

Abstract
Background
The homologous to the E6‐AP carboxyl terminus (HECT)–type ubiquitin E3 ligase ITCH is an enzyme that plays a pivotal role in posttranslational modification by ubiquitin proteasomal protein degradation. Thioredoxin‐interacting protein (TXNIP) is a negative regulator of the thioredoxin system and an endogenous reactive oxygen species scavenger. In the present study, we focused on the functional role of ubiquitin E3 ligase ITCH and its interaction with TXNIP to elucidate the mechanism of cardiotoxicity induced by reactive oxygen species, such as doxorubicin and hydrogen peroxide.
Methods and Results
Protein interaction between TXNIP and ITCH in cardiomyocyte was confirmed by immunoprecipitation assays. Overexpression of ITCH increased proteasomal TXNIP degradation and augmented thioredoxin activity, leading to inhibition of reactive oxygen species generation, p38 MAPK, p53, and subsequent intrinsic pathway cardiomyocyte apoptosis in reactive oxygen species–induced cardiotoxicity. Conversely, knockdown of ITCH using small interfering RNA inhibited TXNIP degradation and resulted in a subsequent increase in cardiomyocyte apoptosis. Next, we generated a transgenic mouse with cardiac‐specific overexpression of ITCH, called the ITCH‐Tg mouse. The expression level of TXNIP in the myocardium in ITCH‐Tg mice was significantly lower than WT littermates. In ITCH‐Tg mice, cardiac dysfunction and remodeling were restored compared with WT littermates after doxorubicin injection and myocardial infarction surgery. Kaplan–Meier analysis revealed that ITCH‐Tg mice had a higher survival rate than WT littermates after doxorubicin injection and myocardial infarction surgery.
Conclusion
We demonstrated, for the first time, that ITCH targets TXNIP for ubiquitin‐proteasome degradation in cardiomyocytes and ameliorates reactive oxygen species–induced cardiotoxicity through the thioredoxin system.
Keywords: ITCH, thioredoxin‐interacting protein, ubiquitin proteasome system
Subject Categories: Basic Science Research, Oxidant Stress, Heart Failure, Animal Models of Human Disease
Introduction
The development of heart failure caused by myocardial infarction (MI) and doxorubicin (Dox)–induced cardiotoxicity is closely associated with progressive left ventricular (LV) remodeling in response to oxidative stress.1 The major cause of excessive oxidative stress in heart failure is considered to be the increase in intracellular reactive oxygen species (ROS) relative to antioxidant defense.2
The thioredoxin system exists ubiquitously. It is expressed in almost eukaryotic cells and maintains homeostasis in response to ROS; it is composed of antioxidant thioredoxin and its endogenous inhibitor thioredoxin‐interacting protein (TXNIP).3 Thioredoxin contains a dithiol/disulfide motif in the redox‐active site and serves as a ROS scavenger.4 In addition, TXNIP acts as a negative regulator of the thioredoxin system through formation of disulfide bonds to thioredoxin active site.5, 6 It was reported that TXNIP expression is suppressed in response to oxidative stress; in turn, this suppression enhances the thioredoxin activity in cardiomyocytes to maintain cellular ROS homeostasis.6 Nevertheless, the mechanism by which oxidative stress suppresses TXNIP in cardiomyocytes is still unclear.
Accumulating evidence demonstrated that posttranslational modifications by the ubiquitin proteasome system play an important role in regulating the complex cell signaling processes fundamental to cardiovascular disease.7, 8, 9 Although the role of the homologous to the E6‐AP carboxyl terminus (HECT)–type E3 ligase in the development of human disease was noted, there has been no basic research to reveal the relationship between HECT‐type E3 ligase and cardiovascular disease, until now. The ubiquitin E3 ligase ITCH was originally identified by genetic analysis of a mutant mouse with aberrant immunological phenotypes and constant scratching of the skin.10 ITCH is a 113‐kDa protein composed of 4 domains: a proline‐rich motif, a Ca2+‐dependent phospholipid‐binding C2 domain, a WW domain, and a HECT domain. The WW domain recognizes the prorich PPXY consensus sequence in substrate proteins, including TXNIP, and the HECT domain attaches ubiquitin to substrate proteins. Polyubiquitylated substrate proteins undergo ubiquitin proteasomal degradation in proteasome.11 An interaction between ITCH and its substrate proteins was reported to be related to cell viability,12, 13 and a recent report demonstrated that ITCH targets TXNIP for ubiquitin proteasomal degradation in 293T and U2OS cells.14
We hypothesized that ITCH maintains cellular ROS homeostasis through TXNIP degradation, which causes an increase in thioredoxin activity. The development of heart failure might be a result of insufficient ITCH‐dependent TXNIP degradation in response to pathological accumulation and excessive production of ROS.
To clarify the potential roles of ITCH in the thioredoxin system and in ROS‐induced cardiotoxicity, we performed overexpression and knockdown studies of ITCH in cardiomyocytes and generated cardiac‐specific ITCH transgenic mice. The aims of the present study were to examine (1) whether ITCH targets TXNIP for ubiquitin proteasomal degradation and protects cells from subsequent apoptosis and cardiotoxicity induced by ROS such as Dox and hydrogen peroxide (H2O2) and (2) whether ITCH transgenic (ITCH‐Tg) mice are able to protect cardiac function and improve survival rate after Dox injection and MI surgery.
Methods
Materials and Reagents
Plasmids and small interfering RNAs
The pRK5‐hem agglutinin–ITCH, or HA‐ITCH, was a kind gift from Dr H. Kamata (Hiroshima University, Japan). Empty vector pcDNA3.1‐positive, Stealth select small interfering RNA (siRNA) ITCH and Stealth RNAi siRNA Negative Control Med GC Duplex #2 were purchased from Invitrogen Corp. The pcDNA3‐FLAG‐thioredoxin was purchased from Addgene.
Antibodies
Mouse monoclonal anti‐TXNIP, rabbit monoclonal anti‐TXNIP, rabbit polyclonal anti‐HA‐probe, mouse monoclonal anti‐HA‐probe, and mouse monoclonal antiubiquitin were purchased from Santa Cruz Biotechnology. Mouse monoclonal anti‐ITCH was purchased from BD Biosciences. Rabbit polyclonal anti–cleaved caspase‐3, rabbit polyclonal anti–precaspase‐3, rabbit polyclonal anti–cleaved caspase‐9, rabbit polyclonal anti–phospho‐p38 MAPK, anti–p38 MAPK, rabbit polyclonal anti–phospho‐p53, mouse monoclonal anti‐p53, rabbit polyclonal anti–Bcl‐2, rabbit polyclonal anti–p22‐phox and rabbit polyclonal anti–β‐tubulin antibody were purchased from Cell Signaling Corp. Anti‐FLAG antibody was purchased from Sigma‐Aldrich.
Reagents
Dox hydrochloride and H2O2 were purchased from Wako Corp. MG132 was obtained from Enzo Life Sciences.
Cardiomyocyte Isolation and Culture and Treatments
The animals were handled according to the animal welfare regulations of Yamagata University, and the study protocol was approved by the animal subjects committee of Yamagata University. The investigation was conducted in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health.
Cultured rat neonatal cardiomyocytes were prepared, as described previously.15 Briefly, ventricles were obtained from Sprague‐Dawley rats aged 1 or 2 days, and cardiomyocytes were isolated by digestion with collagenase. Cardiomyocytes were kept in fatal bovine serum–supplemented medium.
At 4 days after seeding in culture medium, the medium was changed, and treatment with Dox (0.5 μmol/L) or H2O2 (100 μmol/L) was immediately started. The cells were incubated for 3, 6, 12, and 24 hours.
Western Blot Analysis
Cardiomyocytes were lysed in ice‐cold lysis buffer, and the proteins were extracted, as reported previously.15 Equal amounts of protein were subjected to 10% or 14% SDS‐PAGE and transferred to polyvinylidene difluoride membranes. Immunoreactive bands were detected by using an enhanced chemiluminescence kit (Amersham Bioscineces). Protein expression levels were normalized to that of β‐tubulin.
Total RNA Extraction
Total RNA was extracted from cardiomyocytes using TRIzol, as described previously.15 First‐strand cDNA was synthesized from a 1‐μg RNA sample using oligo (dT) primers (Sigma Aldrich, MO, USA) and Superscript III reverse transcriptase.
Real‐Time Reverse Transcription Polymerase Chain Reaction (PCR)
Real‐time PCR was performed with Light Cycler DNA Master SYBR Green I in a 20‐μL volume reaction using Light Cycler (Roche Diagnostics Japan). The following PCR primers were used: for ITCH, 5′‐TAGGCCTCTGAATACTGTA‐3′ (forward) and 5′‐TATCCCATGTTGTCGCT‐3′ (reverse); for TXNIP, 5′‐CAAGTTCGGCTTTGAGCTTC‐3′ (forward) and 5′‐GCCATTGGCAAGGTAAGTGT‐3′ (reverse). Expression levels of mRNA were normalized by glyceraldehyde‐3‐phosphate dehydrogenase.
Immunoprecipitation
Immunoprecipitation was performed, as described previously.16 Protein extracts were prepared in modified RIPA buffer. Cell extracts were precleared with protein A/G beads for 30 minutes. After cell extracts were centrifuged by 4500g for 2 minutes, supernatants were incubated with 2 μL antibody overnight at 4°C. After the incubation, supernatants were added to protein A/G beads and incubated for 2 hours at 4°C. Pellets were washed 4 times with RIPA buffer and resuspended in RIPA lysis buffer. Samples were subjected to 10% SDS‐PAGE.
Overexpression and Knockdown of ITCH in Isolated Rat Neonatal Cardiomyocytes
Plasmids were transfected with Lipofectamine 3000 (Invitrogen), according to the manufacturer's protocol. Knockdown of ITCH was performed by siRNA and an inactivated viral genome‐free HVJ envelope vector (GenomeOne NEO; Ishihara Sangyo Kaisha LTD), according to the manufacturer's instructions.16 Cardiomyocytes were cultured in 6‐well plates and transfected with plasmid or siRNA. After culturing in Opti‐MEM (Invitrogen) for 8 hours, medium was changed and experiments were started.
Assessment of Superoxide Generation
Cardiomyocytes were cultured on glass coverslips and stained with 10 μmol/L DHE (Sigma‐Aldrich) at 37°C for 30 minutes, according to the manufacturer's protocol. DAPI was performed to normalize cell number. The heart tissue was immediately frozen in liquid nitrogen with optimal cutting temperature compound and sectioned at 3‐μm thickness. The section was incubated with 10 μmol/L DHE at 37°C for 30 minutes.17 The mean DHE fluorescent intensity of myocardium in 10 randomly selected fields in each section was quantified with ImageJ software (National Institutes of Health) under a microscope (BX50; Olympus).
Insulin Disulfide Reactivity Assay
Thioredoxin activity was evaluated with insulin disulfide reduction assay, as previously reported.5, 6 Cell extracts were incubated at 37°C for 20 minutes with 2 μL DTT activation buffer composed of 50 mmol/L HEPES (pH 7.6), 1 mmol/L EDTA, 1 mg/mL BSA, and 2 mmol/L DTT. Next, 40 μL reaction buffer containing 200 μL 1 mol/L HEPES (pH 7.6), 40 μL 0.2 mol/L EDTA, 40 μL NADPH (40 mg/mL), and 500 μL insulin (10 mg/mL) was added. The reaction was started by the addition of 10 μL rat thioredoxin reductase and incubated for 20 minutes at 37°C. The reaction was terminated by the addition 0.5 mL 6 mol/L guanidine‐HCl and 1 mmol DTNB, and absorbance at 412 nm was measured spectroscopically.
Cardiac Cell Apoptosis Assay
Terminal deoxynucleotidyl‐transferase‐mediated 2′‐deoxyuridine‐5′‐triphosphate nick‐end labeling (TUNEL) staining was performed with a commercially available kit for detecting end‐labeled DNA, according to the manufacturer's instructions (Roche Applied Science). In in vitro study, cultured cardiomyocytes were fixed by 4% paraformaldehyde 12 hours after Dox stimulation and stained with the TUNEL kit. Following this, DAPI staining was performed to normalize the cell numbers across groups. Approximately 400 to 600 nuclei from random fields were analyzed for each sample. In in vivo study, mouse hearts were excised and fixed with a 10% solution of formalin in PBS 7 days after Dox injection. The hearts were embedded in paraffin and serially cut from the apex to the base. Samples were stained with TUNEL and DAPI, according to the manufacturers' instructions. The percentages of TUNEL‐positive cells were determined by counting 10 random fields per section under a microscope (BX50; Olympus).
Capase‐3 activity in myocardial tissue was measured with an APOPCYTO Capase‐3 Colorimetric Assay kit (MBL) that recognizes the sequence DEVD, according to the manufacturer's instructions.
Dox‐Induced Cardiotoxicity In Vivo
Both wild‐type (WT) and ITCH‐Tg mice were randomly assigned to the control group and the Dox‐treated group. Dox was dissolved in saline and administered by intraperitoneal injection at a dose of 20 mg/kg.18 Control mice received injections of saline of comparable volume.
MI Model In Vivo
Both WT and ITCH‐Tg mice were randomly assigned to the sham‐operated group and the MI group. The same surgeon, who was blinded to the animal genotypes, performed all surgeries. MI was induced in mice aged 10 to 12 weeks, as described previously.19 Briefly, 23 to 27 mg mice were anesthetized by intraperitoneal injection of mixture of ketamine (80 mg/kg per hour) and xylazine (8 mg/kg per hour). Animals were intubated with a 20‐gauge polyethylene catheter and ventilated with a rodent ventilator (Harvard Apparatus). An incision was made along the left sternal border, and the fourth rib was cut proximal to the sternum. The left anterior coronary artery was identified, and an 8‐0 prolene suture was passed around the coronary artery and subsequently tied off. Successful ligation of the coronary artery was visually verified by LV myocardium discoloration. The same procedures were performed in sham‐operated animals, except the coronary artery ligation. Finally, the heart was repositioned in the chest, and the chest wall was closed. The animals were supervised until they regained consciousness.
Measurement of Infarct Size
The infarct length was calculated by measuring the endo‐ and epicardial surface length delimiting the infarct region detected by Masson trichrome staining. Infarct size percentage was calculated as infarct length divided by the total LV circumference.
Transmission Electron Microscopy and Scanning Electron Microscopy Analysis
Hearts were quickly removed and fixed in 2.5% glutaraldehyde buffer. Ultrathin sections (80 nm) were cut on an RMC MTXL ultramicrotome and mounted on 100‐mesh copper grids. Grids were stained with 2% uranyl acetate and Reynold's lead stain. Grids were examined using a transmission electron microscope (Hitachi H‐7100; Hitachi High Technologies Corp) and scanning electron microscope (Hitachi S 5000S; Hitachi).
Analysis of Cardiac Function
The cardiac function was evaluated by 2‐dimensional echocardiography 7 days after Dox or vehicle injection and 28 days after MI surgery with use of an FF Sonic 8900 (Fukudadenshi Co) equipped with a 13‐MHz phase‐array transducer. Mice were intraperitoneally anesthetized with pentobarbital (35 mg/kg), and additional doses were given as needed. LV dimensions at end‐diastole (LVEDD in calculation) and end‐systole (LVESD in calculation) were measured digitally on the M‐mode tracings and were averaged from 3 cardiac cycles. LV fractioning shortening was calculated as ([LVEDD−LVESD]/LVEDD)×100.
Statistical Analysis
All experiments were performed at least 3 times. All values are expressed as mean±SE in figures and as mean±SD in the tables. Comparisons among groups were analyzed by ANOVA with Scheffe post hoc test. Cardiac event‐free curves after Dox injection and MI surgery in vivo were constructed according to the Kaplan–Meier method and were compared using the log‐rank test. P values <0.05 was considered statistically significant. Statistical analyses were performed using a standard software package (JMP version 8; SAS Institute Inc).
Results
In Vitro Study
TXNIP suppression in ROS‐induced cardiotoxicity and the ubiquitin proteasome system
We found that protein and mRNA expression of TXNIP in cardiomyocytes decreased in a time‐dependent manner after Dox stimulation (Figure 1A and 1B). In addition, protein and mRNA levels of TXNIP in cardiomyocytes decreased following H2O2 stimulation, as reported previously (Figure 1C and 1D).6 To elucidate the impact of the ubiquitin proteasome system on TXNIP expression, we confirmed the interaction between TXNIP and ubiquitin by immunoprecipitation. As shown in Figure 1E, TXNIP coupled with ubiquitin, indicating that TXNIP was modulated by the ubiquitin proteasomal system in cardiomyocytes. Furthermore, immunoprecipitation showed that TXNIP ubiquitylation was increased in ROS‐induced cardiotoxicity (Figure 1F). To investigate whether TXNIP suppression was regulated by the ubiquitin proteasome system, neonatal rat cardiomyocytes were incubated with MG132, a nonspecific proteasome inhibitor, for 2 hours before Dox stimulation. As shown in Figure 1G, Dox‐induced TXNIP suppression was reversed by MG132, suggesting that TXNIP suppression was regulated by ubiquitin proteasomal protein degradation.
Figure 1.
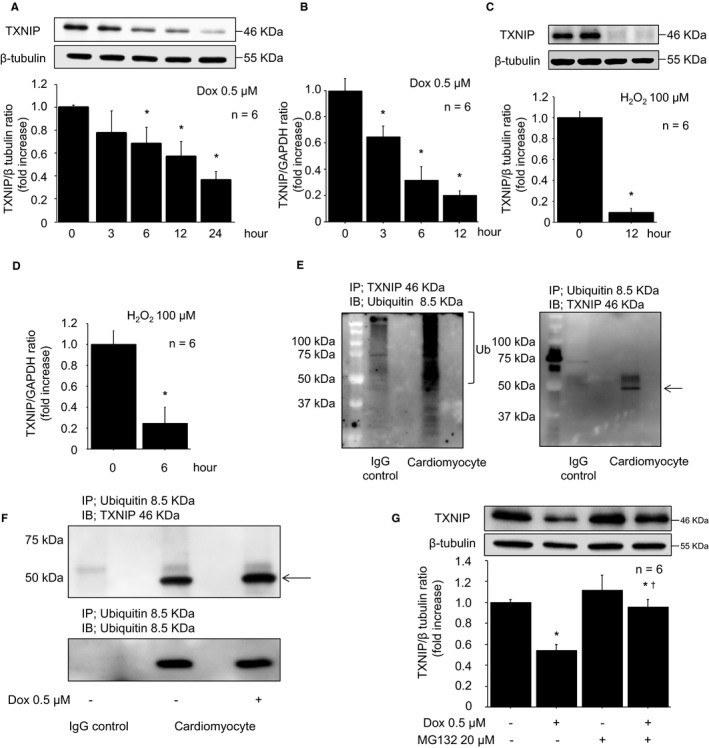
TXNIP suppression in reactive oxygen species–induced cardiotoxicity was modulated by ubiquitin proteasomal protein degradation. A, Representative Western blot of TXNIP in cardiomyocytes after Dox stimulation (0.5 μmol/L). B, TXNIP mRNA levels after Dox stimulation (0.5 μmol/L). C, Representative Western blot of TXNIP in cardiomyocytes after H2O2 stimulation (100 μmol/L, 12 hours). D, TXNIP mRNA levels after H2O2 stimulation (100 μmol/L, 6 hours). Data are expressed as mean±SEM (n=6 per group, *P<0.05 vs 0 hour). E, Neonatal rat cardiomyocyte lysates were immunoprecipitated with rabbit anti‐TXNIP antibody and immunoblotted with mouse anti–ubiquitin antibody (left panel). A long smear band indicates polyubiquitylated TXNIP in cardiomyocytes. Neonatal rat cardiomyocyte lysates were immunoprecipitated with mouse anti–ubiquitin antibody and immunoblotted with rabbit anti‐TXNIP antibody (right panel). An arrow indicated TXNIP. F, Neonatal rat cardiomyocyte lysates subjected to Dox stimulation (0.5 μmol/L, 12 hours) were immunoprecipitated with mouse anti–ubiquitin antibody and immunoblotted with rabbit anti‐TXNIP antibody. G, The TXNIP decrease was reversed by MG132 pretreatment (20 μmol/L, 2 hours) prior to Dox stimulation (0.5 μmol/L, 12 hours). Data are expressed as mean±SEM (n=6 per group, *P<0.05 vs control, † P<0.05 vs Dox stimulation without MG132). Dox indicates doxorubicin; H2O2, hydrogen peroxide; IgG, immunoglobulin G; TXNIP, thioredoxin‐interacting protein.
Interaction between ITCH and TXNIP
The ubiquitin E3 ligase ITCH was reported to play an important role in ubiquitin proteasome degradation of TXNIP in several cells, such as 293T, H1299, and U2OS cells14; however, this interaction had not been studied in cardiomyocytes. To confirm the interaction between ITCH and TXNIP, we performed a protein binding assay, which demonstrated that ITCH and TXNIP interact with each other in cardiomyocytes (Figure 2A). Furthermore, TXNIP ubiquitylation was increased in ITCH‐overexpressing cardiomyocytes compared with empty vector cardiomyocytes (Figure 2B).
Figure 2.
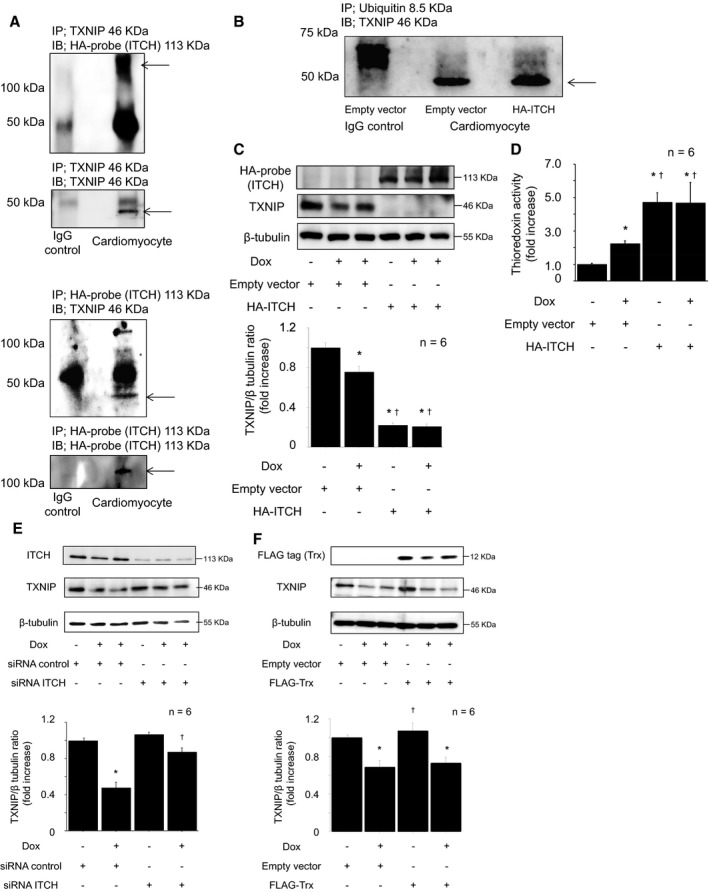
The ubiquitin E3 ligase ITCH targeted TXNIP for ubiquitin proteasomal protein degradation in reactive oxygen species–induced cardiotoxicity. A, Immunoprecipitation confirmed that ITCH interacts with TXNIP. HA‐ITCH–transfected cardiomyocyte lysates were immunoprecipitated and immunoblotted using mouse anti‐TXNIP and rabbit anti–HA‐probe antibodies. B, Ubiquitylated TXNIP was assessed by immunoprecipitation. HA‐ITCH–transfected cardiomyocyte lysates were immunoprecipitated with mouse anti–ubiquitin antibody and immunoblotted with rabbit anti‐TXNIP antibody. C, Representative Western blot of TXNIP in HA‐ITCH–transfected cardiomyocytes after Dox stimulation (0.5 μmol/L, 12 hours). Data are expressed as mean±SEM (n=6 per group, *P<0.05 vs control, † P<0.05 vs control with Dox stimulation). D, Thioredoxin activity determined by insulin disulfide reductase assay after Dox stimulation (0.5 μmol/L, 12 hours). Data are expressed as mean±SEM (n=6 per group, *P<0.05 vs control, † P<0.05 vs control with Dox stimulation). E, Representative Western blot of TXNIP in siRNA ITCH‐transfected cardiomyocytes after Dox stimulation (0.5 μmol/L, 12 hours). Data are expressed as mean±SEM (n=6 per group, *P<0.05 vs siRNA control, † P<0.05 vs siRNA control with Dox stimulation). F, The impact of thioredoxin‐1 on TXNIP expression in cardiomyocytes after Dox stimulation. Representative Western blot of TXNIP in thioredoxin‐1‐transfected cardiomyocytes after Dox stimulation (0.5 μmol/L, 12 hours). Data are expressed as means±SEM (n=6 per group, *P<0.05 vs control with empty vector, † P<0.05 vs Dox stimulation with empty vector). Dox indicates doxorubicin; HA, pRK5‐hem agglutinin; IgG, immunoglobulin G; siRNA, small interfering RNA; TXNIP, thioredoxin‐interacting protein.
Next, we studied the impact of ITCH on TXNIP suppression in cardiomyocytes. As shown in Figure 2C, overexpression of ITCH augmented TXNIP suppression, regardless of Dox‐induced cardiotoxicity. TXNIP was considered a negative regulator of the thioredoxin system,20, 21 so we next performed an insulin disulfide reductase assay to elucidate the impact of ITCH on the thioredoxin system. As shown in Figure 2D, thioredoxin activity was increased in response to Dox stimulation (0.5 μmol/L, 12 hours), but it was significantly much higher in ITCH‐overexpressing cardiomyocytes. In contrast, knockdown of ITCH by siRNA inhibited Dox‐induced TXNIP suppression (Figure 2E). ITCH‐dependent TXNIP degradation augmented thioredoxin activity in response to oxidative stress. Conversely, we examined whether thioredoxin induced ITCH‐dependent TXNIP degradation. As shown in Figure 2F, thioredoxin overexpression did not induce ITCH‐dependent TXNIP degradation in ROS‐induced cardiotoxicity. These results suggest that ROS‐induced TXNIP suppression is related to its interaction with ITCH, and the baseline protein expression level of ITCH was an important factor to regulate TXNIP expression in cardiomyocytes.
Ubiquitin E3 ligase ITCH expression and ROS‐induced cardiotoxicity
Previous reports indicated that HECT‐type E3 ligase was downregulated after ubiquitin transfer,22, 23, 24 thus we examined the serial changes in endogenous expression level of ITCH in response to ROS. At first, ITCH expression was slightly increased but finally decreased after Dox stimulation (Figure 3A). As shown in Figure 3B, mRNA expression level of ITCH was decreased in response to ROS. Protein and mRNA expression levels of ITCH were decreased after H2O2 stimulation (Figure 3C and 3D). Because ITCH was reported to target ITCH itself for ubiquitin proteasomal degradation,22 we used MG132 to investigate whether ITCH was regulated by ubiquitin proteasome system in cardiomyocytes. As shown in Figure 3E and 3F, ITCH interacted with ubiquitin, and expression level of ITCH was modulated by ubiquitin proteasomal degradation in ROS‐induced cardiotoxicity. Relative expression level of ITCH and TXNIP after ROS stimulation was compared. At first, ITCH expression was slightly increased by ROS stimulation, but then gradually decreased after TXNIP suppression (Figure 3G). These data suggested that ITCH decreased by ubiquitin proteasomal protein degradation in ROS‐induced cardiotoxicity after TXNIP ubiquitylation.
Figure 3.
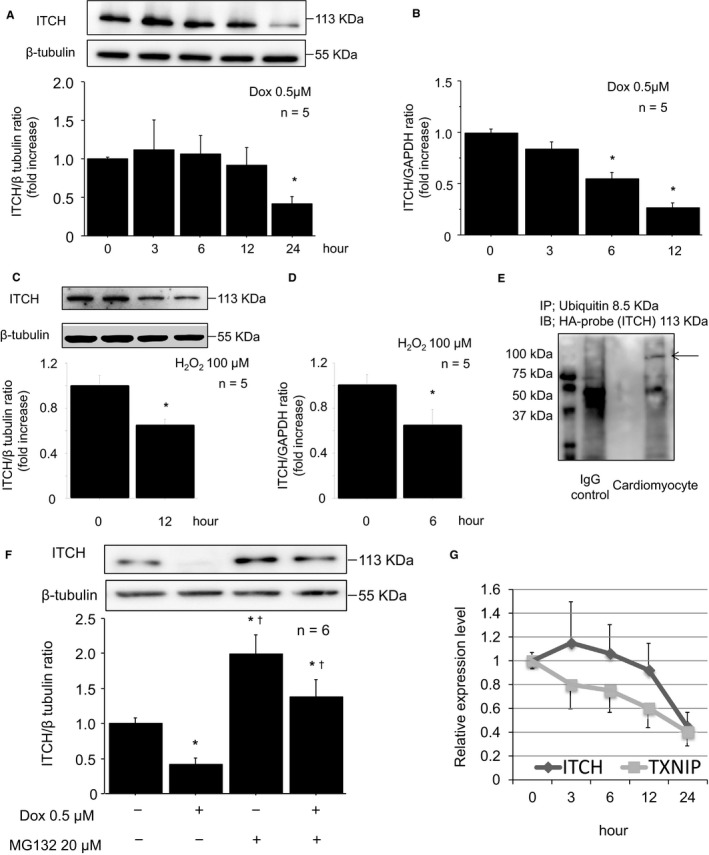
Downregulation of ITCH was regulated by ubiquitin proteasomal degradation in reactive oxygen species–induced cardiotoxicity. A, Representative Western blot of ITCH in cardiomyocytes after Dox stimulation (0.5 μmol/L). B, mRNA levels of ITCH after Dox stimulation (0.5 μmol/L). C, Representative Western blot of ITCH in cardiomyocytes after H2O2 stimulation (100 μmol/L, 12 hours). D, mRNA levels of ITCH after H2O2 stimulation (100 μmol/L, 6 hours). Data are expressed as mean±SEM (n=5 per group, *P<0.05 vs 0 hour). E, Immunoprecipitation showed that ITCH bound with ubiquitin (arrowhead). HA‐ITCH–transfected cardiomyocyte lysates were immunoprecipitated with mouse anti–ubiquitin antibody and immunoblotted with rabbit anti–HA‐probe antibody. F, The ITCH decrease was reversed by pretreatment of MG132 (20 μmol/L, 2 hours). Data are expressed as mean±SEM (n=6 per group, *P<0.05 vs control, † P<0.05 vs Dox stimulation without MG132). G, Relative expression levels of TXNIP and ITCH after Dox stimulation (0.5 μmol/L). Dox indicates doxorubicin; HA, pRK5‐hem agglutinin; H2O2, hydrogen peroxide; IgG, immunoglobulin G; TXNIP, thioredoxin‐interacting protein.
Impact of ITCH on ROS generation
Excess production of ROS relative to antioxidant defenses is a cause of cardiac remodeling. Previous reports indicated that both TXNIP knockdown and thioredoxin overexpression inhibit oxidative stress and subsequent apoptosis,5, 25 thus we performed DHE staining, a marker of superoxide generation.17 Superoxide generation was increased after Dox stimulation, but it was inhibited in ITCH‐overexpressing cardiomyocytes (Figure 4A). In addition, the level of NADPH oxidase subunit p22 phox, a surrogate marker of intracellular ROS generation,26, 27 was reduced in ITCH‐overexpressing cardiomyocytes (Figure 4B). These results indicated the possibility that ITCH ameliorates intracellular ROS generation through both thioredoxin activation and inhibition of TXNIP.
Figure 4.
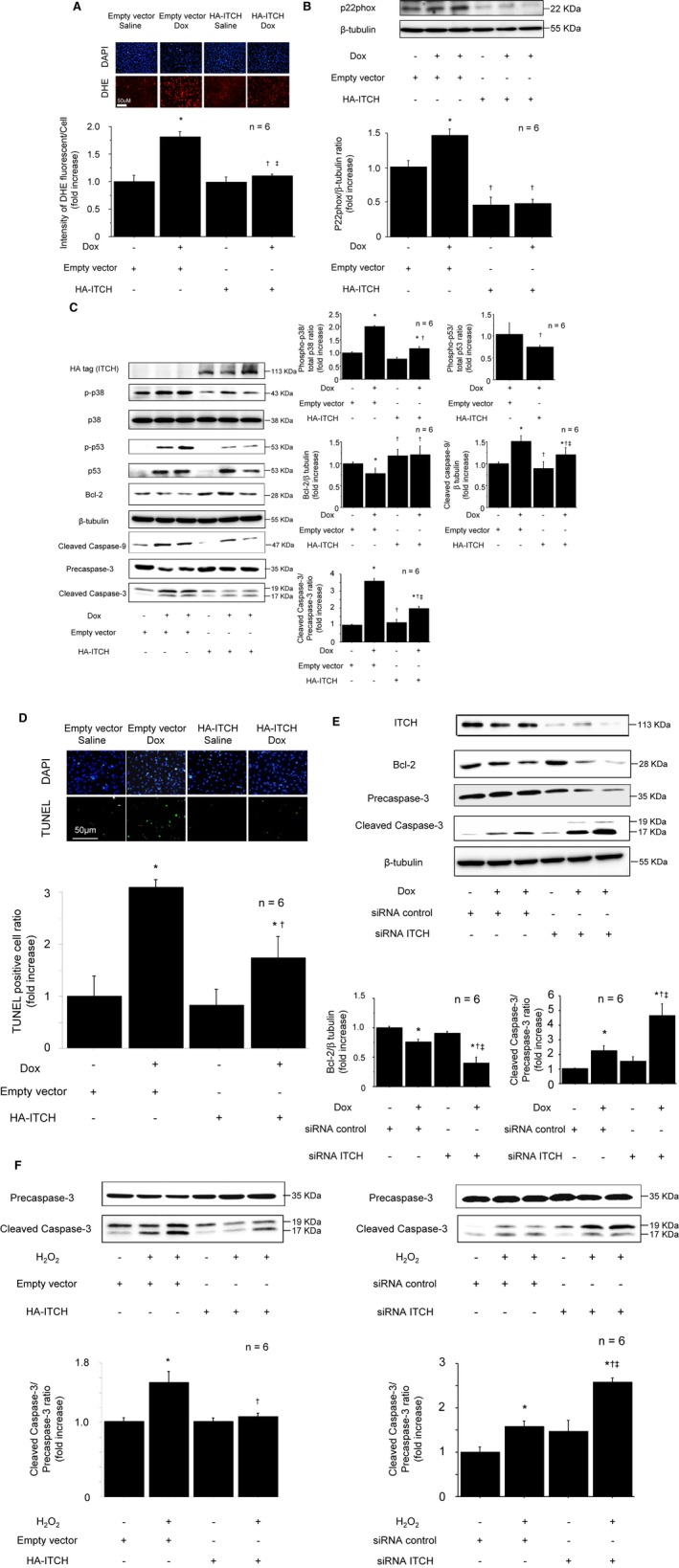
ITCH‐dependent TXNIP degradation ameliorated oxidative stress and cardiomyocyte apoptosis in reactive oxygen species–induced cardiotoxicity. A, Intensity of DHE staining after Dox stimulation (0.5 μmol/L, 12 hours). B, ITCH overexpression in cardiomyocyte inhibited NADPH oxidase subunit p22phox. C, Representative Western blots of p38 MAPK, p53, Bcl‐2, cleaved caspase‐9, and cleaved caspase‐3 in HA‐ITCH‐transfected cardiomyocytes after Dox stimulation (0.5 μmol/L, 12 hours). D, Inhibition of TUNEL‐positive cells by overexpression of HA‐ITCH after Dox stimulation (0.5 μmol/L, 12 hours). Data are expressed as mean±SEM (n=6 per group, *P<0.05 vs control with empty vector, † P<0.05 vs Dox stimulation with empty vector, ‡ P<0.05 vs control with HA‐ITCH vector). E, Representative Western blots of Bcl‐2 and cleaved caspase‐3 in siRNA ITCH‐transfected cardiomyocytes after Dox stimulation (0.5 μmol/L, 12 hours). Data are expressed as mean±SEM (n=6 per group, *P<0.05 vs siRNA control, † P<0.05 vs siRNA control with Dox stimulation, ‡ P<0.05 vs control with siRNA ITCH). F, Representative Western blot of cleaved caspase‐3 after H2O2 stimulation. Left: ITCH overexpression, n=6 per group, *P<0.05 vs control with empty vector, † P<0.05 vs H2O2 stimulation with empty vector. Right: ITCH knockdown using siRNA, n=6 per group, *P<0.05 vs siRNA control, † P<0.05 vs siRNA control with H2O2 stimulation, ‡ P<0.05 vs control with siRNA ITCH. Dox indicates doxorubicin; HA, pRK5‐hem agglutinin; H2O2, hydrogen peroxide; siRNA, small interfering RNA; TUNEL, terminal deoxynucleotidyl‐transferase‐mediated 2′‐deoxyuridine‐5′‐triphosphate nick‐end labeling; TXNIP, thioredoxin‐interacting protein.
ITCH and apoptosis in cardiomyocyte
Apoptosis is a major cause of cardiomyocyte loss in various heart diseases.28 TXNIP activation and subsequent thioredoxin inactivation were reported to induce apoptosis in cardiomyocytes through p38 MAPK, p53, and intrinsic pathway apoptosis.5, 6, 29 Because TXNIP was decreased by overexpression of ITCH, we examined whether ITCH could ameliorate ROS‐induced apoptosis in cardiomyocytes using Dox and H2O2. Overexpression of ITCH inhibited phosphorylation of p38 MAPK and p53 compared with empty vector cardiomyocytes after Dox stimulation. To examine whether ITCH inhibits intrinsic pathway apoptosis, we examined Bcl‐2, cleaved caspase‐9, and cleaved caspase‐3 levels in cardiomyocytes. ITCH‐overexpressing cardiomyocytes preserved Bcl‐2 levels and inhibited cleaved caspase‐9 and cleaved caspase‐3 after Dox stimulation compared with empty vector cardiomyocytes (Figure 4C). Next, we performed TUNEL staining of ITCH‐overexpressing cardiomyocytes. In control cells, the number of TUNEL‐positive nuclei was increased after Dox stimulation; however, the number of TUNEL‐positive nuclei after Dox stimulation were significantly lower in ITCH‐overexpressing cardiomyocytes than in control cells after Dox stimulation (Figure 4D). Conversely, knockdown of ITCH decreased Bcl‐2 levels and augmented cleaved caspase‐3 expression in ROS‐induced cardiotoxicity (Figure 4E). Similarly, H2O2‐induced cleaved caspase‐3 expressions were inhibited in ITCH‐overexpressing cardiomyocytes and augmented in siRNA ITCH‐transfected cardiomyocytes (Figure 4F). These data suggested that ROS‐induced apoptosis was ameliorated by ITCH overexpression in cardiomyocytes.
In Vivo Study
ITCH transgenic mouse generation
Transgenic mice with cardiac‐specific overexpression of ITCH were created at Yamagata University using standard techniques, as reported previously.30 Briefly, a 5.5‐kbp fragment of murine α‐MHC gene promoter (a kind gift from Dr. J. Robbins, Children's Hospital Research Foundation, Cincinnati, OH) and 2.5‐kbp mouse ITCH were subcloned into pBsIISK‐positive plasmids. The plasmid was digested with SpeI to generate a DNA fragment composed of α‐MHC gene promoter, ITCH cDNA, and a poly A tail of the human growth hormone, as illustrated in Figure 5A. We microinjected the construct into the pronuclei of single‐cell fertilized mouse embryos to generate transgenic mice. To detect the exogenous ITCH gene, genomic DNA was extracted from the tail tissues of pups aged 3 to 4 weeks, and PCR was performed with 1 primer specific for the α‐MHC gene promoter and another primer specific for ITCH. After microinjection and embryo implantation, ITCH‐Tg mice were successfully established, and cardiac‐specific expression of transgene was confirmed by reverse transcription PCR and Western blot analysis (Figure 5B and 5C). There were no significant differences in gravimetric data and cardiac function at basal condition between ITCH‐Tg mice and WT littermates.
Figure 5.
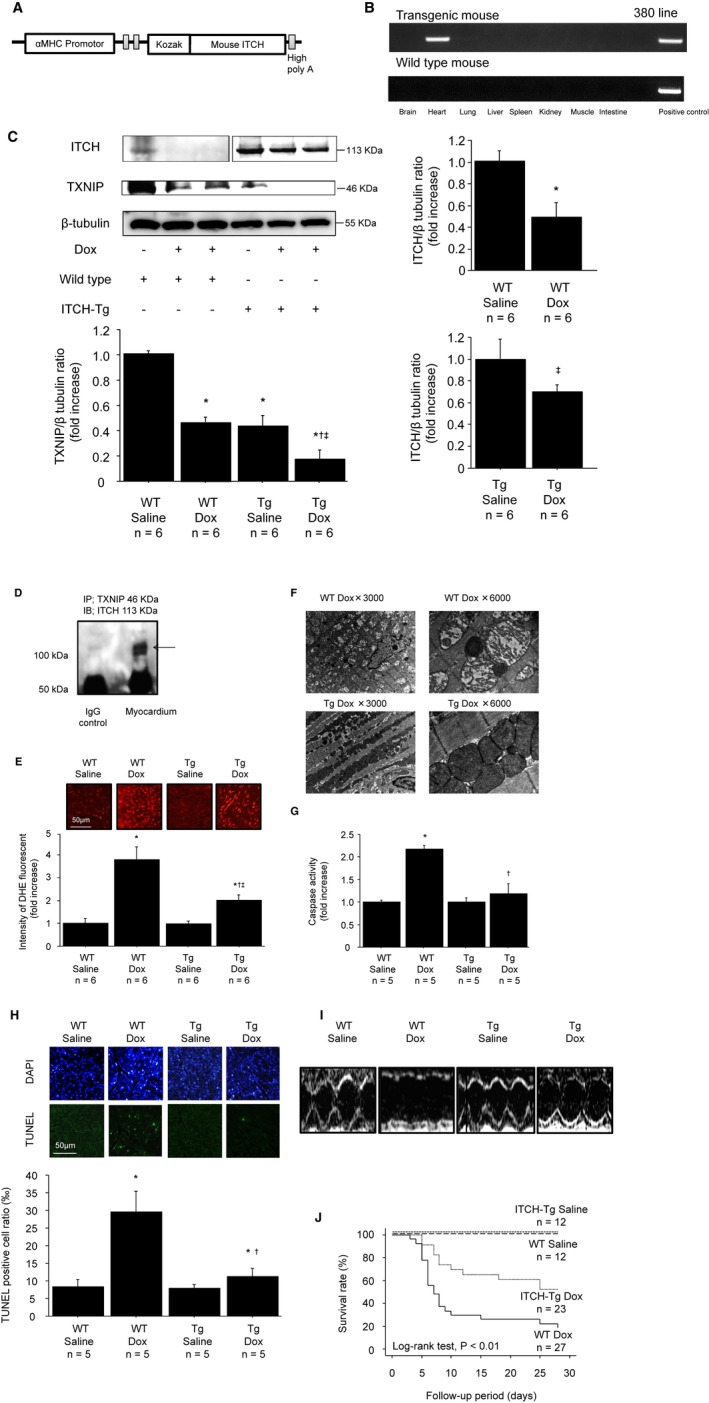
Cardiac‐specific ITCH overexpression protected cardiac function and improved the survival rate in mice with Dox‐induced cardiotoxicity. A, Diagram of the transgene construct used for the generation of ITCH‐Tg mice. The construct contains the α‐MHC promoter, a mouse ITCH cDNA clone, and a human growth hormone (Hgh) polyadenylylation sequence. B, RNA was extracted from ITCH‐Tg mice brain, heart, lung, liver, spleen, kidney, skeletal muscle, and intestine, and cardiac‐specific transgene expression was confirmed by reverse transcription polymerase chain reaction. C, TXNIP and ITCH protein levels were examined by Western blotting 7 days after Dox stimulation. Data are expressed as mean±SEM (n=6 per group, *P<0.05 vs WT saline, † P<0.05 vs WT Dox, ‡ P<0.05 vs ITCH‐Tg saline). D, Immunoprecipitation confirmed that ITCH interacts with TXNIP in vivo. Myocardial lysates in ITCH‐Tg mice were immunoprecipitated with rabbit anti‐TXNIP antibody and immunoblotted with mouse anti‐ITCH antibody. E, Representative images of DHE staining in frozen left ventricular myocardium. Data are expressed as mean±SEM (n=6 per group, *P<0.05 vs WT saline, † P<0.05 vs WT Dox, ‡ P<0.05 vs ITCH‐Tg saline). F, Mitochondrial morphological changes observed by transmission electron microscopy 7 days after Dox injection. G, Caspase‐3 activity in myocardial tissues. Data are expressed as mean±SEM (n=5 per group, *P<0.05 vs WT saline, † P<0.05 vs WT Dox). H, Representative TUNEL and DAPI stained sections from WT and ITCH‐Tg mice. Data are expressed as mean±SEM (n=5 per group, *P<0.05 vs WT saline, † P<0.05 vs WT Dox). I, Representative M‐mode echocardiograms from WT and ITCH‐Tg mice 7 days after saline or Dox injection. J, Survival curves up to 28 days after Dox injection in WT and ITCH‐Tg mice. Dox indicates doxorubicin; Tg, transgenic; TUNEL, terminal deoxynucleotidyl‐transferase‐mediated 2′‐deoxyuridine‐5′‐triphosphate nick‐end labeling; TXNIP, thioredoxin‐interacting protein; WT, wild type.
Dox‐induced cardiotoxicity in vivo
Dox‐induced cardiotoxicity is mainly caused by increased ROS generation in the heart.31, 32 We used ITCH‐Tg mice to investigate whether cardiac‐specific overexpression of ITCH could ameliorate Dox‐induced cardiotoxicity. ITCH was decreased after intraperitoneal Dox injection in both WT and ITCH‐Tg mice. ITCH‐Tg mice had lower TXNIP levels than in WT mice at basal condition (Figure 5C). In accordance with in vitro study, TXNIP expression was suppressed after intraperitoneal Dox injection, and its level was much lower in ITCH‐Tg mice than in WT littermates (Figure 5C). To confirm the interaction between ITCH and TXNIP in vivo, we performed a protein binding assay, which demonstrated that ITCH and TXNIP also interact with each other in vivo (Figure 5D). Next, we performed DHE staining, a marker of superoxide generation, to assess the effect of ITCH expression on myocardial ROS. Superoxide generation was increased after Dox injection; however, it was inhibited in ITCH‐Tg mice (Figure 5E). ROS generation was closely associated with mitochondrial dysfunction.33 As shown in Figure 5F, mitochondrial cristae were fragmented and degraded in WT mice after Dox injection; however, these morphological changes were attenuated in ITCH‐Tg mice. To examine the impact of ITCH on apoptosis in cardiac cells, caspase‐3 activity was measured. As shown in Figure 5G, caspase‐3 activity was significantly increased in WT littermates after Dox injection; however, the activation of caspase‐3 by Dox was inhibited in ITCH‐Tg mice. Furthermore, the number of TUNEL‐positive cells was lower in ITCH‐Tg mice than in WT littermates after Dox injection (Figure 5H). These data indicated that Dox‐induced cardiotoxicity (ie, oxidative stress and subsequent apoptosis) was inhibited in ITCH‐Tg mice.
ITCH protects cardiac atrophy, cardiac function, and survival rate in Dox cardiomyopathy
We examined cardiac function of WT and ITCH‐Tg mice by echocardiography 7 days after Dox injection. As shown in Table 1, body weight, heart weight, and the ratio of heart weight to body weight ratio were significantly reduced after intraperitoneal Dox injection. The decrease in the ratio of heart weight to body weight was significantly attenuated in ITCH‐Tg mice compared with WT littermates. Echocardiographic data showed that WT mice treated with Dox showed larger LV end‐diastole diameter, thicker interventricular septum diameter and LV posterior wall diameter, and lower fractioning shortening than WT mice treated with saline (Table 1 and Figure 5I); however, LV enlargement and cardiac systolic dysfunction induced by Dox were significantly attenuated in ITCH‐Tg mice.
Table 1.
Comparison of Gravimetric and Echocardiographic Data in Doxorubicin Cardiomyopathy
| Variable | WT Saline (n=10) | WT Dox (n=10) | ITCH‐Tg Saline (n=10) | ITCH‐Tg Dox (n=10) |
|---|---|---|---|---|
| BW before, g | 23.6±1.5 | 23.2±1.4 | 23.6±2.1 | 23.9±1.5 |
| BW after, g | 23.7±1.6 | 19.4±2.5a | 23.9±2.1b | 20.9±1.9a, b, c |
| HW after, mg | 115.6±11.8 | 76.5±9.4a | 117.7±13.5b | 95.7±12.4a, b, c |
| HW/BW ratio | 4.88±0.45 | 3.97±0.33a | 4.92±0.35b | 4.56±0.40a, b, c |
| Heart rate, bpm | 548±19 | 542±17 | 567±12 | 554±14 |
| IVSd, mm | 0.82±0.08 | 0.72±0.09a | 0.84±0.06b | 0.74±0.07a, c |
| LVEDD, mm | 2.69±0.11 | 3.04±0.14a | 2.72±0.08b | 2.83±0.13a, b, c |
| LVPWd, mm | 0.87±0.13 | 0.73±0.12a | 0.90±0.13b | 0.77±0.14a, c |
| %FS (%) | 54±5 | 38±7a | 55±4b | 51±6a, b, c |
Mean±SD. %FS indicates fractioning shortening; bpm, beats per minute; BW, body weight; Dox, doxorubicin; HW, heart weight; ITCH‐Tg, cardiac‐specific overexpression of ITCH transgenic mouse; IVSd, interventricular septal diameter; LVEDD, left ventricular end‐diastole dimension; LVPWd, left ventricular posterior wall diameter; WT, wild type.
P<0.05 vs WT saline group.
P<0.05 vs WT Dox group.
P<0.05 vs ITCH‐Tg saline group by ANOVA with Scheffe post hoc test.
We determined the survival rates after Dox injection and compared them between WT littermates and ITCH‐Tg mice. There was no cardiac death in either WT littermates or ITCH‐Tg mice after saline injection. As a consequence of the preserved LV function, the survival rate at 28 days after Dox injection was significantly higher in ITCH‐Tg mice than in WT littermates (Figure 5J).
ITCH protects cardiac remodeling, cardiac function, and survival rate in MI
The infarct size percentage at 28 days, calculated by Masson trichrome staining, was significantly smaller in ITCH‐Tg mice than in WT mice after MI surgery (Figure 6A). Mitochondrial cristae were fragmented and degraded in WT mice 28 days after MI surgery; however, these morphological changes were attenuated in ITCH‐Tg mice (Figure 6B).
Figure 6.
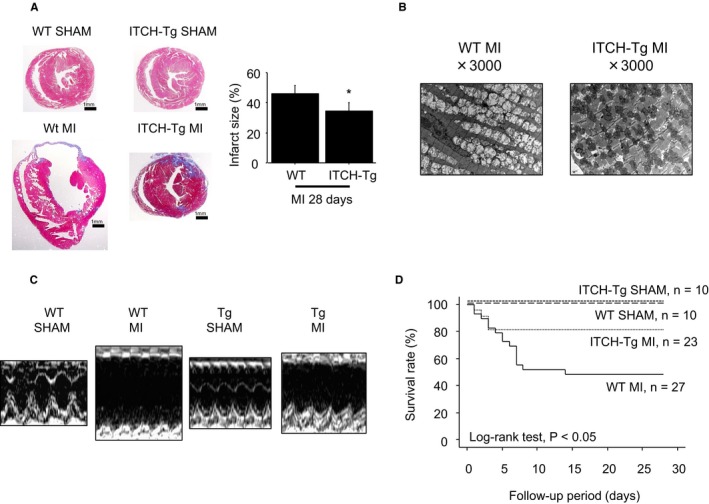
Cardiac‐specific ITCH overexpression protected cardiac function and improved the survival rate in mice with MI surgery. A, Representative Masson trichrome staining in WT and ITCH‐Tg mice after sham and MI surgery. Data are expressed as mean±SEM (n=4 per group, *P<0.05 vs WT MI). B, Mitochondrial morphological changes observed by transmission electron microscopy 28 days after MI surgery. C, Representative M‐mode echocardiograms from WT and ITCH‐Tg mice 28 days after MI surgery. D, Survival curves up to 28 days after MI surgery in WT and ITCH‐Tg mice. Dox indicates doxorubicin; MI, myocardial infarction; Tg, transgenic; TXNIP, thioredoxin‐interacting protein; WT, wild type.
As shown in Table 2, the ratio of heart weight to body weight was significantly increased in WT mice after MI surgery; however, it was significantly attenuated in ITCH‐Tg mice compared with WT mice. Echocardiographic data demonstrated that the LVEDD was smaller and fractioning shortening was higher in ITCH‐Tg mice than in WT mice. In addition, interventricular septum diameter thinning was also attenuated in ITCH‐Tg mice compared with WT mice (Table 2 and Figure 6C).
Table 2.
Comparison of Gravimetric and Echocardiographic Data in Myocardial Infarction
| Variable | WT Sham (n=10) | WT MI (n=10) | ITCH‐Tg Sham (n=10) | ITCH‐Tg MI (n=10) |
|---|---|---|---|---|
| BW before, g | 24.3±1.3 | 25.1±1.5 | 23.8±1.5 | 24.6±2.1 |
| BW after, g | 24.6±1.4 | 26.2±1.9 | 24.3±1.5 | 26.3±2.1 |
| HW after, mg | 118.6±7.1 | 197.2±12.4a | 112.1±5.7b | 156.7±13.5a, b, c |
| HW/BW ratio | 4.82±0.87 | 7.25±0.50a | 4.69±0.12b | 5.82±0.15a, b, c |
| Heart rate, bpm | 561±17 | 552±14 | 566±12 | 558±16 |
| IVSd, mm | 0.88±0.13 | 0.42±0.04a | 0.84±0.03b | 0.61±0.04a, b, c |
| LVEDD, mm | 2.80±0.15 | 4.62±0.10a | 2.74±0.07b | 3.55±0.10a, b, c |
| LVPWd, mm | 0.86±0.13 | 0.82±0.12 | 0.87±0.13 | 0.90±0.13 |
| %FS (%) | 59±6 | 18±6a | 63±6b | 33±10a, b, c |
Mean±SD. %FS indicates fractioning shortening; bpm, beats per minute; BW, body weight; HW, heart weight; ITCH‐Tg, cardiac‐specific overexpression of ITCH transgenic mouse; IVSd, interventricular septal diameter; LVEDD, left ventricular end‐diastole dimension; LVPWd, left ventricular posterior wall diameter; MI, myocardial infarction; WT, wild type.
P<0.05 vs WT sham group.
P<0.05 vs WT MI group.
P<0.05 vs ITCH‐Tg sham group by ANOVA with Scheffe post hoc test.
The survival rates from recovery of MI surgery were compared up to 28 days between WT and ITCH‐Tg mice. There was no cardiac death in either WT littermates or ITCH‐Tg mice after sham operation. The survival rate up to 28 days after MI surgery was significantly higher in ITCH‐Tg mice than in WT mice (Figure 6D).
Discussion
Some findings in this study are new and important. First, the ubiquitin E3 ligase ITCH is expressed in cardiomyocytes and targeted TXNIP for ubiquitin proteasomal protein degradation to maintain cell homeostasis and as response to ROS. Second, ITCH‐dependent TXNIP degradation ameliorates oxidative stress and intrinsic pathway cardiomyocyte apoptosis in ROS‐induced cardiotoxicity through augmentation of thioredoxin activity and inhibition of p38 MAPK and p53. Third, cardiac‐specific overexpression of ITCH preserved cardiac function and improved survival rate for Dox‐induced cardiotoxicity and MI. A schema that includes the suggested pathway from the present study is shown in Figure 7.
Figure 7.
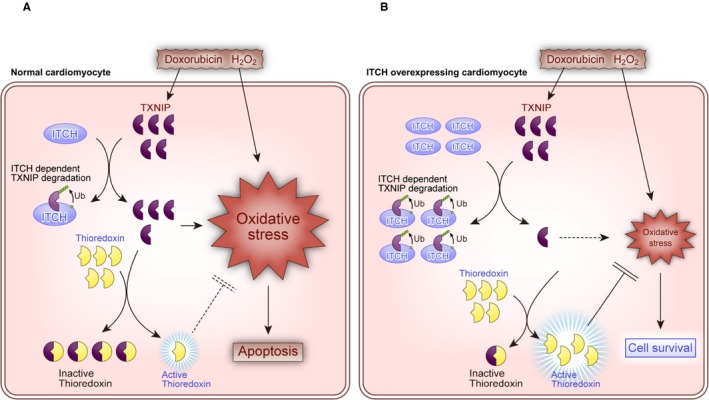
Schematics depicting the proposed pathway of ITCH in reactive oxygen species–induced cardiotoxicity. A, Normal cardiomyocyte. B, ITCH‐overexpressing cardiomyocyte. H2O2, hydrogen peroxide; Tg, transgenic; TXNIP, thioredoxin‐interacting protein; Ub, ubiquitylation.
ITCH‐Dependent TXNIP Ubiquitylation
The ubiquitin proteasome system plays a key role in quality control of intracellular proteins, which is essential for cellular homeostasis.34 Recently, ubiquitin proteasomal degradation was reported to be activated in the failing heart and linked to pathogenesis of cardiovascular disease.35, 36 Ubiquitylation mediates the selective proteasomal degradation of many proteins that are involved in redox status, apoptosis, DNA repair, signal transduction, and organelle biogenesis.37, 38 The amount and catalytic activity of the E3 ligase are considered key factors to augment ubiquitylation, and a previous report showed that the HECT‐type E3 ligase is constitutively active to recognize and transfer ubiquitin to substrate proteins (ie, ubiquitylation).23 In the present study, we showed that overexpression of ITCH augmented ubiquitin proteasomal degradation of TXNIP regardless of ROS‐induced cardiotoxicity in both in vitro and in vivo studies, indicating that the baseline amount of ITCH mainly regulates TXNIP expression through ubiquitylation (Figures 1 and 2).
Extracellular stimuli, such as tumor necrosis factor α, tumor necrosis factor–related apoptosis‐inducing ligand, and epidermal growth factor, were reported to augment catalytic activity of ITCH.22, 23, 39 We showed that Dox and H2O2 promoted ITCH‐dependent TXNIP degradation without a significant increase in ITCH. Time course of ITCH and TXNIP expression showed that relative expression of ITCH compared with TXNIP was high for 12 hours in ROS‐induced cardiotoxicity. Because ITCH is a self‐limiting enzyme, ITCH protein is finally degraded by self‐ubiquitylation 24 hours after its activation (Figure 3). Although a baseline amount of ITCH is important to induce TXNIP degradation, ITCH‐dependent TXNIP degradation in ROS‐induced cardiotoxicity might be modulated by catalytic activation of ITCH by extracellular stimuli and ROS‐induced relative increase in ITCH.
Downregulation of ITCH by Ubiquitin Proteasomal Degradation
Different from other E3 ligases, a HECT‐type E3 ligase was reported to be downregulated by self‐ubiquitylation after substrate ubiquitylation through interaction between its WW domain and HECT PY‐motif.24 Downregulation of ITCH due to self‐ubiquitylation was also reported.22, 23, 40 Similarly, we showed that ITCH was downregulated by ubiquitin proteasomal degradation in ROS‐induced cardiotoxicity (Figure 3).
Downregulation of ITCH was reported to be associated with apoptosis in cancer cells.41 Because the amount of ITCH is an important factor in ubiquitylation, downregulation of ITCH in ROS‐induced cardiotoxicity may lead to insufficient TXNIP degradation, resulting in the disruption of the intracellular thioredoxin system that protects against excessive ROS accumulation and production.
ITCH‐Dependent TXNIP Degradation Restricts ROS‐Induced Cardiotoxicity
It has been reported that ubiquitin E3 ligase ITCH regulates apoptosis through its interaction with substrate proteins such as p53 family member p63 and p73, t‐Bid, c‐jun, and TXNIP.12, 14, 23, 41, 42 TXNIP has attracted much interest in the field of cardiovascular disease because it is a proapoptotic protein and a regulator in the thioredoxin system.43, 44 Previous reports demonstrated, for example, preservation of cardiac function in TXNIP knockout mice and catalytic degradation of TXNIP in rat hearts in a model of ischemic heart disease.43, 44 Inhibition of TXNIP was reported to activate thioredoxin,5, 6, 21 and previous reports demonstrated that TXNIP suppression is important to activate the thioredoxin system against ROS in cardiomyocytes. As shown in Figure 4, we found that ITCH plays a pivotal role in TXNIP suppression, and ITCH‐dependent TXNIP degradation ameliorates superoxide generation by increasing thioredoxin activity in ROS‐induced cardiotoxicity. Furthermore, recent reports showed that TXNIP deteriorates ROS production through NADPH oxidase, a major source of ROS generation in cardiomyocytes.2, 45 In accordance with these reports, we showed that TXNIP suppression by ITCH‐dependent ubiquitin proteasomal degradation increased thioredoxin activity and inhibited NADPH oxidase. Furthermore, mitochondrial damage was attenuated in ITHC‐Tg mice more than in WT mice in ROS‐induced cardiotoxicity. Damaged mitochondria augment mitochondrial ROS production.33 These findings suggested the possibility that ITCH also suppressed mitochondrial ROS production. Considering these findings, ITCH might inhibit ROS generation not only through ROS scavenging but also through inhibition of ROS production.
It is well known that thioredoxin protects cardiomyocytes through ROS scavenging, neovascularization, and inhibition of apoptosis.4, 25, 29 In detail, thioredoxin regulates p38 MAPK and intrinsic pathway apoptosis. Similarly, overexpression of ITCH inhibited phosphorylation of p38 MAPK, p53, and intrinsic pathway apoptosis, indicating that ITCH ameliorates ROS‐induced cardiomyocyte apoptosis (Figure 4).
Oxidative stress and subsequent cardiomyocyte apoptosis are closely associated with cardiac remodeling.1 The thioredoxin system reportedly prevents cardiac remodeling and subsequently enhances cardiac prognosis in ROS‐induced cardiotoxicity.25, 43 Similarly, in the present study, we showed that LV function, cardiac remodeling, and subsequent survival rate were improved in ITCH‐Tg mice compared to WT littermates (Figures 5 and 6). These findings support our hypothesis that the ubiquitin E3 ligase ITCH plays a pivotal role in the pathological processes of ROS‐induced cardiotoxicity.
Limitations
First, because mRNA levels of ITCH and TXNIP were inhibited in ROS‐induced cardiotoxicity, there is likely another mechanism by which ROS inhibits protein expressions of TXNIP and ITCH. Second, because the disulfide bond is unstable for keeping an active form, we could not measure thioredoxin activity in vivo.
Conclusions
The present data showed that, in cardiomyocytes, the HECT‐type ubiquitin E3 ligase ITCH might act as a regulator of the thioredoxin system by targeting TXNIP for ubiquitin proteasomal degradation in vitro and in vivo. ITCH might also play an important role in the maintenance of cell homeostasis in response to ROS by mediating the degradation of TXNIP. Because the thioredoxin system plays a pivotal role in the development of ROS‐induced cardiotoxicity, ITCH and its interaction with substrate proteins might represent a new target for prevention and treatment of ROS‐induced pathological processes.
Sources of Funding
This study was supported, in part, by a grant‐in‐aid for Scientific Research (No. 26893025) from the Ministry of Education Culture, Sport, Science and Technology and a grant‐in‐aid from the global century center of excellence (COE) program of the Japan Society for the Promotion of Science.
Disclosures
None.
Acknowledgment
We thank Emiko Nishidate, Takeshi Nagahashi, and Miyuki Tsuda for their excellent technical assistance.
(J Am Heart Assoc. 2016;5:e002485 doi: 10.1161/JAHA.115.002485)
References
- 1. Hare JM. Oxidative stress and apoptosis in heart failure progression. Circ Res. 2001;89:198–200. [PubMed] [Google Scholar]
- 2. Li JM, Gall NP, Grieve DJ, Chen M, Shah AM. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension. 2002;40:477–484. [DOI] [PubMed] [Google Scholar]
- 3. Burgoyne JR, Mongue‐Din H, Eaton P, Shah AM. Redox signaling in cardiac physiology and pathology. Circ Res. 2012;111:1091–1106. [DOI] [PubMed] [Google Scholar]
- 4. Ahsan MK, Lekli I, Ray D, Yodoi J, Das DK. Redox regulation of cell survival by the thioredoxin superfamily: an implication of redox gene therapy in the heart. Antioxid Redox Signal. 2009;11:2741–2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Junn E, Han SH, Im JY, Yang Y, Cho EW, Um HD, Kim DK, Lee KW, Han PL, Rhee SG, Choi I. Vitamin D3 up‐regulated protein 1 mediates oxidative stress via suppressing the thioredoxin function. J Immunol. 2000;164:6287–6295. [DOI] [PubMed] [Google Scholar]
- 6. Wang Y, De Keulenaer GW, Lee RT. Vitamin D3‐up‐regulated protein‐1 is a stress‐responsive gene that regulates cardiomyocyte viability through interaction with thioredoxin. J Biol Chem. 2002;277:26496–26500. [DOI] [PubMed] [Google Scholar]
- 7. Willis MS, Townley‐Tilson WH, Kang EY, Homeister JW, Patterson C. Sent to destroy: the ubiquitin proteasome system regulates cell signaling and protein quality control in cardiovascular development and disease. Circ Res. 2010;106:463–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Willis MS, Patterson C. Into the heart: the emerging role of the ubiquitin‐proteasome system. J Mol Cell Cardiol. 2006;41:567–579. [DOI] [PubMed] [Google Scholar]
- 9. Patterson C, Ike C, Willis PW IV, Stouffer GA, Willis MS. The bitter end: the ubiquitin‐proteasome system and cardiac dysfunction. Circulation. 2007;115:1456–1463. [DOI] [PubMed] [Google Scholar]
- 10. Perry WL, Hustad CM, Swing DA, O'Sullivan TN, Jenkins NA, Copeland NG. The itchy locus encodes a novel ubiquitin protein ligase that is disrupted in a18 h mice. Nat Genet. 1998;18:143–146. [DOI] [PubMed] [Google Scholar]
- 11. Schwarz SE, Rosa JL, Scheffner M. Characterization of human HECT domain family members and their interaction with UBCH5 and UBCH7. J Biol Chem. 1998;273:12148–12154. [DOI] [PubMed] [Google Scholar]
- 12. Azakir BA, Desrochers G, Angers A. The ubiquitin ligase ITCH mediates the antiapoptotic activity of epidermal growth factor by promoting the ubiquitylation and degradation of the truncated C‐terminal portion of Bid. FEBS J. 2010;277:1319–1330. [DOI] [PubMed] [Google Scholar]
- 13. Ishihara T, Inoue J, Kozaki K, Imoto I, Inazawa J. Hect‐type ubiquitin ligase ITCH targets lysosomal‐associated protein multispanning transmembrane 5 (LAPTM5) and prevents LAPTM5‐mediated cell death. J Biol Chem. 2011;286:44086–44094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang P, Wang C, Gao K, Wang D, Mao J, An J, Xu C, Wu D, Yu H, Liu JO, Yu L. The ubiquitin ligase ITCH regulates apoptosis by targeting thioredoxin‐interacting protein for ubiquitin‐dependent degradation. J Biol Chem. 2010;285:8869–8879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Takahashi H, Takeishi Y, Seidler T, Arimoto T, Akiyama H, Hozumi Y, Koyama Y, Shishido T, Tsunoda Y, Niizeki T, Nozaki N, Abe J, Hasenfuss G, Goto K, Kubota I. Adenovirus‐mediated overexpression of diacylglycerol kinase‐zeta inhibits endothelin‐1‐induced cardiomyocyte hypertrophy. Circulation. 2005;111:1510–1516. [DOI] [PubMed] [Google Scholar]
- 16. Funayama A, Shishido T, Netsu S, Narumi T, Kadowaki S, Takahashi H, Miyamoto T, Watanabe T, Woo CH, Abe JI, Kuwahara K, Nakao K, Takeishi Y, Kubota I. Cardiac nuclear high mobility group box 1 prevents the development of cardiac hypertrophy and heart failure. Cardiovasc Res. 2013;99:657–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Misaka T, Suzuki S, Miyata M, Kobayashi A, Shishido T, Ishigami A, Saitoh S, Hirose M, Kubota I, Takeishi Y. Deficiency of senescence marker protein 30 exacerbates angiotensin ii‐induced cardiac remodelling. Cardiovasc Res. 2013;99:461–470. [DOI] [PubMed] [Google Scholar]
- 18. Nozaki N, Shishido T, Takeishi Y, Kubota I. Modulation of doxorubicin‐induced cardiac dysfunction in toll‐like receptor‐2‐knockout mice. Circulation. 2004;110:2869–2874. [DOI] [PubMed] [Google Scholar]
- 19. Kitahara T, Takeishi Y, Harada M, Niizeki T, Suzuki S, Sasaki T, Ishino M, Bilim O, Nakajima O, Kubota I. High‐mobility group box 1 restores cardiac function after myocardial infarction in transgenic mice. Cardiovasc Res. 2008;80:40–46. [DOI] [PubMed] [Google Scholar]
- 20. Yoshioka J, Schulze PC, Cupesi M, Sylvan JD, MacGillivray C, Gannon J, Huang H, Lee RT. Thioredoxin‐interacting protein controls cardiac hypertrophy through regulation of thioredoxin activity. Circulation. 2004;109:2581–2586. [DOI] [PubMed] [Google Scholar]
- 21. Chung JW, Jeon JH, Yoon SR, Choi I. Vitamin D3 upregulated protein 1 (VDUP1) is a regulator for redox signaling and stress‐mediated diseases. J Dermatol. 2006;33:662–669. [DOI] [PubMed] [Google Scholar]
- 22. Mouchantaf R, Azakir BA, McPherson PS, Millard SM, Wood SA, Angers A. The ubiquitin ligase ITCH is auto‐ubiquitylated in vivo and in vitro but is protected from degradation by interacting with the deubiquitylating enzyme FAM/USP9X. J Biol Chem. 2006;281:38738–38747. [DOI] [PubMed] [Google Scholar]
- 23. Gao M, Labuda T, Xia Y, Gallagher E, Fang D, Liu YC, Karin M. Jun turnover is controlled through JNK‐dependent phosphorylation of the E3 ligase itch. Science. 2004;306:271–275. [DOI] [PubMed] [Google Scholar]
- 24. Bruce MC, Kanelis V, Fouladkou F, Debonneville A, Staub O, Rotin D. Regulation of Nedd4‐2 self‐ubiquitination and stability by a PY motif located within its HECT‐domain. Biochem J. 2008;415:155–163. [DOI] [PubMed] [Google Scholar]
- 25. Adluri RS, Thirunavukkarasu M, Zhan L, Akita Y, Samuel SM, Otani H, Ho YS, Maulik G, Maulik N. Thioredoxin 1 enhances neovascularization and reduces ventricular remodeling during chronic myocardial infarction: a study using thioredoxin 1 transgenic mice. J Mol Cell Cardiol. 2011;50:239–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Octavia Y, Brunner‐La Rocca HP, Moens AL. NADPH oxidase‐dependent oxidative stress in the failing heart: from pathogenic roles to therapeutic approach. Free Radic Biol Med. 2012;52:291–297. [DOI] [PubMed] [Google Scholar]
- 27. Zhao Y, McLaughlin D, Robinson E, Harvey AP, Hookham MB, Shah AM, McDermott BJ, Grieve DJ. Nox2 NADPH oxidase promotes pathologic cardiac remodeling associated with doxorubicin chemotherapy. Cancer Res. 2010;70:9287–9297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, Quaini E, Di Loreto C, Beltrami CA, Krajewski S, Reed JC, Anversa P. Apoptosis in the failing human heart. N Engl J Med. 1997;336:1131–1141. [DOI] [PubMed] [Google Scholar]
- 29. Samuel SM, Thirunavukkarasu M, Penumathsa SV, Koneru S, Zhan L, Maulik G, Sudhakaran PR, Maulik N. Thioredoxin‐1 gene therapy enhances angiogenic signaling and reduces ventricular remodeling in infarcted myocardium of diabetic rats. Circulation. 2010;121:1244–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Arimoto T, Takeishi Y, Takahashi H, Shishido T, Niizeki T, Koyama Y, Shiga R, Nozaki N, Nakajima O, Nishimaru K, Abe J, Endoh M, Walsh RA, Goto K, Kubota I. Cardiac‐specific overexpression of diacylglycerol kinase zeta prevents Gq protein‐coupled receptor agonist‐induced cardiac hypertrophy in transgenic mice. Circulation. 2006;113:60–66. [DOI] [PubMed] [Google Scholar]
- 31. Monti E, Prosperi E, Supino R, Bottiroli G. Free radical‐dependent DNA lesions are involved in the delayed cardiotoxicity induced by adriamycin in the rat. Anticancer Res. 1995;15:193–197. [PubMed] [Google Scholar]
- 32. Rajagopalan S, Politi PM, Sinha BK, Myers CE. Adriamycin‐induced free radical formation in the perfused rat heart: implications for cardiotoxicity. Cancer Res. 1988;48:4766–4769. [PubMed] [Google Scholar]
- 33. Ide T, Tsutsui H, Hayashidani S, Kang D, Suematsu N, Nakamura K, Utsumi H, Hamasaki N, Takeshita A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res. 2001;88:529–535. [DOI] [PubMed] [Google Scholar]
- 34. Hatakeyama S, Nakayama KI. Ubiquitylation as a quality control system for intracellular proteins. J Biochem. 2003;134:1–8. [DOI] [PubMed] [Google Scholar]
- 35. Kumarapeli AR, Horak KM, Glasford JW, Li J, Chen Q, Liu J, Zheng H, Wang X. A novel transgenic mouse model reveals deregulation of the ubiquitin‐proteasome system in the heart by doxorubicin. FASEB J. 2005;19:2051–2053. [DOI] [PubMed] [Google Scholar]
- 36. Ranek MJ, Wang X. Activation of the ubiquitin‐proteasome system in doxorubicin cardiomyopathy. Curr Hypertens Rep. 2009;11:389–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Weissman AM. Themes and variations on ubiquitylation. Nat Rev Mol Cell Biol. 2001;2:169–178. [DOI] [PubMed] [Google Scholar]
- 38. Glickman MH, Ciechanover A. The ubiquitin‐proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373–428. [DOI] [PubMed] [Google Scholar]
- 39. Angers A, Ramjaun AR, McPherson PS. The HECT domain ligase ITCH ubiquitinates endophilin and localizes to the trans‐golgi network and endosomal system. J Biol Chem. 2004;279:11471–11479. [DOI] [PubMed] [Google Scholar]
- 40. Gallagher E, Gao M, Liu YC, Karin M. Activation of the E3 ubiquitin ligase ITCH through a phosphorylation‐induced conformational change. Proc Natl Acad Sci USA. 2006;103:1717–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rossi M, De Laurenzi V, Munarriz E, Green DR, Liu YC, Vousden KH, Cesareni G, Melino G. The ubiquitin‐protein ligase ITCH regulates p73 stability. EMBO J. 2005;24:836–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hansen TM, Rossi M, Roperch JP, Ansell K, Simpson K, Taylor D, Mathon N, Knight RA, Melino G. ITCH inhibition regulates chemosensitivity in vitro. Biochem Biophys Res Commun. 2007;361:33–36. [DOI] [PubMed] [Google Scholar]
- 43. Yoshioka J, Chutkow WA, Lee S, Kim JB, Yan J, Tian R, Lindsey ML, Feener EP, Seidman CE, Seidman JG, Lee RT. Deletion of thioredoxin‐interacting protein in mice impairs mitochondrial function but protects the myocardium from ischemia‐reperfusion injury. J Clin Invest. 2012;122:267–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xiang G, Seki T, Schuster MD, Witkowski P, Boyle AJ, See F, Martens TP, Kocher A, Sondermeijer H, Krum H, Itescu S. Catalytic degradation of vitamin D up‐regulated protein 1 mRNA enhances cardiomyocyte survival and prevents left ventricular remodeling after myocardial ischemia. J Biol Chem. 2005;280:39394–39402. [DOI] [PubMed] [Google Scholar]
- 45. Shah A, Xia L, Goldberg H, Lee KW, Quaggin SE, Fantus IG. Thioredoxin‐interacting protein mediates high glucose‐induced reactive oxygen species generation by mitochondria and the NADPH oxidase, Nox4, in mesangial cells. J Biol Chem. 2013;288:6835–6848. [DOI] [PMC free article] [PubMed] [Google Scholar]