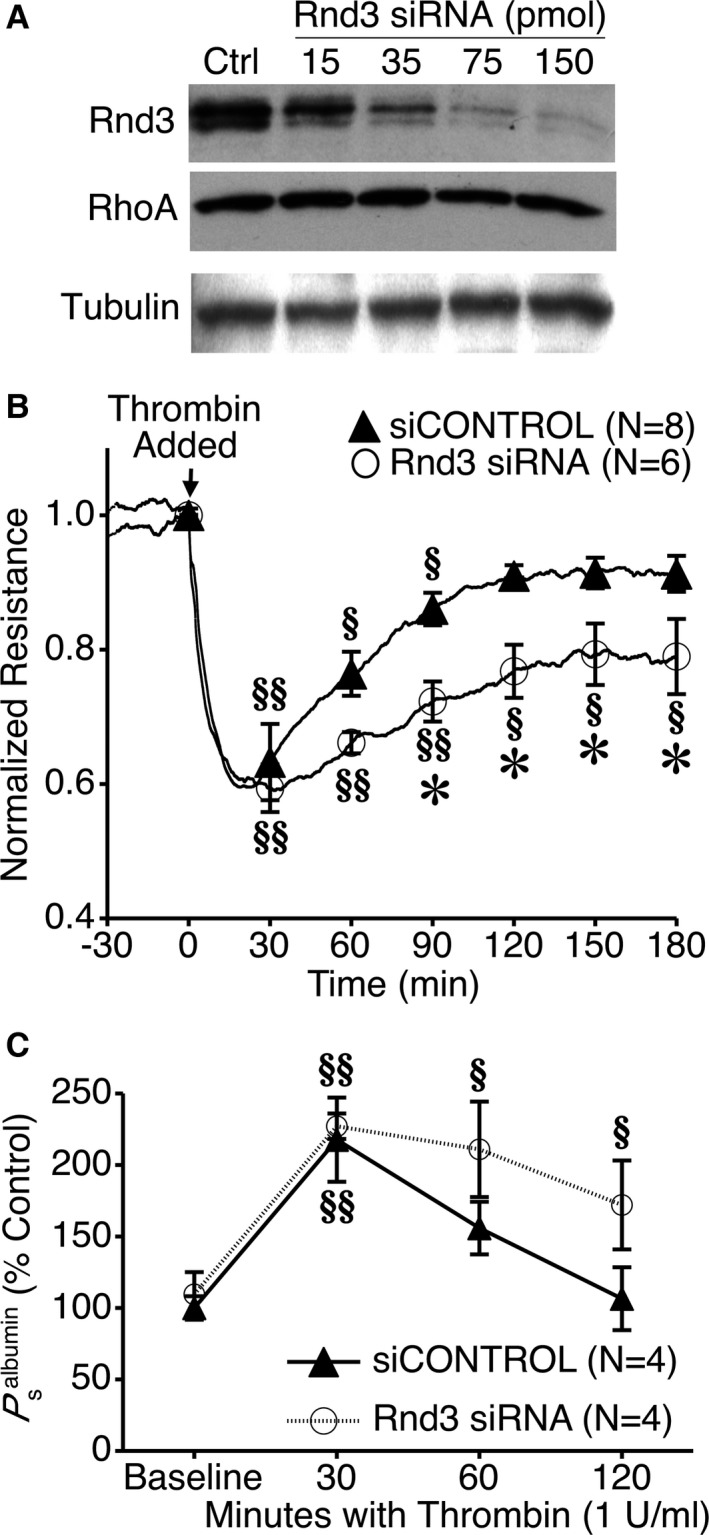
Impact of specific Rnd3 depletion on thrombin‐induced endothelial barrier dysfunction. A, Concentration‐dependent impact of Rnd3 siRNA on Rnd3 expression in HUVEC vs siCONTROL nontargeting RNA (Ctrl). RhoA and β‐tubulin served as loading controls. Blots are representative of 3 experiments. B, Specific Rnd3 depletion with siRNA prolongs thrombin‐induced decreases in HUVEC monolayer TER. C, Rnd3 depletion also prolongs thrombin‐induced increases in P s albumin. § P<0.05, §§ P<0.01 vs baseline. *P<0.05 vs siCONTROL, same time point. HUVEC indicates human umbilical vein endothelial cells; TER, transendothelial electrical resistance.
Table 1.
Baseline TER of Untransfected HUVEC Monolayers Versus Those Transfected With Rnd3 siRNA, siCONTROL RNA, MAT‐FLAG‐Rnd3, or no Vector/siRNA (Mock)
| Group | Mean TER±SE (Ω) | N |
|---|---|---|
| No transfection | 6890±118 | 6 |
| siCONTROL | 6853±369 | 8 |
| Rnd3 siRNA | 6976±384 | 6 |
| Mock | 6947±298 | 9 |
| MAT‐FLAG‐RND3 | 7348±290 | 11 |
HUVEC indicates human umbilical vein endothelial cells; TER, transendothelial electrical resistance.
Rnd3 Overexpression Inhibits Thrombin‐Induced Barrier Dysfunction of Cultured Endothelial Cells
We also performed gain‐of‐function experiments by overexpressing Rnd3 in HUVEC. Successful transfection of MAT‐FLAG‐Rnd3 was confirmed using Western blotting (Figure 3A). The degree of expression within individual cells was variable, with some cells expressing very high amounts of MAT‐FLAG‐Rnd3, and others expressing very little. Dense localization of this ectopic Rnd3 could often be found in perinuclear areas but also occurred near the cell periphery in cells with very high expression (Figure 3B). Transfection of HUVEC with MAT‐FLAG‐Rnd3 had little effect on baseline TER or P a, compared to a mock transfection with no vector (Tables 1 and 2). However, thrombin‐induced barrier dysfunction was significantly inhibited in HUVEC expressing MAT‐FLAG‐Rnd3, compared to mock transfection (Figure 3C). A similar trend was observed using the monolayer permeability model (Figure 3D). These data further support the concept that Rnd3 promotes enhanced barrier function when endothelial cells are challenged with an inflammatory stimulus.
Figure 3.
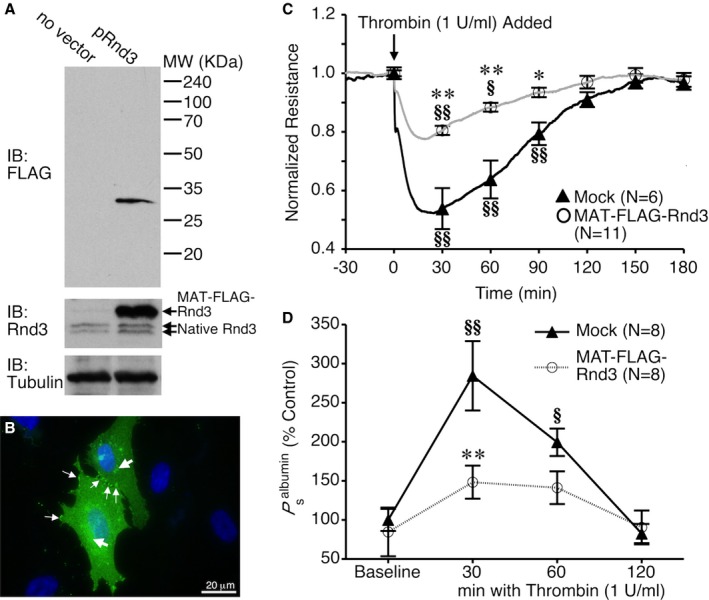
Rnd3 overexpression attenuates thrombin‐induced endothelial barrier dysfunction. A, Western blot showing overexpression of MAT‐FLAG‐Rnd3 in HUVEC. B, Immunofluorescence image showing very high expression of MAT‐FLAG‐Rnd3 (green) throughout the cytoplasm in 2 transfected cells. Nuclei are labeled blue. The large arrows show areas of high expression in the perinuclear area, similar to those seen with endogenous Rnd3. The small arrows show accumulation at the cell borders and cell–cell contacts. C, Rnd3 overexpression in HUVEC attenuates the thrombin‐induced drop in TER and increase in P s albumin (D). *P<0.05, **P<0.01 between groups at the same time point. § P<0.05, §§ P<0.01 vs baseline. HUVEC indicates human umbilical vein endothelial cells; IB, immunoblot; TER, transendothelial electrical resistance.
Table 2.
Baseline P s albumin of Untransfected HUVEC Monolayers Versus Those Transfected With Rnd3 siRNA, siCONTROL RNA, MAT‐FLAG‐Rnd3, or no Vector/siRNA (Mock)
| Group | Mean P s albumin±SE (×10−6 cm/s) | N |
|---|---|---|
| No transfection | 8.1±1.4 | 6 |
| siCONTROL | 6.7±0.6 | 4 |
| Rnd3 siRNA | 7.3±1.0 | 4 |
| Mock | 9.7±1.6 | 8 |
| MAT‐FLAG‐RND3 | 8.2±1.1 | 8 |
HUVEC indicates human umbilical vein endothelial cells.
Influence of Rnd3 on Thrombin‐Induced RhoA, Rac1, and Cdc42 Activation
To better understand the mechanism of the observed changes in thrombin‐induced barrier dysfunction with altered Rnd3 expression, we investigated how Rnd3 overexpression and depletion affect thrombin‐induced changes in RhoA, Rac1, and Cdc42 activities (Figure 4). To our surprise, overexpression with MAT‐FLAG‐Rnd3 had little impact on thrombin‐induced RhoA activation (Figure 4A). However, MAT‐FLAG‐Rnd3 expression did inhibit thrombin‐induced inactivation of Rac1 at 5 minutes (Figure 4B), and had no impact on Cdc42 activation (Figure 4C). Depletion of Rnd3 with siRNA, however, caused a slightly more sustained activation of RhoA (Figure 4D) and inactivation of Rac1 (Figure 4E) by thrombin, with little impact on Cdc42 (Figure 4F).
Figure 4.
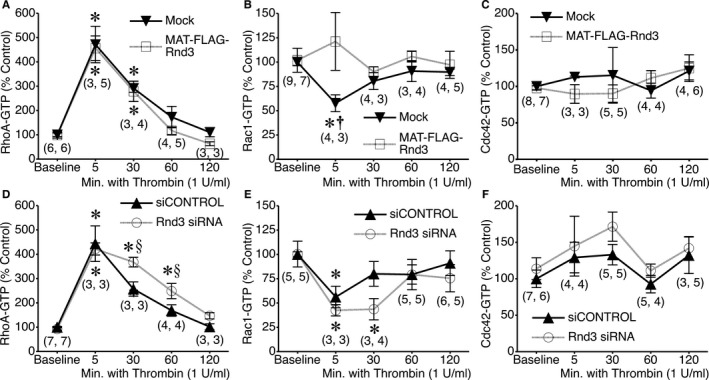
Impact of Rnd3 overexpression or depletion on RhoA, Rac1, and Cdc42 activation by thrombin. A, RhoA‐GTP activation in mock‐transfected or MAT‐FLAG‐Rnd3‐transfected HUVEC monolayers during baseline conditions and 5, 30, 60, and 120 minutes following the addition of 1 U/mL thrombin. B and C, Rac1 and Cdc42 activation, respectively, under the same conditions. D, RhoA activation in HUVEC treated with Rnd3 siRNA or siCONTROL nontargeting RNA under baseline conditions and after treatment with 1 U/mL thrombin for 5, 30, 60, or 120 minutes. E and F, Activation of Rac1 and Cdc42 under the same conditions. *P<0.05 vs baseline. † P<0.05 vs. MAT‐FLAG‐Rnd3, same time point. § P<0.05 between groups, same time point. The numbers in parentheses at each time point indicate the number of replicates for the mock and MAT‐FLAG‐Rnd3 groups, respectively, in (A through C) and for the siCONTROL and Rnd3 siRNA groups, respectively, in (D through F). HUVEC indicates human umbilical vein endothelial cells.
Rnd3 Overexpression Prevents Thrombin‐Induced Cell Retraction
We next investigated the impact of Rnd3 on thrombin‐induced changes in the actin cytoskeleton that contribute to the changes in barrier function. For these studies, we utilized HUVEC expressing GFP‐actin, in order to evaluate dynamic changes in cell shape and the number of actin stress fibers present. Cells expressing either GFP‐actin alone, or co‐expressing mCherry‐Rnd3 were used for study. We first evaluated protrusive activity at cell borders (Figure 5A). In HUVEC expressing only GFP‐actin, thrombin caused a rapid and significant decrease in the frequency of local lamellipodia protrusions, in a similar fashion as previously reported.17 In cells co‐expressing mCherry‐Rnd3, the reduction in the protrusion frequency of local lamellipodia caused by thrombin was less pronounced (Figure 5A). Thrombin also causes increased actin stress fiber formation in endothelial cells. In the current study, the number of actin fibers present in HUVEC expressing only GFP‐actin increased significantly after thrombin was added (Figure 5B). However, this was not the case in cells co‐expressing mCherry‐Rnd3, due in part because these cells already had a slightly elevated number of actin fibers during baseline. Lastly, the ventral surface area covered by HUVEC expressing only GFP‐actin significantly decreased after the addition of thrombin, but in cells co‐expressing mCherry‐Rnd3 this response was blunted (Figure 5C). Collectively, these results show that overexpression of Rnd3 significantly reduces the changes in the actin cytoskeleton elicited by thrombin that facilitate cellular retraction.
Figure 5.
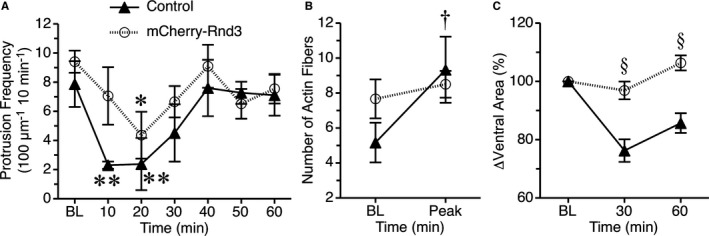
Rnd3 overexpression limits thrombin‐induced actin cytoskeletal changes and endothelial cell retraction. HUVEC expressing GFP‐actin alone (Control) or in combination with mCherry‐Rnd3 were used for these experiments. A, The protrusion frequency of local lamellipodia is shown for baseline (BL) and at 10‐minute intervals after the addition of 1 U/mL thrombin. *P<0.05 and **P<0.01 vs baseline, same treatment group. B, In the same groups of cells, actin fibers were counted at baseline and after addition of thrombin. The mean peak number of fibers after the addition of thrombin is shown. † P<0.05 vs baseline for the nontransfected control group. C, Changes in the ventral surface area covered by each cell were determined at baseline, and 30 and 60 minutes after the addition of thrombin. Data normalized to the mean at baseline for each group are shown. § P<0.05 mCherry‐Rnd3 vs control, same time point. N=6 cells in each group. HUVEC indicates human umbilical vein endothelial cells.
Transfection of Rnd3 Protein Ameliorates Microvascular Hyperpermeability
We next determined to what extent these findings in cells apply to the intact microcirculation. Using a method we previously developed and refined to directly deliver proteins into cells and microvessels,3, 6, 40, 42, 50, 51 we tested the potential therapeutic utility of Rnd3 to reduce microvascular hyperpermeability caused by HSR.52 The mesenteric venules isolated from rats following this injury had a significantly elevated mean P s albumin compared to time‐matched sham controls (Figure 6A). When the CP‐Rnd3 mixture was added to the isolated vessel perfusion bath of venules from rats that had undergone HSR, the mean P s albumin gradually decreased, and after 30 minutes of treatment was significantly lower than that observed for HSR without CP‐Rnd3 treatment (Figure 6A). With this promising result, we assessed the extent to which local CP‐Rnd3 treatment may improve microvascular leakage in vivo. Following HSR, microvascular leakage was evident, with fluorescein isothiocyanate–albumin steadily extravasated from the mesenteric microcirculation over time. However, when CP‐Rnd3 was topically applied onto the mesentery, this extravasation was dramatically decreased (Figure 6B). Combined, these data suggest that transfection of Rnd3 protein into microvessels is an effective short‐term method to reduce microvascular permeability following an ischemic injury in the gut.
Figure 6.
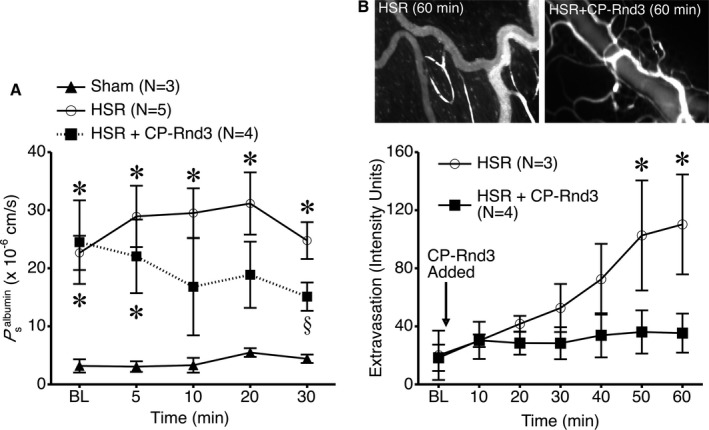
Rnd3 protein delivery ameliorates hemorrhagic shock and resuscitation (HSR)–induced microvascular hyperpermeability. A, The apparent permeability of isolated, perfused mesenteric venules to albumin (P s albumin) was determined. Venules isolated from rats that underwent a 60‐minute fixed pressure hemorrhage (≈40 mm Hg) followed by 60‐minute resuscitation with lactated Ringer's solution (HSR) had a significantly higher P s albumin than time‐matched sham rats that did not undergo hemorrhage (*P<0.05 vs sham, same time point). Addition of the CP‐Rnd3 mixture to the vessel perfusion bath (2 μg/mL of Rnd3 protein and 10 μL/mL of TransIT‐LT1) reduced P s albumin in isolated venules from rats that underwent HSR (§ P<0.05 vs HSR, same time point). B, Extravasation of intravenously infused FITC‐albumin from the mesenteric microcirculation was assessed by intravital microscopy after rats underwent HSR. The intensity in extravascular spaces adjacent to postcapillary venules increased over time; however, this was prevented when the CP‐Rnd3 mixture was applied to the suffusion bathing the mesentery (2 μg/mL of Rnd3 protein and 10 μL/mL of TransIT‐LT1). *P<0.05 HSR vs HSR+CP‐Rnd3, same time point). CP indicates cell permeable; FITC, fluorescein isothiocyanate.
Discussion
In this study, we identified that an atypical Rho family small GTPase, Rnd3, is expressed in endothelial cells. Using a combination of cell culture, ex vivo, and in vivo approaches, we have identified evidence supporting that Rnd3 has barrier‐protective properties under certain conditions, such as following acute inflammatory challenge. The involvement of Rnd3 in the control of endothelial barrier function adds to a growing list of recently described functions ascribed to Rnd family proteins, including Ca2+ homeostasis in the heart,53 regulation of Notch1 signaling,54 control of cell cycle progression,55, 56, 57 development of the central nervous system,58, 59, 60 control of cell motility and metastatic potential of certain cancers,29, 61, 62, 63, 64 and a potential role in cardioprotection against heart failure.65 In addition, we provide novel evidence that delivery of Rnd3 protein can serve as a potential therapeutic tool to ameliorate microvascular hyperpermeability caused by tissue injury.
Rnd3 was initially chosen for study because of its known role as an inhibitor of RhoA/ROCK signaling. RhoA and ROCK have an interesting dual role in the control of endothelial barrier function. Several reports have indicated that activation of RhoA/ROCK is important for inflammatory mediator–induced endothelial barrier dysfunction.5, 6, 22, 23, 66, 67, 68, 69, 70, 71 However, RhoA and ROCK activation are also involved with S1P‐induced endothelial barrier enhancement.17, 19 An elegant study utilizing a fluorescence resonance energy transfer biosensor expressed in endothelial cells to report RhoA activity provided evidence that differential localization of RhoA activation is key for its function to promote or disrupt endothelial barrier function. Activation of RhoA in central regions of endothelial cells, which is elicited by thrombin, is responsible for disruption of the endothelial barrier, while RhoA activation at the cell periphery promotes annealing of disrupted junctions.21 In the current study, we observed that endogenous Rnd3 was localized primarily in perinuclear regions of the cytoplasm (Figure 1). Based on this finding, we would expect that any inhibitory action by endogenous Rnd3 on RhoA/ROCK signaling would likely be localized to the central regions of the cells. This notion is supported by our findings that siRNA‐mediated knockdown of Rnd3 expression caused longer time courses of thrombin‐induced barrier dysfunction in HUVEC monolayers in association with extended periods of RhoA activation. It is worth noting that the general concept of signals localized in the perinuclear or centralized areas of the cytoplasm rapidly leading to elevated permeability of the endothelium has previously been shown in studies of endothelial nitric oxide synthase activation localized to the cytosol or Golgi apparatus,72, 73, 74 leading to the downstream S‐nitrosylation of junction‐associated proteins.75
Focal adhesion kinase–induced activation of p190RhoGAP has been described as part of the feedback mechanism that limits thrombin‐induced activation of RhoA in endothelial cells.76 Knowing this, and that Rnd3 was shown to activate p190RhoGAP in COS7 and 3T3 cells,32 we originally predicted that overexpression of Rnd3 in HUVEC would suppress thrombin‐induced RhoA activation. However, our results indicated this was not the case (Figure 4) and therefore we did not pursue this or the interaction between Rnd3 and p190RhoGAP further. As alternatives, we investigated Rac1 and Cdc42, 2 other key Rho family small GTPases that contribute to the control of endothelial barrier function.22, 24, 77, 78, 79 Thrombin caused a rapid, significant decrease in Rac1‐GTP in agreement with several previous reports.17, 78, 80, 81 However, thrombin did not activate Cdc42 in HUVEC, in contrast to one previous report that described a delayed activation of Cdc42 in immortalized human dermal microvascular endothelial cells.71 It is worth noting that the same immortalized cell line also does not display Rac1 inactivation in response to thrombin.82 When we modulated Rnd3 expression, thrombin‐induced inactivation of Rac1 was affected. Overexpression of Rnd3 abolished thrombin‐induced Rac1 inactivation, while siRNA knockdown of Rnd3 caused prolonged inhibition of Rac1 by thrombin (Figure 4). To our knowledge, this is the first demonstration that modulating expression of Rnd3 can have an impact on Rac1 activity.
Inhibition of Rac1 has previously been shown to increase permeability of venules.17 There is evidence that Rac1 can stabilize cortical actin to promote junctional stability in endothelial cells.22, 23 In addition, coordinated, cyclic activity of Rac1 and RhoA are known to control the protrusion and withdrawal of cell protrusions.83 Recently, we reported that treatment of endothelial cells with either thrombin or a selective inhibitor of Rac1 can decrease the protrusion frequency of small, local lamellipodia in conjunction with disruption of the endothelial barrier.17 These protrusions, also termed junctional‐associated intermittent lamellipodia by other investigators,84, 85 are also disrupted by histamine and appear to control rapid changes in permeability by altering the overlap of adjacent endothelial cells.18 In the current study, cells overexpressing Rnd3 had a less pronounced thrombin‐induced reduction in the frequency of local lamellipodia (Figure 5). This finding supports the notion that the ability of Rnd3 overexpression to decrease thrombin‐induced endothelial barrier dysfunction is in part attributable to an inhibition of those signals that disrupt normal protrusive activity that produces overlap of endothelial cells at intercellular junctions.
Previous reports have indicated that Rnd3 overexpression can profoundly affect actin stress fibers. Overexpression of Rnd3 causes the loss of actin stress fibers in cultured MDCK cells, astrocytes, and Swiss 3T3 fibroblasts.28, 29, 56, 86, 87 In HUVEC, however, Rnd3 expression has been reported to increase actin stress fiber formation, which was attributed to increased transcription and activity of RhoB.36 We have observed similar findings in nonconfluent, fixed HUVEC expressing MAT‐FLAG‐Rnd3 (data not shown). In confluent HUVEC co‐expressing mCherry‐Rnd3 with GFP‐actin, the actin stress fibers appeared more numerous than in cells expressing GFP‐actin alone, although according to strict statistical analysis these differences were not significant. However, the significant increase in actin stress fibers normally observed in response to thrombin was blunted in cells co‐expressing mCherry‐Rnd3 (Figure 5). This was somewhat surprising because in another experiment, Rnd3 overexpression did not prevent thrombin‐induced RhoA activation (Figure 4). This might indicate that the maximal number of actin stress fibers that could be generated was already reached in the cells overexpressing Rnd3, prior to thrombin treatment. In addition, the actin stress fibers observed in cells overexpressing Rnd3 might be qualitatively different from those that arise in response to thrombin activation, possibly more mature and with stronger focal adhesions, because Rnd3 overexpression also prevented thrombin‐induced retraction of cells. Focal adhesions found at the tips of ventral stress fibers help maintain endothelial cell spreading, and their disruption leads to retraction and has been shown to increase permeability of single‐perfused venules.88, 89, 90, 91 Additional work will be needed to elucidate further molecular details at the subcellular level.
To translate the findings from cells to pathophysiologically relevant conditions, we investigated the potential therapeutic use of CP‐Rnd3 to reduce microvascular permeability in a more clinically relevant model. HSR is known to produce systemic inflammation, including elevated microvascular leakage in the gut.2, 52 In our current study, HSR produced a profound increase in both the permeability of isolated venules and in the mesenteric microcirculation. Application of CP‐Rnd3 reversed this effect of HSR. This finding is significant because of the current lack of drugs or biological agents that can reduce microvascular permeability during injury and inflammation. It is important to note that in the current study we have limited our investigation to relatively acute increases in permeability. It is currently unclear how effective this or similar strategies will be when the endothelium is in a more advanced pathophysiologic state, such as with sepsis.
In summary, the current study presents a novel role for Rnd3 in endothelial barrier function. The mechanism involves modulation of the balance between Rac1 and RhoA activation, and alterations in the actin cytoskeleton that are important for determining cell shape and the integrity of intercellular junctions. Importantly, the data show that delivery of Rnd3 protein to intact microvessels or whole tissues can reduce microvascular leakage after injury. Additional studies on this and related potential therapies to reduce inflammation represent an important area of investigation that requires more attention.
Sources of Funding
Research reported in this paper was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under award numbers R01HL098215 (PI: J. Breslin), R01HL070752 (PI: S. Yuan), and R01HL096640 (PI: M. Wu). This project was also supported by an American Heart Association Predoctoral Fellowship (15PRE25710193) awarded to X. Zhang. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosures
None.
(J Am Heart Assoc. 2016;5:e003336 doi: 10.1161/JAHA.116.003336)
References
- 1. Durán WN, Sanchez FA, Breslin JW. Microcirculatory exchange function In: Tuma RF, Durán WN, Ley K, eds. Handbook of Physiology: Microcirculation. San Diego, CA: Academic Press—Elsevier; 2008:81–124. [Google Scholar]
- 2. Yuan SY. Signal transduction pathways in enhanced microvascular permeability. Microcirculation. 2000;7:395–403. [PubMed] [Google Scholar]
- 3. Yuan SY, Wu MH, Ustinova EE, Guo M, Tinsley JH, De Lanerolle P, Xu W. Myosin light chain phosphorylation in neutrophil‐stimulated coronary microvascular leakage. Circ Res. 2002;90:1214–1221. [DOI] [PubMed] [Google Scholar]
- 4. Tinsley JH, Wu MH, Ma W, Taulman AC, Yuan SY. Activated neutrophils induce hyperpermeability and phosphorylation of adherens junction proteins in coronary venular endothelial cells. J Biol Chem. 1999;274:24930–24934. [DOI] [PubMed] [Google Scholar]
- 5. Breslin JW, Yuan SY. Involvement of RhoA and Rho kinase in neutrophil‐stimulated endothelial hyperpermeability. Am J Physiol Heart Circ Physiol. 2004;286:H1057–H1062. [DOI] [PubMed] [Google Scholar]
- 6. Breslin JW, Sun H, Xu W, Rodarte C, Moy AB, Wu MH, Yuan SY. Involvement of ROCK‐mediated endothelial tension development in neutrophil‐stimulated microvascular leakage. Am J Physiol Heart Circ Physiol. 2006;290:H741–H750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhu L, Castranova V, He P. fMLP‐stimulated neutrophils increase endothelial [Ca2+]i and microvessel permeability in the absence of adhesion: role of reactive oxygen species. Am J Physiol Heart Circ Physiol. 2005;288:H1331–H1338. [DOI] [PubMed] [Google Scholar]
- 8. Jiang Y, Wen K, Zhou X, Schwegler‐Berry D, Castranova V, He P. Three‐dimensional localization and quantification of PAF‐induced gap formation in intact venular microvessels. Am J Physiol Heart Circ Physiol. 2008;295:H898–H906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Adamson RH, Curry FE, Adamson G, Liu B, Jiang Y, Aktories K, Barth H, Daigeler A, Golenhofen N, Ness W, Drenckhahn D. Rho and rho kinase modulation of barrier properties: cultured endothelial cells and intact microvessels of rats and mice. J Physiol. 2002;539:295–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zheng HZ, Zhao KS, Zhou BY, Huang QB. Role of Rho kinase and actin filament in the increased vascular permeability of skin venules in rats after scalding. Burns. 2003;29:820–827. [DOI] [PubMed] [Google Scholar]
- 11. Kawano Y, Fukata Y, Oshiro N, Amano M, Nakamura T, Ito M, Matsumura F, Inagaki M, Kaibuchi K. Phosphorylation of myosin‐binding subunit (MBS) of myosin phosphatase by Rho‐kinase in vivo. J Cell Biol. 1999;147:1023–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Amano M, Fukata Y, Kaibuchi K. Regulation and functions of Rho‐associated kinase. Exp Cell Res. 2000;261:44–51. [DOI] [PubMed] [Google Scholar]
- 13. Moy AB, Van Engelenhoven J, Bodmer J, Kamath J, Keese C, Giaever I, Shasby S, Shasby DM. Histamine and thrombin modulate endothelial focal adhesion through centripetal and centrifugal forces. J Clin Invest. 1996;97:1020–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yuan SY. Protein kinase signaling in the modulation of microvascular permeability. Vascul Pharmacol. 2003;39:213–223. [DOI] [PubMed] [Google Scholar]
- 15. Yuan Y, Huang Q, Wu HM. Myosin light chain phosphorylation: modulation of basal and agonist‐stimulated venular permeability. Am J Physiol. 1997;272:H1437–H1443. [DOI] [PubMed] [Google Scholar]
- 16. Garcia JG, Davis HW, Patterson CE. Regulation of endothelial cell gap formation and barrier dysfunction: role of myosin light chain phosphorylation. J Cell Physiol. 1995;163:510–522. [DOI] [PubMed] [Google Scholar]
- 17. Breslin JW, Zhang XE, Worthylake RA, Souza‐Smith FM. Involvement of local lamellipodia in endothelial barrier function. PLoS One. 2015;10:e0117970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Adderley SP, Lawrence C, Madonia E, Olubadewo JO, Breslin JW. Histamine activates p38 MAP kinase and alters local lamellipodia dynamics, reducing endothelial barrier integrity and eliciting central movement of actin fibers. Am J Physiol Cell Physiol. 2015;309:C51–C59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xu M, Waters CL, Hu C, Wysolmerski RB, Vincent PA, Minnear FL. Sphingosine 1‐phosphate rapidly increases endothelial barrier function independently of VE‐cadherin but requires cell spreading and Rho kinase. Am J Physiol Cell Physiol. 2007;293:C1309–C1318. [DOI] [PubMed] [Google Scholar]
- 20. Breslin JW. ROCK and cAMP promote lymphatic endothelial cell barrier integrity and modulate histamine and thrombin‐induced barrier dysfunction. Lymphat Res Biol. 2011;9:3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Szulcek R, Beckers CM, Hodzic J, de Wit J, Chen Z, Grob T, Musters RJ, Minshall RD, van Hinsbergh VW, van Nieuw Amerongen GP. Localized RhoA GTPase activity regulates dynamics of endothelial monolayer integrity. Cardiovasc Res. 2013;99:471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Waschke J, Burger S, Curry FR, Drenckhahn D, Adamson RH. Activation of Rac‐1 and Cdc42 stabilizes the microvascular endothelial barrier. Histochem Cell Biol. 2006;125:397–406. [DOI] [PubMed] [Google Scholar]
- 23. Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279–367. [DOI] [PubMed] [Google Scholar]
- 24. Waschke J, Baumgartner W, Adamson RH, Zeng M, Aktories K, Barth H, Wilde C, Curry FE, Drenckhahn D. Requirement of Rac activity for maintenance of capillary endothelial barrier properties. Am J Physiol Heart Circ Physiol. 2004;286:H394–H401. [DOI] [PubMed] [Google Scholar]
- 25. Boulter E, Garcia‐Mata R, Guilluy C, Dubash A, Rossi G, Brennwald PJ, Burridge K. Regulation of Rho GTPase crosstalk, degradation and activity by RhoGDI1. Nat Cell Biol. 2010;12:477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Riou P, Villalonga P, Ridley AJ. Rnd proteins: multifunctional regulators of the cytoskeleton and cell cycle progression. Bioessays. 2010;32:986–992. [DOI] [PubMed] [Google Scholar]
- 27. Foster R, Hu KQ, Lu Y, Nolan KM, Thissen J, Settleman J. Identification of a novel human Rho protein with unusual properties: GTPase deficiency and in vivo farnesylation. Mol Cell Biol. 1996;16:2689–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nobes CD, Lauritzen I, Mattei MG, Paris S, Hall A, Chardin P. A new member of the Rho family, Rnd1, promotes disassembly of actin filament structures and loss of cell adhesion. J Cell Biol. 1998;141:187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Guasch RM, Scambler P, Jones GE, Ridley AJ. RhoE regulates actin cytoskeleton organization and cell migration. Mol Cell Biol. 1998;18:4761–4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Riento K, Totty N, Villalonga P, Garg R, Guasch R, Ridley AJ. RhoE function is regulated by ROCK I‐mediated phosphorylation. EMBO J. 2005;24:1170–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Madigan JP, Bodemann BO, Brady DC, Dewar BJ, Keller PJ, Leitges M, Philips MR, Ridley AJ, Der CJ, Cox AD. Regulation of Rnd3 localization and function by protein kinase C alpha‐mediated phosphorylation. Biochem J. 2009;424:153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wennerberg K, Forget MA, Ellerbroek SM, Arthur WT, Burridge K, Settleman J, Der CJ, Hansen SH. Rnd proteins function as RhoA antagonists by activating p190 RhoGAP. Curr Biol. 2003;13:1106–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Riento K, Guasch RM, Garg R, Jin B, Ridley AJ. RhoE binds to ROCK I and inhibits downstream signaling. Mol Cell Biol. 2003;23:4219–4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rubenstein NM, Callahan JA, Lo DH, Firestone GL. Selective glucocorticoid control of Rho kinase isoforms regulate cell‐cell interactions. Biochem Biophys Res Commun. 2007;354:603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rubenstein NM, Chan JF, Kim JY, Hansen SH, Firestone GL. Rnd3/RhoE induces tight junction formation in mammary epithelial tumor cells. Exp Cell Res. 2005;305:74–82. [DOI] [PubMed] [Google Scholar]
- 36. Gottesbuhren U, Garg R, Riou P, McColl B, Brayson D, Ridley AJ. Rnd3 induces stress fibres in endothelial cells through RhoB. Biol Open. 2013;2:210–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Parsons BD, Schindler A, Evans DH, Foley E. A direct phenotypic comparison of siRNA pools and multiple individual duplexes in a functional assay. PLoS One. 2009;4:e8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hannus M, Beitzinger M, Engelmann JC, Weickert MT, Spang R, Hannus S, Meister G. siPools: highly complex but accurately defined siRNA pools eliminate off‐target effects. Nucleic Acids Res. 2014;42:8049–8061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Doggett TM, Breslin JW. Study of the actin cytoskeleton in live endothelial cells expressing GFP‐actin. J Vis Exp. 2011;57:3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tinsley JH, Hawker J, Yuan Y. Efficient protein transfection of cultured coronary venular endothelial cells. Am J Physiol. 1998;275:H1873–H1878. [DOI] [PubMed] [Google Scholar]
- 41. Yuan SY, Ustinova EE, Wu MH, Tinsley JH, Xu W, Korompai FL, Taulman AC. Protein kinase C activation contributes to microvascular barrier dysfunction in the heart at early stages of diabetes. Circ Res. 2000;87:412–417. [DOI] [PubMed] [Google Scholar]
- 42. Kurtz KH, Souza‐Smith FM, Moor AN, Breslin JW. Rho kinase enhances contractions of rat mesenteric collecting lymphatics. PLoS One. 2014;9:e94082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Souza‐Smith FM, Molina PE, Breslin JW. Reduced RhoA activity mediates acute alcohol intoxication‐induced inhibition of lymphatic myogenic constriction despite increased cytosolic [Ca(2+)]. Microcirculation. 2013;20:377–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Molina MF, Whitaker A, Molina PE, McDonough KH. Alcohol does not modulate the augmented acetylcholine‐induced vasodilatory response in hemorrhaged rodents. Shock. 2009;32:601–607. [DOI] [PubMed] [Google Scholar]
- 45. Molina PE, Zambell KL, Norenberg K, Eason J, Phelan H, Zhang P, Stouwe CV, Carnal JW, Porreta C. Consequences of alcohol‐induced early dysregulation of responses to trauma/hemorrhage. Alcohol. 2004;33:217–227. [DOI] [PubMed] [Google Scholar]
- 46. Doggett TM, Breslin JW. Acute alcohol intoxication‐induced microvascular leakage. Alcohol Clin Exp Res. 2014;38:2414–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Breslin JW, Gaudreault N, Watson KD, Reynoso R, Yuan SY, Wu MH. Vascular endothelial growth factor‐C stimulates the lymphatic pump by a VEGF receptor‐3‐dependent mechanism. Am J Physiol Heart Circ Physiol. 2007;293:H709–H718. [DOI] [PubMed] [Google Scholar]
- 48. Breslin JW, Wu MH, Guo M, Reynoso R, Yuan SY. Toll‐like receptor 4 contributes to microvascular inflammation and barrier dysfunction in thermal injury. Shock. 2008;29:349–355. [DOI] [PubMed] [Google Scholar]
- 49. Yuan Y, Chilian WM, Granger HJ, Zawieja DC. Permeability to albumin in isolated coronary venules. Am J Physiol. 1993;265:H543–H552. [DOI] [PubMed] [Google Scholar]
- 50. Tinsley JH, De Lanerolle P, Wilson E, Ma W, Yuan SY. Myosin light chain kinase transference induces myosin light chain activation and endothelial hyperpermeability. Am J Physiol Cell Physiol. 2000;279:C1285–C1289. [DOI] [PubMed] [Google Scholar]
- 51. Tinsley JH, Zawieja DC, Wu MH, Ustinova EE, Xu W, Yuan SY. Protein transfection of intact microvessels specifically modulates vasoreactivity and permeability. J Vasc Res. 2001;38:444–452. [DOI] [PubMed] [Google Scholar]
- 52. Childs EW, Tharakan B, Hunter FA, Tinsley JH, Cao X. Apoptotic signaling induces hyperpermeability following hemorrhagic shock. Am J Physiol Heart Circ Physiol. 2007;292:H3179–H3189. [DOI] [PubMed] [Google Scholar]
- 53. Yang X, Wang T, Lin X, Yue X, Wang Q, Wang G, Fu Q, Ai X, Chiang DY, Miyake CY, Wehrens XH, Chang J. Genetic deletion of Rnd3/RhoE results in mouse heart calcium leakage through upregulation of protein kinase A signaling. Circ Res. 2015;116:e1–e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhu Z, Todorova K, Lee KK, Wang J, Kwon E, Kehayov I, Kim HG, Kolev V, Dotto GP, Lee SW, Mandinova A. Small GTPase RhoE/Rnd3 is a critical regulator of Notch1 signaling. Cancer Res. 2014;74:2082–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bektic J, Pfeil K, Berger AP, Ramoner R, Pelzer A, Schafer G, Kofler K, Bartsch G, Klocker H. Small G‐protein RhoE is underexpressed in prostate cancer and induces cell cycle arrest and apoptosis. Prostate. 2005;64:332–340. [DOI] [PubMed] [Google Scholar]
- 56. Villalonga P, Guasch RM, Riento K, Ridley AJ. RhoE inhibits cell cycle progression and Ras‐induced transformation. Mol Cell Biol. 2004;24:7829–7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Poch E, Minambres R, Mocholi E, Ivorra C, Perez‐Arago A, Guerri C, Perez‐Roger I, Guasch RM. RhoE interferes with Rb inactivation and regulates the proliferation and survival of the U87 human glioblastoma cell line. Exp Cell Res. 2007;313:719–731. [DOI] [PubMed] [Google Scholar]
- 58. Mocholi E, Ballester‐Lurbe B, Arque G, Poch E, Peris B, Guerri C, Dierssen M, Guasch RM, Terrado J, Perez‐Roger I. RhoE deficiency produces postnatal lethality, profound motor deficits and neurodevelopmental delay in mice. PLoS One. 2011;6:e19236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pacary E, Heng J, Azzarelli R, Riou P, Castro D, Lebel‐Potter M, Parras C, Bell DM, Ridley AJ, Parsons M, Guillemot F. Proneural transcription factors regulate different steps of cortical neuron migration through Rnd‐mediated inhibition of RhoA signaling. Neuron. 2011;69:1069–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Peris B, Gonzalez‐Granero S, Ballester‐Lurbe B, Garcia‐Verdugo JM, Perez‐Roger I, Guerri C, Terrado J, Guasch RM. Neuronal polarization is impaired in mice lacking RhoE expression. J Neurochem. 2012;121:903–914. [DOI] [PubMed] [Google Scholar]
- 61. Zhao H, Yang J, Fan T, Li S, Ren X. RhoE functions as a tumor suppressor in esophageal squamous cell carcinoma and modulates the PTEN/PI3K/Akt signaling pathway. Tumour Biol. 2012;33:1363–1374. [DOI] [PubMed] [Google Scholar]
- 62. Pinner S, Sahai E. PDK1 regulates cancer cell motility by antagonising inhibition of ROCK1 by RhoE. Nat Cell Biol. 2008;10:127–137. [DOI] [PubMed] [Google Scholar]
- 63. Hidalgo‐Carcedo C, Hooper S, Chaudhry SI, Williamson P, Harrington K, Leitinger B, Sahai E. Collective cell migration requires suppression of actomyosin at cell‐cell contacts mediated by DDR1 and the cell polarity regulators Par3 and Par6. Nat Cell Biol. 2011;13:49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Belgiovine C, Frapolli R, Bonezzi K, Chiodi I, Favero F, Mello‐Grand M, Dei Tos AP, Giulotto E, Taraboletti G, D'Incalci M, Mondello C. Reduced expression of the ROCK inhibitor Rnd3 is associated with increased invasiveness and metastatic potential in mesenchymal tumor cells. PLoS One. 2010;5:e14154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yue X, Yang X, Lin X, Yang T, Yi X, Dai Y, Guo J, Li T, Shi J, Wei L, Fan GC, Chen C, Chang J. Rnd3 haploinsufficient mice are predisposed to hemodynamic stress and develop apoptotic cardiomyopathy with heart failure. Cell Death Dis. 2014;5:e1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sun H, Breslin JW, Zhu J, Yuan SY, Wu MH. Rho and ROCK signaling in VEGF‐induced microvascular endothelial hyperpermeability. Microcirculation. 2006;13:237–247. [DOI] [PubMed] [Google Scholar]
- 67. Carbajal JM, Gratrix ML, Yu CH, Schaeffer RC Jr. ROCK mediates thrombin's endothelial barrier dysfunction. Am J Physiol Cell Physiol. 2000;279:C195–C204. [DOI] [PubMed] [Google Scholar]
- 68. Wojciak‐Stothard B, Ridley AJ. Rho GTPases and the regulation of endothelial permeability. Vascul Pharmacol. 2002;39:187–199. [DOI] [PubMed] [Google Scholar]
- 69. Birukova AA, Birukov KG, Adyshev D, Usatyuk P, Natarajan V, Garcia JG, Verin AD. Involvement of microtubules and Rho pathway in TGF‐beta1‐induced lung vascular barrier dysfunction. J Cell Physiol. 2005;204:934–947. [DOI] [PubMed] [Google Scholar]
- 70. Birukova AA, Smurova K, Birukov KG, Kaibuchi K, Garcia JG, Verin AD. Role of Rho GTPases in thrombin‐induced lung vascular endothelial cells barrier dysfunction. Microvasc Res. 2004;67:64–77. [DOI] [PubMed] [Google Scholar]
- 71. Kouklis P, Konstantoulaki M, Vogel S, Broman M, Malik AB. Cdc42 regulates the restoration of endothelial barrier function. Circ Res. 2004;94:159–166. [DOI] [PubMed] [Google Scholar]
- 72. Sanchez FA, Rana R, Gonzalez FG, Iwahashi T, Duran RG, Fulton DJ, Beuve AV, Kim DD, Duran WN. Functional significance of cytosolic endothelial nitric‐oxide synthase (eNOS): regulation of hyperpermeability. J Biol Chem. 2011;286:30409–30414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sanchez FA, Rana R, Kim DD, Iwahashi T, Zheng R, Lal BK, Gordon DM, Meininger CJ, Duran WN. Internalization of eNOS and NO delivery to subcellular targets determine agonist‐induced hyperpermeability. Proc Natl Acad Sci USA. 2009;106:6849–6853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sanchez FA, Savalia NB, Duran RG, Lal BK, Boric MP, Duran WN. Functional significance of differential eNOS translocation. Am J Physiol Heart Circ Physiol. 2006;291:H1058–H1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Marin N, Zamorano P, Carrasco R, Mujica P, Gonzalez FG, Quezada C, Meininger CJ, Boric MP, Duran WN, Sanchez FA. S‐nitrosation of beta‐catenin and p120 catenin: a novel regulatory mechanism in endothelial hyperpermeability. Circ Res. 2012;111:553–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Holinstat M, Knezevic N, Broman M, Samarel AM, Malik AB, Mehta D. Suppression of RhoA activity by focal adhesion kinase‐induced activation of p190RhoGAP: role in regulation of endothelial permeability. J Biol Chem. 2006;281:2296–2305. [DOI] [PubMed] [Google Scholar]
- 77. Spindler V, Schlegel N, Waschke J. Role of GTPases in control of microvascular permeability. Cardiovasc Res. 2010;87:243–253. [DOI] [PubMed] [Google Scholar]
- 78. Baumer Y, Drenckhahn D, Waschke J. cAMP induced Rac 1‐mediated cytoskeletal reorganization in microvascular endothelium. Histochem Cell Biol. 2008;129:765–778. [DOI] [PubMed] [Google Scholar]
- 79. Breslin JW, Kurtz KM. Lymphatic endothelial cells adapt their barrier function in response to changes in shear stress. Lymphat Res Biol. 2009;7:229–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Birukova AA, Alekseeva E, Mikaelyan A, Birukov KG. HGF attenuates thrombin‐induced endothelial permeability by Tiam1‐mediated activation of the Rac pathway and by Tiam1/Rac‐dependent inhibition of the Rho pathway. FASEB J. 2007;21:2776–2786. [DOI] [PubMed] [Google Scholar]
- 81. Vouret‐Craviari V, Bourcier C, Boulter E, van Obberghen‐Schilling E. Distinct signals via Rho GTPases and Src drive shape changes by thrombin and sphingosine‐1‐phosphate in endothelial cells. J Cell Sci. 2002;115:2475–2484. [DOI] [PubMed] [Google Scholar]
- 82. Qiao J, Huang F, Lum H. PKA inhibits RhoA activation: a protection mechanism against endothelial barrier dysfunction. Am J Physiol Lung Cell Mol Physiol. 2003;284:L972–L980. [DOI] [PubMed] [Google Scholar]
- 83. Machacek M, Hodgson L, Welch C, Elliott H, Pertz O, Nalbant P, Abell A, Johnson GL, Hahn KM, Danuser G. Coordination of Rho GTPase activities during cell protrusion. Nature. 2009;461:99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Abu Taha A, Taha M, Seebach J, Schnittler HJ. ARP2/3‐mediated junction‐associated lamellipodia control Ve‐cadherin‐based cell junction dynamics and maintain monolayer integrity. Mol Biol Cell. 2014;25:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Schnittler H, Taha M, Schnittler MO, Taha AA, Lindemann N, Seebach J. Actin filament dynamics and endothelial cell junctions: the Ying and Yang between stabilization and motion. Cell Tissue Res. 2014;355:529–543. [DOI] [PubMed] [Google Scholar]
- 86. Guasch RM, Blanco AM, Perez‐Arago A, Minambres R, Talens‐Visconti R, Peris B, Guerri C. RhoE participates in the stimulation of the inflammatory response induced by ethanol in astrocytes. Exp Cell Res. 2007;313:3779–3788. [DOI] [PubMed] [Google Scholar]
- 87. Riento K, Ridley AJ. Inhibition of ROCK by RhoE. Methods Enzymol. 2006;406:533–541. [DOI] [PubMed] [Google Scholar]
- 88. Kajimura M, Curry FE. Endothelial cell shrinkage increases permeability through a Ca2+‐dependent pathway in single frog mesenteric microvessels. J Physiol. 1999;518:227–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kajimura M, O'Donnell ME, Curry FE. Effect of cell shrinkage on permeability of cultured bovine aortic endothelia and frog mesenteric capillaries. J Physiol. 1997;503:413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wu MH, Guo M, Yuan SY, Granger HJ. Focal adhesion kinase mediates porcine venular hyperpermeability elicited by vascular endothelial growth factor. J Physiol. 2003;552:691–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Yuan Y, Meng FY, Huang Q, Hawker J, Wu HM. Tyrosine phosphorylation of paxillin/pp125FAK and microvascular endothelial barrier function. Am J Physiol. 1998;275:H84–H93. [DOI] [PubMed] [Google Scholar]