

Abstract
Lafora disease (LD) is a fatal neurodegenerative disorder caused by loss-of-function mutations in either laforin glycogen phosphatase gene (EPM2A) or malin E3 ubiquitin ligase gene (NHLRC1). LD is associated with gradual accumulation of Lafora bodies (LBs). LBs are aggregates of polyglucosan, a long, linear, poorly branched, hyperphosphorylated, insoluble form of glycogen. Loss-of-function mutations either in the EPM2A or in the NHLRC1 gene lead to polyglucosan formation. One hypothesis on LB formation is based on findings that laforin–malin complex downregulates glycogen synthase (GS) through malin-mediated ubiquitination, and the other one is based on findings that laforin dephosphorylates glycogen. According to the first hypothesis, polyglucosan formation is a result of increased GS activity, and according to the second, an increased glycogen phosphate leads to glycogen conformational change, unfolding, precipitation, and conversion to polyglucosan, while GS remains bound to the precipitating glycogen. In this review, we summarize all the recent findings that have important implications for the treatment of LD, all of them showing that partial inhibition of GS activity may be sufficient to prevent the progression of the disease. The current perspective in LD is high-throughput screening for small molecules that act on the disease pathway, that is, partial inhibitors of GS, which opens a therapeutic window for potential treatment of this fatal disease.
Keywords: Lafora disease, glycogen synthase, treatment
Introduction
Lafora disease (LD; OMIM 254780) is an autosomal recessive, progressive metabolic disorder characterized by intractable myoclonus and seizures, inexorable neurological deterioration, cognitive decline, unfavorable clinical course, and poor prognosis. LD usually begins in late childhood or adolescence (9–18 years) after a period of apparent normal development. Although the outcome of LD is always unfavorable, the onset of the symptoms and the progression of the disease may vary. The most common presenting feature is a single seizure followed by progressive myoclonus, generalized and/or focal seizures, cognitive decline, and severe motor and coordination impairments. Myoclonus can be fragmentary, symmetric, or massive and is the primary reason for early wheelchair dependency. Early emotional disturbance, depression, and confusion later evolve into dementia. Rapidly progressive cognitive decline with pyramidal and cerebellar signs lead to a vegetative state. Death usually occurs within 10 years in status epilepticus, most commonly due to pneumonia or complications related to degeneration of the nervous system.1
LD is more prevalent in the Mediterranean basin of Southern Europe, Southern India, Northern Africa, and the Middle East; in isolated populations of southern USA and Quebec, Canada; and in other parts of the world with a high rate of consanguinity.2 It is interesting to stress that LD has not been reported in Finland.3
Molecular genetics of LD
LD is caused by mutations in two genes: EPM2A,4 which encodes laforin, a dual specificity phosphatase that dephosphorylates complex carbohydrates,5 and NHLRC1, which encodes malin,6 an E3-ubiquitin ligase (Figure 1).7 Malin regulates the amount of laforin, and laforin regulates glycogen phosphorylation. To date, more than 50 distinct mutations have been described for each of the two genes. The observed mutations include missense, nonsense, frameshift mutations as well as exon and whole gene deletions. Mutations in the NHLRC1 gene within Japanese, Italian, and Serbian/Montenegrin LD patients are more common than those in the EPM2A gene.6,8,9,10 Conversely, EPM2A mutations are more common in the Spanish and French populations.11 Within the Indian and Arab populations, the distribution of mutations in the two genes is more or less even.12,13
Figure 1.
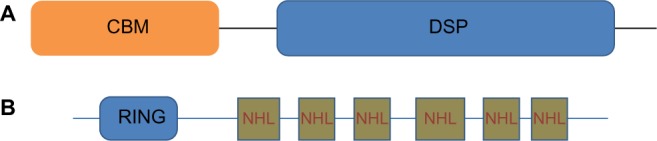
A schematic of laforin and malin.
Notes: (A) Laforin contains a CBM and a DSP domain. (B) Malin contains RING domain and six NHL repeats.
Abbreviations: CBM, carbohydrate-binding module; DSP, dual-specificity phosphatase.
Diagnosis of LD is based on the clinical and electrophysiological findings as well as molecular genetics. On rare occasion, skin biopsy is necessary to detect characteristic Lafora bodies (LBs), periodic acid-Schiff positive structures to confirm the diagnosis. Skin biopsy used to be the primary diagnostic test for LD, but interpretation of findings on skin biopsy involves a risk of false-negative results,13 especially in newly symptomatic individuals, and a risk of false-positive results because of the difficulty in distinguishing LBs from normal periodic acid-Schiff-positive polysaccharides in apocrine glands.14,15 Molecular diagnostics has an absolute priority over skin biopsy and includes targeted gene analysis of the EPM2A or NHLRC1 gene and/or the use of multigene panel, which includes EPM2A, NHLRC1, and other genes of interest. If a diagnosis is not confirmed in a patient with LD features after analysis of the EPM2A and NHLRC1 genes, genomic testing may be considered. Such testing may include whole-exome sequencing, whole-genome sequencing, and whole-mitochondrial sequencing. However, multigene panel strategy and genomic testing should be complemented by exon or whole gene deletion testing in patients with a single heterozygous mutation in one of the genes as well as in patients with an apparently homozygous mutation in one of the genes, but the same mutation is carried by only one parent.
Are defects in malin and laforin gene clinically indistinguishable?
Both in patients and mouse models, defects in laforin and malin lead to clinically indistinguishable phenotypes.4,6,16,17 To date, no correlations between phenotype and mutation type or its location in the gene have been demonstrated. Genotype–phenotype correlations are difficult to establish in LD because of the high allelic heterogeneity and frequent compound heterozygotes in different combinations.18,19 Although a subphenotype consisting of childhood-onset learning disorder followed by epilepsy and neurologic deterioration has been associated with either mutations in exon 1 of EPM2A16,20 or the p.I198N pathogenic variant located in an NHL protein–protein interaction domain of NHLRC1,19 these findings need to be replicated, expanded, and studied further in order to understand their relationship to the underlying pathophysiological processes.
However, a slower course of the disease with a delayed age of death has been reported in some patients with NHLRC1 mutations. This finding has been demonstrated especially for patients with the NHLRC1 c.436G>A (p.D146N) substitution.9,19,21,22 Five of six LD patients who maintained >10 years gait autonomy carried homozygous or compound heterozygous p.D146N mutation and were described as mild.22 In a Turkish family with four brothers affected by homozygous p.D146N mutation in the NHLRC1 gene (three subjects were followed-up for >10 years), late age of onset was reported.23 Franceschetti et al9 speculated that the p.D146N mutation was invariably associated with a slow progression in three unrelated cases with homozygous p.D146N mutation. In the diagnostic workup of slowly progressive adult patients with progressive myoclonic epilepsy, detection of this mutation should be considered.22 But it should be stressed that EEG findings remain typical even in these cases.
But NHLRC1 mutations are not necessarily associated with a slow disease course, as some patients may have extremely severe phenotypes. Brackmann et al24 described a case of a 15-year-old adolescent with NHLRC1 mutation and rapidly progressive phenotype. A Malian family with the NHLRC1 gene mutation presented with severe cognitive decline 4–5 years after the onset of disease.25 Kecmanovi et al26 reported a Montenegrin patient with deletion of the entire NHLRC1 gene and a clinical course more progressive than in most individuals with NHLRC1 mutations. Also, a lower quality of medical care in some of these LD cases with the faster disease progression could not be excluded.
Glycogen metabolism and LD
LD is characterized by the accumulation of LBs and by neurodegeneration.27 LBs are mostly composed of abnormal glycogen, called polyglucosan, which is insoluble and hyperphosphorylated and contains a small amount of protein. A normal glycogen molecule remains soluble because its glucose chains are short, is made of 13 units, and each chain is a branch of another, and as a result of this structure, the whole molecule is spherical. Malformed glycogen lacks glycogen’s normal branching and spherical structure essential to its solubility. Polyglucosans are more similar to plant amylopectin or starch than to glycogen, and like these plant carbohydrates, they are insoluble, precipitate, and accumulate.28 It has been demonstrated that in plants, variants in the starch excess 4 gene (SEX4) result in the accumulation of amylopectin, similar to the way loss of laforin leads to the accumulation of polyglucosans with formation of LBs in humans.29–31 In plants, human laforin can rescue the SEX4-mutated phenotype.30
Glycogen is an efficient energy storage molecule in animals. For de novo glycogen synthesis to proceed, the first glucose residue is attached to a protein known as glycogenin. The attached glucose then serves as the primer required by glycogen synthase (GS), which catalyzes formation of the vast majority of glycogen’s interglucosidic bonds and glycogen branching enzyme, which catalyzes glycogen’s branching. Glycogen is degraded by glycogen phosphorylase (GP) and glycogen debranching enzyme. Protein targeting to glycogen (PTG) is an adaptor protein that mediates dephosphorylation of GS and GP by the pleiotropic phosphatase PP1, which activates GS, inactivates GP, and thus, increases glycogen production.32,33 GS is essential to glucose polymerization by formation of α-1,4-glycosidic bonds whether the final structure is glycogen or polyglucosans. Despite glucose being the preferential energy source for the neurons, neurons synthesize very low glycogen under physiological conditions compared to most other cell types.34 Saez et al34 showed that endogenous neuronal glycogen metabolism participates in the neuronal tolerance to hypoxic stress. Progressive accumulation of glycogen in mouse and Drosophila neurons leads to neuronal loss, locomotion defects, and reduced life span and is responsible for neurodegeneration.35 Laforin and malin were found to promote neuronal survival by restricting glycogen synthesis.36 A complex of these proteins was reported to keep neuronal glycogen synthetic machinery constitutively silent by enforcing GS inactivation and degradation of PTG.36
In brain, LBs form in neuronal perikarya and in neuronal short processes (mostly dendrites), but are never seen in long tract axons. Polyglucosan masses gradually replace the cytoplasms of dendrites, likely underlying onset, progression, and intractability of LD, since dendrites are the chief determinant of a neuron’s excitability state.28,37 Except in the central nervous system, LBs are present in retina, heart, liver, skeletal muscle, and skin but cause no symptoms in these organs.28,37,38 The exact role of the LBs in the pathogenesis of LD has been the subject of intensive research efforts over the past few years.
LBs and polyglucosans do occur in other neurological diseases, including adult polyglucosan body disease, caused by mutations in the glycogen branching enzyme gene.39 But unlike LD, LBs in this disease form exclusively in axons. The affected patients suffer from motor neuron disease and may have mild dementia, but have no epilepsy.39,40 Branching enzyme activity is normal in LD,41 which rules out decreased branching enzyme activity as the mechanism of polyglucosan generation in LD. Further, type VII glycogen storage disease is due to muscle-specific phosphofructokinase deficiency and affects only skeletal muscle.42 Deficiency of this enzyme results in accumulation of its precursor, glucose 6-phosphate (G6P), an allosteric activator of GS. Increased GS activity disturbs the glycogen-branching balance in favor of extension, leading to muscle polyglucosan.43 The ubiquitous presence of polyglucosans in LD rules out phosphofructokinase gene (PFKP) as a LD gene indicating that changes in GS activity result in polyglucosan formation and LD.
Reduced glycogen synthesis could prevent LD
Many studies showed that reducing glycogen synthesis resulted in prevention of formation of LBs, neurodegeneration, and seizure susceptibility, thus preventing LD.44–46 A study by Pederson et al45 showed that genetic elimination of brain GS in laforin-lacking LD mice resulted in long-term correction of the LD phenotype. The same result was obtained through partial reduction of glycogen synthesis by genetically removing PTG in laforin-lacking LD mice.44 These results indicated that in LD caused by laforin deficiency, LBs are due to a disturbance in glycogen metabolism and that loss of laforin’s glycogen-related function underlies this disturbance. Furthermore, in LD caused by malin deficiency, removing PTG resulted in prevention of LBs formation, neurodegeneration, and seizure susceptibility, same as in laforin-deficiency mice.46 These results indicated that malin’s functions that are glycogen related underlie formation of LBs.46
It has been shown that reduction of glycogen synthesis prevents polyglucosan accumulation and neurodegeneration in PTG/laforin, GS/laforin and PTG/malin double knockout mice.44–46 Thus, partial GS inhibition, that is, glycogen synthesis downregulation, may provide a potential treatment for LD. In humans, complete GS elimination cannot be contemplated as this causes significant pathology,47 but partial reduction of GS, or reduction of GS and activation of GP, could be therapeutic. Compounds that are known to partially inhibit GS or PTG, as well as genetic approaches such as antisense oligonucleotides against GS, could prove useful in halting LD early in its course, or preclinically, and prevent its progression.45
Beside impairment in glycogen metabolism, it is thought that LD results in defect in autophagy and that the defect in autophagy could be at the basis of neurodegeneration seen in LD. But a recent study by Duran et al48 demonstrated that glycogen accumulation per se drives neuronal death and that the accumulation of glycogen is not a consequence of autophagy impairment but rather the cause of it. Also, without glycogen accumulation, there is no alteration of autophagy in LD.48 Further, a recent study by Garyali et al49 showed that defects in autophagy may be secondary to glycogen overaccumulation since both laforin and malin knockout cells displayed mammalian target of rapamycin-dependent autophagy defects.
Closing summary
In the new genome era, we are witnessing the progressive development of therapies for many rare diseases. The former concept of gene therapy is abandoned and replaced by a search for small molecules or RNA molecules that act on the disease mechanism. Since the discovery of the first gene associated with LD has passed a little more than 15 years, today we are a step away from the curative therapy. This step means screening for small molecules that act on partial inhibition of GS.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Chan EM, Andrade DM, Franceschetti S, Minassian B. Progressive myoclonus epilepsies: EPM1, EPM2A, EPM2B. In: Delgado-Escueta AV, Guerrini R, Medina MT, Genton P, Bureau M, Dravet C, editors. Myoclonic Epilepsies. Philadelphia: Lippincott, Williams and Wilkins; 2005. pp. 47–58. [PubMed] [Google Scholar]
- 2.Delgado-Escueta AV, Ganesh S, Yamakawa K. Advances in the genetics of progressive myoclonus epilepsy. Am J Med Genet. 2001;106(2):129–138. doi: 10.1002/ajmg.1575. [DOI] [PubMed] [Google Scholar]
- 3.Jansen AC, Andermann E. Progressive myoclonus epilepsy, Lafora type. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews [Internet] Seattle, WA: University of Washington; pp. 1993–2015. [PubMed] [Google Scholar]
- 4.Minassian BA, Lee JR, Herbrick JA, et al. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat Genet. 1998;20(2):171–174. doi: 10.1038/2470. [DOI] [PubMed] [Google Scholar]
- 5.Worby CA, Gentry MS, Dixon JE. Laforin: a dual specificity phosphatase that dephosphorylates complex carbohydrates. J Biol Chem. 2006;281:30412–30418. doi: 10.1074/jbc.M606117200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan EM, Young EJ, Ianzano L, et al. Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat Genet. 2003;35(2):125–127. doi: 10.1038/ng1238. [DOI] [PubMed] [Google Scholar]
- 7.Gentry MS, Worby CA, Dixon JE. Insights into Lafora disease: malin is an E3 ubiquitin ligase that ubiquitinates and promotes the degradation of laforin. Proc Natl Acad Sci U S A. 2005;102:8501–8506. doi: 10.1073/pnas.0503285102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh S, Suzuki T, Uchiyama A, et al. Mutations in the NHLRC1 gene are the common cause for Lafora disease in the Japanese population. J Hum Genet. 2005;50(7):347–352. doi: 10.1007/s10038-005-0263-7. [DOI] [PubMed] [Google Scholar]
- 9.Franceschetti S, Gambardella A, Canafoglia L, et al. Clinical and genetic findings in 26 Italian patients with Lafora disease. Epilepsia. 2006;47(3):640–643. doi: 10.1111/j.1528-1167.2006.00479.x. [DOI] [PubMed] [Google Scholar]
- 10.Kecmanovi M, Jovi N, Keckarević-Marković M, et al. Clinical and genetic data on Lafora disease patients of Serbian/Montenegrin origin. Clin Genet. 2016;89(1):104–108. doi: 10.1111/cge.12570. [DOI] [PubMed] [Google Scholar]
- 11.Singh S, Sethi I, Francheschetti S, et al. Novel NHLRC1 mutations and genotype-phenotype correlations in patients with Lafora’s progressive myoclonic epilepsy. J Med Genet. 2006;43(9):e48. doi: 10.1136/jmg.2005.039479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singh S, Ganesh S. Lafora progressive myoclonus epilepsy: a meta-analysis of reported mutations in the first decade following the discovery of the EPM2A and NHLRC1 genes. Hum Mutat. 2009;30(5):715–723. doi: 10.1002/humu.20954. [DOI] [PubMed] [Google Scholar]
- 13.Lesca G, Boutry-Kryza N, de Toffol B, et al. Novel mutations in EPM2A and NHLRC1 widen the spectrum of Lafora disease. Epilepsia. 2010;51(9):1691–1698. doi: 10.1111/j.1528-1167.2010.02692.x. [DOI] [PubMed] [Google Scholar]
- 14.Drury I, Blaivas M, Abou-Khalil BW, Beydoun A. Biopsy results in a kindred with Lafora disease. Arch Neurol. 1993;50(1):102–105. doi: 10.1001/archneur.1993.00540010096025. [DOI] [PubMed] [Google Scholar]
- 15.Andrade DM, Ackerley CA, Minett TS, et al. Skin biopsy in Lafora disease: genotype-phenotype correlations and diagnostic pitfalls. Neurology. 2003;61(11):1611–1614. doi: 10.1212/01.wnl.0000096017.19978.cb. [DOI] [PubMed] [Google Scholar]
- 16.Ganesh S, Delgado-Escueta AV, Suzuki T, et al. Genotype-phenotype correlations for EPM2A mutations in Lafora’s progressive myoclonus epilepsy: exon 1 mutations associate with an early-onset cognitive deficit subphenotype. Hum Mol Genet. 2002;11(11):1263–1271. doi: 10.1093/hmg/11.11.1263. [DOI] [PubMed] [Google Scholar]
- 17.DePaoli-Roach AA, Tagliabracci VS, Segvich DM, Meyer CM, Irimia JM, Roach PJ. Genetic depletion of the malin E3 ubiquitin ligase in mice leads to lafora bodies and the accumulation of insoluble laforin. J Biol Chem. 2010;285(33):25372–25381. doi: 10.1074/jbc.M110.148668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chan EM, Andrade DM, Franceschetti S, Minassian B. Progressive myoclonus epilepsies: EPM1, EPM2A, EPM2B. Adv Neurol. 2005;95:47–57. [PubMed] [Google Scholar]
- 19.Gómez-Abad C, Gómez-Garre P, Gutiérrez-Delicado E, et al. Lafora disease due to EPM2B mutations: a clinical and genetic study. Neurology. 2005;64(6):982–986. doi: 10.1212/01.WNL.0000154519.10805.F7. [DOI] [PubMed] [Google Scholar]
- 20.Annesi G, Sofia V, Gambardella A, et al. A novel exon 1 mutation in a patient with atypical lafora progressive myoclonus epilepsy seen as childhood-onset cognitive deficit. Epilepsia. 2004;45(3):294–295. doi: 10.1111/j.0013-9580.2004.33203.x. [DOI] [PubMed] [Google Scholar]
- 21.Lanoiselée HM, Genton P, Lesca G, Brault F, De Toffol B. Are c.436G>A mutations less severe forms of Lafora disease? A case report. Epilepsy Behav Case Rep. 2014;2:19–21. doi: 10.1016/j.ebcr.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferlazzo E, Canafoglia L, Michelucci R, et al. Mild Lafora disease: clinical, neurophysiologic, and genetic findings. Epilepsia. 2014;55(12):e129–e133. doi: 10.1111/epi.12806. [DOI] [PubMed] [Google Scholar]
- 23.Baykan B, Striano P, Gianotti S, et al. Late-onset and slow-progressing Lafora disease in four siblings with EPM2B mutation. Epilepsia. 2005;46(10):1695–1697. doi: 10.1111/j.1528-1167.2005.00272.x. [DOI] [PubMed] [Google Scholar]
- 24.Brackmann FA, Kiefer A, Agaimy A, Gencik M, Trollmann R. Rapidly progressive phenotype of Lafora disease associated with a novel NHLRC1 mutation. Pediatr Neurol. 2011;44(6):475–477. doi: 10.1016/j.pediatrneurol.2011.01.012. [DOI] [PubMed] [Google Scholar]
- 25.Traoré M, Landouré G, Motley W, et al. Novel mutation in the NHLRC1 gene in a Malian family with a severe phenotype of Lafora disease. Neurogenetics. 2009;10(4):319–323. doi: 10.1007/s10048-009-0190-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kecmanovi M, Jovi N, Cuki M, et al. Lafora disease: severe phenotype associated with homozygous deletion of the NHLRC1 gene. J Neurol Sci. 2013;325(1–2):170–173. doi: 10.1016/j.jns.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 27.Girard JM, Turnbull J, Ramachandran N, Minassian BA. Progressive myoclonus epilepsy. Handb Clin Neurol. 2013;113:1731–1736. doi: 10.1016/B978-0-444-59565-2.00043-5. [DOI] [PubMed] [Google Scholar]
- 28.Minassian BA. Lafora’s disease: towards a clinical, pathologic, and molecular synthesis. Pediatr Neurol. 2001;25(1):21–29. doi: 10.1016/s0887-8994(00)00276-9. [DOI] [PubMed] [Google Scholar]
- 29.Niittylä T, Comparot-Moss S, Lue WL, et al. Similar protein phosphatases control starch metabolism in plants and glycogen metabolism in mammals. J Biol Chem. 2006;281(17):11815–11818. doi: 10.1074/jbc.M600519200. [DOI] [PubMed] [Google Scholar]
- 30.Gentry MS, Dowen RH, 3rd, Worby CA, Mattoo S, Ecker JR, Dixon JE. The phosphatase laforin crosses evolutionary boundaries and links carbohydrate metabolism to neuronal disease. J Cell Biol. 2007;178(3):477–488. doi: 10.1083/jcb.200704094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gentry MS, Dixon JE, Worby CA. Lafora disease: insights into neurodegeneration from plant metabolism. Trends Biochem Sci. 2009;34(12):628–639. doi: 10.1016/j.tibs.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Printen JA, Brady MJ, Saltiel AR. PTG, a protein phosphatase 1-binding protein with a role in glycogen metabolism. Science. 1997;275(5305):1475–1478. doi: 10.1126/science.275.5305.1475. [DOI] [PubMed] [Google Scholar]
- 33.Fong NM, Jensen TC, Shah AS, Parekh NN, Saltiel AR, Brady MJ. Identification of binding sites on protein targeting to glycogen for enzymes of glycogen metabolism. J Biol Chem. 2000;275(45):35034–35039. doi: 10.1074/jbc.M005541200. [DOI] [PubMed] [Google Scholar]
- 34.Saez I, Duran J, Sinadinos C, et al. Neurons have an active glycogen metabolism that contributes to tolerance to hypoxia. J Cereb Blood Flow Metab. 2014;34(6):945–955. doi: 10.1038/jcbfm.2014.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duran J, Tevy MF, Garcia-Rocha M, Calbo J, Milan M, Guinovart JJ. Deleterious effects of neuronal accumulation of glycogen in flies and mice. EMBO Mol Med. 2012;4:719–729. doi: 10.1002/emmm.201200241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vilchez D, Ros S, Cifuentes D, et al. Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat Neurosci. 2007;10(11):1407–1413. doi: 10.1038/nn1998. [DOI] [PubMed] [Google Scholar]
- 37.Lafora GR. Uber das Corkommen amyloider Korperchen im Innern der Ganglienzellen; zugliech Ein zum Studium der amyloiden Substanz im Nervensystem. [About the presence of amyloid corpuscles inside the ganglion cells; a simultaneously study of amyloid substance in the nervous system] Virchows Arch Pathol Anat. 1911;205:295–303. German. [Google Scholar]
- 38.Van Heycop Ten, Ham MW. Lafora disease, a form of progressive myoclonus epilepsy. In: Vinken PJ, Bruyn GW, editors. The Epilepsies. Handbook of Clinical Neurology. Vol. 15. Amsterdam: North-Holland Publishers; 1975. pp. 382–422. [Google Scholar]
- 39.Lossos A, Meiner Z, Barash V, et al. Adult polyglucosan body disease in Ashkenazi Jewish patients carrying the Tyr329Ser mutation in the glycogen-branching enzyme gene. Ann Neurol. 1998;44(6):867–872. doi: 10.1002/ana.410440604. [DOI] [PubMed] [Google Scholar]
- 40.Robitaille Y, Carpenter S, Karpati G, DiMauro SD. A distinct form of adult polyglucosan body disease with massive involvement of central and peripheral neuronal processes and astrocytes: a report of four cases and a review of the occurrence of polyglucosan bodies in other conditions such as Lafora’s disease and normal ageing. Brain. 1980;103(2):315–336. doi: 10.1093/brain/103.2.315. [DOI] [PubMed] [Google Scholar]
- 41.Zimmerman CP, Gold AM. Glycogen branching enzyme in Lafora myoclonus epilepsy. Biochem Med. 1982;28(1):83–93. doi: 10.1016/0006-2944(82)90058-8. [DOI] [PubMed] [Google Scholar]
- 42.Tarui A, Okuno G, Ikura Y, Tanaka T, Suda M, Nishikawa M. Phosphofructokinase deficiency in skeletal muscle. A new type of glycogenosis. Biochem Biophys Res Commun. 1965;19:517–523. doi: 10.1016/0006-291x(65)90156-7. [DOI] [PubMed] [Google Scholar]
- 43.Hays AP, Hallett M, Delfs J, et al. Muscle phosphofructokinase deficiency: abnormal polysaccharide in a case of late-onset myopathy. Neurology. 1981;31(9):1077–1086. doi: 10.1212/wnl.31.9.1077. [DOI] [PubMed] [Google Scholar]
- 44.Turnbull J, DePaoli-Roach AA, Zhao X, et al. PTG depletion removes Lafora bodies and rescues the fatal epilepsy of Lafora disease. PLoS Genet. 2011;7(4):e1002037. doi: 10.1371/journal.pgen.1002037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pederson BA, Turnbull J, Epp JR, et al. Inhibiting glycogen synthesis prevents Lafora disease in a mouse model. Ann Neurol. 2013;74(2):297–300. doi: 10.1002/ana.23899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Turnbull J, Epp JR, Goldsmith D, et al. PTG protein depletion rescues malin-deficient Lafora disease in mouse. Ann Neurol. 2014;75(3):442–446. doi: 10.1002/ana.24104. [DOI] [PubMed] [Google Scholar]
- 47.Kollberg G, Tulinius M, Gilljam T, et al. Cardiomyopathy and exercise intolerance in muscle glycogen storage disease 0. N Engl J Med. 2007;357(15):1507–1514. doi: 10.1056/NEJMoa066691. [DOI] [PubMed] [Google Scholar]
- 48.Duran J, Gruart A, García-Rocha M, Delgado-García JM, Guinovart JJ. Glycogen accumulation underlies neurodegeneration and autophagy impairment in Lafora disease. Hum Mol Genet. 2014;23(12):3147–3156. doi: 10.1093/hmg/ddu024. [DOI] [PubMed] [Google Scholar]
- 49.Garyali P, Segvich DM, DePaoli-Roach AA, Roach PJ. Protein degradation and quality control in cells from laforin and malin knockout mice. J Biol Chem. 2014;289(30):20606–20614. doi: 10.1074/jbc.M114.580167. [DOI] [PMC free article] [PubMed] [Google Scholar]