

Abstract
Background
Limited information exists on the role of B‐cell‐dependent mechanisms in the progression of heart failure (HF). However, in failing human myocardium, there is evidence of deposition of activated complement components as well as anticardiac antibodies. We aimed to determine the contribution of B‐cells in HF progression using a nonsurgical mouse model of nonischemic cardiomyopathy (CMP).
Methods and Results
CMP protocol involved the use of l‐NAME and NaCl in the drinking water and angiotensin‐II infusion for 35 days. At day 35, mice were analyzed by cardiac magnetic resonance imaging, gene expression, and histology. Mice (12 weeks old) were divided into 4 groups, all in C57BL/6 background: wild‐type (WT) CMP; severe combined immunodeficiency (SCID) CMP (T‐ and B‐cell deficient); CD22− CMP (B‐cell depleted); and Nude CMP (T‐cell deficient), with their respective controls. We performed B‐cell depletion and reconstitution protocols. The protective effect of B‐cell depletion was demonstrated by a significant reduction of cell hypertrophy and collagen deposition and a preserved ejection fraction in the CD22− CMP group compared to WT CMP. Once SCID mice underwent B‐cell reconstitution with isolated CMP B‐cells, the CMP phenotype was restored. Furthermore, deposition of IgG3 and apoptosis in the myocardium follows the development of CMP; in addition, in vitro studies demonstrated that activated B‐cells stimulate collagen production by cardiac fibroblasts.
Conclusions
The absence of B‐cells in this model of HF resulted in less hypertrophy and collagen deposition, preservation of left ventricular function, and, in association with these changes, a reduction in expression of proinflammatory cytokines, immunoglobulin G deposition, and apoptosis in the myocardium. Taken together, these data suggest that B‐cells play a contributory role in an angiotensin‐II‐induced HF model.
Keywords: antibodies, cardiomyopathy, immune system, lymphocytes, remodeling
Subject Categories: Animal Models of Human Disease, Fibrosis, Growth Factors/Cytokines, Inflammation, Cardiomyopathy
Introduction
A number of inflammatory pathways are activated in the course of myocardial injury and in the setting of heart failure (HF).1, 2, 3, 4 Of interest are recent experimental and clinical observations that suggest activation of humoral immune responses after myocardial injury occurs. Indeed, if B‐cell‐produced cytokines or antibodies are directed against the myocardium, as it results from myocardial injury, this pathway of immune activation may contribute to disease progression and thus represent a new target for therapy.
The evidence in support of B‐cell activation after myocardial injury is as follows: First, in patients with acute HF, there is activation of B‐cells. Markers of B‐cell activation increase in hospitalized patients with HFrEF and return to normal after treatment.5 Second, a variety of anticardiac antibodies have been found in patients after myocardial infarction (MI) and in patients with various forms of HF6, 7, 8, 9; in these settings, the initial myocardial injury was not immunologically mediated, suggesting that after myocardial ischemia or necrosis new antigens are exposed that trigger an immune response and antibody production. Third, a large proportion of patients with end‐stage cardiomyopathy, regardless of the etiology, have anticardiac antibodies deposited in the myocardium. In a study of 100 samples of failing myocardium, it was found that 70% were positive for immunoglobulin (Ig)G3 with an equal proportion among ischemic and nonischemic patients. Interestingly, at least one of the antibodies was directed against a mitochondrial protein.10 Fourth, increased levels of activated complement components are found in the circulation of patients with advanced disease and, more important, present in failing myocardium,11, 12 a finding that gives credence to the potential pathological role of antibody deposition in failing myocardium. And, finally, limited clinical observations suggest that strategies to remove antibodies may have an impact on the course of HF.5, 13 For example, studies using immune adsorption in patients with nonischemic cardiomyopathy that have circulating anti‐beta‐receptor antibodies appear to improve clinically as well as recover cardiac function.14 Also, as a proof of principle, therapeutic plasma exchange in patients with HF has been performed and appears to be safe.5
These observations, taken together, are consistent with the hypothesis that after myocardial injury, B‐cell activation occurs that triggers downstream effects that result in anticardiac antibody formation, complement deposition, and further myocardial injury. Accordingly, the purpose of these studies was to test, in an animal model of nonischemic cardiomyopathy (CMP), the role of B‐cells in the progression of HF.
Methods
Mouse Model of CMP Induction
Male wild‐type (WT; strain C57BL/6J; Harlan Laboratories, Indianapolis, IN), Nude (strain J:NU, Foxn1nu/Foxn1nu; The Jackson Laboratory, Bar Harbor, ME), and severe combined immunodeficient (SCID; strain B6.CB17‐Prkdcscid/SzJ C57BL/6 background; The Jackson Laboratory) mice were used in a CMP model modified from Oestreicher et al.15 CMP groups were given drinking water containing 0.1 mg/mL of l‐NAME (Sigma‐Aldrich, St. Louis, MO) and 1% NaCl (Sigma‐Aldrich). After 1 week of acclimatization to water, mice were anesthetized with inhaled isoflurane and implanted with subcutaneous osmotic minipumps (ALZET model 1004; DURECT Corporation ALZET Osmotic Pumps, Cupertino, CA) that delivered angiotensin‐II (Ang‐II; Sigma‐Aldrich) at a rate of 0.7 mg/kg per day for an additional 4 weeks. All mice were sacrificed at the end of the fifth week. All experiments used 3‐month‐old mice, which demonstrated decreased function, but a higher survival rate, compared to older mice (5–8 months old). All experiments were performed under approval of the Houston Methodist Hospital Research Institute Institutional Animal Care and Use Committee (Houston, TX).
B‐Cell Depletion
Male WT mice were treated with 2 doses of a mouse CD22 antibody (Bio X Cell, West Lebanon, NH) 3 days before initiation of the CMP protocol and again 21 days into the protocol. The antibody was injected intraperitoneally on both occassions with a final concentration of 0.2 mg/33 μL and diluted in PBS for a total volume of 200 μL. A nonspecific IgG1 antibody (Bio X Cell) was used as a control. Initial B‐cell depletion of 90% was confirmed by flow cytometric analysis of mouse spleens, whereas the other cellular compartments (ie, T‐cells) remained unchanged.
B‐Cell Reconstitution
Mice underwent the CMP protocol as previously described, and at day 35, they were sacrificed and the spleens removed. A single‐cell suspension from spleens was prepared using 100 μL of ACK lysis buffer followed by 300 μL of PBS and then filtered through a 70‐micron cell strainer. B‐cell isolation was performed from this cell suspension using a B‐cell isolation kit (Miltenyi Biotec, Cambridge, MA), following the manufacturer's instructions. Briefly, the cell suspension was centrifuged at 300g for 10 minutes, supernatant removed, and pellet resuspended in buffer. Biotin‐antibody cocktail was added at 10 μL per 107 total cells and the solution incubated for 15 minutes at 2 to 8°C. After incubation, 30 μL of buffer and 20 μL of anti‐biotin microbeads per 107 total cells were added. The incubation process was repeated, and cells were washed and magnetically separated to obtain the unstimulated, purified B‐cells. These purified B‐cells were then diluted in PBS and injected intraperitoneally in SCID mice. Three days after IP injection, the HF protocol was initiatied in WT mice, SCID mice, and SCID mice with reconstituted B‐cells (SCID+B‐cells). B‐cell reconstitution was confirmed by flow cytometric analysis of mouse spleens.
Histological Analysis
Mouse hearts were removed and sectioned midheart, with apex portions used for polymerase chain reaction (PCR) studies and base portions fixed in 2% paraformaldehyde, processed, paraffin embedded, and cut into 5‐micron sections. To measure fibrosis, sections were stained using a trichrome kit (Sigma‐Aldrich), according to manufacturer's instructions. Slides were then cover slipped and analyzed at ×20 magnification using an Olympus AX70 microscope (Olympus, Tokyo, Japan). Pictures were taken of all regions of the left ventricle and analyzed for fibrosis using Image Pro Plus v4.0 analysis software (Media Cybernetics, Silver Spring, MD). Color cube‐based selection criteria were used to denote positive staining (within the color spectrum of blue dye) and stained/unstained areas were measured. The results expressed are the average percent tissue area (pixels) stained by the dye. Analysis was performed by an observer blinded to the sample identities. Myocyte size was measured as previously described by measuring myocyte diameter at the level of the nucleus in hematoxylin and eosin–stained sections.16
Immunohistochemistry and Immunofluorescence
Briefly, we performed antigen retrieval in rehydrated sections with 1% sodium citrate, after which sections were blocked for 30 minutes using 1% horse serum in PBS, followed by washing in PBS alone for 15 minutes. Samples were then incubated at a 1:100 dilution for 30 minutes against antibody subclasses: IgG3‐FITC (Abcam, Cambridge, MA); IgG1‐FITC (eBioscience, San Diego, CA), IgG2a‐FITC (eBioscience), IgG2b‐FITC (eBioscience), and IgM‐FITC (eBioscience). Then, samples were washed 3 times with PBS for 10 minutes. Finally, each section was incubated for 5 minutes in 3% Sudan Black to eliminate endogenous fluorescence and cover slipped in aqueous mounting media. For dual fluorescence, staining was performed using IgG3‐FITC (Abcam) and B‐cell lymphoma‐2‐associated X protein (BAX)‐TRITC (Santa Cruz Biotechnology, Santa Cruz, CA). Photomicrographs were taken using a Diagnostic Instruments SPOT II digital camera (Diagnostic Instruments, Inc., Sterling Heights, MI) mounted on an Olympus AX70 fluorescent microscope by an observer blinded to the source of each specimen. Preset exposure settings were unchanged for all photomicrographs. Two blinded observers analyzed the photomicrographs, which were later decoded for analysis. Samples were considered positive or negative based on the presence of fluorescence in the sarcolemma. Apoptosis was assessed by immunohistochemistry staining using an anti‐ssDNA/Apostain monoclonal antibody assay (eBiosciences), according to the manufacturer's instructions.
Flow Cytometry Analysis
Blood was obtained by cardiac puncture at the end of each experiment and centrifuged on a Ficoll gradient, and the buffy coat layer was washed twice in PBS. Spleens were removed and prepared as described above (B‐cell reconstitution protocol) for cell labeling. Cells were then stained and incubated for 30 minutes with antibodies to CD22‐PE, CD45‐FITC, CD3‐PE (Santa Cruz Biotechnology), and CD69‐FITC (BioLegend, San Diego, CA), CD19‐FITC, CD86‐PE, and CD80‐FITC (eBioscience). Samples were analyzed by a BD LSRII flow cytometer using FACSDiva software (BD Biosciences, San Jose, CA).
Mean Arterial Pressure Measurement
Blood pressure measurements were taken on conscious animals 3 times each week throughout the protocol using a tail‐cuff CODA noninvasive blood monitoring system (Kent Scientific Corporation, Torrington, CT). This system used 10 cuff inflations and internal software to calculate and record average mean arterial pressures (MAPs). Initial experiments used telemetry monitoring of MAP to validate tail‐cuff measurements, but telemetry was not used routinely because of the invasiveness and inflammatory effects of the implantation.
Magnetic Resonance Imaging
Magnetic resonance imaging (MRI) was performed using a 9.4 Tesla (89 mm bore) superconducting magnet with a Bruker AVANCE console (Bruker Corporation, Billerica, MA) that has live animal imaging capabilities. For mouse imaging, the system is equipped for isoflurane anesthesia administration and cardiac gating, which is performed as a service by the imaging core at our facility. The identities of all animals were blinded before imaging and uncoded after analysis. MRI analysis was performed at the completion of the 35‐day CMP protocol, just before sacrifice.
Analysis of Gene Expression
Total mRNA was isolated from cardiac tissues using RNA‐binding columns (RNaqueous‐4‐PCR; Ambion, Austin, TX), according to the manufacturer's protocols. RNA was immediately reverse transcribed into cDNA using a kit employing random primers (iScript; Bio‐Rad, Hercules, CA), according to the manufacturer's directions. Real‐time reaction mixtures contained 11 μL of H2O, 12 μL of IQ SYBR Green Supermix (Bio‐Rad), 1 μL of primers, and 1 μL of cDNA template (25 μL total). Sense and antisense primers were chosen from the RTPrimerDB public primer database for mouse. Reactions were aliquoted into 96‐well plates, plates were sealed, centrifuged at 500g for 60 seconds, and then samples were amplified for 40 cycles of 10 seconds at 95°C, 30 seconds at 55°C, and 10 seconds at 72°C. Real‐time PCR was performed in a MyIQ5 iCycler (Bio‐Rad). All samples were measured in triplicates and normalized to GAPDH controls run on the same plate for each mouse cDNA sample that was tested. Gene expression levels were calculated by using (2Ct (gene)/2Ct (GAPDH))×1000 to standardize to GAPDH and expressed as relative transcript numbers. Positive gene expression was additionally visualized by conventional PCR and ethidium bromide stained agarose gel electrophoresis using appropriate primer sets.
Plasma Immunoassays
At day 35, mice were sacrificed and blood collected in tubes containing EDTA. Blood was then centrifuged at 5 000 g for 10 minutes, and plasma collected and frozen at −80°C. Cytokine profile was then measured using a ProcartaPlex multiplex immunoassay (eBioscience), according to the manufacturer's instructions, and analyzed with a Luminex 200 instrument (Luminex Corporation, Austin, TX).
In Vitro Experiments
Control and HF mice were produced using the protocol outlined above. At day 35, mice from both groups were sacrificed and B‐cells obtained from spleens as described in the B‐cell reconstitution protocol section. Control B‐cells were stimulated with LPS and CpG ODN (ODN M362) for 12 hours whereas HF‐derived B‐cells sat in media without stimulation. After 12 hours, all tubes were centrifuged and the supernatants collected. These supernatants were cocultured with previously isolated and cultured cardiac fibroblasts, which had reached confluence and were maintained in media (RPMI 1641 with penicillin/streptomycin) supplemented with 10% FCS. All 3 supernatants were added to separate fibroblast cultures along with their respective controls. The supernatant‐stimulated fibroblast reaction was stopped at 6 hours using lysis buffer provided with the RNAqueous RNA isolation kit (Ambion), according to the manufacturer's instructions. cDNA was obtained and reverse‐transcriptase (RT)‐PCR was run for genes associated with proliferation, collagen production, and nuclear activation.
Statistical Analysis
All experiments had 5 animals per group and each experiment was repeated 3 times, making a total of 15 animals per group once all experiments were completed. Data are reported as mean±SD. Analysis was done using GraphPad Prism software (GraphPad Software Inc., La Jolla, CA). ANOVA with Tukey comparison as a post‐test with a 95% CI were used when analyzing more than 3 groups. A P value <0.05 was considered significant.
Results
Cytokine and B‐Cell Activation in a Mouse Model of Nonischemic CMP
As shown in Figure 1A, with the expression of a variety of inflammatory cytokines in failing myocardium and controls, there is increased myocardial expression of proinflammatory cytokines, such as interleukin (IL)‐1β, interferon‐gamma (IFN)‐γ, tumor necrosis factor alpha (TNF‐α) and IL‐6, as well as a reduction in the anti‐inflammatory cytokine IL‐10. Figure 1B depicts the cytokine profile in peripheral blood samples from HF and normal mice. As shown, the only cytokine significantly upregulated is B‐cell activating factor (BAFF). Figure 1C shows the expression of a variety of B‐cell activation markers in splenic B‐cell isolates obtained from HF mice and controls. There was a marked increase in expression of CD69, and CD22, whereas the total B‐cell compartment was unchanged in CMP mice. Figure 1D illustrates myocardial tissue from HF mice and controls that was stained for the presence of immunoglobulins. As shown, antibody deposition in myocardial tissue was primarily of the IgG3 subclass, whereas the other subclasses (IgG1, IgG2a, and IgG2b) were not present.
Figure 1.
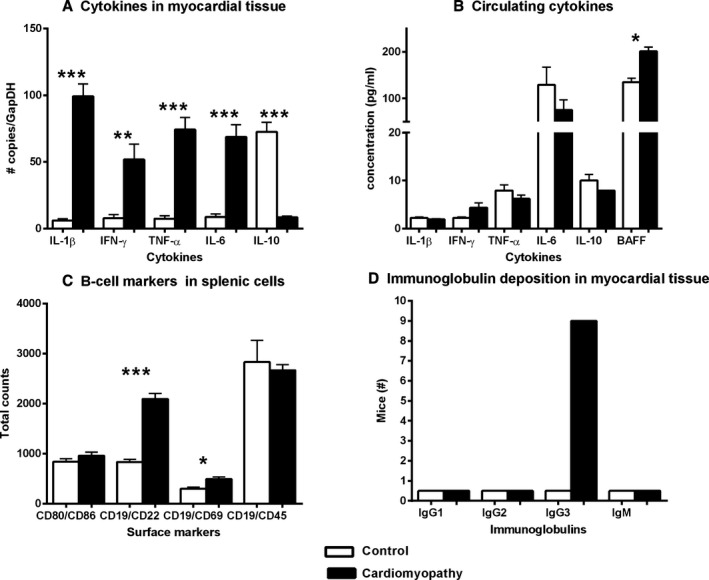
Immune system changes in a mouse model of nonischemic cardiomyopathy. A, Proinflammatory cytokines are upregulated in the myocardial tissue milieu, whereas (B) cytokines in the circulation only show a significant increase in BAFF expression. C, Surface marker profiles of splenic B‐cells of CMP mice compared to controls show an overall increase in activated B‐cells (CD19/CD69), but not total B‐cell numbers, and/or total leucocytes (CD19/CD45) side‐scatter horizontal was used for B‐cell markers (CD19) and side‐scatter vertical was used for other markers (CD86, CD69, and CD45). D, Immunofluorescence assays determined the deposition of immunoglobulins in myocardial tissue: only IgG3 was present and distributed along the sarcolemma. All P values compare controls versus all other groups: *P<0.05; **P<0.01; ***P<0.001. BAFF indicates B‐cell activating factor; CMP indicates cardiomyopathy; IFN‐γ, interferon‐gamma; Ig, immunoglobulin; IL, interleukin; TNF‐α, tumor necrosis factor alpha.
B‐Cells Are Not Required for Hypertensive Response, But Are Necessary to Produce CMP Phenotype
Figure 2 shows the hemodynamic response of control untreated mice and CMP‐treated mice in the following groups: wild type (WT CMP); mice that lack B‐ and T‐cells (SCID CMP); mice treated with an antibody to eliminate CD22+ cells (CD22− CMP); or mice that lack T‐cells (Nude CMP). As shown in Figure 2A, hypertensive response was equal among all the treated groups (controls: mean 75 mm Hg vs 160 mm Hg for all CMP‐treated groups at day 21; P<0.001). Figure 2B shows the percent change in heart weight per tibia length among the CMP‐treated groups. As shown, greater increases in heart weight were observed in the WT CMP and Nude CMP groups (32.2±2.7% and 40.9±15%, respectively) compared to the SCID CMP and CD22− CMP groups (14.6±8% and 15.5±6.6%, respectively). Figure 2C shows left ventricular (LV) ejection fraction (LVEF), as measured by cardiac MRI (cMRI), at the end of the cardiomyopathy protocol. As shown, there was a significant reduction in LVEF in WT CMP and Nude CMP mice compared to controls (49.4±7% and 53±10%, respectively, vs 67±3% in untreated control). The SCID CMP and CD22− CMP groups did not have reduction in LVEF compared to controls (63±4% and 59±7%, respectively, vs 67±3% in untreated controls). Figure 2D shows the changes in LV mass, which followed the same pattern.
Figure 2.
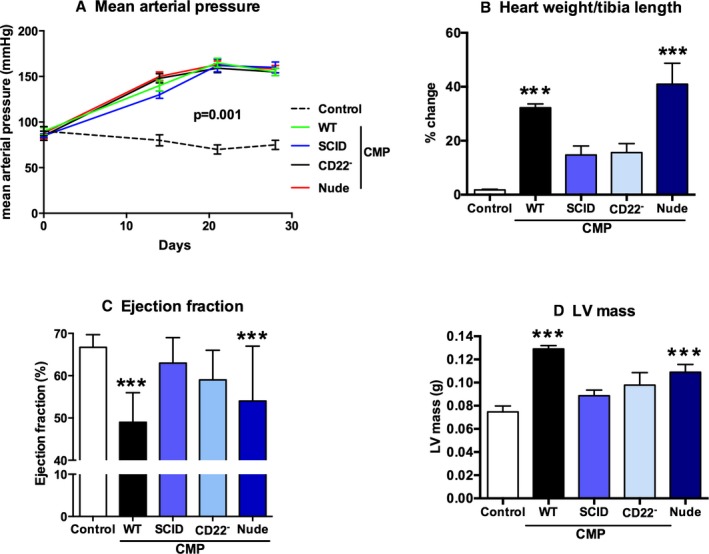
B‐cells are not required for hypertension, but for cardiomyopathy. Shown is the hemodynamic response of untreated mice and cardiomyopathy treated mice in the following groups: wild type (WT CMP); mice that lack B‐ and T‐cells (SCID CMP); mice treated with an antibody to eliminate CD22+ cells (CD22− CMP); or mice that lack T‐cells (Nude CMP). A, Hypertensive response was equal among all the CMP‐treated groups, independent of B‐ and T‐cell compartments, compared to controls. B, As shown by the percent change in heart weight per tibia length, greater increases were observed in the WT CMP and Nude CMP groups compared to the SCID CMP and CD22− CMP groups. C, LVEF, as measured by cMRI at the end of the cardiomyopathy protocol, is significantly reduced in WT CMP and Nude CMP mice compared to controls. The SCID CMP and CD22− CMP groups have a significantly higher LVEF than the WT CMP mice. D, Changes in LV mass followed an inverse pattern to the changes in LVEF, with LV mass elevated in WT CMP compared to controls (n=15 per group). All P values compare controls versus all other groups: *P<0.05; **P<0.01; ***P<0.001.
Figure 3A shows the change in myocyte size in untreated controls and in WT CMP, SCID CMP, CD22− CMP, and Nude CMP groups. Myocyte size for controls was 26.0±3.7 μm. The greatest increase in myocyte size was observed in the WT CMP group (32.9±1.1 μm) and the Nude CMP group (32.3±2.1 μm). For mice in the CD22− CMP and in SCID CMP groups, the increase in myocyte size was significantly lower than that observed in the WT CMP group (28.0±1.4 and 27.8±0.5 μm for CD22− CMP and SCID CMP, respectively) and not significantly different than that of controls. Figure 3B shows the change in collagen content in untreated controls and in WT CMP, SCID CMP, CD22− CMP, and Nude CMP groups. As shown, a large increase in collagen content was observed in WT CMP and Nude CMP (21.8±2.5% and 26.0±7.1% area stained, respectively), whereas only a modest increase in collagen content was observed in the SCID CMP and CD22− CMP groups (12.5±2.8% and 14.7±2.6% area stained, respectively) compared to untreated controls.
Figure 3.
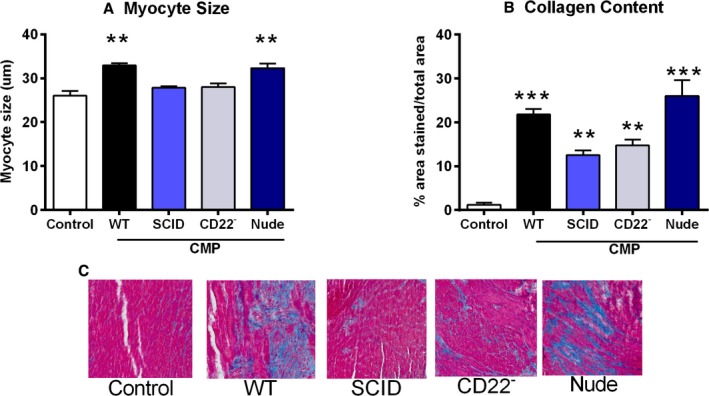
Lack of B‐cells attenuates adverse remodeling in cardiomyopathy. A, The greatest increase in myocyte size compared to untreated controls was observed in the WT CMP and Nude CMP groups, whereas in mice of the CD22− CMP and SCID CMP groups, the increase in myocyte size was insignificant compared to controls. B, A significant increase in collagen content was observed in WT CMP and Nude CMP mice compared to controls, whereas only a modest increase in collagen content was observed in the SCID CMP and CD22− CMP groups (n=15 per group). C, Representative pictures of myocardial tissue sections stained for trichrome in all 5 groups. All P values compare controls versus all other groups: *P<0.05; **P<0.01; ***P<0.001.
The Cardiomyopathy Phenotype Is Restored After B‐Cell Reconstitution
Figure 4 shows the effect of B‐cell reconstitution on SCID mice treated to develop CMP. The hypertensive response was the same in the reconstitution group (SCID+B‐cells CMP) as the other CMP groups (Figure 4A). The figure shows untreated controls or mice treated to produce CMP in the following backgrounds: WT; SCID; and SCID with B‐cell reconstitution. We observed that once we reconstituted B‐cells in SCID mice and induced CMP, myocyte size in the SCID+B‐cells CMP group (28.2±2.6 μm) was increased compared to SCID CMP (23.8±1.8 μm) and similar to WT CMP (28.8±2.1 μm), whereas controls showed minimal change (23.7±0.9 μm) (Figure 4C). This effect was also observed with the percent change in the heart weight to tibia length ratio (SCID+B‐cells CMP: 47±6.1%; SCID CMP: 8±3.2%; WT CMP: 50±4%; and controls: 5±0.1%; Figure 4B) and collagen content (SCID+B‐cells CMP: 29.4±2.7%; SCID CMP: 11.2±1.4%; WT CMP: 36.2±7.8%; and controls: 3.4±1.3% area stained; Figure 4D). Finally, brain natriuretic peptide (BNP) expression was high in the WT CMP group, low in B‐cell‐deficient mice (CD22− or SCID CMP), and elevated in SCID with B‐cell reconstitution compared to controls (Figure 4E).
Figure 4.
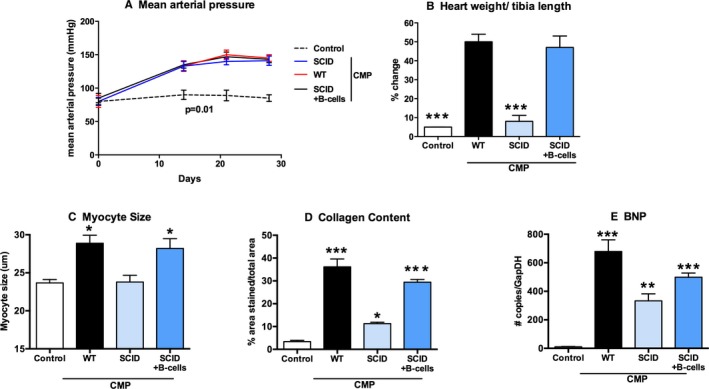
Reconstitution of B‐cells in SCID mice restores the heart failure phenotype. The figure shows untreated controls and mice treated to produce cardiomyopathy in the following backgrounds: WT; SCID; and SCID with B‐cell reconstitution (SCID+B‐cells). Once we reconstituted B‐cells in SCID mice and induced CMP, (A) hypertensive response was similar, (C) myocyte size was increased, and this effect was also observed with the (B) percent change in heart weight to tibia length and (D) collagen content. E, BNP expression was high in WT CMP group, lower in B‐cell‐deficient mice (CD22− CMP [not shown] or SCID CMP), and elevated in SCID mice with B‐cell reconstitution (n=15 per group). All P values compare controls versus all other groups: *P<0.05; **P<0.01; ***P<0.001. BNP indicates brain natriuretic peptide; CMP, cardiomyopathy; SCID, severe combined immune deficiency; WT, wild type.
Intact B‐Cell Function in CMP Is Associated With Increased Levels of Inflammatory Cytokines, IgG Deposition, and Bax Expression in Myocardium
Figure 5 shows gene expression profiling of inflammatory markers in cardiac tissue of control and CMP‐treated mice measured at the end of the treatment protocol. As shown, the WT CMP group was characterized by increases in IL‐1β (Figure 5A), TNF‐α (Figure 5B), IL‐6 (Figure 5C), and IL‐10 (Figure 5D) expression, relative to the control group. In the groups with B‐cell‐deficient mice, SCID CMP, and CD22− CMP, there was reduced expression of these genes. The SCID+B‐cell CMP group showed increased expression of TNF‐α, IL‐6, and IL‐1β. IL‐10 expression was significantly lower in the WT CMP group and elevated to levels comparable to the WT control in the SCID CMP and CD22− CMP groups (Figure 5D).
Figure 5.
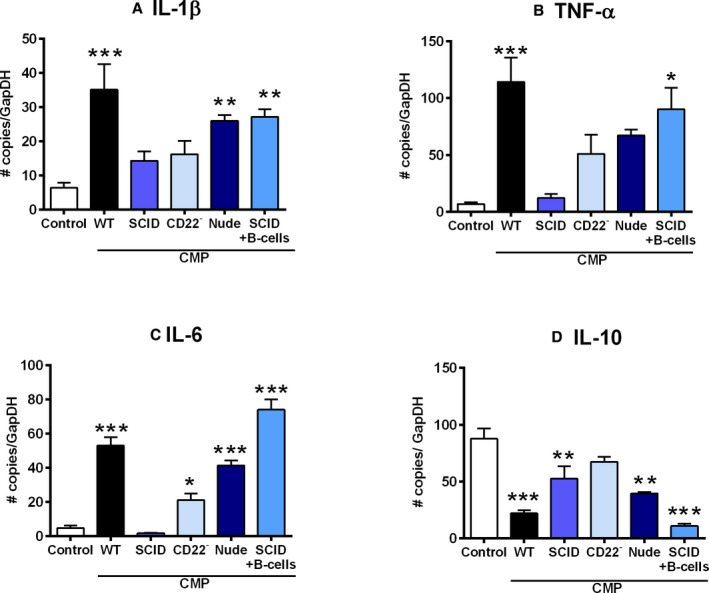
Inflammatory cytokine profile in B‐cell‐deficient mice and B‐cell‐reconstituted mice compared to controls with CMP. Quantitative gene expression of inflammatory markers in cardiac tissues at the end of the treatment protocol, the WT CMP group was characterized by increase in (A) IL‐1β, (B) TNF‐α, and (C) IL‐6 expression compared to untreated controls. In the groups with B‐cell‐deficient mice (SCID CMP, CD22− CMP, and Nude CMP), there was reduced expression of these genes. After B‐cell reconstitution in the SCID CMP group, expression of TNF‐α, IL‐6, and IL‐1β increased. D, IL‐10 expression was significantly reduced in the WT CMP group and increased to levels comparable to the untreated controls in the SCID CMP and CD22− CMP groups. All P values compare controls versus all other groups: *P<0.05; **P<0.01; ***P<0.001. CMP indicates cardiomyopathy; IL, interleukin; SCID, severe combined immune deficiency; TNF‐α, tumor necrosis factor alpha; WT, wild type.
Figure 6A shows analysis of IgG3 and Bax in myocardium of untreated control mice and CMP groups. IgG3 protein expression was present in most mice in the WT CMP group (7 of 8). Fewer mice in the B‐cell‐deficient groups were positive for IgG3: 0 of 8 of SCID CMP and 3 of 8 CD22− CMP mice. In contrast, higher numbers of mice were positive for IgG3 in the T‐cell‐deficient Nude CMP (6 of 8) and SCID+B‐cell CMP (5 of 8) groups. Bax expression was present in most mice in the WT CMP group (7 of 8). Fewer mice in the B‐cell‐deficient groups were positive for Bax: 0 of 8 of SCID CMP and 1 of 8 of CD22− CMP mice. In contrast, higher numbers of mice in the T‐cell‐deficient Nude CMP (6 of 8) and SCID+B‐cell (5 of 8) groups were positive for Bax. In Figure 6B, the association of anti‐ssDNA and IgG3 deposition is depicted. These findings support the hypothesis that cardiomyocyte cell death was occurring by apoptotic mechanisms and not necrosis. In Figure 6C, 2×2 tables demonstrate the association of IgG3 staining with Bax and anti‐ssDNA in the myocardial tissue samples of all groups studied, which emphasizes that almost all had concomitant staining, but apoptosis was never present without IgG3, suggesting the important role that IgG3 deposition plays in this CMP model. Figure 7 shows representative images where BAX‐positive areas stain red and IgG3‐positive areas stain green, with dual staining in the bottom panel in yellow.
Figure 6.
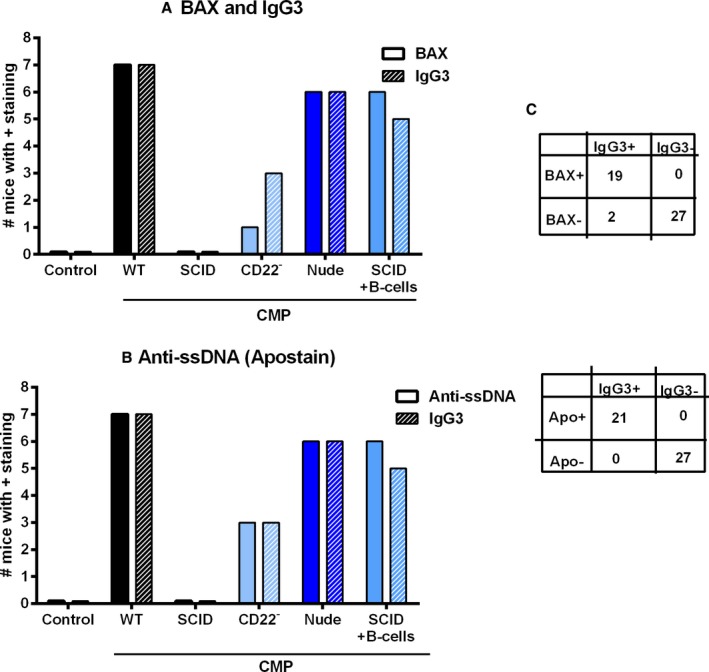
Markers of antibody deposition and activation of apoptosis in myocardial tissue of control and CMP mice. A, IgG3 expression was present in most mice in the WT CMP group. Fewer mice in the B‐cell‐deficient groups, SCID CMP and CD22− CMP, were positive for IgG3. In contrast, higher numbers of mice in the T‐cell‐depleted but with intact B‐cell function groups, Nude CMP or SCID with B‐cell reconstitution CMP, were positive for IgG3. Bax expression in myocardium of control mice and various CMP‐treated groups is also shown, with a pattern of expression similar to IgG3. C, The 2×2 tables show the distribution of IgG3 and Bax or IgG3 and anti‐ssDNA staining in all the pooled analyzed myocardial tissue samples (n=8/group). B. A similar pattern was observed when compared to Ant‐ssDNA staining.
Figure 7.
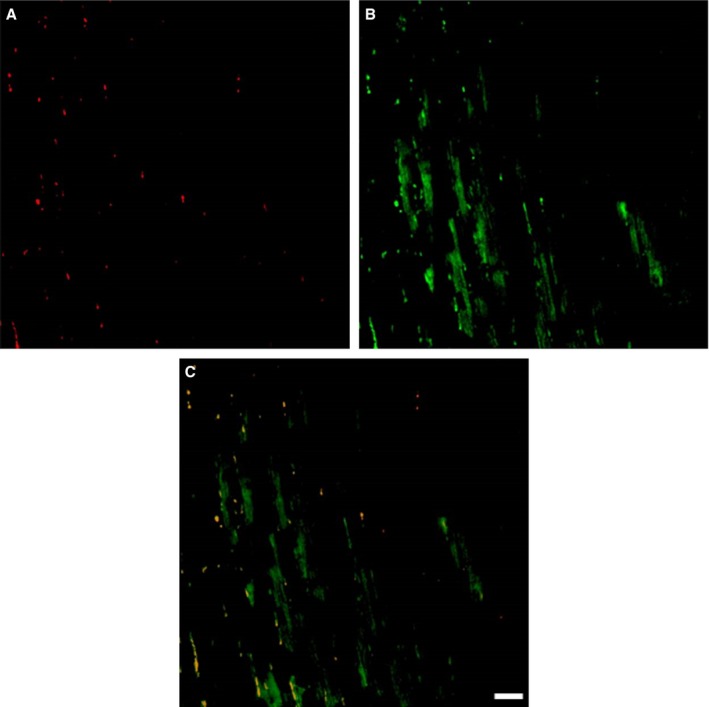
Colocalization of IgG3 with markers of apoptosis. Figure shows representative images where BAX‐positive areas stained red (A) and IgG3‐positive areas stained green (B), with dual staining in the bottom panel in yellow (C). Scale bar, 100 μm. BAX indicates B‐cell lymphoma‐2‐associated X protein; Ig, immunoglobulin.
B‐Cell‐Mediated Mechanisms Are Associated With Increased Fibroblast Proliferation
Figure 8 shows the change in gene expression of a variety of markers in cardiac fibroblasts after stimulation with supernatants of either untreated B‐cells or activated B‐cells with LPS or CpG ODN or untreated B‐cells obtained from HF mice (HF B‐cells). As shown, B‐cell supernatants of activated B‐cells and HF B‐cells were capable of stimulating cardiac fibroblast to increase expression of nuclear factor kappa B (NF‐κB; 2‐fold), proliferating cell nuclear antigen (PCNA; 4‐fold), Ki‐67 (6‐fold), IL‐6 (12‐fold), and collagen (2‐fold). Of interest, collagen expression was highest after fibroblast stimulation with HF B‐cell supernatant (4‐fold increase) compared to control.
Figure 8.

Markers of proliferation and collagen expression in isolated cardiac fibroblasts after stimulation with activated B‐cell products. Figure shows gene expression profile in cultured cardiac fibroblasts where there is increased expression of NF‐κB, PCNA, ki‐67, IL‐6, and collagen (Coll) I and III with either HF B‐cell supernatant or stimulated B‐cell (LPS and CpG‐ODN) supernatant. HF indicates heart failure; IL, interleukin; NF‐κB, nuclear factor kappa B; PCNA, proliferating cell nuclear antigen; RT‐PCR, reverse‐transcriptase polymerase chain reaction.
Discussion
This study demonstrates that in a mouse model based on Ang‐II and l‐NAME, there is a consistent CMP phenotype characterized by hypertension and LV dysfunction with an associated activation of the inflammatory cascade, as shown by myocardial expression of inflammatory cytokines, IgG3 antibody deposition, upregulation of apoptotic signals in myocardial tissue, and peripheral B‐cell activation. By utilizing mice with different genetic modifications as well as selectively depleting B‐cells from the periphery, we demonstrated that, for full expression of the CMP phenotype, B‐cells are required. The role of B‐cells in CMP development was independent of the hypertensive response and associated with attenuation of the inflammatory response. These observations demonstrate that, in this model of nonischemic cardiomyopathy, B‐cells play a central role in the establishment of cardiac injury, independent of the hypertensive response.
Cardiac injury, as induced in this model, is characterized by myocyte hypertrophy, extensive collagen deposition, LV dilatation with reduced ejection fraction, and increases in markers of disease (ie, BNP). In the initial phase, there is a compensatory hypertrophy, followed by LV dilatation and a marked decrease in ejection fraction at week 5 of the CMP protocol. This model with the use of l‐NAME and Ang‐II demonstrates a severe phenotype of CMP with a nonsurgical approach and a human‐like nonischemic CMP. Other characteristics of this model, including edema, decreased. In addition, the profile of inflammatory cytokines was consistent with observations in humans, where there is elevation of TNF‐α, INF‐γ, IL‐1β, and IL‐6 and a decrease in IL‐10, an anti‐inflammatory cytokine, in HF. In this animal model, the initial injury was not infectious, inflammatory, or ischemic, and thus the observations suggest that, after hypertensive injury, a series of mechanisms are activated that ultimately lead to CMP. But of great interest was the observation that mice depleted of B‐cells that developed an intact hypertensive response did not have the same degree of adverse remodeling observed in immune‐competent mice. The implications of this observation are important because it will direct research to further characterize and intervene in these inflammatory responses, but also suggest that hypertension, as a sole hemodynamic insult, may not be sufficient to fully promote cardiac injury. Alternatively, injury mediated by a hypertension‐signaling pathway is magnified by the addition of a second pathway that induces increased expression of BAFF, which, in turn, leads to B‐cell activation and a cascade of B‐cell responses.
With regard to the role of B‐cells in the development of nonischemic CMP, the findings of the present study are important because we demonstrated that, even when the initial injury is not immunologically mediated, activation of B‐cells was necessary to fully express the extent of myocardial injury. This suggests that B‐cell‐mediated responses, whether they result from antibody production or B‐cell‐produced cytokines and B‐cell interactions, cause cardiac injury or amplify an inflammatory response that is detrimental to cardiac function. In fact, the recognition that, in animals with intact B‐cell function and the phenotypic expression of HF, there was deposition of IgG3 in the myocardium and expression of Bax, a proapoptotic molecule, suggests that a pathway of cell injury, in addition to fibrotic response, accompanies the remodeling process.
Limited information exists on the role of B‐cells in the development of CMP, with the exception of murine and human models, where the mechanism of cardiac injury is immunologically mediated, such as animal models of virally mediated injury or cases of acute myocarditis and some forms of dilated CMP in humans.17, 18 But new observations provide credence to the role of B‐cells in pathological processes in the cardiovascular system. Zouggari et al. demonstrated, in a murine model of MI, that B‐cells were activated and responsible for monocyte migration and amplification of myocardial injury.19 Additionally, in models of atherosclerosis, B‐cells contribute to plaque formation by antibody production, cytokine release, and cell‐mediated interactions.20 The observations described above, as well as the one described in these studies, are consistent with the contribution of B‐cells in conditions wherein the initial cardiac injury is not immunologically mediated and represent a major contribution of this investigational area. In addition, we now provide evidence that soluble factors produced from B‐cells obtained from HF mice increase collagen expression in cardiac fibroblasts.
In this study, we utilized SCID mice, which lack T‐ and B‐cells, as well as Nude mice, which lack T‐cells, and treated them to induce CMP. Furthermore, we treated WT mice with an antibody to eliminate CD22+‐positive cells, a marker expressed on mature B‐cells, as a way to eliminate B‐cells from the periphery. The consistent findings presented in this study demonstrate that mice without B‐cells, whether genetically determined as in the SCID mice or by antibody depletion of CD22+ cells, did not develop LV dysfunction, myocyte hypertrophy, and fibrosis; nor did they have evidence of IgG3 deposition or Bax expression in the myocardium. Additionally, experiments wherein we reconstituted SCID mice with CMP B‐cells and subsequently treated to produce CMP demonstrated that the failing phenotype was present.
There are several potential contributions of B‐cells to cardiac injury in this model. First, dysregulation of B‐cell subpopulations (B1 vs B2 and Bregs) may occur, therefore activating proinflammatory pathways that promote cell hypertrophy and fibrosis with increased expression of TNF‐α, INF‐γ, IL‐1β, and IL‐6 and blunting of anti‐inflammatory pathways with reductions in IL‐10.21, 22 Consistent with this hypothesis are the observations from our studies with B‐cell supernatants obtained from activated B‐cells and from HF B‐cells wherein after incubation with cardiac fibroblasts, there was increased expression of collagen gene. Second, the presence of IgG3 in failing myocardium is consistent with the possibility of cell injury being mediated by nonspecific activation of Fc gamma receptor (FcgR), a receptor for the Fc portion of IgG, and triggering of apoptotic pathways,23 as suggested by an increase in Bax expression. Finally, IgG3 may recognize specific receptors of antigenic determinants that decrease cardiac cell function or activate functional receptors that promote cell death.24 Germane to this discussion are the observations of a study in human subjects with advanced HF in which we found that 70% of the myocardial samples studied, regardless of the etiology, were positive for anticardiac antibodies and at least 50% also were positive for activated complement components, an association suggesting that antibody formation in advanced HF, regardless of the initial mechanisms of cardiac injury, may participate in the progression of the disease state.10 The consistency of the observations in failing human myocardium and in this murine model of nonischemic CMP strongly suggests that B‐cells play a central role in disease progression.
In this murine model of nonsurgically induced nonischemic CMP, intact B‐cells were required for full expression of injury. This implies that B‐cell‐mediated responses, whether attributable to antibody formation or B‐cell‐produced cytokines, are activated after myocardial injury, which, in turn, contributes to myocyte hypertrophy, fibrosis, and activation of apoptotic pathways that amplify the initial cardiac injury. The precise mechanism by which B‐cell activation occurs is unknown and remains the focus of continued investigations. These findings support the idea that modulation of B‐cell‐mediated responses, as, for example, by the administration of an anti‐B‐cell antibody as performed in these experiments, may prevent the progression of HF and give credence to continued clinical investigations aimed at preventing B‐cell activation in HF.
Disclosures
None.
Acknowledgments
The authors thank Gerd Brunner, PhD and Mohamad Ghosn, PhD (both supported by T32 HL07812), for acquiring the mouse cMRI images. This work was supported, in part, by RO1 HL63090 (Joel Morrisett, PhD).
(J Am Heart Assoc. 2016;5:e002484 10.1161/JAHA.115.002484)
References
- 1. Suzuki H, Sato R, Sato T, Shoji M, Iso Y, Kondo T, Shibata M, Koba S, Katagiri T. Time course of changes in the levels of interleukin 6 in acutely decompensated heart failure. Int J Cardiol. 2005;100:415–420. [DOI] [PubMed] [Google Scholar]
- 2. Milani RV, Mehra MR, Endres S, Eigler A, Cooper ES, Lavie CJ Jr, Ventura HO. The clinical relevance of circulating tumor necrosis factor‐alpha in acute decompensated chronic heart failure without cachexia. Chest. 1996;110:992–995. [DOI] [PubMed] [Google Scholar]
- 3. Peschel T, Schonauer M, Thiele H, Anker SD, Schuler G, Niebauer J. Invasive assessment of bacterial endotoxin and inflammatory cytokines in patients with acute heart failure. Eur J Heart Fail. 2003;5:609–614. [DOI] [PubMed] [Google Scholar]
- 4. Sato Y, Takatsu Y, Kataoka K, Yamada T, Taniguchi R, Sasayama S, Matsumori A. Serial circulating concentrations of C‐reactive protein, interleukin (IL)‐4, and IL‐6 in patients with acute left heart decompensation. Clin Cardiol. 1999;22:811–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Torre‐Amione G, Orrego CM, Khalil N, Kottner‐Assad C, Leveque C, Celis R, Youker KA, Estep JD. Therapeutic plasma exchange a potential strategy for patients with advanced heart failure. J Clin Apher. 2010;25:323–330. [DOI] [PubMed] [Google Scholar]
- 6. Limas CJ, Goldenberg IF, Limas C. Autoantibodies against beta‐adrenoceptors in human idiopathic dilated cardiomyopathy. Circ Res. 1989;64:97–103. [DOI] [PubMed] [Google Scholar]
- 7. Staudt A, Mobini R, Fu M, Grosse Y, Stangl V, Stangl K, Thiele A, Baumann G, Felix SB. Beta(1)‐adrenoceptor antibodies induce positive inotropic response in isolated cardiomyocytes. Eur J Pharmacol. 2001;423:115–119. [DOI] [PubMed] [Google Scholar]
- 8. Doesch AO, Mueller S, Nelles M, Konstandin M, Celik S, Frankenstein L, Goeser S, Kaya Z, Koch A, Zugck C, Katus HA. Impact of troponin I‐autoantibodies in chronic dilated and ischemic cardiomyopathy. Basic Res Cardiol. 2011;106:25–35. [DOI] [PubMed] [Google Scholar]
- 9. Baba A, Yoshikawa T, Mitamura H, Akaishi M, Ogawa S. [Autoantibodies against sarcolemmal Na‐K‐ATPase in patients with dilated cardiomyopathy: autoimmune basis for ventricular arrhythmias in patients with congestive heart failure]. J Cardiol. 2002;39:50–51. [PubMed] [Google Scholar]
- 10. Youker KA, Assad‐Kottner C, Cordero‐Reyes AM, Trevino AR, Flores‐Arredondo JH, Barrios R, Fernandez‐Sada E, Estep JD, Bhimaraj A, Torre‐Amione G. High proportion of patients with end‐stage heart failure regardless of aetiology demonstrates anti‐cardiac antibody deposition in failing myocardium: humoral activation, a potential contributor of disease progression. Eur Heart J. 2014;35:1061–1068. [DOI] [PubMed] [Google Scholar]
- 11. Oliveira GH, Brann CN, Becker K, Thohan V, Koerner MM, Loebe M, Noon GP, Torre‐Amione G. Dynamic expression of the membrane attack complex (MAC) of the complement system in failing human myocardium. Am J Cardiol. 2006;97:1626–1629. [DOI] [PubMed] [Google Scholar]
- 12. Aukrust P, Gullestad L, Lappegard KT, Ueland T, Aass H, Wikeby L, Simonsen S, Froland SS, Mollnes TE. Complement activation in patients with congestive heart failure: effect of high‐dose intravenous immunoglobulin treatment. Circulation. 2001;104:1494–1500. [DOI] [PubMed] [Google Scholar]
- 13. Gullestad L, Aass H, Fjeld JG, Wikeby L, Andreassen AK, Ihlen H, Simonsen S, Kjekshus J, Nitter‐Hauge S, Ueland T, Lien E, Froland SS, Aukrust P. Immunomodulating therapy with intravenous immunoglobulin in patients with chronic heart failure. Circulation. 2001;103:220–225. [DOI] [PubMed] [Google Scholar]
- 14. Dandel M, Wallukat G, Englert A, Lehmkuhl HB, Knosalla C, Hetzer R. Long‐term benefits of immunoadsorption in beta(1)‐adrenoceptor autoantibody‐positive transplant candidates with dilated cardiomyopathy. Eur J Heart Fail. 2012;14:1374–1388. [DOI] [PubMed] [Google Scholar]
- 15. Oestreicher EM, Martinez‐Vasquez D, Stone JR, Jonasson L, Roubsanthisuk W, Mukasa K, Adler GK. Aldosterone and not plasminogen activator inhibitor‐1 is a critical mediator of early angiotensin II/NG‐nitro‐l‐arginine methyl ester‐induced myocardial injury. Circulation. 2003;108:2517–2523. [DOI] [PubMed] [Google Scholar]
- 16. Bruckner BA, Stetson SJ, Perez‐Verdia A, Youker KA, Radovancevic B, Connelly JH, Koerner MM, Entman ME, Frazier OH, Noon GP, Torre‐Amione G. Regression of fibrosis and hypertrophy in failing myocardium following mechanical circulatory support. J Heart Lung Transplant. 2001;20:457–464. [DOI] [PubMed] [Google Scholar]
- 17. Latva‐Hirvela J, Kyto V, Saraste A, Eriksson S, Vuorinen T, Pettersson K, Saukko P. Development of troponin autoantibodies in experimental coxsackievirus b3 myocarditis. Eur J Clin Invest. 2009;39:457–462. [DOI] [PubMed] [Google Scholar]
- 18. Caforio AL, Tona F, Bottaro S, Vinci A, Dequal G, Daliento L, Thiene G, Iliceto S. Clinical implications of anti‐heart autoantibodies in myocarditis and dilated cardiomyopathy. Autoimmunity. 2008;41:35–45. [DOI] [PubMed] [Google Scholar]
- 19. Zouggari Y, Ait‐Oufella H, Bonnin P, Simon T, Sage AP, Guerin C, Vilar J, Caligiuri G, Tsiantoulas D, Laurans L, Dumeau E, Kotti S, Bruneval P, Charo IF, Binder CJ, Danchin N, Tedgui A, Tedder TF, Silvestre JS, Mallat Z. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat Med. 2013;19:1273–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tsiantoulas D, Diehl CJ, Witztum JL, Binder CJ. B cells and humoral immunity in atherosclerosis. Circ Res. 2014;114:1743–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yokoyama T, Nakano M, Bednarczyk JL, McIntyre BW, Entman M, Mann DL. Tumor necrosis factor‐alpha provokes a hypertrophic growth response in adult cardiac myocytes. Circulation. 1997;95:1247–1252. [DOI] [PubMed] [Google Scholar]
- 22. Lund FE. Cytokine‐producing B lymphocytes‐key regulators of immunity. Curr Opin Immunol. 2008;20:332–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Staudt Y, Mobini R, Fu M, Felix SB, Kuhn JP, Staudt A. Beta1‐adrenoceptor antibodies induce apoptosis in adult isolated cardiomyocytes. Eur J Pharmacol. 2003;466:1–6. [DOI] [PubMed] [Google Scholar]
- 24. Jane‐wit D, Altuntas CZ, Johnson JM, Yong S, Wickley PJ, Clark P, Wang Q, Popovic ZB, Penn MS, Damron DS, Perez DM, Tuohy VK. Beta 1‐adrenergic receptor autoantibodies mediate dilated cardiomyopathy by agonistically inducing cardiomyocyte apoptosis. Circulation. 2007;116:399–410. [DOI] [PubMed] [Google Scholar]