

Abstract
Background
Stroke is a leading cause of death in the world. In >80% of strokes, the initial acute phase of ischemic injury is due to the occlusion of a blood vessel resulting in severe focal hypoperfusion, excitotoxicity, and oxidative damage. Interferon‐β (IFNβ), a cytokine with immunomodulatory properties, was approved by the US Food and Drug Administration for the treatment of relapsing‐remitting multiple sclerosis for more than a decade. Its anti‐inflammatory properties and well‐characterized safety profile suggest that IFNβ has therapeutic potential for the treatment of ischemic stroke.
Methods and Results
We investigated the therapeutic effect of IFNβ in the mouse model of transient middle cerebral artery occlusion/reperfusion. We found that IFNβ not only reduced infarct size in ischemic brains but also lessened neurological deficits in ischemic stroke animals. Further, multiple molecular mechanisms by which IFNβ modulates ischemic brain inflammation were identified. IFNβ reduced central nervous system infiltration of monocytes/macrophages, neutrophils, CD4+ T cells, and γδ T cells; inhibited the production of inflammatory mediators; suppressed the expression of adhesion molecules on brain endothelial cells; and repressed microglia activation in the ischemic brain.
Conclusions
Our results demonstrate that IFNβ exerts a protective effect against ischemic stroke through its anti‐inflammatory properties and suggest that IFNβ is a potential therapeutic agent, targeting the reperfusion damage subsequent to the treatment with tissue plasminogen activator.
Keywords: CD4+ T cells, Interferon‐β, ischemic stroke, microglia, monocytes/macrophages, neuroinflammation, reperfusion, γδ T cells
Subject Categories: Ischemic Stroke, Translational Studies, Inflammation, Treatment
Introduction
Stroke is a leading cause of death and results in permanent disability in up to 30% of survivors. In >80% of strokes, the initial acute phase of ischemic injury is due to the occlusion of a blood vessel, resulting in severe focal hypoperfusion, excitotoxicity, and oxidative damage.1, 2, 3 The inflammatory response that characterizes the subacute phase of ischemia is initiated by resident microglia (MG) and peripheral inflammatory cells infiltrating the region surrounding the infarct core and leading to secondary neurodegeneration.4
The initiation of cerebral ischemia is associated with blood–brain barrier (BBB) breakdown, extravasation of plasma protein and bioactive phospholipids, rapid activation of resident MG resulting in migration to the ischemic site, and the release of endogenous nitric oxide, proinflammatory cytokines such as tumor necrosis factor (TNF)α and interleukin (IL)‐1β, and chemokines such as CCL2 and CCL3.4, 5 TNFα induces the upregulation of adhesion molecules on cerebral endothelial cells and, in conjunction with CCL3 and CXCL3, promotes the adhesion and directed transmigration of blood‐borne leukocytes. This results in infiltration of peripheral immune cells including neutrophils, monocytes/macrophages, and T cells into the infarcted tissue. Invading monocytes/macrophages were detected in the ischemic brain as early as day 1, reaching a peak on day 3. The numbers of neutrophils and T lymphocytes including CD4+ T cells, natural killer T cells, and γδ T cells were increased on day 3 and declined on day 7 postinjury.1, 6
Current clinical therapies using thrombolytic agents are limited by temporal restrictions and do not prevent the secondary, inflammation‐related central nervous system (CNS) damage. Therefore, the development of new anti‐inflammatory therapies in ischemic stroke is necessary and urgent. Interferon‐β (IFNβ), a cytokine with immunomodulatory properties, was approved by US Food and Drug Administration for the treatment of relapsing‐remitting multiple sclerosis for more than a decade.7, 8 Its anti‐inflammatory properties,9, 10, 11, 12, 13 and well‐characterized safety profile suggest that IFNβ has therapeutic potential for the treatment of ischemic stroke.
In this study, we evaluated the therapeutic effect of IFNβ in the mouse model of transient middle cerebral artery occlusion/reperfusion (MCAO/R). Our results show that IFNβ not only reduced infarct size in ischemic brains but also lessened neurological deficits in ischemic stroke animals. Moreover, the reduction of brain infarct in the IFNβ‐treated mice was associated with decreased infiltration of monocytes/macrophages, neutrophils, γδ T cells, and CD4+ T cells. Further investigation of molecular mechanisms revealed that IFNβ treatment inhibited the production of inflammatory mediators, suppressed the expression of adhesion molecules, and repressed the activation of MG in the ischemic brain. Thus, our results suggest that IFNβ has a protective effect in ischemic stroke through its anti‐inflammatory properties.
Materials and Methods
Mice
C57BL/6 wild‐type mice were obtained from Jackson Laboratories. Ifnar1 tm1Agt/Mmjax (Ifnar1 −/− mice) were purchased from Mutant Mouse Regional Resources Center. All mice were kept and bred in the animal facility of Indiana University School of Medicine–Fort Wayne. All animal procedures in this study were conducted in strict compliance with the National Institutes of Health “Guide for the Care and Use of Laboratory Animals” and approved by Purdue Animal Care and Use Committee.
Reagents
Lipopolysaccharide (LPS; Escherichia coli O55:B5) and triphenyltetrazolium chloride (TTC) were purchased from Sigma‐Aldrich. IFNβ was purchased from PBL Interferon Source. Recombinant murine granulocyte‐macrophage colony‐stimulating factor and TNFα were purchased from Peprotech Inc. Prostaglandin E2 was purchased from Cayman Chemical Company. Alexa Fluor 488–conjugated anti‐mouse CD4 (clone: RM4‐5), CD45 (clone: 30‐F11), and CD31 (clone: MEC13.3); phycoerythrin‐conjugated anti‐mouse CD3 (clone: 145‐2C11) and CD11b (clone: M1/70); phycoerythrin/Cy7–conjugated anti‐mouse TCRγδ (clone: GL3); and Allophycocyanin‐conjugated anti‐mouse CD54/intercellular adhesion molecule (ICAM)‐1 (clone: YN1/1.7.4), F4/80 (clone: BM8), and Ly6G (clone: 1A8) were purchased from BioLegend. Fluorescein isothiocyanate–conjugated anti‐mouse CD62P/P‐selectin (clone: RB40.34) and phycoerythrin‐conjugated anti‐mouse CD62E/E‐selectin (clone: 10E9.6) were purchased from BD PharMingen. Alexa Fluor 488–cojugated donkey anti‐rabbit IgG (H+L) was purchased from Life Technologies. Rabbit polyclonal anti‐mouse Iba1 was purchased from Wako Pure Chemical Industries, Ltd.
Mouse Focal Brain Ischemia Model
Male mice, aged 8 to 12 weeks and weighing 20 to 30 g, were subjected to MCAO/R for induction of focal brain ischemia. Briefly, the mice were initially anesthetized with 2% and maintained on 1.5% isoflurane in a mixture of 70% air and 30% O2 with the use of a vaporizer. Body temperature was maintained at ≈37°C with a warming lamp and heating pad. Cerebral blood flow was measured before, during, and after ischemia by using laser Doppler flowmetry at the parietal bone (2 mm posterior and 3 mm lateral from bregma). Each mouse's resting CBF value was taken as the baseline value, and changes in cerebral blood flow after the induction of brain ischemia were expressed as percentages of the resting value. The right common carotid artery and the right external carotid artery were exposed through a midline neck incision. The external carotid artery was dissected distally, ligated, and coagulated along with the terminal lingual and maxillary artery branches. A minimal incision was made in the external carotid artery stump, with iridectomy scissors. After the incision, occlusion of the middle cerebral artery was performed by using a silicon‐coated 6‐0 nylon monofilament (Doccol Corp; 0.23 mm). The distance from the suture tip to the right common carotid artery bifurcation was ≈9 mm. During right middle cerebral artery occlusion, a reduction in cerebral blood flow of >60% was confirmed with laser Doppler flowmetry. Forty minutes after MCAO, the filament was withdrawn to allow blood flow reperfusion. After surgery, mice recovered in cages, where the temperature was kept at 34°C for 1 hour. For IFNβ treatment, 10 000 U of recombinant murine IFNβ suspended in 100 μL of PBS were administered via intravenous injection to MCAO/R mice. The concentration of IFNβ used in this study was based on the previous study established in experimental autoimmune encephalomyelitis models.14, 15, 16
Infarct Volume Measurements
Forty‐eight hours after reperfusion, the mice were sacrificed and their brains were removed. Next, 2‐mm coronal slices were obtained with use of a rodent brain matrix. The sections were stained with 1% TTC for 10 minutes at 37°C for measurements of infarct volume. Infarction volume was calculated according to Swanson and colleagues to compensate for brain swelling in the ischemic hemisphere17 and analyzed by using ImageJ (National Institutes of Health).
Neurological Assessment
Neurological function was evaluated by using a 6‐point score as previously described18 and performed at 48 hours after reperfusion. An expanded 6‐point scale is used (0, normal; score 1, mild circling behavior with or without inconsistent rotation when picked up by the tail, <50% attempts to rotate to the contralateral side; score 2, mild consistent circling, >50% attempts to rotate to contralateral side; score 3, consistent strong and immediate circling, the mouse holds a rotation position for more than 1 to 2 seconds, with its nose almost reaching its tail; score 4, severe rotation progressing into barreling, loss of walking, or righting reflex; and score 5, comatose or moribund).
Mononuclear Cell Isolation and Fluorescence‐Activated Cell Sorter Analysis
Mice were perfused with ice‐cold PBS; brains were freed from meninges; and the olfactory bulb and cerebellum were discarded. The harvested brains were separated into hemispheres. The brain was homogenized with 1× Hanks balanced saline solution buffer and filtered through a 40‐μm nylon cell strainer. After centrifugation, cells were resuspended in 10 mL of 30% Percoll and underlayered with 5 mL of 70% Percoll. The samples were then centrifuged at room temperature for 25 minutes at 1000g. The cells at the 30:70 interface were isolated, washed, and stained with anti‐CD45, anti‐CD11b, and anti‐Ly6G, followed by fluorescence‐activated cell sorter (FACS) analysis of 20 000 counts per sample. For CD4+ T‐cell and γδ T‐cell analysis, the cells collected from the 30:70 interface were stained with anti‐CD3, anti‐CD4, and anti‐TCRγδ, followed by FACS analysis.
Real‐Time Reverse Transcription–Polymerase Chain Reaction
Expression of different genes was detected by quantitative polymerase chain reaction as previous described.19 The sequences of primers are as following: E‐selectin: 5′‐CATCCTGCAGTGGTCATGGT‐3′ and 5′‐GCAAGTCACAGCTTGCTCAC3′; ICAM‐1: 5′‐CACGTGCTGTATGGTCCTCG‐3′ and 5′‐TAGGAGATGGGTTCCCCCAG‐3′; CXCL3: 5′‐CCCAGACAGAAGTCATAGCCA‐3′ and 5′‐GTGAGGGGCTTCCTCCTTTC‐3′; CCL3: 5′‐CCCAGCCAGGTGTCATTTTC‐3′ and 5′‐CTCAAGCCCCTGCTCTACAC‐3′; MMP‐9: 5′‐AAAACCTCCAACCTCACGGA‐3′ and 5′‐GCGGTACAAGTATGCCTCTGC‐3′; IL‐1β: 5′‐CCCTGCAGCTGGAGAGTGTGGA‐3′ and 5′‐TGTGCTCTGCTTGTGAGGTGCTG‐3′; IL‐6: 5′‐TCCTCTCTGCAAGAGACTTCCATCC‐3′ and 5′‐GGGAAGGCCGTGGTTGTCACC‐3′; IL‐23p19: 5′‐TGCTGGATTGCAGAGCAGTAA‐3′ and 5′‐GCATGCAGAGATTCCGAGAGA‐3′; TNFα: 5′‐ATGGCCTCCCTCTCATCAGT‐3′ and 5′‐CTTGGTGGTTTGCTACGACG‐3′; and IL‐12p40: 5′‐TGGTTTGCCATCGTTTTGCTG‐3′ and 5′‐ACAGGTGAGGTTCACTGTTTCT‐3′.
Cell Culture
Mouse brain endothelial cells (bEnd.3; American Type Culture Collection) were grown in 25‐cm2 flasks. Once the cells were grown to confluence, they were trypsinized and seeded onto tissue culture plates. The cells were subjected to FACS analysis for brain endothelial cell–specific marker CD31 (platelet endothelial cell adhesion molecule‐1) expression. More than 94% of cells were positive for platelet endothelial cell adhesion molecule‐1 expression.
MG were generated from neonatal mice. In brief, cerebral cortical cells from 1‐ to 2‐day‐old mice were dissociated and plated in 75‐cm2 culture flasks. The medium was removed and replenished with medium containing 10 ng/mL granulocyte‐macrophage colony‐stimulating factor on days 5 and 10 after plating. On day 15, MG were harvested by shaking the flasks at 300 rpm at 37°C for 30 minutes. More than 90% of harvested cells were positive for CD11b and F4/80 expression by FACS analysis.
Immunohistochemistry
Mice were transcardiacally perfused with PBS and brains were extracted. Coronal brain blocks (2 mm) were cut, postfixed in 4% paraformaldehyde, and cryoprotected in 6% followed by 30% sucrose‐PBS. Brain blocks were subsequently embedded in optimal cutting temperature compound and frozen with isopentane cooled by liquid nitrogen. Serial 15‐μm frozen sections were cut in a cryostat, mounted on superfrost plus slides, and stored at −20°C until staining. After washing with PBS, slides were permeabilized with 0.5% Triton X‐100/PBS for 30 minutes and then incubated with blocking solution (0.25% Triton X‐100/PBS, 3% normal donkey serum in PBS) for 1 hour at room temperature. The primary antibody (rabbit anti‐Iba1, 1:800; Wako) was added on slides for 2‐hour incubation at room temperature. Slides were washed with PBS and incubated with fluorescent secondary antibody (donkey anti‐rabbit IgG, 1:400 Alexa Fluor 488; Invitrogen) for 1 hour at room temperature. After washing with PBS, slides were mounted and coverslipped with ProLong Gold anti‐fade mountant with DAPI (Invitrogen). Brain regions of interest were defined in relation to anatomical landmarks by using the Franklin and Paxinos Mouse Brain in Stereotaxic Guidance. The Nuance FX multispectral imaging system and Nuance software (PerkinElmer) were used for image collection and analysis. Each stained section was processed under identical gain and laser power setting. Two or 3 different sections from each mouse at the bregma 0.98 to 0.86 mm were stained and analyzed (n=6 mice in the vehicle‐ and IFNβ‐treated groups). Activation of MG was evaluated on the basis of Iba1 intensity exceeding the set threshold and of cellular morphology. Image of single‐cell morphology was acquired by using the Fluoview FV10i confocal microscope (Olympus) and analyzed with the use of ImageJ.
Immunocytochemistry
MG were fixed in 2% paraformaldehyde for 15 minutes at room temperature. After being washed with PBS, cells were permeabilized with 0.3% Triton X‐100/PBS for 10 minutes and then blocked in 3% normal donkey serum for 30 minutes at room temperature. Cells were incubated with primary antibody (rabbit anti‐Iba1, 1:800) for 1 hour at room temperature. After being washed with PBS, cells were incubated with fluorescent secondary antibody (donkey anti‐rabbit, 1:400 Alexa Fluor 488) for 1 hour at room temperature. After being washed with PBS, cells were coverslipped with ProLong Gold antifade mountant with DAPI (Invitrogen). Confocal images were acquired by using Fluoview FV10i confocal microscope (Olympus) and analyzed by using ImageJ.
Statistical Analysis
Results are given as mean±SEM. Comparisons between 2 groups were done by using the paired t test, whereas comparisons between multiple groups were done by using 1‐way ANOVA followed by Bonferroni post hoc test. For in vivo study with the groups containing ≥7 mice, a Shapiro–Wilk normality test was performed to ensure a normal distribution of the data. For neurological scores, comparisons between 2 groups were calculated with use of the χ2 test of proportions. For quantitative cell counts of activated MG and measurements of MG soma size in the immunohistochemistry brain samples, comparisons between multiple groups were calculated with the use of Kruskal–Wallis test followed by Dunn post hoc test. Statistical significance was determined with P values <0.05.
Results
IFNβ Confers a Protective Effect Against Ischemic Stroke
In this study, we used the mouse MCAO/R model. Physiological parameters for the MCAO/R mice are listed in Table. All mice had similar body temperatures and blood oxygen levels during surgery, and a blood flow drop of >60% between the initial reading and after suture insertion. We first evaluated whether pretreatment with IFNβ would offer a protective effect in ischemic stroke. Mice were administered IFNβ or vehicle 3 hours before the induction of MCAO/R. Forty‐eight hours after reperfusion, infarct volumes were assessed by using TTC staining. Pretreatment with IFNβ significantly reduced the infarct volume (5.4±0.8 IFNβ versus 15.2±1.9 vehicle; Figure 1A and 1B). Lower neurological scores were also observed in IFNβ‐treated mice (1.2±0.2 IFNβ versus 2.1±0.3 vehicle; Figure 1C). We then evaluated the therapeutic effect of IFNβ treatment in ischemic stroke. IFNβ was administrated to MCAO/R mice 3 hours after reperfusion, and the ischemic brain infarction was accessed by using TTC at 48 hours postreperfusion. Similar to the results of IFNβ pretreatment, IFNβ posttreatment reduced infarct volumes (7.3±0.8 IFNβ versus 15.9±1.2 vehicle; Figure 1D and 1E) as well as the neurological score (1.4±0.2 IFNβ versus 2.2±0.3 vehicle; Figure 1F). The protective effect of IFNβ in ischemic stroke is mediated through type I IFN receptor because IFNβ failed to reduce infarct volume in Ifnar1 −/− MCAO/R mice compared with vehicle‐treated controls (Figure 1G and 1H). Altogether, our results demonstrated IFNβ attenuated ischemic brain infarct and lessened ischemia‐induced neurological deficits, suggesting its therapeutic potential for ischemic brain injury.
Table 1.
Physiological Parameters of Mice During MCAO/R
| Strain | Parameter | Body Weight, g | During MCAO | ||
|---|---|---|---|---|---|
| Body Temperature (°C) | Blood O2 (%) | Drop in Doppler Signal (%) | |||
| C57BL/6 Wild‐type | Vehicle (N=15) | 24.8±1.7 | 37.2±0.3 | 97.2±1.7 | 72.5±10.8 |
| IFNβ (post 3h) (N=15) | 25.7±1.5 | 37.1±0.2 | 97.6±0.7 | 78.7±9.2 | |
| Vehicle (N=10) | 27.8±4.3 | 37.2±0.2 | 97.7±0.6 | 80.7±7.1 | |
| IFNβ (pre 3h) (N=10) | 26.6±1.9 | 37.2±0.2 | 97.5±1.0 | 74.4±6.7 | |
| Ifnar1 −/− | Vehicle (N=8) | 23.2±1.1 | 37.3±0.2 | 97.2±1.4 | 72.2±8.8 |
| IFNβ (post 3h) (N=8) | 23.2±1.3 | 37.3±0.2 | 97.1±1.0 | 79.6±4.6 | |
IFN indicates interferon; MCAO/R, middle cerebral artery occlusion/reperfusion; Post 3h, 3 hours after reperfusion; Pre 3h, 3 hours before MCAO/R induction.
Figure 1.
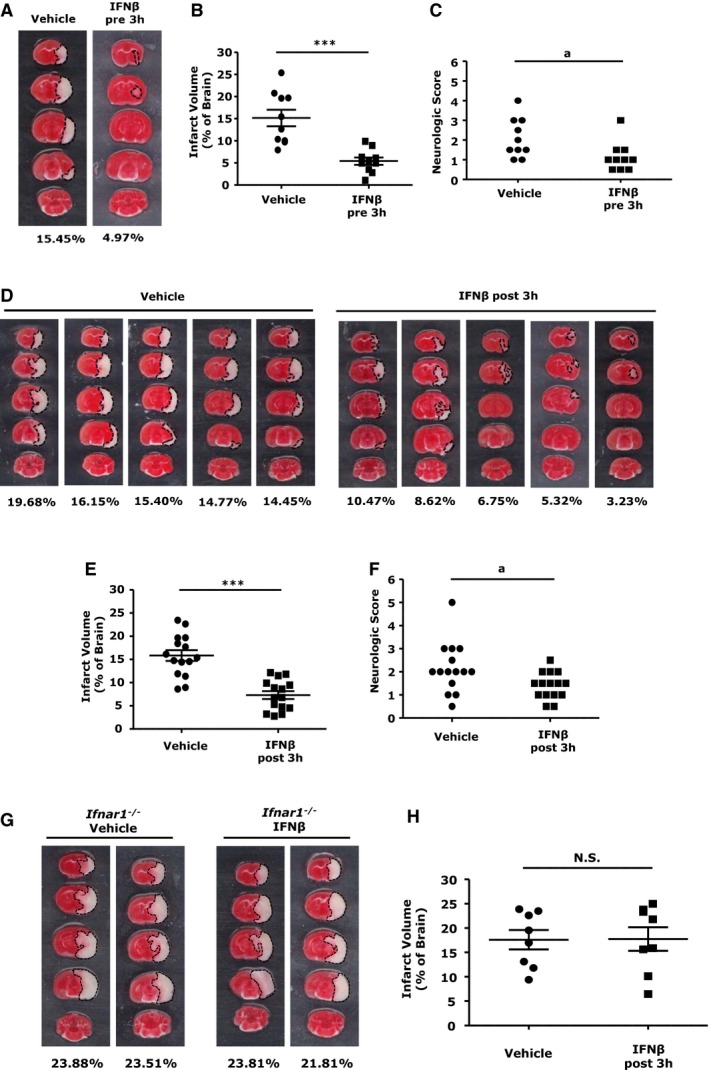
Interferon (IFN)β‐treated middle cerebral artery occlusion/reperfusion (MCAO/R) mice are protected from cerebral ischemic injury. C57BL/6 mice (n=10/group) intravenously treated with vehicle or IFNβ were subjected to MCAO/R 3 hours later. A, Forty‐eight hours after MCAO/R, mice were sacrificed, and ischemic brains were harvested and sliced (2 mm), followed by TTC staining. B, The infarct size of ischemic brains of vehicle‐ and IFNβ‐pretreated mice was measured. C, Neurological scores of vehicle‐ and IFNβ‐treated MCAO/R mice were evaluated. C57BL/6 mice (n=15/group) subjected to MCAO/R were treated with vehicle or IFNβ 3 hours after reperfusion. D, Ischemic brains of vehicle‐ and IFNβ‐treated MCAO/R mice were subjected to TTC staining, and 5 representative TTC‐stained samples are shown. E and F, The infarct volume was measured and the neurological scores were determined. Type I IFN receptor–deficient (Ifnar1 −/−) mice (n=8/group) were subjected to MCAO/R and treated with vehicle or IFNβ 3 hours after reperfusion. At 48 hours later, the mice were sacrificed, and brains were sliced (2 mm), followed by TTC staining. G, Two representative TTC‐stained samples are shown. H, The infarct volume for vehicle‐ and IFNβ‐treated Ifnar1 −/− MCAO mice was measured. *** P<0.001, N.S., no significance by paired t test. a P<0.05 by the χ2 test.
IFNβ Suppresses Inflammation in Ischemic Brains
Ischemia‐induced CNS inflammation exerts a fundamental role in stroke pathophysiology and promotes infarct formation in the ischemic brain.20, 21, 22 Because IFNβ markedly attenuated infarct size 48 hours post injury (Figure 1), we investigated whether IFNβ inhibits the production of inflammatory cytokines in the ischemic brains at an early phase (24 hours post injury). We collected ischemic brains from MCAO/R mice 24 hours after reperfusion and extracted RNA followed by quantitative PCR analysis for inflammatory cytokine expression. Our results show that inflammatory cytokines, including IL‐1β, IL‐6, IL‐23p19, and TNFα, were upregulated in the ipsilateral hemispheres of vehicle‐treated MCAO/R mice and that IFNβ treatment significantly reduced proinflammatory cytokine expression (Figure 2). Thus, our results suggest that IFNβ‐mediated reduction of inflammatory cytokine expression in the ischemic brain might contribute to its protective effects in ischemic stroke.
Figure 2.
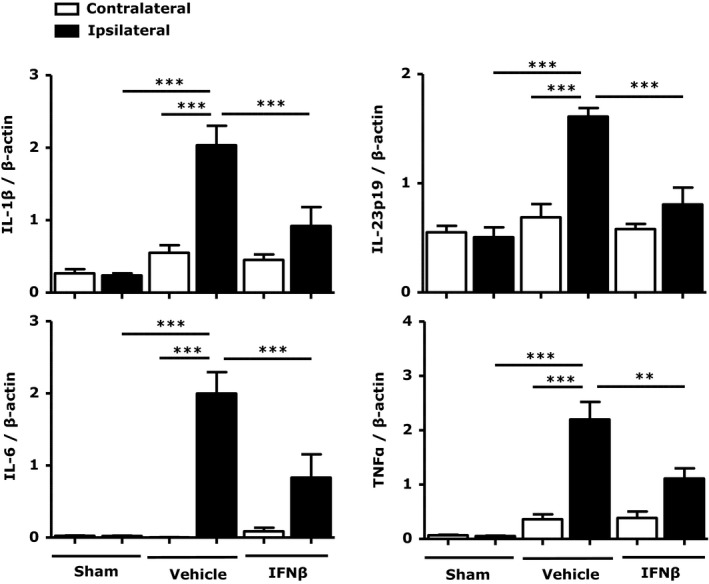
Interferon (IFN)β inhibits the production of proinflammatory cytokines in the ischemic brain. Ipsilateral and contralateral brain hemispheres from sham control, vehicle‐, and IFNβ‐treated middle cerebral artery occlusion/reperfusion (MCAO/R) mice (n=7/group) were harvested 24 hours after reperfusion. mRNA expression of interleukin (IL)‐1β, IL‐6, IL‐23p19, and TNF‐α was determined by quantitative polymerase chain reaction (Q‐PCR). Data are presented as the group mean±SEM. **P<0.01, ***P<0.001 by 1‐way ANOVA with Bonferroni's multiple comparison test.
IFNβ Suppresses MG Activation in Ischemic Brains
In response to ischemic injury, rapidly activated MG are the major source of inflammatory mediators that cause cytotoxic effects in the ischemic brain.22, 23 Because the upregulation of inflammatory cytokines in the ischemic brain was inhibited by IFNβ (Figure 2), we speculated that IFNβ might suppress MG activation. Brain slices were prepared from 24‐hour ischemic brains followed by Iba1 staining. Cerebral ischemia significantly increased Iba1 expression in the cortex of ipsilateral, but not contralateral hemispheres (Figure 3A, top). When comparing MG activation in the ipsilateral hemispheres of vehicle‐ and IFNβ‐treated MCAO/R mice, IFNβ treatment markedly suppressed Iba1 expression (Figure 3A, right). In addition, confocal images showed a round and amoeboid morphology with shorter processes and enlarged soma in vehicle controls, suggestive of activated MG, whereas MG from IFNβ‐treated animals displayed ramified morphology with long branching processes and smaller soma, indicative of a resting phenotype (Figure 3A, insets). Quantitative analysis of MG activation revealed a significant reduction in Iba1+ cell numbers (Figure 3B, top) as well as soma size (Figure 3B, bottom) for IFNβ‐treated MCAO/R mice.
Figure 3.
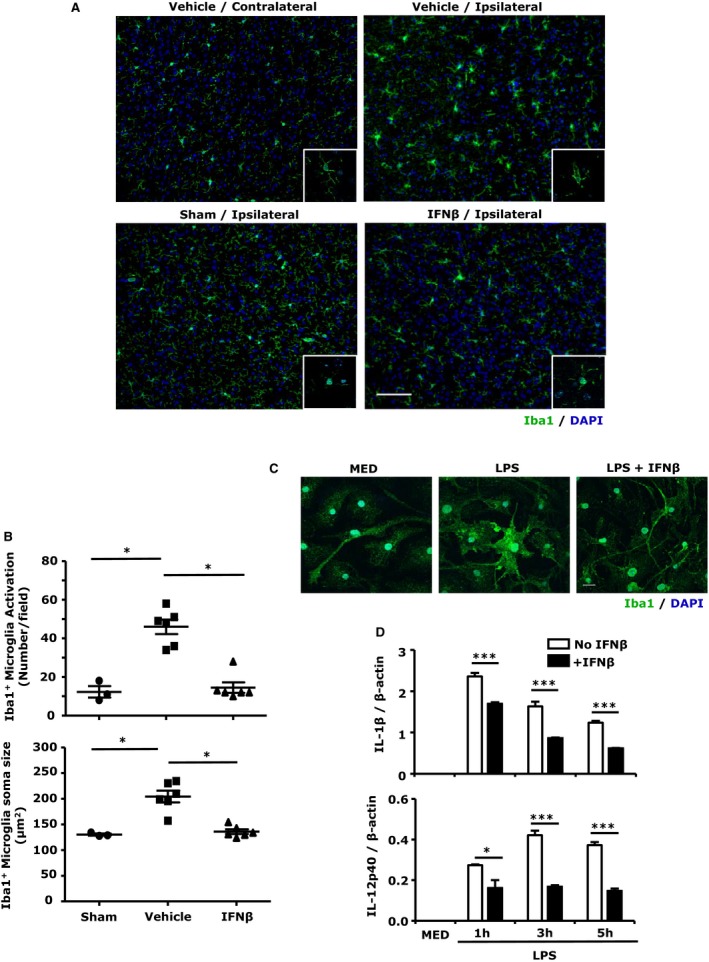
Interferon (IFN)β suppresses microglia (MG) activation in the ischemic brain as well as in primary MG. A, Representative images of Iba1‐positive MG in the contralateral and ipsilateral cortex of vehicle‐treated and in the ipsilateral cortex of sham and IFNβ‐treated middle cerebral artery occlusion/reperfusion (MCAO/R) mice. Insets: confocal images of Iba1‐positive MG. DAPI (blue) stains for cell nucleus. Scale bar, 50 μm. B, Quantitative cell counts of activated MG and measurements of MG soma size in the ipsilateral cortex of sham (n=3), vehicle‐ (n=6), and IFNβ‐treated (n=6) MCAO/R mice. Data are presented as the group mean±SEM. *P<0.05 by Kruskal–Wallis test with post hoc Dunn multiple comparison test. C and D, Primary MG were pretreated with IFNβ for 1 hour followed by liposaccharide (LPS) treatment. Twenty hours after LPS stimulation, cells were fixed and subjected to Iba1 immunocytochemistry staining. Representative confocal images of Iba1‐labeled (green) primary MG. DAPI‐stained cell nuclei are depicted in blue. Scale bar, 20 μm (C). At 1, 3, or 5 hours after LPS stimulation, cells were collected and subjected to RNA extraction followed by quantitative polymerase chain reaction (Q‐PCR) for IL‐1β and IL‐12p40 expression (D). Data are the representative results of 3 independent experiments. *P<0.05, ***P<0.001 by 1‐way ANOVA with Bonferroni's multiple comparison test.
The inhibitory effect of IFNβ on MG activation was further confirmed in primary MG in vitro. Cells were treated with LPS in the presence or absence of IFNβ for 24 hours followed by Iba1 staining. MG treated with LPS had increased Iba1 expression and changed morphology from ramified to the amoeboid phenotype. IFNβ suppressed LPS‐induced Iba1 expression and preserved the ramified phenotype similar to the phenotype of resting MG (MED; Figure 3C). In addition, IFNβ inhibited LPS‐induced IL‐1β and IL‐12p40 expression (Figure 3D). Taken together, our results demonstrate that IFNβ suppressed ischemia‐induced MG activation in vivo and LPS‐induced MG activation in vitro.
IFNβ Diminishes Peripheral Immune Cell Infiltration Into the Ischemic Brain
Cerebral ischemia followed by reperfusion promotes CNS infiltration of peripheral immune cells including neutrophils, monocytes/macrophages, and T cells, leading to secondary brain injury characterized by enlarged brain infarction.22, 24 To investigate whether IFNβ affects inflammatory cell infiltration, we characterized infiltrating cell populations in the ischemic brains of vehicle‐ and IFNβ‐treated MCAO/R mice at 48 hours after reperfusion. We observed a significant increase in cell numbers of CD45hiCD11b+ monocytes/macrophages and CD11b+Ly6G+ neutrophils in ipsilateral hemispheres of vehicle‐treated mice, with only a few infiltrating cells in the contralateral hemispheres (Figure 4A through 4C). We then compared cell infiltrates in the ischemic brains of vehicle‐ and IFNβ‐treated animals. Our results show significantly lower numbers of infiltrating CD45hiCD11b+ monocytes/macrophages and CD11b+Ly6G+ neutrophils in the ipsilateral hemispheres of IFNβ‐treated mice (Figure 4B and 4C). In contrast to the CD45hiCD11b+ infiltrating peripheral immune cells, IFNβ did not affect the numbers of CD45intCD11b+ resident MG (results not shown).
Figure 4.
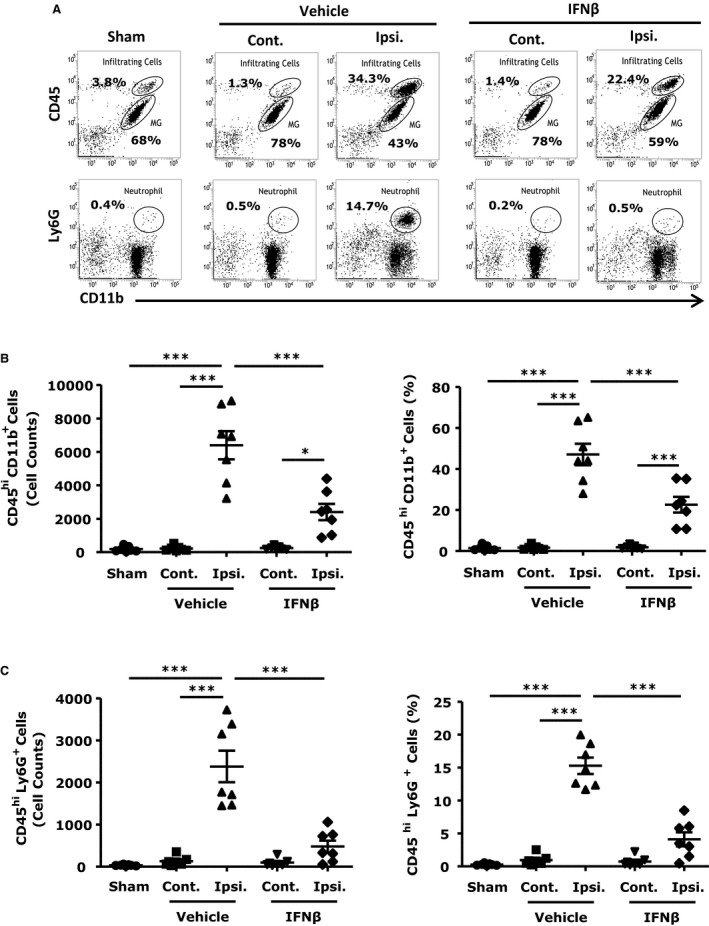
Interferon (IFN)β interferes with the recruitment of inflammatory peripheral immune cells into the ischemic brain. C57BL/6 mice (n=7/group) subjected to middle cerebral artery occlusion/reperfusion (MCAO/R) were treated with vehicle or IFNβ 3 hours after reperfusion. Forty‐eight hours later, ischemic brains of vehicle and IFNβ‐treated MCAO/R mice and sham control brains were harvested. The contralateral and ipsilateral hemispheres of vehicle‐ and IFNβ‐treated mice as well as sham control brains were subjected to mononuclear cell isolation by 30%:70% Percoll gradient. Cells were then stained with antibodies against CD45, CD11b, and Ly6G. A, top, central nervous system–infiltrating immune cells (CD45hi CD11b+) and MG (CD45int CD11b+). A, bottom, neutrophils (CD11b+Ly6G+). B and C, Numbers and percentages of peripheral infiltrating cells (CD45hi CD11b+) and neutrophils (CD11b+Ly6G+) were determined. ***P<0.001 by 1‐way ANOVA with Bonferroni's multiple comparison test.
Both CD4+ T and γδ T cells have been reported to play a pathogenic role in ischemic stroke.25, 26 In line with previous study,26 we found increased numbers of CD4+ T and γδ T cells in the ischemic brains and enlarged infarct size on day 4 post injury. More importantly, IFNβ treatment diminished both CD4+ T‐ and γδ T‐cell numbers (Figure 5C and 5D) and attenuated infarct size (Figure 5A and 5B). Taken together, our results demonstrate that IFNβ treatment suppressed the infiltration of monocytes/macrophages, neutrophils, and CD4+ T and γδ T cells, suggesting that inhibition of CNS infiltration of inflammatory cells represents one of the mechanisms for the protective effect of IFNβ in ischemic stroke.
Figure 5.
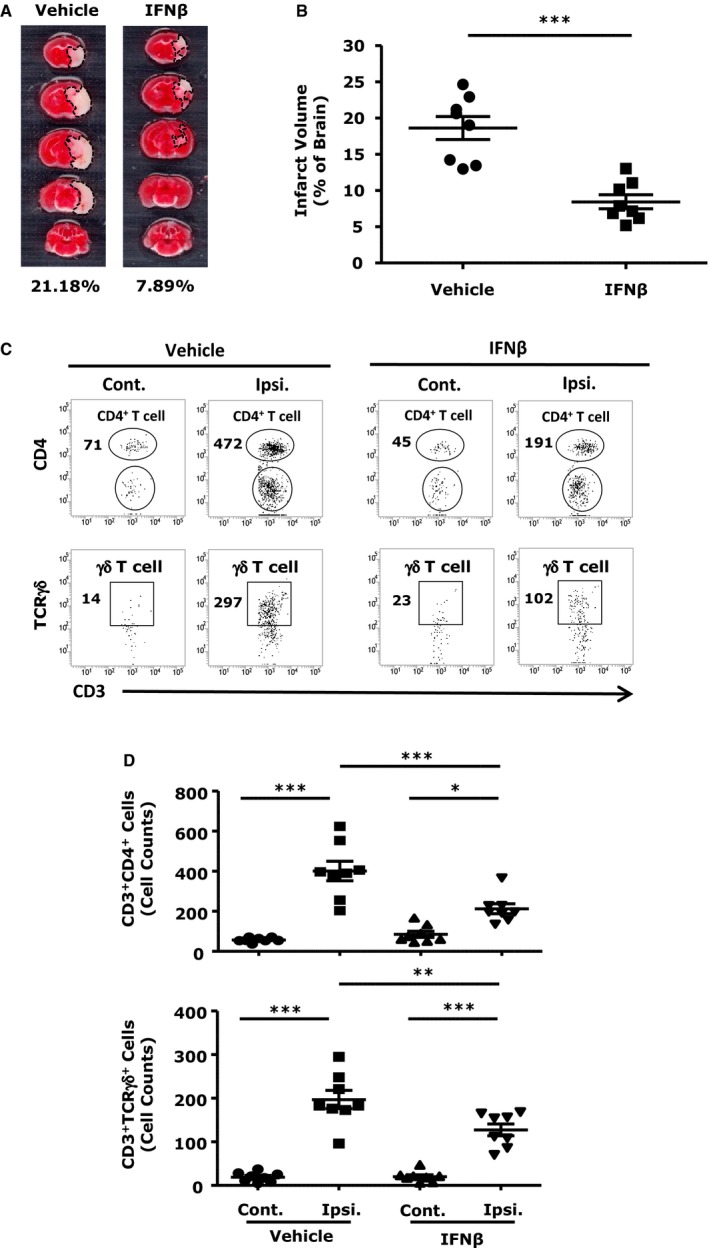
Interferon (IFN)β suppresses CNS infiltration of CD4+ T and γδ T cells in the ischemic brain. A, C57BL/6 mice (n=8/group) subjected to middle cerebral artery occlusion/reperfusion (MCAO/R) were treated with either vehicle or IFNβ 3 hours after reperfusion. Ninety‐six hours later, mice were sacrificed, and ischemic brains were harvested and sliced (2 mm), followed by TTC staining. B, The infarct volume of ischemic brains was measured. ***P<0.001 by paired t test. C, Mononuclear cells were isolated from the contralateral and ipsilateral hemispheres of vehicle‐ and IFNβ‐treated mice as well as sham control brains. Cells were stained with antibodies against CD3, CD4, and TCRγδ. CD4+ T and γδ T cells were determined as CD3+ CD4+ and CD3+ TCRγδ+, respectively. D, Numbers of CD3+ CD4+ T cells and CD3+ TCRγδ+ T cells were determined. *P<0.05, **P<0.01, ***P<0.001 by 1‐way ANOVA with Bonferroni's multiple comparison test.
IFNβ Inhibits Adhesion Molecule Expression in the Ischemic Brain
The influx of inflammatory cells into ischemic brains is facilitated by adhesion molecules expressed on the surface of brain endothelial cells. E‐selectin, P‐selectin, ICAM‐1, and vascular cell adhesion molecule (VCAM)‐1 were reported to be upregulated in brain endothelial cells within hours of reperfusion in the MCAO/R model.27, 28 Our results also demonstrate a marked increase in ICAM‐1 and E‐selectin in the ipsilateral hemispheres at 24 hours post ischemia (Figure 6A). Further comparison of ICAM‐1 and E‐selectin expression in IFNβ‐ and vehicle‐treated MCAO/R mice revealed that IFNβ abolished ischemia‐induced upregulation of ICAM‐1 and E‐selectin in the ipsilateral hemispheres (Figure 6A). Although it was previously reported that the expression of VCAM‐1 was increased in ischemic brains, we did not observe an increase of VCAM‐1 expression in ipsilateral compared with contralateral hemispheres of stroke mice, and IFNβ treatment did not alter VCAM‐1 expression in the ischemic brain (results not shown). The effect of IFNβ on the expression of adhesion molecules was also studied in the brain endothelial cell line bEnd.3. bEnd.3 cells were activated with TNFα in the absence or presence of IFNβ, and the expression of adhesion molecules was measured at mRNA and protein levels. TNFα upregulated ICAM‐1 and E‐selectin expression at both mRNA and protein level and IFNβ inhibited TNFα‐induced ICAM‐1 and E‐selectin upregulation (Figure 6B and 6C). In addition, IFNβ reduced TNFα‐induced surface P‐selectin expression (Figure 6D). Although TNFα upregulated VCAM‐1, IFNβ did not affect VCAM‐1 expression (data not shown). Altogether, our results demonstrate that IFNβ inhibited the upregulation of adhesion molecules in the ischemic brain and in brain endothelial cells.
Figure 6.
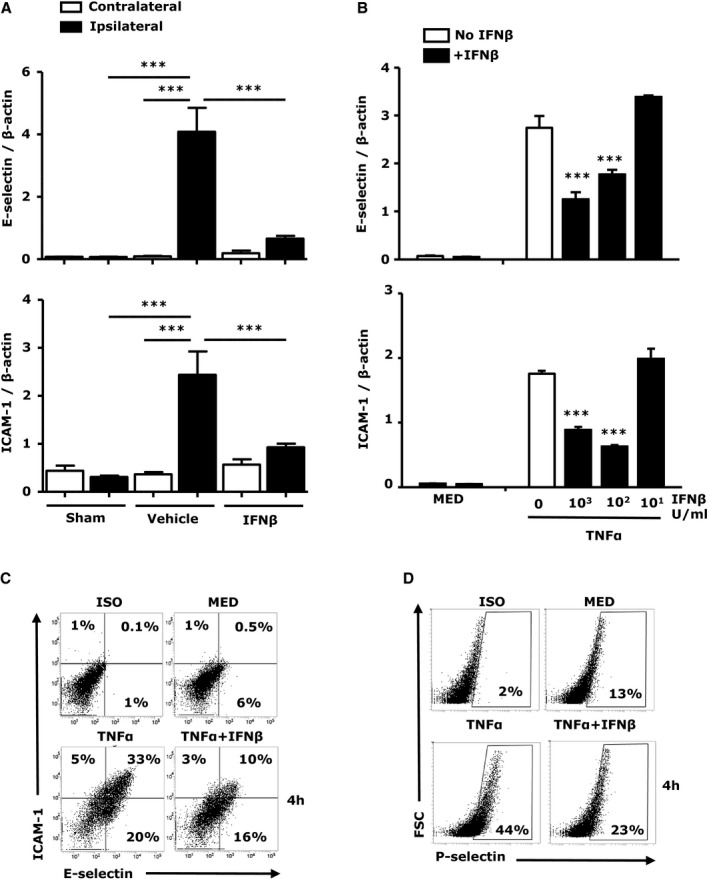
Interferon (IFN)β inhibits the upregulation of adhesion molecules in vivo and in vitro. A, RNA was extracted 24 hours after reperfusion from sham control, vehicle‐, and IFNβ‐treated middle cerebral artery occlusion/reperfusion (MCAO/R) mice (n=7/group) and analyzed for expression of E‐selectin and ICAM‐1 by quantitative polymerase chain reaction (Q‐PCR). Data are presented as the group mean±SEM. B through D, bEnd.3 cells were pretreated with IFNβ with different concentration (10 to 1000 U/mL) for 1 hour followed by tumor necrosis factor (TNF)α (50 ng/mL) for 1 hour. mRNA expression of E‐selectin and ICAM‐1 was determined by Q‐PCR (B). bEnd.3 cells were pretreated with IFNβ (1000 U/mL) for 1 hour followed by TNFα (50 ng/mL) for 4 hours. The surface expression of ICAM‐1, E‐selectin, and P‐selectin was determined by FACS (C and D). Data are the representative results of 3 independent experiments. ***P<0.001 by 1‐way ANOVA with Bonferroni's multiple comparison test.
IFNβ Suppresses Chemokine and Matrix Metallopeptidase 9 Production in Ischemic Brains
Chemokines produced in the ischemic brain facilitate the recruitment of peripheral immune cells to the site of injury. Chemokines such as CCL2/3, essential for monocyte/macrophage recruitment, and CXCL3, essential for neutrophils recruitment, are highly induced in the ischemic brain.29, 30, 31 In addition, matrix metallopeptidase (MMP)‐9 released during ischemic injury compromises BBB integrity and facilitates CNS infiltration of inflammatory immune cells.32 To investigate whether IFNβ regulates chemokines and MMP‐9 production in the ischemic brain, we compared the expression of CCL3, CXCL3, and MMP‐9 in vehicle‐ and IFNβ‐treated MCAO/R mice. The expression of CCL3, CXCL3, and MMP‐9 was dramatically increased in the ipsilateral but not contralateral hemispheres (Figure 7A). Importantly, IFNβ treatment impaired ischemia‐induced CCL3, CXCL3, and MMP‐9 expression in the ischemic brain (Figure 7A). We further investigated the effect of IFNβ on chemokines and MMP‐9 production in MG, major producers of these inflammatory mediators during brain ischemia. Similar to the in vivo results, IFNβ reduced CCL3, CXCL3, and MMP‐9 expression in LPS‐activated MG (Figure 7B and 7C).
Figure 7.
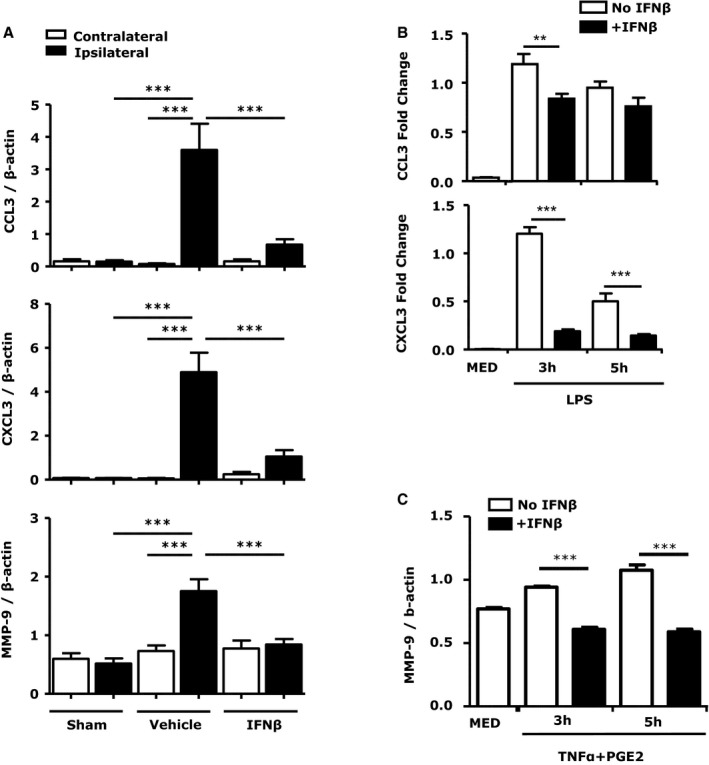
Interferon (IFN)β suppresses chemokine and matrix metallopeptidase (MMP)‐9 expression in the ischemic brain and in primary microglia (MG). A, RNA from brains of sham controls, vehicle‐, and IFNβ‐treated middle cerebral artery occlusion/reperfusion (MCAO/R) mice (n=7/group) was extracted. The expression of CCL3, CXCL3, and MMP‐9 was determined by quantitavie polymerase chain reaction (Q‐PCR). Data are presented as the group mean±SEM. B and C, Primary MG were pretreated with IFNβ (1000 U/mL) for 1 hour and stimulated with LPS (100 ng/mL) or TNFα (20 ng/mL)+PGE2 (10−6 mol/L) followed by Q‐PCR analysis for CCL3/CXCL3, and MMP‐9. Data are the representative results of 3 independent experiments. **P<0.01, ***P<0.001 by 1‐way ANOVA with Bonferroni's multiple comparison test.
Discussion
The effect of IFNβ in cerebral ischemia, reported in a rather limited number of publications, is still controversial. In a study of transient focal ischemia in rat, IFNβ failed to show a protective effect,33 whereas 3 other studies reported beneficial effects on systemic or intracerebroventricular administration of IFNβ in a rabbit model of thromboembolic cerebral ischemia34 and in rat and mouse models of transient MCAO/R.35, 36 The only mechanism identified to date is the inhibitory effect of IFNβ on MMP‐9 production resulting in reduced CNS infiltration of monocytes and neutrophils.36 In this study, our aim was to better understand the protective effects of IFNβ in ischemic stroke, with the ultimate goal of developing an IFNβ‐based therapeutic strategy for the treatment of ischemic stroke.
We used the mouse model of MCAO/R to study the effect of IFNβ treatment in cerebral ischemia. IFNβ was administered systemically either before or after MCAO/R and the effect was evaluated on days 2 and 4 post ischemia. IFNβ administration 3 hours before MCAO/R resulted in a ≈64% reduction in infarct volume. IFNβ administration 3 hours post ischemia, which is the therapeutically relevant regimen, still reduced infarct volume on day 2 by ≈54%. Moreover, postischemia administration of IFNβ also prevented the increase in infarct volume at later time points, with ≈55% reduction in infarct volume on day 4. The protective effect of IFNβ was mediated through IFNAR since attenuation of the ischemic infarct by IFNβ was not observed in Ifnar1 −/− mice. Interestingly, in contrast to a previous report,35 in our experiments, Ifnar1 −/− mice subjected to MCAO/R displayed a slightly larger infarct size than did WT mice (17.6±2.0 versus 15.9±1.2), suggesting that endogenous IFNβ might play a protective role in ischemia‐induced brain injury. Collectively, our results demonstrate that IFNβ‐mediated type I IFN receptor activation confers both short‐term and long‐term protective effects against ischemic stroke. The short‐term effects are probably mediated through inhibition of MG activation, whereas reduction in the recruitment of peripheral immune cells such as neutrophils, monocytes/macrophages, CD4+ T cells, and γδ T cells could mediate the long‐term beneficial effects of IFNβ.
MG, the resident immune cells in brain, are rapidly activated in response to ischemic injury and trigger an innate immune response characterized by increased phagocytosis and production of inflammatory mediators and MMPs, leading to BBB damage.37 Subsequently, peripheral inflammatory immune cells infiltrate the ischemic brain, resulting in secondary damage. We observed MG activation 24 hours post injury and increased inflammatory cell infiltration into the ischemic brain 48 hours post reperfusion. In the ischemic brain, MG exhibited a high level of Iba1 expression and a large size of soma, characteristic of the activated MG phenotype. In sharp contrast, MG from IFNβ‐treated MCAO/R mice displayed lower levels of Iba1 and a smaller size of soma, similar to sham controls. The inhibitory effect of IFNβ on MG activation is related to a significant reduction in the expression of proinflammatory cytokines TNFα, IL‐1β, IL‐23p19, and IL‐6 in the ischemic brain. We documented a direct effect of IFNβ on MG‐derived proinflammatory cytokines in cultures of LPS‐stimulated primary MG, which is consistent with a previous study showing IFNβ reduced MG activation in cerebellar organotypic cultures.38
The reperfusion‐induced secondary brain injury is mediated primarily by peripheral immune cells infiltrating the ischemic CNS. The influx of inflammatory immune cells is facilitated by the upregulation of adhesion molecules including E‐selectin, P‐selectin, ICAM‐1, and VCAM‐1 on brain endothelial cells after brain ischemia.27, 28 Our results show that treatment with IFNβ significantly reduces inflammatory cell infiltrates, suggesting that IFNβ might inhibit the upregulation of adhesion molecule expression on endothelial cells in the ischemic brain. Thus far, the effect of IFNβ on the expression of adhesion molecules has been only partially studied with inconsistent results. Makar et al39 showed that IFNβ suppressed ICAM‐1 expression in experimental autoimmune encephalomyelitis. In contrast, Veldhuis et al36 showed that subcutaneous IFNβ administration did not suppress ischemia‐induced ICAM‐1 upregulation on brain endothelial cells in a rat stroke model. Our results show that IFNβ intravenous administration suppresses ischemia‐induced upregulation of ICAM‐1 and E‐selectin in the ischemic brain of MCAO/R mice. The regulatory effect of IFNβ on the expression of adhesion molecules is further supported by our in vitro study with the bEnd.3 cell line, in which TNFα‐induced upregulation of ICAM‐1, E‐selectin, and P‐selectin is suppressed by IFNβ. The discrepancy between our results and those of Veldhuis et al might be because of differences in the routes of IFNβ administration. Presumably, intravenous administration would deliver IFNβ more efficiently than s.c. administration allowing sufficient IFNβ to access the brain microvasculature and suppress ischemia‐induced upregulation of adhesion molecules.
Chemokines, in addition to adhesion molecules, are required for recruiting leukocytes. During ischemic injury, chemokines such as CCL2/3 and CXCL3 are released and promotes leukocyte recruitment.40, 41, 42, 43, 44 In line with previous findings, our results show that CCL3 and CXCL3 are upregulated in the ischemic brain. Moreover, we show for the first time that IFNβ inhibits CCL3 and CXCL3 expression in the ischemic brain. CCL3 and CXCL3 function in the recruitment of monocytes/macrophages and neutrophils, respectively, and the IFNβ‐induced decrease in CCL3 and CXCL3 expression correlates with reduced infiltration of monocytes/macrophages and neutrophils in the ischemic brain of IFNβ‐treated MCAO/R mice.
The study by Shichita and colleagues demonstrated that IL‐17‐producing γδ T cells play a pivotal role in ischemic stroke pathogenesis.26 Although IL‐17–deficient mice developed brain infarct at the early phase (day 1), the infarct did not enlarge during the delayed phase (day 4) of ischemic stroke, suggesting that IL‐17 plays a pathogenic role during the delayed phase. Moreover, the study also demonstrated that γδ T cells activated by IL‐1β and IL‐23 were the major producers of IL‐17 during ischemic stroke and were responsible for the exacerbation of brain injury during the delayed phase of ischemia. Our results show that IFNβ attenuates brain infarct size at the delayed phase (day 4), suggesting that IFNβ might suppress γδ T‐cell infiltration. We observed a large increase in γδ T‐cell numbers in the ischemic brain on day 4. More importantly, IFNβ significantly decreased the number of γδ T cells in the ischemic brain. Although we did not specifically measure IL‐17 production by CNS‐infiltrating γδ T cells, the expression of IL‐1β and IL‐23 in the ischemic brain was inhibited by IFNβ, which would ultimately result in decreased recruitment to and activation of IL‐17–producting γδ T cells in the ischemic brain. To the best of our knowledge, this is the first report that one of the mechanisms for the protective effect of IFNβ in ischemic stroke involves the reduction in γδ T‐cell infiltration during brain ischemia.
In summary, here we demonstrate that IFNβ reduces ischemic brain infarct through its anti‐inflammatory effects. Several molecular mechanisms were identified in our current study. First, IFNβ reduces CNS infiltration of inflammatory cells, including monocytes/macrophages, neutrophils, CD4+ T cells, and γδ T cells. Second, IFNβ suppressed the expression of inflammatory cytokines, chemokines, MMP‐9, and adhesion molecules in the ischemic brain. Finally, IFNβ inhibited ischemia‐induced MG activation. Current American Heart Association Stroke Council guidelines for the treatment of patients with acute ischemic stroke include administration of tissue plasminogen activator intravenously within 3 hours of the ischemic attack. Although this treatment reduces neurological deficits and improves functional outcomes,45 it also increases reperfusion damage. This is primarily due to the recruitment of inflammatory immune cells to the CNS through the combined activity of MMPs, chemokines, and inflammatory cytokines that affect both the BBB and the peripheral immune cells.46, 47 Therefore, through its multiple effects, including inhibition of proinflammatory cytokines, chemokines, and MMP‐9, reduction in inflammatory cell recruitment to the CNS, and inhibition of microglia activation, IFNβ is a potential therapeutic agent targeting the reperfusion damage subsequent to the treatment with tissue plasminogen activator.
Sources of Funding
This work was supported by American Heart Association grant 12SDG8170005 to Dr Yen.
Disclosures
The authors have no financial conflict of interest.
(J Am Heart Assoc. 2016;5:e002610 doi: 10.1161/JAHA.115.002610)
References
- 1. Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol. 2010;87:779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lakhan SE, Kirchgessner A, Hofer M. Inflammatory mechanisms in ischemic stroke: therapeutic approaches. J Transl Med. 2009;7:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Muir KW, Tyrrell P, Sattar N, Warburton E. Inflammation and ischaemic stroke. Curr Opin Neurol. 2007;20:334–342. [DOI] [PubMed] [Google Scholar]
- 4. Weinstein JR, Koerner IP, Moller T. Microglia in ischemic brain injury. Future Neurol. 2010;5:227–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Denes A, Thornton P, Rothwell NJ, Allan SM. Inflammation and brain injury: acute cerebral ischaemia, peripheral and central inflammation. Brain Behav Immun. 2010;24:708–723. [DOI] [PubMed] [Google Scholar]
- 6. Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe CU, Siler DA, Arumugam TV, Orthey E, Gerloff C, Tolosa E, Magnus T. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009;40:1849–1857. [DOI] [PubMed] [Google Scholar]
- 7. Carroll WM. Clinical trials of multiple sclerosis therapies: improvements to demonstrate long‐term patient benefit. Mult Scler. 2009;15:951–958. [DOI] [PubMed] [Google Scholar]
- 8. Vosoughi R, Freedman MS. Therapy of MS. Clin Neurol Neurosurg. 2010;112:365–385. [DOI] [PubMed] [Google Scholar]
- 9. Kieseier BC. The mechanism of action of interferon‐beta in relapsing multiple sclerosis. CNS Drugs. 2011;25:491–502. [DOI] [PubMed] [Google Scholar]
- 10. Yen JH, Ganea D. Interferon beta induces mature dendritic cell apoptosis through caspase‐11/caspase‐3 activation. Blood. 2009;114:1344–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yen JH, Kong W, Ganea D. IFN‐beta inhibits dendritic cell migration through STAT‐1‐mediated transcriptional suppression of CCR7 and matrix metalloproteinase 9. J Immunol. 2010;184:3478–3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yen JH, Kong W, Hooper KM, Emig F, Rahbari KM, Kuo PC, Scofield BA, Ganea D. Differential effects of IFN‐beta on IL‐12, IL‐23, and IL‐10 expression in TLR‐stimulated dendritic cells. J Leukoc Biol. 2015;98:689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang X, Markovic‐Plese S. Interferon beta inhibits the Th17 cell‐mediated autoimmune response in patients with relapsing‐remitting multiple sclerosis. Clin Neurol Neurosurg. 2010;112:641–645. [DOI] [PubMed] [Google Scholar]
- 14. Martin‐Saavedra FM, Flores N, Dorado B, Eguiluz C, Bravo B, Garcia‐Merino A, Ballester S. Beta‐interferon unbalances the peripheral T cell proinflammatory response in experimental autoimmune encephalomyelitis. Mol Immunol. 2007;44:3597–3607. [DOI] [PubMed] [Google Scholar]
- 15. Cheng W, Zhao Q, Xi Y, Li C, Xu Y, Wang L, Niu X, Wang Z, Chen G. IFN‐beta inhibits T cells accumulation in the central nervous system by reducing the expression and activity of chemokines in experimental autoimmune encephalomyelitis. Mol Immunol. 2015;64:152–162. [DOI] [PubMed] [Google Scholar]
- 16. Yasuda CL, Al‐Sabbagh A, Oliveira EC, Diaz‐Bardales BM, Garcia AA, Santos LM. Interferon beta modulates experimental autoimmune encephalomyelitis by altering the pattern of cytokine secretion. Immunol Invest. 1999;28:115–126. [DOI] [PubMed] [Google Scholar]
- 17. Swanson RA, Shiraishi K, Morton MT, Sharp FR. Methionine sulfoximine reduces cortical infarct size in rats after middle cerebral artery occlusion. Stroke. 1990;21:322–327. [DOI] [PubMed] [Google Scholar]
- 18. Liu DZ, Xie KQ, Ji XQ, Ye Y, Jiang CL, Zhu XZ. Neuroprotective effect of paeoniflorin on cerebral ischemic rat by activating adenosine A1 receptor in a manner different from its classical agonists. Br J Pharmacol. 2005;146:604–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yen JH, Kocieda VP, Jing H, Ganea D. Prostaglandin E2 induces matrix metalloproteinase 9 expression in dendritic cells through two independent signaling pathways leading to activator protein 1 (AP‐1) activation. J Biol Chem. 2011;286:38913–38923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Amantea D, Micieli G, Tassorelli C, Cuartero MI, Ballesteros I, Certo M, Moro MA, Lizasoain I, Bagetta G. Rational modulation of the innate immune system for neuroprotection in ischemic stroke. Front Neurosci. 2015;9:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kawabori M, Yenari MA. Inflammatory responses in brain ischemia. Curr Med Chem. 2015;22:1258–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Benakis C, Garcia‐Bonilla L, Iadecola C, Anrather J. The role of microglia and myeloid immune cells in acute cerebral ischemia. Front Cell Neurosci. 2014;8:461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xia W, Han J, Huang G, Ying W. Inflammation in ischaemic brain injury: current advances and future perspectives. Clin Exp Pharmacol Physiol. 2010;37:253–258. [DOI] [PubMed] [Google Scholar]
- 24. Wang Q, Tang XN, Yenari MA. The inflammatory response in stroke. J Neuroimmunol. 2007;184:53–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yilmaz G, Arumugam TV, Stokes KY, Granger DN. Role of T lymphocytes and interferon‐gamma in ischemic stroke. Circulation. 2006;113:2105–2112. [DOI] [PubMed] [Google Scholar]
- 26. Shichita T, Sugiyama Y, Ooboshi H, Sugimori H, Nakagawa R, Takada I, Iwaki T, Okada Y, Iida M, Cua DJ, Iwakura Y, Yoshimura A. Pivotal role of cerebral interleukin‐17‐producing gammadelta T cells in the delayed phase of ischemic brain injury. Nat Med. 2009;15:946–950. [DOI] [PubMed] [Google Scholar]
- 27. Lindsberg PJ, Carpen O, Paetau A, Karjalainen‐Lindsberg ML, Kaste M. Endothelial ICAM‐1 expression associated with inflammatory cell response in human ischemic stroke. Circulation. 1996;94:939–945. [DOI] [PubMed] [Google Scholar]
- 28. Zhang R, Chopp M, Zhang Z, Jiang N, Powers C. The expression of P‐ and E‐selectins in three models of middle cerebral artery occlusion. Brain Res. 1998;785:207–214. [DOI] [PubMed] [Google Scholar]
- 29. Hori M, Nakamachi T, Rakwal R, Shibato J, Nakamura K, Wada Y, Tsuchikawa D, Yoshikawa A, Tamaki K, Shioda S. Unraveling the ischemic brain transcriptome in a permanent middle cerebral artery occlusion mouse model by DNA microarray analysis. Dis Model Mech. 2012;5:270–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zaremba J, Ilkowski J, Losy J. Serial measurements of levels of the chemokines CCL2, CCL3 and CCL5 in serum of patients with acute ischaemic stroke. Folia Neuropathol. 2006;44:282–289. [PubMed] [Google Scholar]
- 31. Wolinski P, Glabinski A. Chemokines and neurodegeneration in the early stage of experimental ischemic stroke. Mediators Inflamm. 2013;2013:727189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chaturvedi M, Kaczmarek L. MMP‐9 inhibition: a therapeutic strategy in ischemic stroke. Mol Neurobiol. 2014;49:563–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Maier CM, Yu F, Nishi T, Lathrop SJ, Chan PH. Interferon‐beta fails to protect in a model of transient focal stroke. Stroke. 2006;37:1116–1119. [DOI] [PubMed] [Google Scholar]
- 34. Liu H, Xin L, Chan BP, Teoh R, Tang BL, Tan YH. Interferon‐beta administration confers a beneficial outcome in a rabbit model of thromboembolic cerebral ischemia. Neurosci Lett. 2002;327:146–148. [DOI] [PubMed] [Google Scholar]
- 35. Marsh B, Stevens SL, Packard AE, Gopalan B, Hunter B, Leung PY, Harrington CA, Stenzel‐Poore MP. Systemic lipopolysaccharide protects the brain from ischemic injury by reprogramming the response of the brain to stroke: a critical role for IRF3. J Neurosci. 2009;29:9839–9849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Veldhuis WB, Derksen JW, Floris S, Van Der Meide PH, De Vries HE, Schepers J, Vos IM, Dijkstra CD, Kappelle LJ, Nicolay K, Bar PR. Interferon‐beta blocks infiltration of inflammatory cells and reduces infarct volume after ischemic stroke in the rat. J Cereb Blood Flow Metab. 2003;23:1029–1039. [DOI] [PubMed] [Google Scholar]
- 37. Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17:796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. di Penta A, Moreno B, Reix S, Fernandez‐Diez B, Villanueva M, Errea O, Escala N, Vandenbroeck K, Comella JX, Villoslada P. Oxidative stress and proinflammatory cytokines contribute to demyelination and axonal damage in a cerebellar culture model of neuroinflammation. PLoS One. 2013;8:e54722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Makar TK, Trisler D, Bever CT, Goolsby JE, Sura KT, Balasubramanian S, Sultana S, Patel N, Ford D, Singh IS, Gupta A, Valenzuela RM, Dhib‐Jalbut S. Stem cell based delivery of IFN‐beta reduces relapses in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2008;196:67–81. [DOI] [PubMed] [Google Scholar]
- 40. Clark AW, Krekoski CA, Bou SS, Chapman KR, Edwards DR. Increased gelatinase a (MMP‐2) and gelatinase B (MMP‐9) activities in human brain after focal ischemia. Neurosci Lett. 1997;238:53–56. [DOI] [PubMed] [Google Scholar]
- 41. Fujimura M, Gasche Y, Morita‐Fujimura Y, Massengale J, Kawase M, Chan PH. Early appearance of activated matrix metalloproteinase‐9 and blood‐brain barrier disruption in mice after focal cerebral ischemia and reperfusion. Brain Res. 1999;842:92–100. [DOI] [PubMed] [Google Scholar]
- 42. Lai AY, Todd KG. Microglia in cerebral ischemia: molecular actions and interactions. Can J Physiol Pharmacol. 2006;84:49–59. [DOI] [PubMed] [Google Scholar]
- 43. Minami M, Satoh M. [Chemokines as mediators for intercellular communication in the brain]. Nihon Yakurigaku Zasshi. 2000;115:193–200. [DOI] [PubMed] [Google Scholar]
- 44. Romanic AM, White RF, Arleth AJ, Ohlstein EH, Barone FC. Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: inhibition of matrix metalloproteinase‐9 reduces infarct size. Stroke. 1998;29:1020–1030. [DOI] [PubMed] [Google Scholar]
- 45. Hacke W, Donnan G, Fieschi C, Kaste M, von Kummer R, Broderick JP, Brott T, Frankel M, Grotta JC, Haley EC Jr, Kwiatkowski T, Levine SR, Lewandowski C, Lu M, Lyden P, Marler JR, Patel S, Tilley BC, Albers G, Bluhmki E, Wilhelm M, Hamilton S; Investigators AT, Investigators ET, Investigators Nr‐PSG . Association of outcome with early stroke treatment: pooled analysis of ATLANTIS, ECASS, and NINDS rt‐PA stroke trials. Lancet. 2004;363:768–774. [DOI] [PubMed] [Google Scholar]
- 46. Hermann DM, Bassetti CL. Implications of ATP‐binding cassette transporters for brain pharmacotherapies. Trends Pharmacol Sci. 2007;28:128–134. [DOI] [PubMed] [Google Scholar]
- 47. Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354:610–621. [DOI] [PubMed] [Google Scholar]