

Abstract
Mutations in the progranulin gene cause frontotemporal dementia (FTD), a debilitating neurodegenerative disease that involves atrophy of the frontal and temporal lobes and affects personality, behavior, and language. Progranulin-deficient mouse models of FTD exhibit deficits in compulsive and social behaviors reminiscent of patients with FTD, and develop excessive microgliosis and increased release of inflammatory cytokines. Activation of nicotinic acetylcholine receptors (nAChRs) by nicotine or specific α7 nAChR agonists reduces neuroinflammation. Here, we investigated whether activation of nAChRs by nicotine or α7 agonists improved the excessive inflammatory and behavioral phenotypes of a progranulin-deficient FTD mouse model. We found that treatment with selective α7 agonists, PHA-568487 or ABT-107, strongly suppressed the activation of NF-κB in progranulin-deficient cells. Treatment with ABT-107 also reduced microgliosis, decreased TNFα levels, and reduced compulsive behavior in progranulin-deficient mice. Collectively, these data suggest that targeting activation of the α7 nAChR pathway may be beneficial in decreasing neuroinflammation and reversing some of the behavioral deficits observed in progranulin-deficient FTD.
Keywords: frontotemporal dementia, progranulin, nicotine, inflammation, microglial activation
1. Introduction
Frontotemporal dementia (FTD) is a clinical presentation that encompasses a broad group of disorders that result in atrophy of the frontal and temporal lobes of the brain—areas associated with personality, behavior, and language. Mutations in the progranulin gene (GRN), which cause haploinsufficiency of progranulin levels, are a major cause of FTD [1–3]. Progranulin-deficient mice develop excessive microgliosis and secrete increased levels of inflammatory cytokines, key elements of neuroinflammation [4–6]. Preclinical studies suggest a compelling role for the nicotinic cholinergic system in reducing inflammation in the brain, implicating it as a potential therapeutic target for alleviating progranulin deficiency-associated deficits in FTD.
Nicotinic acetylcholine receptors (nAChRs) are ligand-gated channels composed of a pentameric complex of a possible 12 different subunits. In the brain, nAChRs are widely distributed in both neurons and glia, and predominantly comprise the α7 and α4β2 subtypes [7–8]. In neurons, nAChRs are involved in a number of physiological functions. α7–containing nAChRs in particular are highly permeable to calcium, implicating them as significant modulators of intracellular signaling and neurotransmitter release and, consequently, in the pathophysiology of a variety of neurological diseases. Indeed, loss of basal forebrain cholinergic neurons and decreased production of ACh significantly contributes to early Alzheimer’s disease dementia. In animal models, nicotine enhances long-term potentiation [9] and episodic and working memory [10]. Conversely, anti–α7 nAChR antibodies induced inflammation and increased amyloid accumulation in mouse models of Alzheimer’s disease [11]. Recent studies have demonstrated a protective effect of α7 nAChRs in decreasing L-Dopa-induced dyskinesias in Parkinson’s disease as well, implicating an emerging role for α7 nAChRs in multiple therapeutic areas [12].
nAChRs have also been implicated in the cholinergic anti-inflammatory pathway, as they are also expressed in non-neuronal cells of the brain. The α7 subunit–containing receptors in particular modulate innate immunity and inflammatory responses by regulating the release of inflammatory cytokines and chemokines [13–14]. Administration of α7 nAChR agonists inhibited release of TNFα, IL-1, IL-6, and IL-8 [15]. Furthermore, activation of α7 nAChRs resulted in decreased translocation of NF-κB to the nucleus [15], a critical event in triggering downstream inflammatory pathways.
Thus, activating nAChRs, especially α7 subtypes, may attenuate the increased microgliosis and inflammatory cytokine release observed in progranulin-deficient mice. In the current study, we aimed to determine whether nicotine, or specific α7 agonists of nAChRs, could indeed reverse the excessive neuroinflammation and behavioral deficits observed in a mouse model of progranulin-deficient FTD.
2. Materials and Methods
2.1. Mice
For all experiments, male and female mice were used in gender-balanced groups. Grn−/− mice were obtained from the laboratory of Robert V. Farese, Jr [16]. All mice were housed in a pathogen-free barrier facility with a 12-h light/dark cycle and ad libitum access to food and water. All behavior experiments were conducted during daylight hours unless otherwise noted. All animal procedures were carried out under University of California, San Francisco, Institutional Animal Care and Use Committee-approved guidelines.
2.2. Chemicals
LPS and nicotine were purchased from Sigma (St. Louis, MO). PHA-568487 and recombinant TNFα were purchased from R&D Systems (Minneapolis, MN). ABT-107, a full α7 agonist 5-(6-[(3R)-1-azabicyclo[2.2.2]oct-3-yloxy]pyridazin-3-yl)-1H-indole, was obtained from AbbVie Inc. (North Chicago, IL).
2.3. Generation of Bone Marrow–Derived Macrophages and Microglia for NF-κB Reporter Assay
Bone marrow–derived cells were obtained from 2-month-old Grn−/− mice. To immortalize myeloid cells, Ficoll-purified progenitors were infected with ER-Hoxb8 retrovirus and maintained in granulocyte macrophage colony-stimulating factor (GM-CSF) medium with estrogen as described [17]. Cells were transduced with pGreenFire-5xκB lentivirus expressing both GFP and luciferase as reporters to generate immortalized myeloid cell lines. For experiments, macrophages were differentiated from ER-Hoxb8 GM-CSF myeloid progenitor cells by plating in medium without estrogen. For primary microglia experiments, cortices from postnatal day 3 Grn+/+ or Grn−/− mice were isolated and plated in T75 flasks in DMEM, 10% FBS, GlutaMAX, 2% B16-conditioned medium, 100 units/ml penicillin, and 100 μg/ml streptomycin. At DIV 7, flasks were shaken for 2 h and detached microglia were replated in 96-well plates in B16-conditioned medium-free medium. Microglia were treated with Lenti-pGreenFire-5xκB on the same day, and experiment was conducted 48 h following infection. Myeloid or microglia were pre-treated with nicotine or PHA for indicated time, followed by treatment with recombinant TNFα or LPS for 2 h. Cells were lysed in luciferase assay buffer (Promega, Madison, WI), and luciferase activity was read on a Victor luminometer (Perkin Elmer, Waltham, MA).
2.4. Drug Dosing
In all experiments, mice were treated for 14 days by either Alzet osmotic mini-pumps or daily intraperitoneal (I.P.) injection. For Alzet mini-pump experiments, mice received a total per day of 4.2 mg/kg nicotine, 0.42 mg/kg PHA-568487, or 0.45 mg/kg ABT-107 at a rate of 0.25 μl/h. Osmotic pumps were implanted subcutaneously under the skin on the dorsum while mice were anesthetized with Avertin (Sigma). For I.P. experiments, mice received a once daily injection of 0.6 mg/kg nicotine or 0.064 mg/kg ABT-107. For both I.P. and osmotic pump experiments, nicotine and PHA-568487 were solubilized in saline, and ABT-107 was solubilized with 10 N HCl, followed by further dilution with water.
2.5. Brain Harvest and Quantification of Microglia
Mice were transcardially perfused with 0.9% saline after deep anesthesia with Avertin. One hemi-brain was fixed with 4% PFA for 24 h, incubated in 30% sucrose for 24 h, and sectioned on a Leica freezing microtome (30 μm-thick sections). The other hemi-brain was snap-frozen in dry ice for mRNA analyses. Experimenters were blind to the genotypes or treatment of the mice for all immunohistochemical analyses. Sections were washed and incubated with anti-CD68 (MCA1957GA, 1:500, Serotec, Raleigh, NC) after quenching endogenous peroxidase activity. Staining of CD68 was detected with 0.25 mg/ml DAB with 0.01% H2O2. Images were acquired on a brightfield Leica DM5000B microscope with a Leica DFC310 FX camera and staining was quantified using NIH ImageJ software.
For immunofluorescence staining, sections were incubated with anti-Iba1 (#019-19741, 1:500, Wako, Osaka, Japan) after permeabilization in Tris-buffered saline with 0.5% Triton X-100 (TBST) and blocking with 10% normal donkey serum. Immunoreactive structures were detected with Alexa Fluor donkey anti-rabbit 488 secondary antibody (Invitrogen, Carlsbad, CA). Images were acquired on a Leica DM5000B microscope and quantified with Volocity software (Perkin Elmer, Waltham, MA). All DAB and fluorescent imaging and quantification were performed in the cortical region of brains.
2.6. RNA Isolation and Quantitative Real-Time PCR
To measure mRNA of cytokines, cortical brain tissue was homogenized with a 21G needle in RLT buffer with 1% β-ME. RNA was isolated per the RNeasy isolation kit instructions (Qiagen, Valencia, CA), and the remaining DNA in the sample was removed by incubation with RNase-free DNase (Ambion, Austin, TX). RNA was then converted to cDNA by the TaqMan reverse transcription (RT) kit (Applied Biosystems, Foster City, CA). Real-time RT-PCR was performed on the ABI 7900 HT sequence detector (Applied Biosystems) with SYBR Green PCR master mix (Applied Biosystems). The relative gene expression was quantified as 2−ΔCt. The mean value of replicates of each sample was expressed as the threshold cycle (Ct) at which the level of fluorescence begins to increase rapidly. The amount of gene expression was calculated as the difference (ΔCT) between the CT value of the sample for target gene and the CT value of that sample for the endogenous control GAPDH (ΔCt = Ct(target gene) −Ct(GAPDH)). The relative amount of gene expression for each target gene was determined by 2−ΔCt and expressed as the fold change as compared with a control. The primers used were as follows: GAPDH sense GGG AAG CCC ATC ACC ATC TT, antisense GCC TTC TCC ATG GTG GTG AA; TNFα sense CAT CAG TTC TAT GGC CCA GA, antisense TGC TCC TCC ACT TGG TGG TT; IL-1α sense CAC CTT ACA CCT ACC AGA GTG ATT TG, antisense TGT TGC AGG TCA TTT AAC CAA GTG; IL-1β sense TGA AAG ACG GCA CAC CCA CCC, antisense TGC CCT GGG GAA GGC ATT AGA; IL6 sense ACC ACT TCA CAA GTC GGA GGC T, antisense TCT CTG AAG GAC TCT GGC TTT GTC T; TGFβ sense AAC CCC CAT TGC TGT CCC GTG, antisense GCG CTG AAT CGA AAG CCC TGT; IL4 sense TGC GAA GCA CCT TGG AAG CCC, antisense GGG ACG CCA TGC ACG GAG ATG; CD11b sense GTG TGA CTA CAG CAC AAG CCG, antisense CCC AAG GAC ATA TTC ACA GCC T.
2.7. Behavior tests
2.7.1. Three-Chambered Social Test
For the habituation phase, mice were allowed to freely explore a three-chambered box including two empty wire cups in the outer chambers for 10 min under dim light. Mice were then closed into the center chamber, and a conspecific stranger mouse was placed under one wire cup and a green Lego toy was placed under the other wire cup. Barriers were released, and the test mice were allowed to freely explore the box, now including a stranger mouse and an object, for 10 min. In both phases, the amount of time spent sniffing (interaction time) was measured for the mouse cup and the object cup. Data were presented as ratio of sniffing time for the mouse cup over sniffing time for the object cup. The boxes were thoroughly cleaned with 70% ethanol between mouse sessions.
2.7.2. Elevated Plus Maze
The elevated plus maze consisted of two open (without walls) and two enclosed (with walls) arms elevated 63 cm above the ground (Hamilton-Kinder, Poway, CA). Mice were first allowed to habituate in the testing room under dim light for 30 min before testing. During testing, mice were placed at the junction between the open and closed arms of the plus maze and allowed to explore for 10 min. The distance traveled and the time spent in the open and closed arms were determined. The maze was thoroughly cleaned with 70% ethanol between testing sessions.
2.7.3. Burrowing Test
All burrowing tests were performed at the same time of the day to ensure consistency [18]. Briefly, at 6:00 pm, mice were placed in a clean cage with a burrow (plastic canister with one end open) containing 300 g of food pellets. The open end of the burrow was raised slightly above the cage floor. At 9:00am the next morning, the burrows were weighed, and the difference in weight was recorded as the amount of food burrowed.
2.8. Statistical Analysis
Data were analyzed with GraphPad Prism v.5 (GraphPad, San Diego, CA). Differences between means were assessed with paired Student’s t test, or with one-way or two-way analysis of variance followed by post hoc testing of all pairwise comparisons among groups with Tukey-Kramer correction for multiple comparisons. Pearson’s correlation coefficients were used to quantify the linear relationship between two variables. The Shapiro-Wilk test of normality was applied to all data sets, and in cases where the data did not demonstrate a normal distribution, non-parametric tests were used to analyze statistical differences. The Wilcoxon matched pairs test was used for paired comparisons. An F test or Bartlett’s test was performed to determine significant differences in variances between groups for t-tests and ANOVAs, respectively. All values are given as mean ± S.E.M.
3. Results
Upon activation, Grn−/− microglia are susceptible to a hyperactive proinflammatory state and release increased levels of proinflammatory cytokines than Grn+/+ microglia [16]. NF-κB signaling is a master regulator of inflammatory mediators. To determine the effects of nicotine on NF-κB signaling in progranulin-deficient cells, we established an NF-κB reporter macrophage line by immortalizing bone marrow–derived macrophages stably expressing luciferase under 5 repeats of κB enhancer element (5xκB) that binds to NF-κB p65/p50 dimers [19]. Cells were treated with nicotine before stimulation with lipopolysaccharide (LPS). LPS treatment of Grn−/− cells markedly increased their NF-κB-dependent luciferase expression, which was significantly reduced by nicotine pretreatment in a dose-dependent manner (Figure 1A, B).
Fig. 1.
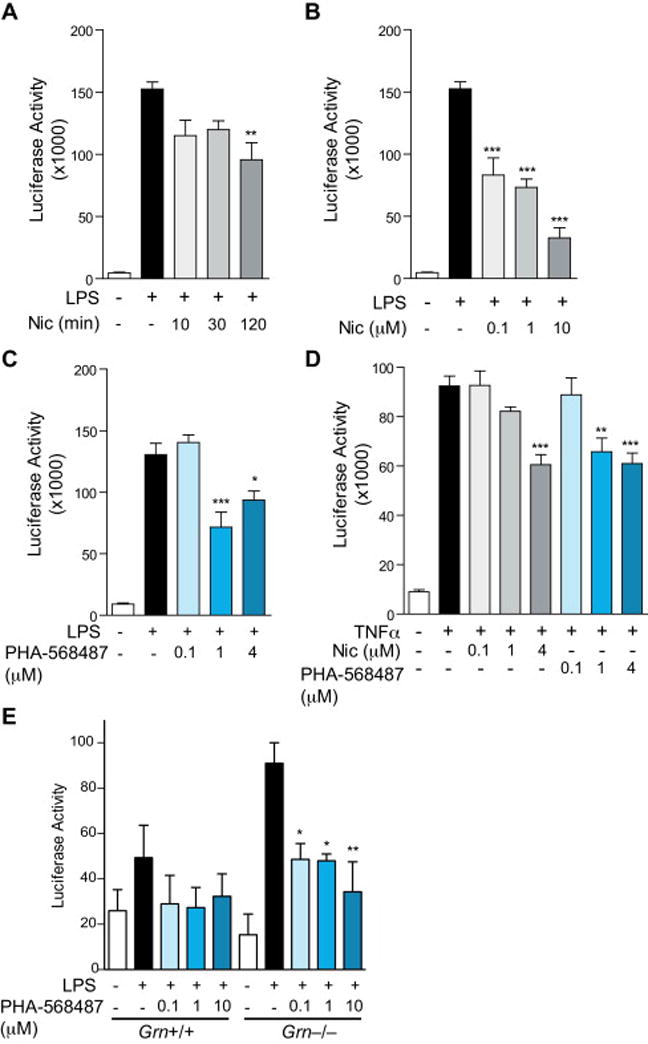
Nicotine inhibits NF-κB activation. (A) Increasing time of pre-treatment with 4 μM nicotine decreased the luciferase signal from progranulin knock-out myeloid cells expressing pGreenFire-5xκB reporter construct and treated with 10 ng/ml LPS for 2 h. n = 5–6 wells per condition. **P < 0.01 by ANOVA followed by Tukey-Kramer post-hoc test. (B) Dose-dependent decrease of luciferase signal in response to nicotine in progranulin knock-out myeloid cells expressing pGreenFire-5xκB reporter construct and treated with 10 ng/ml LPS for 2 h. n = 6 wells per condition. ***P < 0.001 by ANOVA followed by Tukey-Kramer post-hoc test. (C) Pre-treatment with increasing concentrations of PHA-568487 decreased the luciferase signal from progranulin knock-out myeloid cells expressing pGreenFire-5xκB reporter construct and treated with 10 ng/ml LPS for 2 h. n = 4 wells per group. *P < 0.05, ***P < 0.001 by ANOVA followed by Tukey-Kramer post-hoc test. (D) Pre-treatment with increasing concentrations of nicotine or PHA-568487 decreased, in a dose-dependent manner, the luciferase signal from progranulin knock-out myeloid cells expressing pGreenFire-5xκB reporter construct and treated with 100 ng/ml TNFα for 2 h. n = 4 wells per group. **P < 0.01, ***P < 0.001 by ANOVA followed by Tukey-Kramer post-hoc test. (E) Pre-treatment with increasing concentrations of PHA-568487 decreased, in a dose-dependent manner, the luciferase signal from progranulin knock-out microglia infected with pGreenFire-5xκB reporter construct and treated with 10 ng/ml LPS for 2 h. n = 3 wells per group. *P < 0.05, **P < 0.01 by ANOVA followed by Tukey-Kramer post-hoc test.
Nicotine is a non-selective agonist of nAChRs. Since α7 nAChRs in particular have been implicated in the cholinergic anti-inflammatory pathway [20], we examined whether specific agonists of the α7 nAChR could attenuate LPS-induced NF-κB in Grn−/− macrophages. PHA-568487 was developed to activate α7 nAChR [21]. PHA-568487 (at 1 or 4 μM) decreased NF-κB-dependent luciferase signal in response to LPS stimulation (Figure 1C). We then induced NF-κB activation by TNFα signaling since excessive TNFα signaling has been implicated in GRN haploinsufficiency [22]. Increasing concentrations of either nicotine or PHA-568487 resulted in a dose-dependent reduction of the TNFα-stimulated increase in NF-κB activation in Grn−/− macrophages (Figure 1D). We then directly compared LPS-induced NF-κB activation in Grn+/+ and Grn−/− microglia and their responses to PHA-568487. Microglia from Grn−/− mice exhibited significant NF-κB activation upon stimulation, as expected. The modest induction of NF-κB activation induced by LPS stimulation in wildtype microglia did not reach statistical significance, most likely due to low concentration of LPS. Notably, treatment with PHA-568487 led to a dose-dependent suppression of NF-κB activation in Grn−/− microglia (Figure 1E).
Grn−/− mice exhibit elevated microgliosis in multiple brain regions, including cortex and thalamus [4, 16, 23]. To determine the in vivo effects of nicotine, we injected Grn−/− mice with 0.6 mg/kg of nicotine once a day, I.P., for 14 days (Figure 2). Brain sections were immunostained for Iba1, a microglial marker. Vehicle-injected Grn−/− mice had significantly more Iba1 immunoreactivity than age-matched wild-type controls (Figure 2A, B). Correspondingly, mRNA levels of CD11b, another marker for microglia, were increased in vehicle-injected Grn−/− mice (Figure 2C). Injection of nicotine induced a trend of reduction in Iba1 immunoreactivity or levels of CD11b control, which did not reach statistical significance (Figure 2B, C).
Fig. 2.
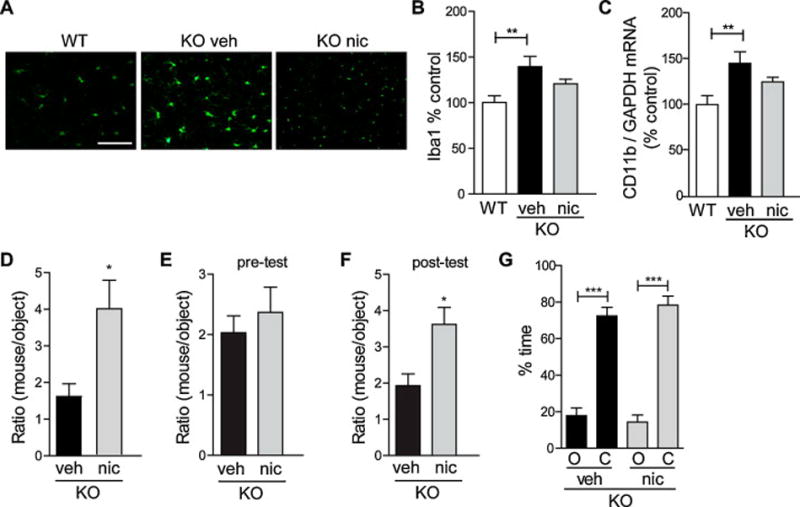
Nicotine reduces progranulin-deficiency-induced microgliosis and rescues social behavior deficits. (A–D, G) Six-month-old progranulin knock-out mice were injected daily (I.P.) with vehicle control or 0.6 mg/kg nicotine for 14 days. (A) Representative images of cryosections immunostained for Iba1-positive microglia from wild-type or progranulin knock-out mice injected with vehicle or nicotine. (B) Quantification of Iba1-positive immunoreactivity (right). n = 7–10 mice per group. **P < 0.01 by ANOVA followed by Tukey-Kramer post-hoc test. (C) CD11b mRNA levels were measured by qRT-PCR from brains and normalized to GAPDH mRNA levels. n = 7–9 mice per group. **P < 0.01 by ANOVA followed by Tukey-Kramer post-hoc test. (D) One day after the last injection, mice were tested in the three-chambered social test. The amount of time spent interacting with a stranger mouse was normalized to the amount of time spent interacting with an object, shown as a ratio of mouse/object. n = 6–7 mice per group. *P < 0.05 by student’s t-test. (E–F) Twelve-month-old progranulin knock-out mice were injected daily (I.P.) with vehicle control or 0.6 mg/kg nicotine for 14 days. Mice were tested in the three-chambered social test before (E) and after (F) treatment. n = 6 mice per group. *P < 0.05 by student’s t-test. (G) Six-month-old progranulin knock-out mice were tested in the elevated plus maze. Amount of time spent in the open arms (O) and closed arms (C) is shown. n = 7–8 mice per group. ***P < 0.001 by paired t-test. Scale bar: 100 μm.
Decreased sociability is the primary behavioral deficit observed in Grn−/− mice [5]. Compared to wild-type controls, knock-out mice spend less time interacting with a conspecific mouse than with an object in the three-chamber social test [5]. Grn−/− mice injected with nicotine preferred to spend more time with the mouse than those injected with vehicle (Figure 2D), suggesting beneficial effects of nicotine. Importantly, the sociability deficits of 12 month-old Grn−/− mice were also reversed with daily nicotine treatment for 14 days, suggesting beneficial effects of nicotine even when disease phenotype has already been established (Figure 2E, F). Nicotine treatment did not seem to affect the degree of anxiety in Grn−/− mice. In the elevated plus maze, vehicle- and nicotine-injected Grn−/− mice spent a similar percentage of time in the open or closed arms (Figure 2G).
Next, we examined the anti-inflammatory effects of α7 nAChR signaling by directly comparing nicotine and PHA-568487 in Grn−/− mice. We implanted Alzet osmotic mini-pumps in Grn−/− mice to infuse vehicle, nicotine, or PHA-568487 for 14 days. Additionally, we tested the efficacy of a second specific agonist of the α7 nAChR, ABT-107, which has previously been shown to decrease secretion of inflammatory cytokines [24–25]. ABT-107 was administered daily for 14 days (I.P). Compared with vehicle treatment, nicotine or PHA-568487 treatment significantly suppressed CD68 immunoreactivity, a marker for activated microglia (Figure 3A, B). ABT-107 also significantly decreased CD68 immunoreactivity in Grn−/− mice (Figure 3C, D).
Fig. 3.
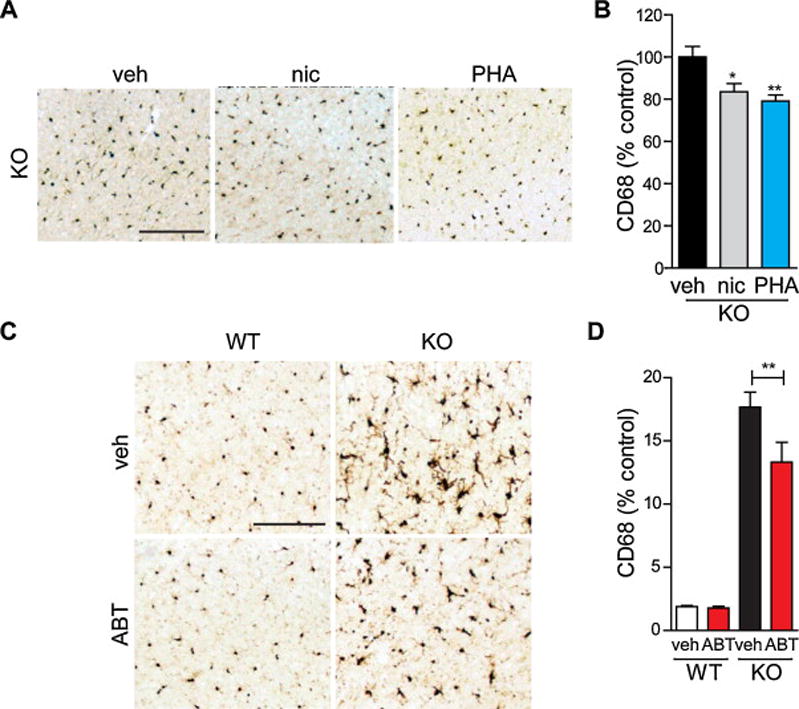
Selective α7 nAChR agonists PHA-568487 and ABT-107 decrease microgliosis. (A–B) Nine-month-old progranulin knock-out mice were treated with vehicle, nicotine, or PHA-568487 by Alzet osmotic mini-pumps for 14 days. (A) Representative images of cortical cryosections immunostained for CD68-positive microglia from vehicle, nicotine, or PHA-568487 treated mice. (B) Quantification of CD68 immunoreactivity. n = 10 mice per group. *P < 0.05, **P < 0.01 by ANOVA followed by Tukey-Kramer post-hoc test. (C–D) Sixteen-month-old wild-type or progranulin knock-out mice were injected daily (I.P.) with vehicle or ABT-107 for 14 days. (C) Representative images of cortical cryosections immunostained for CD68-positive microglia from wild-type or progranulin knock-out mice treated with vehicle or ABT-107. (D) Quantification of CD68 immunoreactivity. n = 5–7 mice per group. **P < 0.01 by two-way ANOVA followed by Bonferroni post-hoc test. Scale bars: 200 μm.
To further confirm the importance of α7 nAChRs in progranulin-deficiency-induced inflammation, we tested the efficacy of α7 nAChR agonists on inflammatory cytokine expression. Nicotine or PHA-568487 treatment by Alzet osmotic pump significantly reduced levels of the pro-inflammatory cytokine IL-1β (Figure 4A), but not TNFα (Figure 4B). Infusion of ABT-107 and nicotine also induced a similar reduction in the levels of IL-1β (Figure 4C). Interestingly, levels of TNFα were significantly reduced by ABT-107, but not nicotine, suggesting that ABT-107 may be a more potent inhibitor of excessive TNFα signaling in Grn−/− mice (Figure 4D).
Fig. 4.
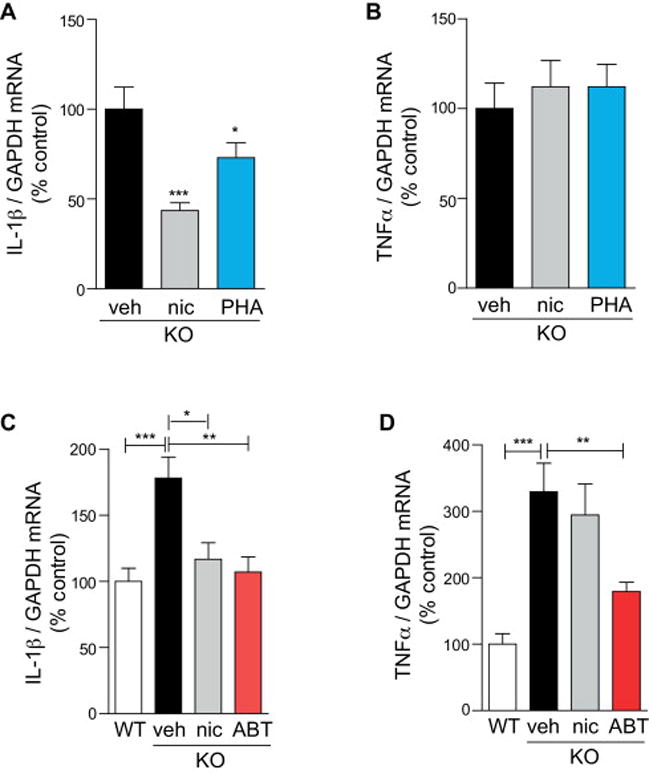
Selective α7 nAChR agonists decrease IL-1β and TNFα. (A–B) Nine-month-old progranulin knock-out mice were treated with vehicle, nicotine, or PHA-568487 by Alzet osmotic mini-pumps for 14 days. mRNA levels of IL-1β (A) or TNFα (B) were measured by qRT-PCR from brains and normalized to GAPDH mRNA levels. n = 9–10 mice per group. *P < 0.05, ***P < 0.001 by ANOVA followed by Tukey-Kramer post-hoc test. (C–D) Twelve-month-old progranulin knock-out mice were treated with vehicle, nicotine, or ABT-107 by Alzet osmotic mini-pumps for 14 days. mRNA levels of IL-1β (C) and TNFα (D) were measured by qRT-PCR from brains and normalized to GAPDH mRNA levels. Age-matched non-treated wild-type mice were used as control. n = 8–13 mice per group. *P < 0.05, **P < 0.01, ***P < 0.001 by ANOVA followed by Tukey-Kramer post-hoc test.
We further examined the in vivo effects of ABT-107 by treating wild-type or Grn−/− mice with vehicle or ABT-107 daily, I.P., for 14 days. ABT-107 treatment reduced TNFα levels in Grn−/− mice, but had little effects in wild-type mice (Figure 5A). To determine whether the anti-inflammatory effects of ABT-107 were associated with behavioral improvement, we tested these mice in a test of compulsivity, a symptom often associated with FTLD [26]. We measured the amount of food that mice burrowed out of a canister placed in their home cage overnight, and found that Grn−/− mice burrowed significantly more than wild-type mice (Figure 5B). However, treatment with ABT-107 significantly reduced the amount of food burrowed by Grn−/− mice compared with vehicle treatment, suggesting protection against FTD-related compulsivity (Figure 5C). Notably, there was a significant positive correlation between the amount burrowed and TNFα levels in the brains of these mice, supporting a role for TNFα in progranulin deficiency-induced behavioral impairment (Figure 5D).
Fig. 5.

Selective α7 nAChR agonist ABT-107 decreases TNFα and rescues compulsive behavior. (A–D) Sixteen-month-old wild-type or progranulin knock-out mice were injected daily (I.P.) with vehicle or ABT-107 for 14 days. (A) mRNA levels of TNFα were measured by qRT-PCR from brains and normalized to GAPDH mRNA levels. n = 5–7 mice per group. ***P < 0.001 by two-way ANOVA followed by Bonferroni post-hoc test. (B–C) Mice were tested for burrowing behavior. The amount of food burrowed after 15 h was measured. Progranulin knock-out mice burrowed more than wild-type mice (B). n = 5–6 mice per group. *P < 0.05 by student’s t-test. (C) Vehicle or ABT-107-treated mice were tested for burrowing behavior. n = 5–8 mice per group. *P < 0.05 by two-way ANOVA followed by Bonferroni post-hoc test. (D) The amount of food burrowed was correlated with increased TNFα mRNA levels. n = 25 mice, **P < 0.01 with Gaussian approximation, r2 = 0.2730.
4. Discussion
Consistent with previous studies [27–28], we found that nicotine or α7 nAChR agonist treatment decreased NF-κB activation in progranulin-deficient macrophages. Specific α7 nAChR agonist PHA-568487 appeared to be more potent than nicotine in attenuating NF-κB activation induced by TNFα in cultured progranulin-deficient macrophages, supporting beneficial effects of selective activation of α7 nAChRs. Interestingly, the two α7 nAChR agonists we used exhibited differences as well. While both selective agonists significantly decreased microgliosis, PHA-568487 only decreased levels of IL-1β, but not TNFα, in progranulin-deficient mice (similar to nicotine), whereas ABT-107 appeared to decrease both. Although the three compounds could have differences in brain availability, some of the differential effects on cytokine release may also be reflective of the difference in the in vitro profiles of the compounds, with ABT-107 being 100-fold more potent to bind to α7 nAChRs (Ki of 0.2–0.6 nM vs 44 nM) and ~5-fold more potent to activate α7 receptors with EC50 of 50 nM vs 258 nM [29–30]. Both compounds also exhibit in vivo efficacy with PHA-568487 attenuating spatial memory deficits in a mouse model reflective of the cognitive impairment associated with schizophrenia [31], and ABT-107 exhibiting pre-clinical in vivo efficacy across a battery of assays associated with discrete cognitive domains [32]. In this latter study, ABT-107 was also shown to be neuroprotective via GSKβ inhibition and distinguished itself from nicotine as it did not induce behavioral sensitization or induce psychomotor stimulation in rats [32]. An additional α7 agonist, A-833834, potently suppressed TNFα release via in vitro LPS induction or in vivo by zymosan-induced peritonitis [33]. The current data supports a role for α7 nAChR activation in decreasing pro-inflammatory cytokines.
It has been previously demonstrated that modulation of TNFα release and subsequent downstream NF-κB activation is mediated by agonist activation of α7 nAChRs [34]. Selective α7 nAChR agonists can also inhibit pro-inflammatory cytokines by blocking phosphorylation of STAT3, which in turn prevents NF-κB activity [35]. In addition, α7 agonists were shown to be neuroprotective in an ischemic stroke injury model, where PHA-568487 reversed sensory motor deficits and attenuated lesion volume and neuronal apoptosis via reduction of CD68+ macrophages, activation of anti-oxidant genes, and reduced NF-κB activity in the affected infarct brain region [36]. In an Alzheimer’s disease mouse model, microglial activation caused an upregulation of α7 nAChRs, which could contribute to suppression of TNFα production [37]. This would support previous studies demonstrating that activation of α7 nAChRs on microglia is neuroprotective in brain ischemia via induction of Nrf2 anti-oxidant genes [38]. Collectively, these reports combined with the current study using selective α7 agonists continue to support the neuroprotective and anti-inflammatory properties of these compounds.
Here, we demonstrate a new phenotype in progranulin-deficient mice in the burrowing test, a measure of repetitive and compulsive activities and stereotyped behavior that has been used to characterize activities of daily living (ADLs) in mice [18, 39–40]. Thus far, the primary behavior test that has been used to characterize FTD-associated behavior deficits in mice has been the three-chambered social test, which is a complex test that can be susceptible to numerous variables including lighting, time of day, age and sex of the stranger mouse, and experimenter error [5, 23, 41]. In contrast, mice display natural burrowing behavior that can be captured in a simple test that requires minimal experimenter handling. Of note, burrowing is commonly used to assess obsessive compulsive disorder (OCD)-like behaviors in rodents [42], and OCD-like symptoms are common and constitute a subset of criteria for diagnosis in behavioral variant FTD (bvFTD) [26, 43]. Indeed, progranulin-deficient mice exhibited an increased burrowing phenotype, which was reversed by ABT-107. Although previous studies indicated decreased burrowing in mice in response to LPS administration, our data support that a chronic inflammatory state may actually lead to increases in compulsive behaviors [44–45].
The selective effect of ABT-107 on TNFα levels is intriguing—TNFα is an important inflammatory factor, but it has also been implicated in modulating neuronal and synaptic function [46–48]. TNFα is consistently and dramatically increased in progranulin-deficient mice [4, 6, 16, 23], suggesting that it may play an integral role in mediating synaptic deficits underlying behavioral changes in these mice. Here, we provide evidence that ABT-107 markedly decreases TNFα levels, and this decrease is significantly correlated with improved burrowing behavior, demonstrating for the first time a link between inflammation and FTD-like behavior deficits. However, we cannot discount the possibility that the anti-inflammatory effects of cholinergic agonists are distinct from the effects on neuronal function that drive behavioral changes. Since α7 nAChRs are present on both neurons and microglia, activating the cholinergic system may benefit both pathways separately and, furthermore, this two-pronged approach may attenuate the reciprocal detrimental effects that each has on the other. Future studies will be necessary to establish the causality between microglial inflammation and neuronal dysfunction and behavioral outcome, especially in the context of progranulin-deficiency-associated FTD.
Acknowledgments
We thank Michael E. Ward for immortalized cell lines, Gary Howard for editorial review, Robert V. Farese, Jr. for generation of progranulin-deficient mice, and Erica Nguyen for administrative assistance. This work was supported in part by the Consortium for Frontotemporal Dementia (L.G.), NIH (1R01AG036884 and R01AG030207 to L.G.), S.D Bechtel Jr. Foundation, and NIH/NCRR CO6 RRO18928 (a facility grant to J. David Gladstone Institutes). S.S.M. is supported by NIH fellowship F32NS076239.
Abbreviations
- nAChR
nicotinic acetylcholine receptor
- FTD
frontotemporal dementia
- PGRN
progranulin
- LPS
lipopolysaccharide
- PFA
paraformaldehyde
- IP
intraperitoneal
- DAB
3,3′-diaminobenzidine
- GM-CSF
granulocyte macrophage colony-stimulating factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–9. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- 2.Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–4. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- 3.Gass J, Cannon A, Mackenzie IR, Boeve B, Baker M, Adamson J, et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet. 2006;15:2988–3001. doi: 10.1093/hmg/ddl241. [DOI] [PubMed] [Google Scholar]
- 4.Yin F, Banerjee R, Thomas B, Zhou P, Qian L, Jia T, et al. Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J Exp Med. 2010;207:117–28. doi: 10.1084/jem.20091568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yin F, Dumont M, Banerjee R, Ma Y, Li H, Lin MT, et al. Behavioral deficits and progressive neuropathology in progranulin-deficient mice: a mouse model of frontotemporal dementia. FASEB J. 2010 doi: 10.1096/fj.10-161471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Minami SS, Min SW, Krabbe G, Wang C, Zhou Y, Asgarov R, et al. Progranulin protects against amyloid beta deposition and toxicity in Alzheimer’s disease mouse models. Nat Med. 2014;20:1157–64. doi: 10.1038/nm.3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Millar NS, Harkness PC. Assembly and trafficking of nicotinic acetylcholine receptors (Review) Mol Membr Biol. 2008;25:279–92. doi: 10.1080/09687680802035675. [DOI] [PubMed] [Google Scholar]
- 8.Wessler I, Kirkpatrick CJ. Acetylcholine beyond neurons: the non-neuronal cholinergic system in humans. Br J Pharmacol. 2008;154:1558–71. doi: 10.1038/bjp.2008.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Welsby P, Rowan M, Anwyl R. Nicotinic receptor-mediated enhancement of long-term potentiation involves activation of metabotropic glutamate receptors and ryanodine-sensitive calcium stores in the dentate gyrus. Eur J Neurosci. 2006;24:3109–18. doi: 10.1111/j.1460-9568.2006.05187.x. [DOI] [PubMed] [Google Scholar]
- 10.Wallace TL, Porter RH. Targeting the nicotinic alpha7 acetylcholine receptor to enhance cognition in disease. Biochem Pharmacol. 2011;82:891–903. doi: 10.1016/j.bcp.2011.06.034. [DOI] [PubMed] [Google Scholar]
- 11.Lykhmus O, Voytenko L, Koval L, Mykhalskiy S, Kholin V, Peschana K, et al. alpha7 Nicotinic Acetylcholine Receptor-Specific Antibody Induces Inflammation and Amyloid beta42 Accumulation in the Mouse Brain to Impair Memory. PLoS One. 2015;10:e0122706. doi: 10.1371/journal.pone.0122706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang D, McGregor M, Decker MW, Quik M. The alpha7 nicotinic receptor agonist ABT-107 decreases L-Dopa-induced dyskinesias in parkinsonian monkeys. J Pharmacol Exp Ther. 2014;351:25–32. doi: 10.1124/jpet.114.216283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olofsson PS, Rosas-Ballina M, Levine YA, Tracey KJ. Rethinking inflammation: neural circuits in the regulation of immunity. Immunol Rev. 2012;248:188–204. doi: 10.1111/j.1600-065X.2012.01138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gahring LC, Enioutina EY, Myers EJ, Spangrude GJ, Efimova OV, Kelley TW, et al. Nicotinic receptor alpha7 expression identifies a novel hematopoietic progenitor lineage. PLoS One. 2013;8:e57481. doi: 10.1371/journal.pone.0057481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang H, Liao H, Ochani M, Justiniani M, Lin X, Yang L, et al. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med. 2004;10:1216–21. doi: 10.1038/nm1124. [DOI] [PubMed] [Google Scholar]
- 16.Martens LH, Zhang J, Barmada SJ, Zhou P, Kamiya S, Sun B, et al. Progranulin deficiency promotes neuroinflammation and neuron loss following toxin-induced injury. J Clin Invest. 2012;122:3955–9. doi: 10.1172/JCI63113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang GG, Calvo KR, Pasillas MP, Sykes DB, Hacker H, Kamps MP. Quantitative production of macrophages or neutrophils ex vivo using conditional Hoxb8. Nat Methods. 2006;3:287–93. doi: 10.1038/nmeth865. [DOI] [PubMed] [Google Scholar]
- 18.Deacon RM. Burrowing in rodents: a sensitive method for detecting behavioral dysfunction. Nat Protoc. 2006;1:118–21. doi: 10.1038/nprot.2006.19. [DOI] [PubMed] [Google Scholar]
- 19.Badr CE, Niers JM, Tjon-Kon-Fat LA, Noske DP, Wurdinger T, Tannous BA. Real-time monitoring of nuclear factor kappaB activity in cultured cells and in animal models. Mol Imaging. 2009;8:278–90. [PMC free article] [PubMed] [Google Scholar]
- 20.Tracey KJ. Physiology and immunology of the cholinergic antiinflammatory pathway. J Clin Invest. 2007;117:289–96. doi: 10.1172/JCI30555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walker DP, Wishka DG, Piotrowski DW, Jia S, Reitz SC, Yates KM, et al. Design, synthesis, structure-activity relationship, and in vivo activity of azabicyclic aryl amides as alpha7 nicotinic acetylcholine receptor agonists. Bioorg Med Chem. 2006;14:8219–48. doi: 10.1016/j.bmc.2006.09.019. [DOI] [PubMed] [Google Scholar]
- 22.Miller ZA, Rankin KP, Graff-Radford NR, Takada LT, Sturm VE, Cleveland CM, et al. TDP-43 frontotemporal lobar degeneration and autoimmune disease. J Neurol Neurosurg Psychiatry. 2013;84:956–62. doi: 10.1136/jnnp-2012-304644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Filiano AJ, Martens LH, Young AH, Warmus BA, Zhou P, Diaz-Ramirez G, et al. Dissociation of frontotemporal dementia-related deficits and neuroinflammation in progranulin haploinsufficient mice. J Neurosci. 2013;33:5352–61. doi: 10.1523/JNEUROSCI.6103-11.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Radek RJ, Robb HM, Stevens KE, Gopalakrishnan M, Bitner RS. Effects of the novel alpha7 nicotinic acetylcholine receptor agonist ABT-107 on sensory gating in DBA/2 mice: pharmacodynamic characterization. J Pharmacol Exp Ther. 2012;343:736–45. doi: 10.1124/jpet.112.197970. [DOI] [PubMed] [Google Scholar]
- 25.Kohlhaas KL, Bitner RS, Gopalakrishnan M, Rueter LE. Effects of alpha7 nicotinic acetylcholine receptor agonists on antipsychotic efficacy in a preclinical mouse model of psychosis. Psychopharmacology (Berl) 2012;220:823–33. doi: 10.1007/s00213-011-2535-6. [DOI] [PubMed] [Google Scholar]
- 26.Perry DC, Whitwell JL, Boeve BF, Pankratz VS, Knopman DS, Petersen RC, et al. Voxel-based morphometry in patients with obsessive-compulsive behaviors in behavioral variant frontotemporal dementia. Eur J Neurol. 2012;19:911–7. doi: 10.1111/j.1468-1331.2011.03656.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paris D, Beaulieu-Abdelahad D, Abdullah L, Bachmeier C, Ait-Ghezala G, Reed J, et al. Anti-inflammatory activity of anatabine via inhibition of STAT3 phosphorylation. Eur J Pharmacol. 2013;698:145–53. doi: 10.1016/j.ejphar.2012.11.017. [DOI] [PubMed] [Google Scholar]
- 28.Dowling O, Rochelson B, Way K, Al-Abed Y, Metz CN. Nicotine inhibits cytokine production by placenta cells via NFkappaB: potential role in pregnancy-induced hypertension. Mol Med. 2007;13:576–83. doi: 10.2119/2007-00067.Dowling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Malysz J, Anderson DJ, Gronlien JH, Ji J, Bunnelle WH, Hakerud M, et al. In vitro pharmacological characterization of a novel selective alpha7 neuronal nicotinic acetylcholine receptor agonist ABT-107. J Pharmacol Exp Ther. 2010;334:863–74. doi: 10.1124/jpet.110.167072. [DOI] [PubMed] [Google Scholar]
- 30.Hurst R, Rollema H, Bertrand D. Nicotinic acetylcholine receptors: from basic science to therapeutics. Pharmacol Ther. 2013;137:22–54. doi: 10.1016/j.pharmthera.2012.08.012. [DOI] [PubMed] [Google Scholar]
- 31.Karamihalev S, Prickaerts J, van Goethem NP. Donepezil and the alpha-7 agonist PHA 568487, but not risperidone, ameliorate spatial memory deficits in a subchronic MK-801 mouse model of cognitive impairment in schizophrenia. Behav Brain Res. 2014;272:248–51. doi: 10.1016/j.bbr.2014.07.017. [DOI] [PubMed] [Google Scholar]
- 32.Bitner RS, Bunnelle WH, Decker MW, Drescher KU, Kohlhaas KL, Markosyan S, et al. In vivo pharmacological characterization of a novel selective alpha7 neuronal nicotinic acetylcholine receptor agonist ABT-107: preclinical considerations in Alzheimer’s disease. J Pharmacol Exp Ther. 2010;334:875–86. doi: 10.1124/jpet.110.167213. [DOI] [PubMed] [Google Scholar]
- 33.Li J, Mathieu SL, Harris R, Ji J, Anderson DJ, Malysz J, et al. Role of alpha7 nicotinic acetylcholine receptors in regulating tumor necrosis factor-alpha (TNF-alpha) as revealed by subtype selective agonists. J Neuroimmunol. 2011;239:37–43. doi: 10.1016/j.jneuroim.2011.08.007. [DOI] [PubMed] [Google Scholar]
- 34.Parrish WR, Rosas-Ballina M, Gallowitsch-Puerta M, Ochani M, Ochani K, Yang LH, et al. Modulation of TNF release by choline requires alpha7 subunit nicotinic acetylcholine receptor-mediated signaling. Mol Med. 2008;14:567–74. doi: 10.2119/2008-00079.Parrish. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peña G, Cai B, Liu J, van der Zanden EP, Deitch EA, de Jonge WJ, et al. Unphosphorylated STAT3 modulates alpha7 nicotinic receptor signaling and cytokine production in sepsis. Eur J Immunol. 2010 doi: 10.1002/eji.201040540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han Z, Shen F, He Y, Degos V, Camus M, Maze M, et al. Activation of alpha-7 nicotinic acetylcholine receptor reduces ischemic stroke injury through reduction of pro-inflammatory macrophages and oxidative stress. PLoS One. 2014;9:e105711. doi: 10.1371/journal.pone.0105711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsumura A, Suzuki S, Iwahara N, Hisahara S, Kawamata J, Suzuki H, et al. Temporal changes of CD68 and alpha7 nicotinic acetylcholine receptor expression in microglia in Alzheimer’s disease-like mouse models. J Alzheimers Dis. 2015;44:409–23. doi: 10.3233/JAD-141572. [DOI] [PubMed] [Google Scholar]
- 38.Parada E, Egea J, Buendia I, Negredo P, Cunha AC, Cardoso S, et al. The microglial alpha7-acetylcholine nicotinic receptor is a key element in promoting neuroprotection by inducing heme oxygenase-1 via nuclear factor erythroid-2-related factor 2. Antioxid Redox Signal. 2013;19:1135–48. doi: 10.1089/ars.2012.4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deacon R. Assessing burrowing, nest construction, and hoarding in mice. J Vis Exp. 2012:e2607. doi: 10.3791/2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deacon RM, Raley JM, Perry VH, Rawlins JN. Burrowing into prion disease. Neuroreport. 2001;12:2053–7. doi: 10.1097/00001756-200107030-00052. [DOI] [PubMed] [Google Scholar]
- 41.Ghoshal N, Dearborn JT, Wozniak DF, Cairns NJ. Core features of frontotemporal dementia recapitulated in progranulin knockout mice. Neurobiol Dis. 2012;45:395–408. doi: 10.1016/j.nbd.2011.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deacon RM. Digging and marble burying in mice: simple methods for in vivo identification of biological impacts. Nat Protoc. 2006;1:122–4. doi: 10.1038/nprot.2006.20. [DOI] [PubMed] [Google Scholar]
- 43.Ames D, Cummings JL, Wirshing WC, Quinn B, Mahler M. Repetitive and compulsive behavior in frontal lobe degenerations. J Neuropsychiatry Clin Neurosci. 1994;6:100–13. doi: 10.1176/jnp.6.2.100. [DOI] [PubMed] [Google Scholar]
- 44.Tarr AJ, Chen Q, Wang Y, Sheridan JF, Quan N. Neural and behavioral responses to low-grade inflammation. Behav Brain Res. 2012;235:334–41. doi: 10.1016/j.bbr.2012.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hart AD, Wyttenbach A, Perry VH, Teeling JL. Age related changes in microglial phenotype vary between CNS regions: grey versus white matter differences. Brain Behav Immun. 2012;26:754–65. doi: 10.1016/j.bbi.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lewitus GM, Pribiag H, Duseja R, St-Hilaire M, Stellwagen D. An adaptive role of TNFalpha in the regulation of striatal synapses. J Neurosci. 2014;34:6146–55. doi: 10.1523/JNEUROSCI.3481-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, et al. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–5. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- 48.Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–9. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]