

Abstract
Background
The small GTPase Rac1 regulates diverse cellular functions, including both apicobasal and planar cell polarity pathways; however, its role in cardiac outflow tract (OFT) development remains unknown. In the present study, we aimed to examine the role of Rac1 in the anterior second heart field (SHF) splanchnic mesoderm and subsequent OFT development during heart morphogenesis.
Methods and Results
Using the Cre/loxP system, mice with an anterior SHF‐specific deletion of Rac1 (Rac1 SHF) were generated. Embryos were collected at various developmental time points for immunostaining and histological analysis. Intrauterine echocardiography was also performed to assess aortic valve blood flow in embryos at embryonic day 18.5. The Rac1 SHF splanchnic mesoderm exhibited disruptions in SHF progenitor cellular organization and proliferation. Consequently, this led to a spectrum of OFT defects along with aortic valve defects in Rac1 SHF embryos. Mechanistically, it was found that the ability of the Rac1 SHF OFT myocardial cells to migrate into the proximal OFT cushion was severely reduced. In addition, expression of the neural crest chemoattractant semaphorin 3c was decreased. Lineage tracing showed that anterior SHF contribution to the OFT myocardium and aortic valves was deficient in Rac1 SHF hearts. Furthermore, functional analysis with intrauterine echocardiography at embryonic day 18.5 showed aortic valve regurgitation in Rac1 SHF hearts, which was not seen in control hearts.
Conclusions
Disruptions of Rac1 signaling in the anterior SHF results in aberrant progenitor cellular organization and defects in OFT development. Our data show Rac1 signaling to be a critical regulator of cardiac OFT formation during embryonic heart development.
Keywords: cellular organization, congenital heart defect, outflow tract development, Rac1
Subject Categories: Basic Science Research, Developmental biology, Animal Models of Human Disease, Congenital Heart Disease
Introduction
Cardiac outflow tract (OFT) defects account for approximately one‐third of all congenital heart defects (CHDs) reported in humans births and often require intervention within the first year of life. Even after surgical correction, risk of morbidity and mortality from OFT defects remains high.1, 2 Nevertheless, the molecular mechanisms underlying OFT defects are not well defined. Understanding the developmental mechanisms of OFT formation is crucial for new insights into improving diagnostics and designing therapeutic approaches for CHD patients.
Cell polarity is the asymmetrical organization of cell membrane proteins, intracellular organelles, and actin cytoskeleton that can influence cell fate and specialized functions such as migration and proliferation.3 Establishment of polarity is a critical step in a multitude of developmental events, including formation of the OFT.4, 5 The intricate process of OFT development involves coordination and interactions between 2 distinct cell types, second heart field (SHF) progenitors and cardiac neural crest cells.6 The SHF progenitors give rise to the myocardial and endothelial cells of the OFT, semilunar valves, along with vascular smooth muscle cells at the base of the aorta and pulmonary trunk. Neural crest cells contribute to septation of the OFT and remodeling of the aortic arches and semilunar valves.7, 8 At approximately embryonic day 8.5 (E8.5), cardiac neural crest cells will delaminate from the neural tube, migrate into the pharyngeal region, and become closely apposed with SHF progenitors in the dorsal pericardial wall.9 SHF cells form an apicobasally polarized epithelium in the splanchnic mesoderm, and maintenance of this apicobasal polarity is crucial for heart tube elongation and OFT morphogenesis.10 Planar cell polarity, which is orthogonal to apicobasal polarity, has also been shown to be a critical regulator of the SHF because tissue‐specific deletion of core planar cell polarity genes Vangl2 and Dishevelled1/2 recapitulates the spectrum of OFT defects reported in the full‐body mutants.11, 12 Recent studies have suggested that apicobasal polarity and planar cell polarity signaling are interconnected where crosstalk occurs to maintain cellular polarity and overall tissue architecture.13, 14
Rac1 is a small signaling GTPase from the Rac subfamily of Rho GTPases and has been implicated in both apicobasal polarity and planar cell polarity signaling pathways.15, 16 Rac1 acts as a pleiotropic effector of numerous cellular processes including regulation of the actin cytoskeleton and overall cell shape and morphology, which are critical components of cell polarity.17 The role of Rac1 in cell polarity has been shown by orientation of hair cells in the cochlea, convergent extension cellular movements, and anterior–posterior body axis specification during development.18, 19, 20 We recently demonstrated Rac1 to be a critical regulator of cardiomyocyte polarity and cardiac septation during heart development21; however, the role of Rac1 in OFT development is unknown. The SHF progenitors in the dorsal splanchnic mesoderm can be further subdivided into 2 subdomains: the anterior and the posterior SHF.22, 23 A Mef2c‐Cre transgenic mouse has been developed that drives expression of Cre recombinase solely in the anterior subdomain of the SHF, which contributes to the right ventricle and the OFT. Specifically, the Cre recombinase sequence in the Mef2c‐Cre transgenic mouse is under regulatory control of an anterior heart field–specific enhancer and promoter, which was first discovered by Dodou et al.8, 24 To study the role of Rac1 in OFT formation, we generated an anterior SHF‐specific deletion of Rac1 using the Mef2c‐Cre transgenic mouse, as described previously.21 We showed that Rac1 signaling in the anterior SHF is critical for progenitor cellular organization in the splanchnic mesoderm and that disruptions in Rac1 signaling result in a spectrum of OFT defects, along with aortic valve defects. Furthermore, we showed that a Rac1 deficiency restricted to the anterior SHF leads to neural crest cell migration defects.
Methods
Mice
Rac1 f/f (stock 5550) and mT/mG mice (stock 7676) were purchased from Jackson Laboratory (Bar Harbor, ME).25, 26 The anterior SHF‐specific Mef2c‐Cre transgenic mice were rederived from embryos obtained from the Mutant Mouse Regional Resource Centers (Chapel Hill, NC).8 A breeding program to generate Mef2c‐Cre;Rac1 f/f (Rac1 SHF) mice was carried out and genotyping was performed, as described previously.21 All mouse experiments and procedures were approved by the animal use subcommittee at the University of Western Ontario in accordance with the guidelines of the Canadian Council of Animal Care.
Fate Mapping Analysis
Fate mapping analysis was performed using Mef2c‐Cre and the mT/mG global double‐fluorescent Cre reporter mice to trace the anterior SHF and derivatives.8, 26 A breeding strategy was carried out to generate Mef2c‐Cre;mT/mG and Mef2c‐Cre;Rac1 f/f ;mT/mG (Rac1 SHF ;mT/mG) mice. The mT/mG mice possess loxP sites on either side of the membrane‐targeted Tomato (mT) cassette with a membrane‐targeted green fluorescent protein (GFP) cassette downstream. Before Mef2c‐Cre mediated excision, mT (red fluorescence) is expressed in all cells. After Mef2c‐Cre–mediated excision, the mT cassette is cleaved out, and membrane‐targeted GFP (mG) is expressed in all SHF progenitors and SHF‐derived tissues.
Histological Analysis
Neonatal and embryonic samples were fixed overnight in 4% paraformaldehyde at 4°C, dehydrated, and embedded in paraffin. Embryos were serially sectioned as 5‐μm‐thick sections with a Leica RM2255 microtome, and sections were mounted onto positively charged albumin/glycerin‐coated slides, as described previously.21 Slides underwent a dewaxing process and were stained with hematoxylin and eosin or picrosirius red for morphological analysis. Images were captured with a Zeiss Observer D1 light microscope.
Immunohistochemistry
For immunohistochemical staining on paraffin sections, samples underwent a deparaffin process, and antigen retrieval was performed in sodium citrate buffer, as described previously.21 The primary antibodies used for immunostaining included phosphohistone‐H3 (phospho S10; Abcam), GFP (Abcam), α‐actinin (Sigma‐Aldrich), semaphorin 3c (Sema3c; Santa Cruz Biotechnology), and activating enhancer binding protein 2 alpha (AP2α) (Santa Cruz Biotechnology). Slides were then incubated with biotinylated secondary antibody followed by incubation with avidin and biotinylated horseradish peroxidase from the Santa Cruz ImmunoCruz ABC staining system kit. Antigen was visualized with diaminobenzidine substrate solution, and slides were counterstained with hematoxylin. All images were captured with a Zeiss Observer D1 microscope using AxioVision release 4.7 software. For frozen sections, embryos were fixed for 1 hour in 4% paraformaldehyde, cryoprotected in 30% sucrose, and embedded in FSC22 frozen section media (Leica). Embryos were sectioned in a sagittal orientation at 10 μm thick and immunostained with the primary antibodies for Scrib (Santa Cruz Biotechnology), atypical protein kinase C zeta (aPKCζ) (Santa Cruz Biotechnology), and active (nonphosphorylated) β‐catenin (Cell Signaling). Additional staining was also performed with Alexa Fluor 488 phalloidin (Life Technologies), Alexa Fluor 647 wheat germ agglutinin (Invitrogen), and Hoechst 33342 (Invitrogen). A Zeiss LSM 510 Duo microscope with ZEN 2012 software (Zeiss) located at the Biotron Experimental Climate Change Research Center at Western University (London, Ontario, Canada) was used to obtain confocal images.
Intrauterine Echocardiography
Echocardiography was performed on pregnant mice using the Vevo 2100 system (VisualSonics).27 Briefly, pregnant mice were anesthetized with isoflurane, and fur was shaved from the abdomen to better image embryos. A map was drawn of the location of embryos relative to each other to facilitate harvesting of embryos after intrauterine echocardiography. A dynamically focused 40‐MHz probe was used to obtain 2‐dimensional images of embryonic hearts in short‐axis view. Diastolic and systolic left ventricular internal diameters (LVID) were measured from M‐mode recordings. Calculations were performed as followed: EF(%)=[(LVIDd)3−(LVIDs)3]/(LVIDd)3×100 and FS(%)=(LVIDd−LVIDs)/LVIDd×100. EF indicates ejection fraction, d indicates diastolic, s indicates systolic, and FS indicates fractional shortening.
Statistical Analysis
All data are presented as median and interquartile range. Because the sample size was 4 to 8 per group and was not normally distributed, a nonparametric Mann–Whitney test was performed to compare the difference between the 2 groups. A 2‐tailed P value <0.05 was considered statistically significant.
Results
Early Defects in the Splanchnic Mesoderm of Rac1 SHF
Lineage tracing analysis using Mef2c‐Cre and mT/mG global double‐fluorescent reporter mice revealed that lack of Rac1 in the anterior SHF resulted in defects in splanchnic mesoderm at E9.5. Large acellular spaces were observed between GFP+ SHF progenitor cells, and fewer cells resided in the overall splanchnic mesoderm region of E9.5 Rac1 SHF ;mTmG embryos (Figure 1C and 1D) compared with littermate Mef2c‐Cre;mTmG;Rac1 f/+ controls (Figure 1A and 1B). The number of GFP+ SHF progenitor cells in the splanchnic mesoderm was significantly less in Rac1 SHF embryos compared with controls (Figure 1E). Before differentiation, the SHF progenitor population in the splanchnic mesoderm is highly proliferative.28 To analyze SHF proliferation, phosphohistone‐H3 immunostaining was performed on sagittal sections of E9.5 embryos (Figure 1F and 1G). The proliferation rate of SHF progenitors in E9.5 Rac1 SHF was significantly reduced compared with littermate Rac1 f/f controls (Figure 1H). This reduced proliferation rate likely resulted in a reduced number of SHF progenitors within the Rac1 SHF splanchnic mesoderm. These results define a requirement for Rac1 signaling in SHF progenitor cell proliferation.
Figure 1.
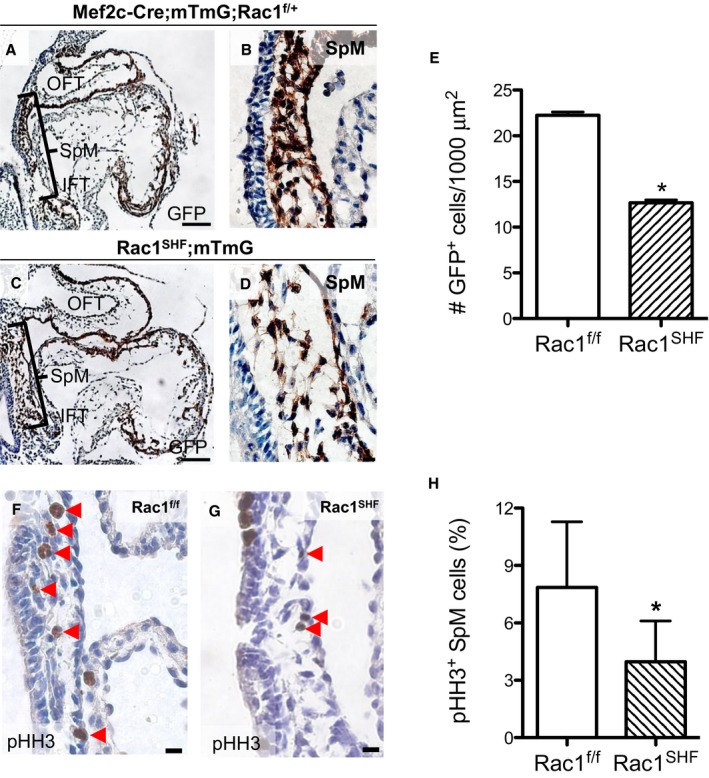
Early defects in embryonic day 9.5 (E9.5) Rac1 SHF splanchnic mesoderm. Lineage tracing with Mef2c‐Cre;mT/mG showed a decreased number of GFP + SHF cells along with large acellular spaces in the splanchnic mesoderm of Rac1 SHF ;mT/mG (C and D) compared with littermate control embryos (A and B). The number of GFP + SHF progenitor cells in the splanchnic mesoderm was significantly reduced in E9.5 Rac1 SHF ;mT/mG compared with controls (E). n=4 embryos per group. Immunostaining for pHH3 in the splanchnic mesoderm (F and G) showed a reduced proliferation rate in Rac1 SHF SHF progenitors compared to Rac1 f/f controls (H). n=4 (Rac1 SHF) and n=5 (Rac1 f/f) embryos. Scale bars: 100 μm (A and C), 10 μm (B and D), 20 μm (F and G). *P<0.05 by Mann–Whitney test. GFP indicates green fluorescent protein; IFT, inflow tract; OFT, outflow tract; pHH3, phosphohistone H3; SHF, second heart field; SpM, splanchnic mesoderm.
Disruption of Cell Shape and Organization in Rac1 SHF Splanchnic Mesoderm
SHF progenitor cells possess apicobasal polarity that forms a polarized epithelial layer in the dorsal pericardial wall.10 Our data show that cellular organization was severely disrupted in the E9.5 Rac1 SHF anterior SHF progenitors (Figure 2). The basolateral and apical domains of the Rac1 f/f splanchnic mesoderm were marked by Scribble and aPKCζ, respectively. In addition, the SHF progenitors have a cuboidal shape, with neighboring cells aligned with one another to form the epithelial layer (Figure 2A through 2C). In comparison, organization of the Rac1 SHF SHF epithelial layer was lost, and SHF progenitor cells exhibited a rounded morphology. Cellular organization in the E9.5 Rac1 SHF splanchnic mesoderm was disturbed where the basal domains of neighboring SHF progenitors were no longer aligned with one another, and the cells displayed a rounded morphology (Figure 2D through 2F). Orientation of the long axis of the anterior SHF progenitor cells were measured relative to the axis of the dorsal pericardial wall (Figure 2G), and it was found that E9.5 Rac1 SHF SHF progenitor cells had a random, unorganized orientation compared with Rac1 f/f controls. Rac1 f/f SHF progenitors had an orientation range of 62° to 89.5°, averaging 78.5° relative to the dorsal pericardial wall axis. In comparison, Rac1 SHF SHF progenitors oriented with a significantly smaller angle averaging 41.8° relative to the dorsal pericardial wall axis (Figure 2H). Cell–cell junctions, as marked by active (nonphosphorylated) β‐catenin expression, were disrupted in the E9.5 Rac1 SHF splanchnic mesoderm compared with Rac1 f/f controls, further supporting a disruption in SHF progenitor cell organization (Figure 2I and 2J). Taken together, these findings support a critical role of Rac1 signaling in regulation of overall SHF progenitor cell shape and maintenance of cell organization.
Figure 2.
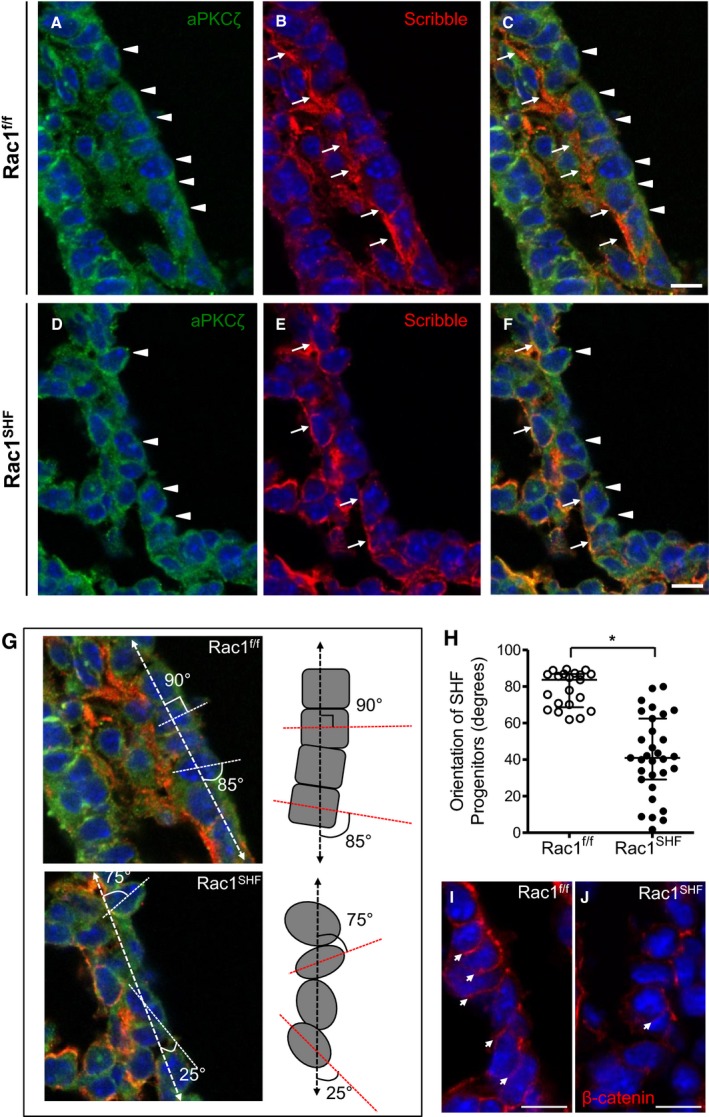
Disrupted apicobasal cell polarity and orientation in Rac1 SHF splanchnic mesoderm. A and B, In embryonic day 9.5 (E9.5) Rac1 f/f SHF progenitors, the basolateral domain is marked by Scribble (B, arrows), and the apical domain is marked by aPKCζ (A, arrowheads). The SHF progenitors have a distinct cuboidal shape, forming an organized epithelial layer (C). Polarity is disrupted in Rac1 SHF SHF progenitors in which the Scribble‐positive basal domains (E, arrows) and aPKCζ‐positive apical domains (D, arrowheads) of individual cells are no longer aligned with neighboring cells due to the rounded morphology of the SHF progenitors (F). C, Overlay of panels A and B. F, Overlay of panels D and E. The angle of the E9.5 SHF progenitor cell long axis (dashed line) was measured relative to the axis of the dorsal pericardial wall (dashed arrow line) to obtain the degree of orientation (G). Images in panel G are from panels C and F with an overlay of schematic axis and angle measurements. H, The angle of each SHF progenitor cell in the splanchnic mesoderm was measured and plotted. *P<0.05 by Mann–Whitney test, n=4 embryos per group. Each data point represents 1 SHF progenitor cell. Active (nonphosphorylated) β‐catenin marked cell–cell junctions in E9.5 Rac1 f/f SHF progenitors (I, arrows). In comparison, cell–cell junctions were disrupted in E9.5 Rac1 SHF SHF progenitors (J). Scale bars: 10 μm (A through F, I and J). SHF indicates second heart field.
Loss of Rac1 in the SHF Results in Shortened OFT and Myocardial Defects of the OFT
Sufficient lengthening of the heart tube from SHF progenitors ensures convergence of the inflow and outflow poles during cardiac looping. A shortened OFT alters looping and leads to arterial pole misalignment defects such as double‐outlet right ventricle and overriding aorta.29, 30 The sagittal length of the OFT was measured at E10.5 (Figure 3A and 3B), and it was found that the Rac1 SHF OFT was significantly shorter compared with controls (Figure 3C). Closer analysis of the SHF‐derived OFT myocardium revealed a disorganized myocardial layer in E10.5 Rac1 SHF OFT compared with controls (Figure 3D through 3G). Myocardial cells in the Rac1 f/f OFT were aligned with one another and had a general oval or cuboidal cell shape (Figure 3D and 3F). In contrast, myocardial cells in the Rac1 SHF OFT were not as well organized, and the shapes of the cells were not as distinctly oval or cuboidal (Figure 3E and 3G). In addition, cell–cell adhesion junction marked by β‐catenin was generally disrupted in the myocardium of E10.5 Rac1 SHF OFT compared with Rac1 f/f controls (Figure 3H and 3I). These results suggest an important role of Rac1 in generating the cellular organization of the myocardial layer and lengthening of the OFT.
Figure 3.
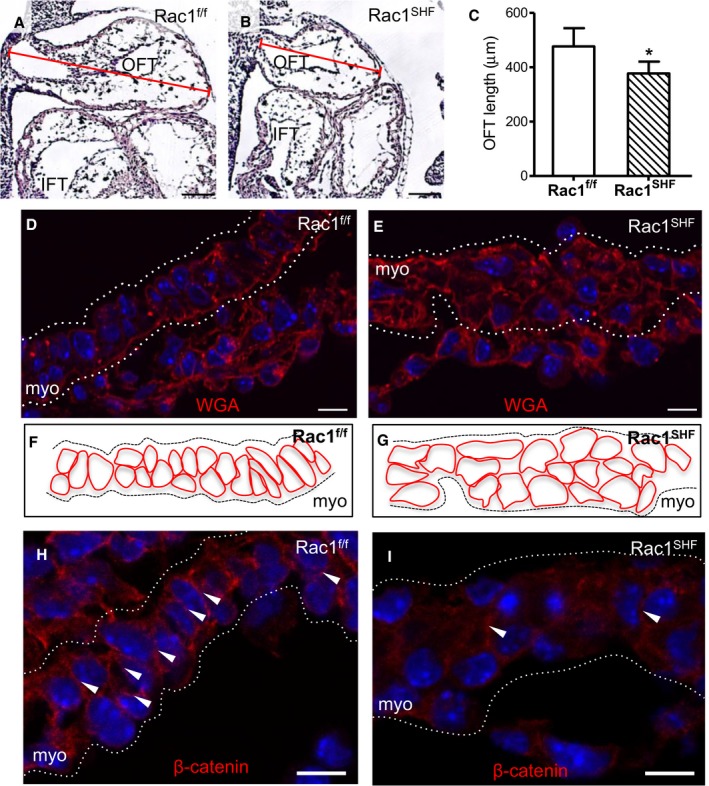
Defects in early Rac1 SHF outflow tract development. A through C, Length of the OFT at embryonic day 10.5 (E10.5) was measured in sagittal sections (A and B) and was found to be significantly shorter in Rac1 SHF compared with Rac1 f/f controls (C). * P<0.05 by Mann–Whitney test, n=4 (Rac1 SHF) and n=5 (Rac1 f/f) embryos. D and E, WGA staining to mark cell membranes of SHF cells in the OFT at E10.5 show a disorganized myocardial layer in Rac1 SHF compared with Rac1 f/f littermate controls. F and G, Schematic diagrams of panels D and E, respectively. H and I, Active (nonphosphorylated) β‐catenin staining is reduced at cell–cell junctions in E10.5 Rac1 SHF OFT myocardial cells. White arrows indicate cell–cell adhesion sites. Dotted white lines indicate boundaries of the OFT myocardial layer in panels D, E H, and I. Scale bars: 100 μm (A and B), 10 μm (D, E, H, and I). IFT indicates inflow tract; myo indicates myocardium; OFT, outflow tract; SHF, second heart field; WGA, wheat germ agglutinin.
OFT Defects in Rac1 SHF Hearts
Examination of Rac1 SHF hearts revealed a spectrum of OFT defects from E14.5 to postnatal day 0 (P0) that was 100% penetrant (Table 1). The majority of the defects found were OFT alignment defects. More than one‐third of Rac1 SHF hearts (23 of 64) displayed a double‐outlet right ventricle (Figure 4A through 4D), and more than another third (24 of 64) exhibited an overriding aorta (Figure 4E and 4F). Transposition of the great arteries was found in 1 P0 Rac1 SHF sample (Figure 4G and 4H). In addition to defects in OFT alignment, defects in septation of the OFT resulting in persistent truncus arteriosis or common arterial trunk was observed in 9.4% (6 of 64) of Rac1 SHF hearts (Figure 4I and 4J). Narrowing of the great arteries was also observed in Rac1 SHF hearts including stenosis of the pulmonary artery (Figure 4K and 4L) and aortic atresia (Figure 4M and 4N). In 1 case of aortic atresia in Rac1 SHF hearts, aortic valves were also severely malformed (Figure 4O and 4P). In addition, we observed abnormalities in aortic arch artery remodeling resulting in a retroesophageal right subclavian artery or a vascular ring, a congenital defect in which vascular structures surround and constrict the esophagus and trachea. This was evident in 18.8% (12 of 64) of Rac1 SHF hearts, and of the 12 samples that had a vascular ring, 3 had a right‐sided aortic arch (Figure 5A through 5F and Videos S1 and S2). Taken together, these data strongly support a critical requirement for Rac1 signaling in anterior SHF progenitors for normal OFT morphogenesis.
Table 1.
Outflow Tract Defects in Rac1 SHF Hearts (E14.5‐P0)
| TGA | PTA | DORV | Overriding Aorta | Aortic Atresia | Vascular Ring | |
|---|---|---|---|---|---|---|
| n=64 | 1 | 6 | 23 | 32 | 5 | 12 |
| % | 1.6 | 9.4 | 35.9 | 50 | 7.8 | 18.8 |
All 64 Rac1 SHF hearts had ≥1 OFT defect. No OFT defects were found in Rac1 f/f hearts (n=36). DORV indicates double‐outlet right ventricle; PTA, persistent truncus arteriosus; TGA, transposition of great arteries.
Figure 4.
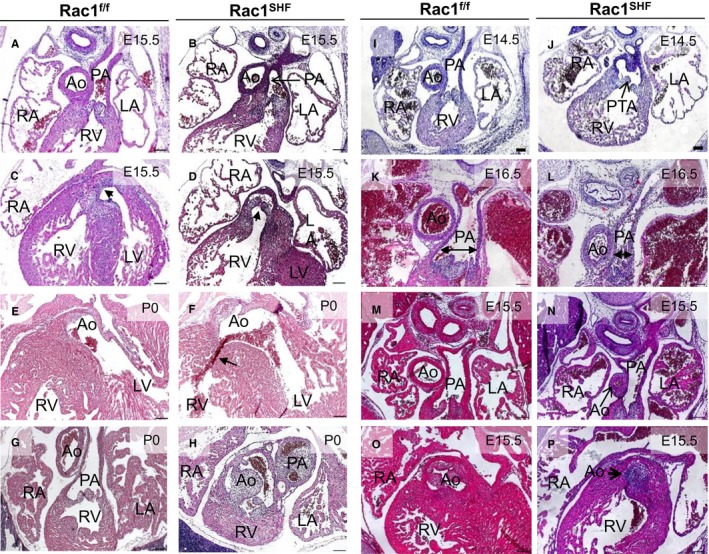
Spectrum of OFT defects found in E14.5‐P0 Rac1 SHF hearts. Double‐outlet right ventricle was found in E15.5 Rac1 SHF hearts (A through D). The PA connects to the RV in both Rac1 SHF and Rac1 f/f hearts (A and B). The Rac1 SHF Ao (arrow) incorrectly connects to the RV compared with Rac1 f/f controls, in which the Ao connects to the LV (C and D). Overriding Ao, in which the Ao is positioned directly over a ventricular septal defect (arrow), was observed in Rac1 SHF hearts (E and F). Transposition of the great arteries was found in 1 Rac1 SHF sample in which the Ao and PA openings were switched, which resulted in the Ao connecting to the RV in Rac1 SHF and the PA connecting to the LV (G and H), respectively. PTA, in which the common OFT is not divided into aorta and pulmonary, was observed in Rac1 SHF hearts (I and J). Stenosis of the PA was observed in Rac1 SHF hearts (K and L). Aortic atresia was observed in Rac1 SHF hearts (M and N), and in 1 sample with aortic atresia, distinct aortic valves were absent (O and P). Scale bars: 100 μm. Ao indicates aorta; E, embryonic day; LA, left atrium; LV, left ventricle; OFT, outflow tract; PA, pulmonary artery; PTA, persistent truncus arteriosus; RA, right atrium; RV, right ventricle; SHF, second heart field.
Figure 5.
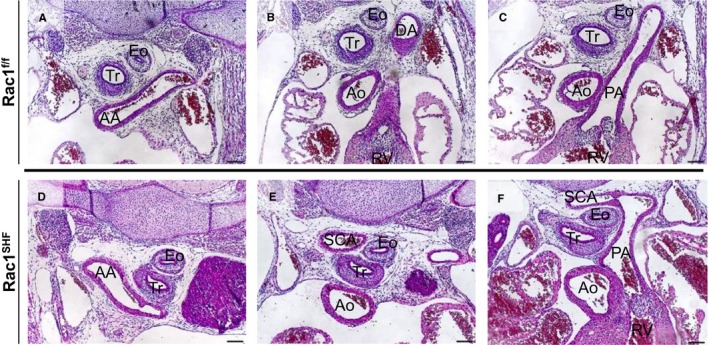
Vascular rings in embryonic day 15.5 Rac1 SHF hearts. A right‐sided AA was observed in Rac1 SHF (D) hearts compared with Rac1 f/f controls (A). An aberrant retroesophageal right SCA, which arose from AA, was observed next to the Tr and Eo in Rac1 SHF hearts (E) compared with Rac1 f/f controls (B). The SCA in Rac1 SHF hearts joined with the PA, forming a vascular ring (F) in Rac1 SHF hearts compared with controls (C). Scale bars: 100 μm. AA indicates aortic arch; Ao, aorta; DA, dorsal aorta; Eo, esophagus; PA, pulmonary artery; SCA, subclavian artery; Tr, trachea.
Myocardialization Defects in Rac1 SHF Hearts
Myocardialization is a process in which myocardial cells migrate and grow into the flanking mesenchyme. At E12, myocardial cells of the OFT begin to invade the neighboring cushion mesenchyme. This leads to a gradual replacement of the mesenchymal tissue with myocardium to form the muscular outlet septum by E17.31 Abnormalities in muscularization of the OFT cushions was observed in E12.5 Rac1 SHF hearts. The Rac1 SHF OFT myocardium lacked a polarized morphology and did not extend as far into the cushion mesenchyme compared with controls (Figure 6A and 6B). Muscularization of the proximal OFT septum is complete in P0 Rac1 f/f control hearts, as shown by α‐actinin–positive muscle tissue separating the aorta from the right ventricle (Figure 6C). In P0 Rac1 SHF hearts, the proximal OFT septum remained nonmuscularized, and remnant collagen‐rich mesenchymal tissue remained (Figure 6D), staining positive for picrosirius red (Figure 6E and 6F). Rac1 SHF samples with defects in muscularization of the proximal OFT septum also presented with OFT misalignment defects including overriding aorta and double‐outlet right ventricles. These results further support a crucial role for Rac1 signaling in regulating polarity, along with elongation and migration of cardiomyocytes during the process of OFT myocardialization.
Figure 6.
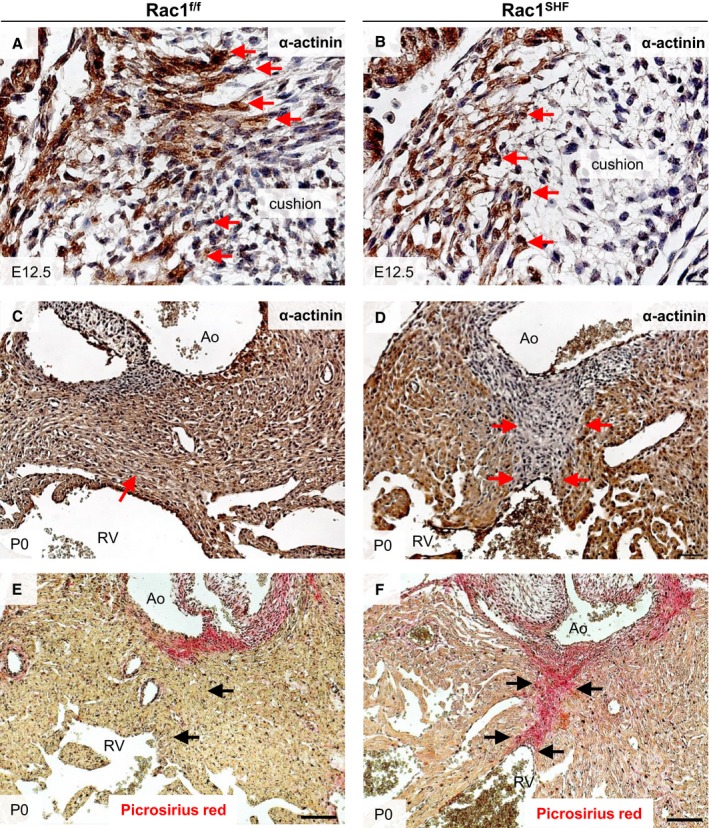
Abnormalities in Rac1 SHF OFT myocardialization. E12.5 Rac1 SHF OFT cardiomyocytes, marked by α‐actinin immunostaining (B), exhibited a blunted morphology instead of a polarized morphology and did not extend as far into the OFT cushions compared with controls (A). Red arrows indicate invading cardiomyocytes. The proximal OFT septum remained nonmuscularized in P0 Rac1 SHF hearts (D), indicated by an absence of α‐actinin staining (arrows in D) compared with control (C), in which the septum is muscularized (arrow in C). The nonmuscularized proximal OFT septum in Rac1 SHF hearts (F) stained positive for picrosirius red (arrows in F), indicating that this tissue remained mesenchymal compared with controls (E), which had become muscularized (arrow in E). n=3 for each staining per group. Scale bars: 10 μm (A and B), 50 μm (C through F). Ao indicates aorta; E, embryonic day; OFT, outflow tract; RV, right ventricle; SHF, second heart field.
Decreased Cardiac Neural Crest Cell Migration in Rac1 SHF
The aortic arch artery and OFT septation defects observed in Rac1 SHF hearts suggest abnormal formation or remodeling of the arteries involving cardiac neural crest cells. Sema3c is a secreted glycoprotein part of the semaphorin protein family, which plays a role in axon guidance.32 Sema3c is expressed in the OFT myocardium and acts as a chemoattractant, navigating neural crest cells to the OFT. Neural crest cells express the multimeric complexes of plexins and neuropilin, which are the receptors that recognize semaphorin ligands.33 Interactions between SHF progenitors and cardiac neural crest cells have been shown to be critical for development of the aortic arches and semilunar valves.6, 7 Sema3c expression was analyzed in Rac1 SHF hearts, and intensity of Sema3c protein staining in E11.5 Rac1 SHF OFT myocardium was found to be decreased compared with Rac1 f/f controls (Figure 7C, 7D, and 7F). Along with this, the mRNA level of Sema3c, as measured by real‐time polymerase chain reaction, was significantly reduced in Rac1 SHF hearts compared with littermate controls at E13.5 (Figure 7G). Consequently, this would be predicted to result in a reduction in the chemoattractant signal attracting the migrating neural crest cells into the developing heart. The number of migrating cardiac neural crest cells, marked by the transcription factor AP2α, was significantly decreased in the region between the foregut and pericardial cavity in E10.5 Rac1 SHF embryos (Figure 7A, 7B, and 7E). These results suggest that disruption of Rac1 signaling in the SHF‐derived OFT myocardium leads to reduced expression of axonal guidance signals that attract migrating neural crest cells into the developing OFT.
Figure 7.
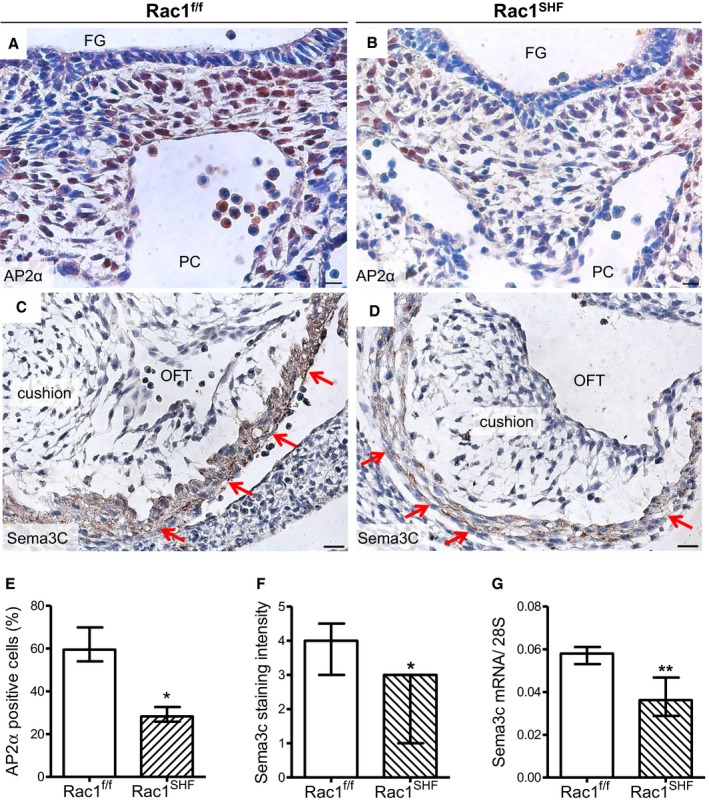
Defects in neural crest cell contribution to OFT development in Rac1 SHF hearts. A and B, AP2α immunostaining was performed to mark neural crest cells in the pharyngeal region of embryonic day 10.5 (E10.5) samples. C and D, Sema3c immunostaining (brown color) in Rac1 SHF and Rac1 f/f OFT myocardium at E11.5. E, The number of neural crest cells was significantly reduced in E10.5 Rac1 SHF pharyngeal arches compared with controls. F, Intensity of Sema3c staining in the E11.5 OFT myocardium was ranked on a scale from 1 to 5. Overall intensity of Sema3c staining was reduced in Rac1 SHF compared with Rac1 f/f controls. G, The level of Sema3c mRNA in Rac1 SHF hearts at E13.5 was significantly reduced compared with controls. * P<0.05 Mann–Whitney test. n=4 to 5 hearts (E and F) and n=7 to 8 hearts (G) per group. Scale bars: 20 μm (A and B), 10 μm (C and D). FG indicates foregut; OFT, outflow tract; PC, pericardial cavity; Sema3c, semaphorin 3c; SHF, second heart field.
Aortic Valve Defects in Rac1 SHF Hearts
Formation of valves involves a complex process of cushion formation, elongation, valve remodeling, and maturation.34 Both SHF progenitors and cardiac neural crest cells contribute to aortic valve development. Neural crest cells are known to be involved in late gestation remodeling and maturation of the aortic valves.7, 8 We predicted that the decreased number of migrated cardiac neural crest cells into Rac1 SHF hearts would also affect development of Rac1 SHFaortic valves. P0 Rac1 SHF heart sections were analyzed and found to have large, thickened aortic valve leaflets compared with controls that, in contrast, had undergone remodeling and matured into thin, elongated leaflets (Figure 8A and 8B). Cell density of the aortic valve leaflets was also significantly decreased in P0 Rac1 SHF hearts compared with P0 Rac1 f/f hearts (14.3±1.0 versus 17.8±0.8 cells/1000 μm2, P<0.05, n=5–7), suggesting defects in extracellular matrix remodeling, which is necessary for the dense packing of cells in the elongating valves.
Figure 8.
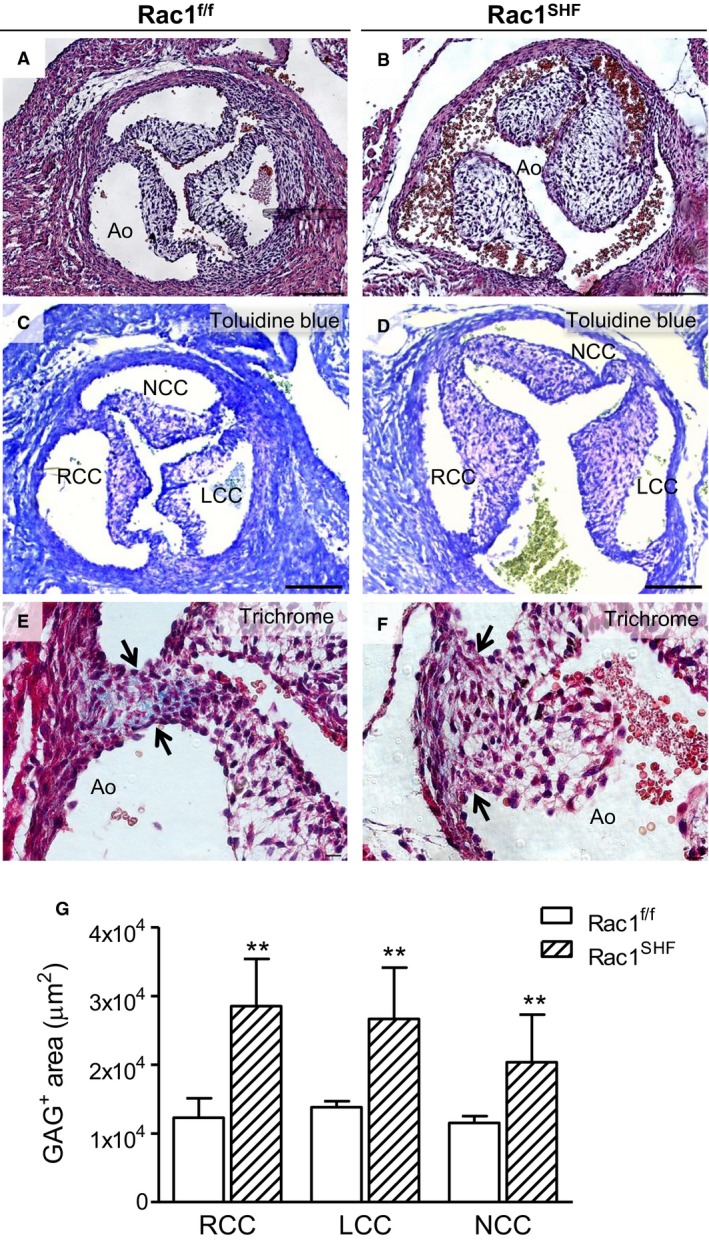
Aortic valve defects in P0 Rac1 SHF hearts. More than 30% (9 of 28) of P0 Rac1 SHF hearts exhibited thick aortic valve leaflets (B) compared with the thin, remodeled valves of controls (A). Toluidine blue staining showed that GAG (light purple color) occupies the acellular space of Rac1 SHF and littermate valve leaflets (C and D). Masson's trichrome staining in Rac1 f/f aortic valves showed collagen in the commissure of valve leaflets (E, arrows), which was absent in Rac1 SHF aortic valves (F, arrows). The GAG‐positive area (light purple color) in each valve leaflet (C and D) was quantified in (G). **P<0.01 by Mann–Whitney test, n=5 to 6 hearts per group. Scale bars: 100 μm (A through D), 10 μm (E and F). Ao indicates aorta; GAG, glycosaminoglycans; LCC, left coronary cusp; NCC, noncoronary cusp; RCC, right coronary cusp; SHF, second heart field.
To assess glycosaminoglycans, a component of extracellular matrix in the aortic valve, toluidine blue staining was performed.35 The acellular space of the valve leaflets stained light purple, indicating the presence of glycosaminoglycans (Figure 8C and 8D). Notably, a significantly larger amount of glycosaminoglycans was present in Rac1 SHF aortic valves compared with littermate Rac1 f/f controls at P0 (P<0.01) (Figure 8G).
During the early stages of development, there is little to no collagen present in the semilunar valves. As the valves mature, collagen content increases, especially at the commissure of the valves.36 Analysis of P0 Rac1 f/f aortic valves after Masson's trichrome staining revealed early signs of maturation, with collagen present at the valve commissure (Figure 8E). In comparison, P0 Rac1 SHF aortic valves were less mature and had little to no collagen present at the valve commissure (Figure 8F).
Lineage tracing with mT/mG at E12.5 revealed decreased SHF progenitor contribution to the OFT cushions in Rac1 SHF hearts compared with control hearts, which had numerous SHF‐derived GFP+ cells in the cushion mesenchyme (Figure 9A and 9B, black arrows). At E14.5, SHF contribution to the aortic valve leaflets continued to be severely reduced in Rac1 SHF hearts (Figure 9C and 9D, black arrows). In addition, at both E12.5 and E14.5, contribution of SHF progenitors to the Rac1 SHF OFT myocardium seemed to be decreased compared with controls (Figure 9A through 9D, red arrows). This lineage tracing data strongly suggest an overall impairment in overall SHF progenitor cell contribution to the developing heart in Rac1 SHF embryos and a critical role for SHF Rac1 in maturation and remodeling of the aortic valves.
Figure 9.
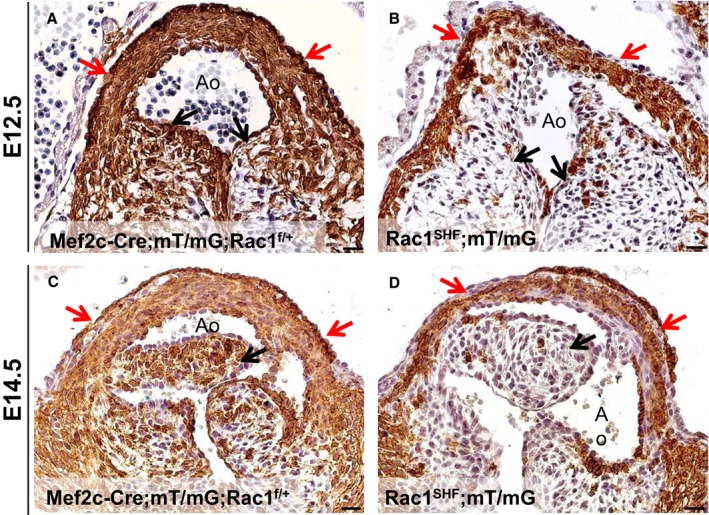
Decreased SHF contribution to Rac1 SHF OFT. Fate mapping with mT/mG reporter shows a decreased SHF progenitor contribution to the OFT myocardium (red arrows in B and D) in E12.5 and E14.5 Rac1 SHF hearts compared with controls (red arrows in A and C). SHF contribution to the valve leaflets was also severely reduced in Rac1 SHF hearts compared with controls (black arrows, A through D). Paraffin sections were immunostained with anti‐GFP. Scale bars: 20 μm. n=3 for each time point per group. Ao indicates aorta; E, embryonic day; OFT, outflow tract; SHF, second heart field.
Abnormal Aortic Valve Function in Rac1 SHF Hearts
The observed aortic valve defects would be predicted to result in functional impairment of the valves. To assess the function of the malformed Rac1 SHF aortic valves, intrauterine echocardiography at E18.5 was performed. Embryos from timed pregnant female mice were analyzed prior to genotyping, and thus echo analysis was performed in a blinded manner. All E18.5 Rac1 f/f control embryos did not demonstrate any evidence of aortic valve regurgitation (Figure 10A). In contrast, E18.5 Rac1 SHF embryos were found to have severe aortic regurgitation during diastole (Figure 10B). In addition, LVID during systole was significantly increased in Rac1 SHF hearts, whereas no significant differences were observed in LVID during diastole. Both ejection fraction and fractional shortening in E18.5 Rac1 SHF embryos were also significantly reduced compared with controls (Table 2). Decreased left ventricular function in Rac1 SHF hearts is likely related to the defects in myocardial development reported in our previous study.21 These echocardiography data further define a requirement for SHF Rac1 in development and functional maturation of the aortic valves.
Figure 10.
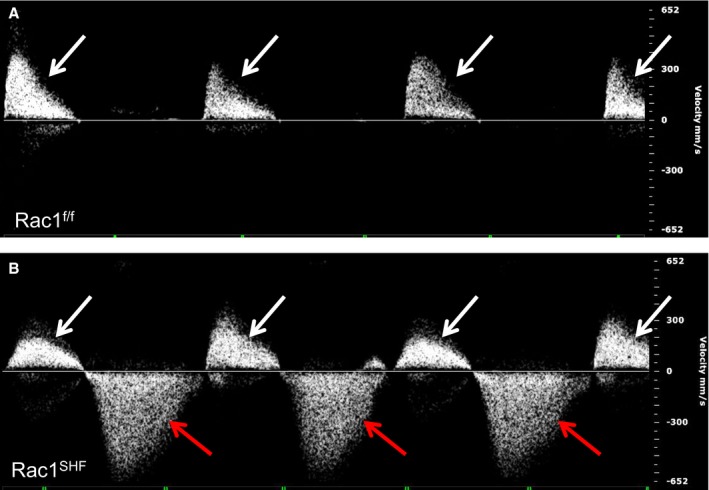
Aortic valve regurgitation in Rac1 SHF hearts. Intrauterine echocardiography at embryonic day 18.5 showed that Rac1 SHF hearts had severe aortic regurgitation during diastole (B, red arrows), which is not observed in Rac1 f/f littermates (A). White arrows in (A and B) indicate forward blood flow during systole. Shown are representatives of 5 fetuses per group. SHF indicates second heart field.
Table 2.
Intrauterine Echocardiography in Embryonic Day 18.5 Rac1 SHF Hearts
| Rac1f/f (n=5) | Rac1 SHF (n=5) | |
|---|---|---|
| Duration of backflow, ms | 0 (0) | 183 (66) |
| Velocity of backflow, mm/s | 0 (0) | 164 (19) |
| LVIDd, mm | 0.90 (0.03) | 0.91 (0.18) |
| LVIDs, mm | 0.57 (0.06) | 0.67 (0.11)* |
| Ejection fraction (%) | 32.5 (4.6) | 26.1 (5.7)** |
| Fractional shortening (%) | 69.3 (6.0) | 59.6 (9.5)** |
LVIDd indicates left ventricular internal diameter in diastole; LVIDs, left ventricular internal diameter in systole.
*P<0.05, **P<0.01 by unpaired Student t test.
Discussion
Recent studies have established cell and tissue polarity as important regulators of cardiac OFT development, especially factors involved in the planar cell polarity pathway11, 12; however, the role of Rac1 in cardiac OFT development is not clear. In the present study, we demonstrated that a deficiency of Rac1 signaling in the anterior SHF disrupted progenitor cell shape and overall epithelial organization in the splanchnic mesoderm. In addition, the proliferation rate of SHF progenitors was decreased with a loss of Rac1 signaling. Subsequently, as heart development progressed, these early defects in the SHF splanchnic mesoderm resulted in a spectrum of OFT and aortic valve defects. We also showed that Rac1 deficiency disrupted the levels of Sema3c, a chemoattractant for cardiac neural crest cells, generated by the SHF‐derived OFT myocardium, that would provide a mechanism for our observed reduction in migrating neural crest cells observed in the developing OFT (Figure 11).
Figure 11.
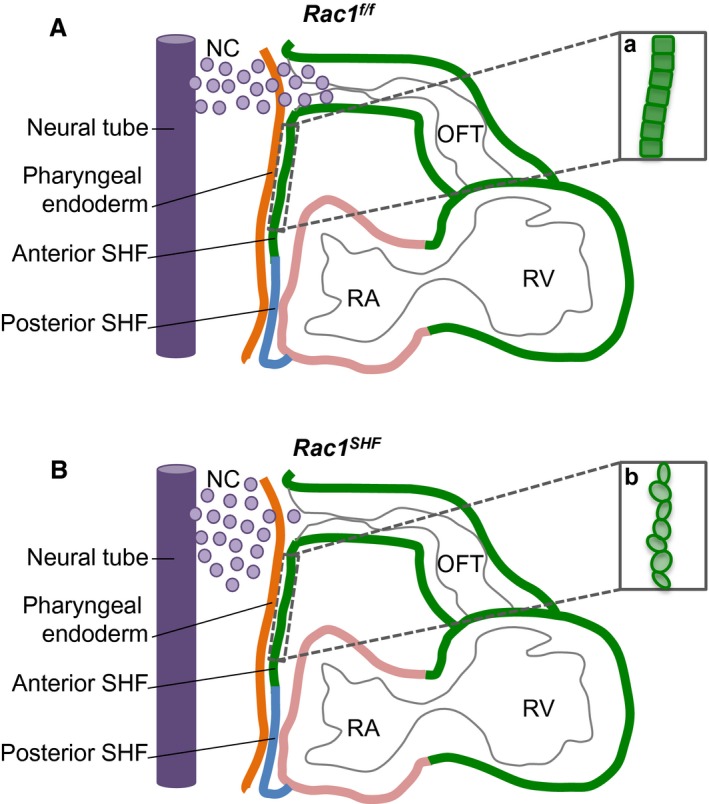
Schematic diagram of midsagittal section of embryonic day 9.5 (E9.5) splanchnic mesoderm. At E9.5, the Rac1 f/f anterior SHF progenitors in the splanchnic mesoderm are organized into a polarized epithelium (a). Chemotactic signals, such as semaphorin 3c, secreted by the OFT myocardium act as axonal guidance cues for migration of cardiac NC cells from the neural tube (A). In Rac1 SHFembryos, the anterior SHF progenitors are rounded and disorganized, displaying a loss of polarized epithelium characteristics (b). Expression of chemotactic signals is reduced, resulting in decreased migration of neural cells into the OFT (B). NC indicates neural crest; OFT, outflow tract; RA, right atrium; RV, right ventricle; SHF, second heart field.
Intricate signaling cascades exist between the cardiac neural crest and the SHF. Defects in the cardiac neural crests can affect the SHF progenitors and vice versa. Ablation of cardiac neural crest cells led to a failure of the SHF progenitors to lengthen the heart tube and contribute to the OFT myocardium.30, 37 In addition to ablation, tissue‐specific genetic deletions in the cardiac neural crest cells, which affect signaling pathways including Bmp and Smad, and in the transcription factor Tbx3 all have secondary effects on the SHF and result in defects in OFT remodeling, alignment, and elongation.38, 39, 40 Conversely, tissue‐specific genetic deletions in the SHF tissue, including in Notch and Tbx1, can also have a noncell autonomous effect on migration of cardiac neural crest cells into the developing heart.7, 41 Our study showed that conditional deletion of Rac1 in the SHF reduced the migration of cardiac neural crest cells into the pharyngeal region, suggesting a role for Rac1 in supporting the intercellular signaling that exists between the SHF and neural crest. The mechanism by which Rac1 signaling regulates expression of the neural crest chemoattractant Sema3c in the SHF‐derived OFT myocardium remains to be determined. Studies have shown that Rac1 activates c‐jun N‐terminal kinase (JNK) to increase gene expression, including expression of the zinc‐finger transcription factor Gata6.42, 43 Sema3c can be transcriptionally regulated by Gata6, and in humans, Gata6 mutations can cause persistent truncus arteriosis through disruptions in the semaphorin–plexin signaling pathway.44, 45 Whether this Rac1–JNK–Gata6–semaphorin signaling pathway is conserved in the SHF‐derived OFT myocardium and subsequently signals to the migrating cardiac neural crest during cardiac morphogenesis remains to be determined in future studies.
The OFT defects observed in the Rac1 SHF embryos in the present study are likely due to a combination of abnormalities in several development events that are linked to initial establishment of cell polarity, including proliferation and migration. First, proliferation of the Rac1 SHF SHF progenitors in the splanchnic mesoderm is dramatically reduced, decreasing the overall number of anterior SHF cells contributing to OFT development. The two biological processes of cell polarity and proliferation are coordinated during development, during which the polarized state of a progenitor affects its proliferation rate. This linkage has been clearly observed in development of the nervous system.46, 47 In this study, we showed that a deficiency of Rac1 signaling in the anterior SHF progenitors resulted in a loss of cellular organization and is correlated with a decrease in proliferation rate, further supporting a link between these two developmental processes. Francou et al has also demonstrated a correlation with direct alteration of the epithelial properties of the anterior SHF progenitors, resulting in reduced progenitor proliferation rate and ectopic differentiation of the SHF progenitor cells10; however, the direct mechanistic link between proliferation and cell polarity in the SHF progenitors remains unclear. In neural progenitors, apically localized aPKCζ attenuates the activity of p27Xic1, which is an inhibitor of cyclin‐dependent kinase‐2 (Cdk2). This leads to increased Cdk2 activity and shortening of the G1 and S phases, increasing the rate of proliferation.48 Whether a similar mechanism exists in anterior SHF progenitors remains to be determined. Furthermore, concomitant to a decrease in SHF progenitor cell proliferation, a decreased migratory ability of the Rac1 SHF anterior SHF progenitors to add to the arterial pole of the heart tube likely contributed to the observed OFT defects. Establishment of cell polarity is intrinsically linked to migratory ability. Both polarization and migration are events dependent on dynamic changes to the actin cytoskeleton, which is regulated by Rac1.49 Evidence of impaired migratory ability of the Rac1 SHF SHF during heart development was recently demonstrated in our previous study.21 Migration defects were also observed in the present study based on mT/mG lineage tracing, which showed reduced contribution of SHF cells to the aortic valves and OFT and impaired migration of cardiomyocytes into the Rac1 SHF proximal outlet septum during the myocardialization process. These observations support establishment of SHF progenitor epithelial organization as critical to the developmental process.
Valve remodeling and development is regulated by coordinated actions between matrix metalloproteinase and a disintegrin and metalloproteinase with thrombospondin motifs families of zinc metalloproteinases.50 Studies have shown a role for Rac1 in the regulation of extracellular matrix degradation along with matrix metalloproteinase expression and activity.51, 52 The thickened Rac1 SHF aortic valve leaflets along with decreased valve cell density and increased deposition of glycosaminoglacans observed in this study strongly suggest defects in valvular extracellular matrix degradation and remodeling. Whether Rac1 signaling in the SHF‐derived cells residing in the aortic valve leaflets also regulate the extracellular matrix through matrix metalloproteinases remains to be determined in future studies. Along with this, blood flow–induced hemodynamics can influence valve morphogenetic cues.53 Our previous study reported atrial and ventricular septal defects along with abnormalities in development of the ventricles in Rac1 SHF hearts.21 Whether these structural defects altered the hemodynamic flow and subsequently affected remodeling of the Rac1 SHF aortic valves remains to be determined.
In summary, our study is the first to show that Rac1 signaling in the anterior SHF is critical to normal cell organization in the splanchnic mesoderm. Loss of Rac1 leads to disruptions in anterior SHF progenitor cellular organization early in development and affects subsequent steps of heart development, including OFT and aortic valve morphogenesis, along with signaling to the neural crest cells. Our study suggests that perturbed Rac1 signaling in the anterior SHF could account for some of the OFT defects observed in humans.
Sources of Funding
This work was funded by operating grants (to Feng and Drysdale) from Canadian Institutes of Health Research (CIHR) and Heart & Stroke Foundation of Ontario (HSFO). Leung was supported by a Natural Sciences and Engineering Research Council (NSERC) Scholarship. Feng was a HSFO Career Investigator.
Disclosures
None.
Supporting information
Video S1. Three‐dimensional reconstruction of normal morphology of aortic arch and pulmonary artery in an embryonic day 15.5 Rac1f/f heart. In Rac1f/f hearts, the aortic arch (pink) is located on the left side. Ascending and descending aorta are in red. The descending aorta runs parallel to the esophagus (green) and trachea (yellow). Pulmonary artery is in blue.
Video S2. Three‐dimensional reconstruction of vascular ring in an embryonic day 15.5 Rac1SHF heart. In Rac1SHF hearts, the aortic arch (pink) is located incorrectly on the right side. The ascending and descending aorta are in red. The descending aorta wraps around the esophagus (green) and trachea (yellow), forming a vascular ring. Pulmonary artery is in blue.
Acknowledgments
The authors would like to thank Murong Liu for her assistance in mouse breeding and genotyping.
(J Am Heart Assoc. 2016;5:e002508 doi: 10.1161/JAHA.115.002508)
Accompanying Videos S1 and S2 are available at http://jaha.ahajournals.org/content/5/1/e002508/suppl/DC1
References
- 1. Bruneau BG. The developmental genetics of congenital heart disease. Nature. 2008;451:943–948. [DOI] [PubMed] [Google Scholar]
- 2. Jenkins KJ, Gauvreau K, Newburger JW, Spray TL, Moller JH, Iezzoni LI. Consensus‐based method for risk adjustment for surgery for congenital heart disease. J Thorac Cardiovasc Surg. 2002;123:110–118. [DOI] [PubMed] [Google Scholar]
- 3. Bryant DM, Mostov KE. From cells to organs: building polarized tissue. Nat Rev Mol Cell Biol. 2008;9:887–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Phillips HM, Rhee HJ, Murdoch JN, Hildreth V, Peat JD, Anderson RH, Copp AJ, Chaudhry B, Henderson DJ. Disruption of planar cell polarity signaling results in congenital heart defects and cardiomyopathy attributable to early cardiomyocyte disorganization. Circ Res. 2007;101:137–145. [DOI] [PubMed] [Google Scholar]
- 5. Henderson DJ, Phillips HM, Chaudhry B. Vang‐like 2 and noncanonical Wnt signaling in outflow tract development. Trends Cardiovasc Med. 2006;16:38–45. [DOI] [PubMed] [Google Scholar]
- 6. High FA, Jain R, Stoller JZ, Antonucci NB, Lu MM, Loomes KM, Kaestner KH, Pear WS, Epstein JA. Murine Jagged1/Notch signaling in the second heart field orchestrates Fgf8 expression and tissue‐tissue interactions during outflow tract development. J Clin Invest. 2009;119:1986–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jain R, Engleka KA, Rentschler SL, Manderfield LJ, Li L, Yuan L, Epstein JA. Cardiac neural crest orchestrates remodeling and functional maturation of mouse semilunar valves. J Clin Invest. 2011;121:422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Verzi MP, McCulley DJ, De Val S, Dodou E, Black BL. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev Biol. 2005;287:134–145. [DOI] [PubMed] [Google Scholar]
- 9. Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development. 2000;127:1607–1616. [DOI] [PubMed] [Google Scholar]
- 10. Francou A, Saint‐Michel E, Mesbah K, Kelly RG. TBX1 regulates epithelial polarity and dynamic basal filopodia in the second heart field. Development. 2014;141:4320–4331. [DOI] [PubMed] [Google Scholar]
- 11. Sinha T, Wang B, Evans S, Wynshaw‐Boris A, Wang J. Disheveled mediated planar cell polarity signaling is required in the second heart field lineage for outflow tract morphogenesis. Dev Biol. 2012;370:135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ramsbottom SA, Sharma V, Rhee HJ, Eley L, Phillips HM, Rigby HF, Dean C, Chaudhry B, Henderson DJ. Vangl2‐regulated polarisation of second heart field‐derived cells is required for outflow tract lengthening during cardiac development. PLoS Genet. 2014;10:e1004871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tao H, Suzuki M, Kiyonari H, Abe T, Sasaoka T, Ueno N. Mouse prickle1, the homolog of a PCP gene, is essential for epiblast apical‐basal polarity. Proc Natl Acad Sci USA. 2009;106:14426–14431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Djiane A, Yogev S, Mlodzik M. The apical determinants aPKC and dPatj regulate Frizzled‐dependent planar cell polarity in the Drosophila eye. Cell. 2005;121:621–631. [DOI] [PubMed] [Google Scholar]
- 15. Henderson DJ, Chaudhry B. Getting to the heart of planar cell polarity signaling. Birth Defects Res A Clin Mol Teratol. 2011;91:460–467. [DOI] [PubMed] [Google Scholar]
- 16. Mack NA, Georgiou M. The interdependence of the Rho GTPases and apicobasal cell polarity. Small GTPases. 2014;5:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bosco EE, Mulloy JC, Zheng Y. Rac1 GTPase: a “Rac” of all trades. Cell Mol Life Sci. 2009;66:370–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Migeotte I, Grego‐Bessa J, Anderson KV. Rac1 mediates morphogenetic responses to intercellular signals in the gastrulating mouse embryo. Development. 2011;138:3011–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Roszko I, Sawada A, Solnica‐Krezel L. Regulation of convergence and extension movements during vertebrate gastrulation by the Wnt/PCP pathway. Semin Cell Dev Biol. 2009;20:986–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grimsley‐Myers CM, Sipe CW, Geleoc GS, Lu X. The small GTPase Rac1 regulates auditory hair cell morphogenesis. J Neurosci. 2009;29:15859–15869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Leung C, Lu X, Liu M, Feng Q. Rac1 signaling is critical to cardiomyocyte polarity and embryonic heart development. J Am Heart Assoc. 2014;3:e001271 doi: 10.1161/JAHA.114.001271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kelly RG, Brown NA, Buckingham ME. The arterial pole of the mouse heart forms from Fgf10‐expressing cells in pharyngeal mesoderm. Dev Cell. 2001;1:435–440. [DOI] [PubMed] [Google Scholar]
- 23. Cai CL, Liang X, Shi Y, Chu PH, Pfaff SL, Chen J, Evans S. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev Cell. 2003;5:877–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dodou E, Verzi MP, Anderson JP, Xu SM, Black BL. Mef2c is a direct transcriptional target of ISL1 and GATA factors in the anterior heart field during mouse embryonic development. Development. 2004;131:3931–3942. [DOI] [PubMed] [Google Scholar]
- 25. Glogauer M, Marchal CC, Zhu F, Worku A, Clausen BE, Foerster I, Marks P, Downey GP, Dinauer M, Kwiatkowski DJ. Rac1 deletion in mouse neutrophils has selective effects on neutrophil functions. J Immunol. 2003;170:5652–5657. [DOI] [PubMed] [Google Scholar]
- 26. Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double‐fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. [DOI] [PubMed] [Google Scholar]
- 27. Liu Y, Lu X, Xiang FL, Poelmann RE, Gittenberger‐de Groot AC, Robbins J, Feng Q. Nitric oxide synthase‐3 deficiency results in hypoplastic coronary arteries and postnatal myocardial infarction. Eur Heart J. 2014;35:920–931. [DOI] [PubMed] [Google Scholar]
- 28. van den Berg G, Abu‐Issa R, de Boer BA, Hutson MR, de Boer PA, Soufan AT, Ruijter JM, Kirby ML, van den Hoff MJ, Moorman AF. A caudal proliferating growth center contributes to both poles of the forming heart tube. Circ Res. 2009;104:179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kirby ML, Waldo KL. Neural crest and cardiovascular patterning. Circ Res. 1995;77:211–215. [DOI] [PubMed] [Google Scholar]
- 30. Yelbuz TM, Waldo KL, Kumiski DH, Stadt HA, Wolfe RR, Leatherbury L, Kirby ML. Shortened outflow tract leads to altered cardiac looping after neural crest ablation. Circulation. 2002;106:504–510. [DOI] [PubMed] [Google Scholar]
- 31. Kruithof BP, van den Hoff MJ, Wessels A, Moorman AF. Cardiac muscle cell formation after development of the linear heart tube. Dev Dyn. 2003;227:1–13. [DOI] [PubMed] [Google Scholar]
- 32. Pasterkamp RJ, Kolodkin AL. Semaphorin junction: making tracks toward neural connectivity. Curr Opin Neurobiol. 2003;13:79–89. [DOI] [PubMed] [Google Scholar]
- 33. Brown CB, Feiner L, Lu MM, Li J, Ma X, Webber AL, Jia L, Raper JA, Epstein JA. PlexinA2 and semaphorin signaling during cardiac neural crest development. Development. 2001;128:3071–3080. [DOI] [PubMed] [Google Scholar]
- 34. Hinton RB, Yutzey KE. Heart valve structure and function in development and disease. Annu Rev Physiol. 2011;73:29–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Simionescu DT, Lovekamp JJ, Vyavahare NR. Degeneration of bioprosthetic heart valve cusp and wall tissues is initiated during tissue preparation: an ultrastructural study. J Heart Valve Dis. 2003;12:226–234. [PubMed] [Google Scholar]
- 36. Colvee E, Hurle JM. Maturation of the extracellular material of the semilunar heart values in the mouse. A histochemical analysis of collagen and mucopolysaccharides. Anat Embryol (Berl). 1981;162:343–352. [DOI] [PubMed] [Google Scholar]
- 37. Waldo KL, Hutson MR, Stadt HA, Zdanowicz M, Zdanowicz J, Kirby ML. Cardiac neural crest is necessary for normal addition of the myocardium to the arterial pole from the secondary heart field. Dev Biol. 2005;281:66–77. [DOI] [PubMed] [Google Scholar]
- 38. Stottmann RW, Choi M, Mishina Y, Meyers EN, Klingensmith J. BMP receptor IA is required in mammalian neural crest cells for development of the cardiac outflow tract and ventricular myocardium. Development. 2004;131:2205–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jia Q, McDill BW, Li SZ, Deng C, Chang CP, Chen F. Smad signaling in the neural crest regulates cardiac outflow tract remodeling through cell autonomous and non‐cell autonomous effects. Dev Biol. 2007;311:172–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mesbah K, Harrelson Z, Theveniau‐Ruissy M, Papaioannou VE, Kelly RG. Tbx3 is required for outflow tract development. Circ Res. 2008;103:743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vitelli F, Morishima M, Taddei I, Lindsay EA, Baldini A. Tbx1 mutation causes multiple cardiovascular defects and disrupts neural crest and cranial nerve migratory pathways. Hum Mol Genet. 2002;11:915–922. [DOI] [PubMed] [Google Scholar]
- 42. Tan NY, Li JM, Stocker R, Khachigian LM. Angiotensin II‐inducible smooth muscle cell apoptosis involves the angiotensin II type 2 receptor, GATA‐6 activation, and FasL‐Fas engagement. Circ Res. 2009;105:422–430. [DOI] [PubMed] [Google Scholar]
- 43. Coso OA, Chiariello M, Yu JC, Teramoto H, Crespo P, Xu N, Miki T, Gutkind JS. The small GTP‐binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell. 1995;81:1137–1146. [DOI] [PubMed] [Google Scholar]
- 44. Lepore JJ, Mericko PA, Cheng L, Lu MM, Morrisey EE, Parmacek MS. GATA‐6 regulates semaphorin 3C and is required in cardiac neural crest for cardiovascular morphogenesis. J Clin Invest. 2006;116:929–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kodo K, Nishizawa T, Furutani M, Arai S, Yamamura E, Joo K, Takahashi T, Matsuoka R, Yamagishi H. GATA6 mutations cause human cardiac outflow tract defects by disrupting semaphorin‐plexin signaling. Proc Natl Acad Sci USA. 2009;106:13933–13938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sabherwal N, Papalopulu N. Apicobasal polarity and cell proliferation during development. Essays Biochem. 2012;53:95–109. [DOI] [PubMed] [Google Scholar]
- 47. Sabherwal N, Tsutsui A, Hodge S, Wei J, Chalmers AD, Papalopulu N. The apicobasal polarity kinase aPKC functions as a nuclear determinant and regulates cell proliferation and fate during Xenopus primary neurogenesis. Development. 2009;136:2767–2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sabherwal N, Thuret R, Lea R, Stanley P, Papalopulu N. aPKC phosphorylates p27Xic1, providing a mechanistic link between apicobasal polarity and cell‐cycle control. Dev Cell. 2014;31:559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Cell migration: integrating signals from front to back. Science. 2003;302:1704–1709. [DOI] [PubMed] [Google Scholar]
- 50. Lockhart M, Wirrig E, Phelps A, Wessels A. Extracellular matrix and heart development. Birth Defects Res A Clin Mol Teratol. 2011;91:535–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tovell VE, Chau CY, Khaw PT, Bailly M. Rac1 inhibition prevents tissue contraction and MMP mediated matrix remodeling in the conjunctiva. Invest Ophthalmol Vis Sci. 2012;53:4682–4691. [DOI] [PubMed] [Google Scholar]
- 52. Hamamura K, Zhang P, Zhao L, Shim JW, Chen A, Dodge TR, Wan Q, Shih H, Na S, Lin CC, Sun HB, Yokota H. Knee loading reduces MMP13 activity in the mouse cartilage. BMC Musculoskelet Disord. 2013;14:312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hove JR, Koster RW, Forouhar AS, Acevedo‐Bolton G, Fraser SE, Gharib M. Intracardiac fluid forces are an essential epigenetic factor for embryonic cardiogenesis. Nature. 2003;421:172–177. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Three‐dimensional reconstruction of normal morphology of aortic arch and pulmonary artery in an embryonic day 15.5 Rac1f/f heart. In Rac1f/f hearts, the aortic arch (pink) is located on the left side. Ascending and descending aorta are in red. The descending aorta runs parallel to the esophagus (green) and trachea (yellow). Pulmonary artery is in blue.
Video S2. Three‐dimensional reconstruction of vascular ring in an embryonic day 15.5 Rac1SHF heart. In Rac1SHF hearts, the aortic arch (pink) is located incorrectly on the right side. The ascending and descending aorta are in red. The descending aorta wraps around the esophagus (green) and trachea (yellow), forming a vascular ring. Pulmonary artery is in blue.