

Abstract
Androgens play an important role in prostate cancer (PCa) development and progression. Accordingly, androgen deprivation therapy remains the front-line treatment for locally recurrent or advanced PCa, but patients eventually relapse with the lethal form of the disease termed castration resistant PCa (CRPC). Importantly, castration does not eliminate androgens from the prostate tumor microenvironment which is characterized by elevated tissue androgens that are well within the range capable of activating the androgen receptor (AR). In this mini-review, we discuss emerging data that suggest a role for the enzymes mediating pre-receptor control of dihydrotestosterone (DHT) metabolism, including AKR1C2, HSD17B6, HSD17B10, and the UGT family members UGT2B15 and UGT2B17, in controlling intratumoral androgen levels, and thereby influencing PCa progression. We review the expression of steroidogenic enzymes involved in this pathway in primary PCa and CRPC, the activity and regulation of these enzymes in PCa experimental models, and the impact of genetic variation in genes mediating pre-receptor DHT metabolism on PCa risk. Finally, we discuss recent data that suggests several of these enzymes may also play an unrecognized role in CRPC progression separate from their role in androgen inactivation.
Keywords: Androgen, Androgen Receptor, Androgen Deprivation Therapy, LNCaP Cell, Androgen Level
Introduction
Androgens regulate normal prostate growth and function by interacting with the androgen receptor (AR), and play a central role in prostate cancer (PCa) progression [1]. As such, PCa biology may be influenced by mechanisms that modulate the intracellular production or accumulation of androgens. As early as sixty years ago, steroid hormones were recognized to exist in either active or inactive forms which could be enzymatically interconverted in a tissue-specific manner. This concept in steroid-hormone physiology was called pre-receptor control and implied that inactive metabolites could serve as precursors for metabolic conversion to active ligands, thereby complementing the pool of ligands available for receptor binding in a tissue-specific manner [2, 3].
Here, we review the emerging evidence that suggests pre-receptor metabolic control of dihydrotestosterone (DHT) levels, specifically, those enzymes that mediate the catabolism of DHT or reverse this process, may be a critical determinant of androgen levels in castration resistant PCa (CRPC) tumors. We review the enzymes involved in pre-receptor control of DHT metabolism, their altered expression in the progression of primary PCa to CRPC, and the evidence for their relevance in treatment resistance and disease progression from clinical studies and experiments in PCa cell lines as well as patient-derived xenografts. We discuss their impact on androgen levels in vitro and the effect of drugs and other factors that modulate their activity, including genetic variation in these enzymes and link to PCa risk. We conclude by discussing recent data that suggest UGT enzymes and members of the ARK1C family may also have a role in CRPC progression separate from their role in androgen inactivation [4].
Pre-Receptor Control of DHT Metabolism in Prostate
In prostate tissue, intracellular levels of DHT are regulated by phase I (reducing) and phase II (conjugating) enzymes that mediate DHT catabolism and thereby regulate access of DHT to the AR [5] (Fig. 1). AKR1C2 is the primary enzyme responsible for the reversible reduction of DHT to 5α-androstane-3,17-diol (3α-androstanediol or 3α-diol, a low affinity AR ligand), which is subsequently glucuronidated to 3α-diol glucuronide (3α-diol G), and released into circulation. While AKR1C2 is capable of bidirectional activity (i.e., catalyzing conversion of 3α-diol back to DHT), intracellularly it functions primarily to reduce DHT [3, 6]. The reductase activity of AKR1C2, together with the reverse oxidative activity of 3α-HSDs, including HSD17B6, HSD17B10, and RDH5, is a critical molecular switch that regulates tissue androgen levels [3, 6–8].
Fig. 1.
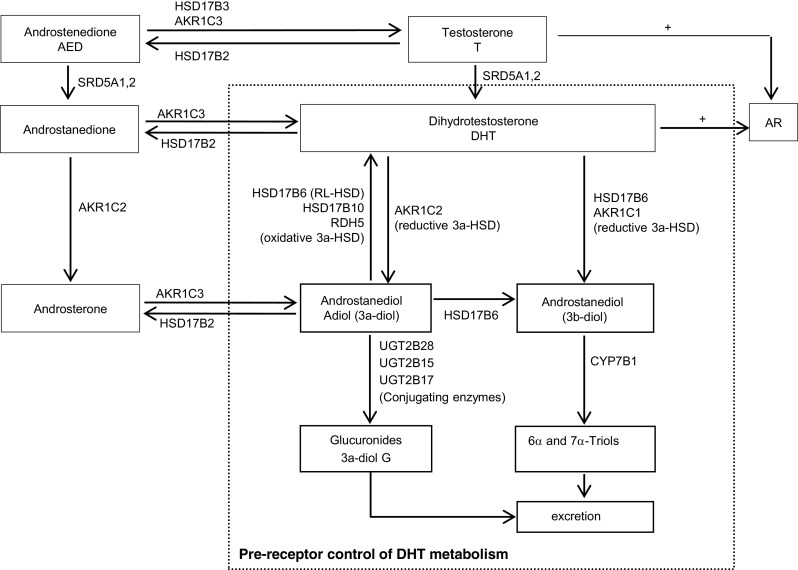
Pre-receptor control of DHT metabolism in prostate (dashed square). Both reducing and conjugating DHT catabolizing enzymes together with DHT oxidases regulate intracellular levels of DHT and hereby, its access to the androgen receptor (AR). AKR1C2 catalyzes the reversible reduction of DHT to 3α-diol. HSD17B6 (RL-HSD), HSD17B10, and RDH5 oxidize 3α-diol back to DHT. The activities of both reductive 3α-HSDs and oxidative 3α-HSDs determine the intracellular levels of DHT. HSD17B6 also converts physiological concentration of DHT to 3β-diol. AKR1C1 catalyzes the irreversible conversion of DHT to 3β-diol. UGT2B28, UGT2B17, and UGT2B15 are all involved of glucuronidation of androgen metabolites; UGT2B15 and UGT2B17 can also directly glucuronidate DHT (not shown)
Transcripts of both HSD17B6 (also called RL-HSD) and HSD17B10 are highly expressed in the prostate; however, several studies suggest HSD17B6 is more active in converting 3α-diol to DHT in prostate cells [9, 10]. Basal epithelial cell expression of HSD17B6 is present at the protein level, while transcript profiling of cultured epithelial and stromal cells detects stromal expression as well [9, 11]. AKR1C1 catalyzes the irreversible conversion of DHT to 5α-androstane-3 β,17 β-diol (3β-diol), a possible endogenous ligand for the estrogen receptor β (ER β, in prostate) [3]. Interestingly, besides its oxidative activity, HSD17B6 can also convert physiological concentration of DHT to 3β-diol [11] and acts as an epimerase to convert 3α-diol to 3β-diol, although at much higher substrate concentrations [12].
The phase II conjugating enzymes UGT2B17 and UGT2B15 are highly expressed in the prostate and irreversibly terminate the androgen signal by glucuronidation of 3α-diol (as well as testosterone (T), DHT, and other metabolites) [13–16]. (UGT2B28 is present in the prostate but does not directly glucuronidate DHT [17]; UGT2B7 is able to glucuronidate DHT, but its activity is only 1–10 % of UGT2B15 or 17, and it is not expressed in the prostate [18, 19]). Therefore, the relative activity of the reductive 3α-HSDs AKR1C2 and AKR1C1 in converting DHT to 3α-diol or 3β-diol, respectively, vs activity of the oxidative 3α-HSDs HSD17B6 and HSD17B10 in converting 3α-diol back to DHT, and activity of the conjugating enzymes UGT2B15 and UGT2B17 in glucuronidating DHT, collectively govern the levels of active androgen in the prostate.
Altered Expression of Enzymes Involved in Pre-Receptor DHT Metabolism in PCa Progression to CRPC
Altered Expression of Genes Mediating DHT Production and Catabolism in Primary PCa
Differential changes in the expression of reductive and oxidative enzyme pairs favoring the conversion of inactive diones to active androgens (e.g., androstenedione (AED) to testosterone (T) or androstanedione to DHT) have been observed between normal prostate and primary PCa. These include increased tumor expression of the reductive enzymes HSD17B3 (31-fold) [20] and AKR1C3 (2–5-fold) [21–24], and decreased expression of the oxidative enzyme catalyzing the reverse reaction, HSD17B2 (7-fold) [20, 25], suggesting a shift in tumoral androgen metabolism to the formation of T and DHT from their inactive dione precursors (Fig. 2a, b). SRD5A is responsible for conversion of T and AED to DHT and androstanedione, respectively. A consistent observation in primary PCa is a decrease in the expression of tumoral SRD5A2 (2–4-fold) [26–29], the principle isoform of this enzyme expressed in benign prostate tissue [30] and a relative shift in primary and recurrent prostate tumors to increased expression of SRD5A1 (2-fold) [27, 31] (although some studies have shown Gleason grade-related increases in both SRD5A1 and SRD5A2 [32]).
Fig. 2.
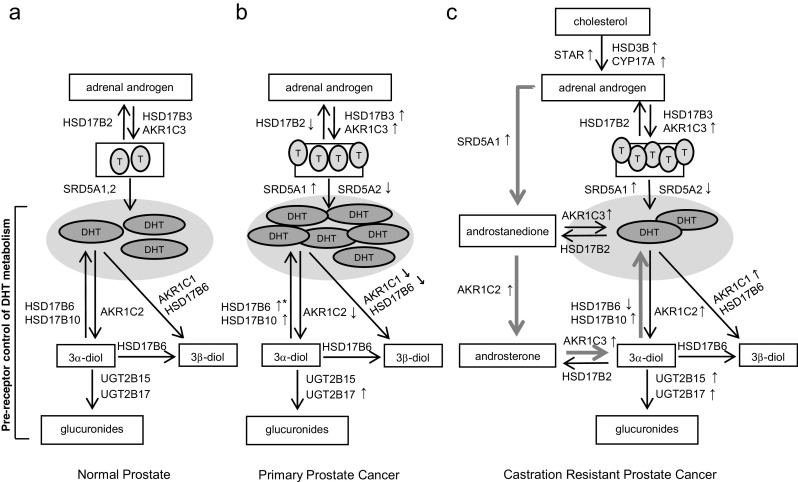
Altered pre-receptor metabolism of DHT in prostate cancer progression. a Schematic of androgen synthesis and pre-receptor control of DHT metabolism in normal prostate tissue. b Differential changes in steroidogenic enzymes in primary prostate cancer vs normal prostate tissue: increased tumor expression of reductive enzymes HSD17B3 and AKR1C3 and decreased expression of the oxidative enzyme HSD17B2, favoring production of active androgens from inactive diones; a decrease in tumoral SRD5A2 and increase in SRD5A1; and decreased expression of both AKR1C2 and AKR1C1 with increased expression of HSD17B10, favoring production of DHT. Expression of HSD17B6 is low in untreated primary PCa, but increased in men treated with androgen deprivation (upward arrow with*). c Altered expression of genes mediating DHT production and catabolism in CRPC: variably increased expression of STAR, CYP17A, and HSD3β2, involved in de novo steroid synthesis; consistently increased expression of AKR1C3 and SRD5A1, mediating conversion of adrenal androgens to downstream steroids; paradoxical increase in expression of genes involved in DHT catabolism including AKR1C1, AKR1C2, and UGT2B15 and 17; and increased expression of HSD17B10 in conjunction with increased AKR1C2, suggesting activity via a testosterone bypass pathway (thick arrows) with steroid flux directed to DHT via androstanedione, androsterone, and 3α-diol. (Upward arrows denote increased gene expression, and downward arrows denote decreased gene expression)
Primary PCa also demonstrates a selective loss of both AKR1C2 (16-fold) and AKR1C1 (2–4-fold) vs paired benign tissues [8, 26, 33, 34]; as a consequence of this defect in DHT catabolism, DHT levels were significantly higher in primary PCa tumors [26]. In contrast, increased expression of HSD17B10 (3-fold), one of the oxidative enzyme capable of mediating the back conversion of 3α-diol to DHT, was observed in malignant epithelial cells compared to normal, which would also support an increased capacity to generate DHT in tumor tissue [35]. This drive to maintain DHT levels is consistent with the twofold increase in prostate expression of HSD17B6 (another oxidative enzyme capable of mediating the back conversion of 3α-diol to DHT) observed in a study of men treated with androgen deprivation therapy (ADT), which was associated with a higher HSD17B6 score in those who showed biochemical progression [36]. However, epithelial expression of HSD17B6 (which can also mediate the conversion of DHT to 3β-diol) is low in untreated primary PCa, which is hypothesized to reflect loss of the 3β-diol/ERβ mediated growth inhibition pathway during malignant transformation [11].
These findings suggest that by favoring DHT production, the decreased expression of reductive enzymes that mediate catabolism of DHT together with the increased expression of oxidative enzymes that mediate conversion of 3α-diol back to DHT represent an important control point for growth regulation that is lost in primary PCa (Fig. 2b). Notably, however, UGT2B17 levels are increased (2-fold) in PCa vs benign tissue [37], and some reports have suggested an association of low androgens with aggressive disease [38]. As such, nuances to role of tissue androgen levels in mediating PCa risk remain to be elucidated.
Altered Expression of Genes Mediating DHT Production and Catabolism in CRPC
CRPC is characterized by altered expression of numerous genes involved in steroid synthesis and metabolism, including enzymes directly impacting pre-receptor DHT metabolism such as SRD5A, AKR1C2, and UGT2B15 and 17 (Fig. 2c). While increased expression of genes involved in de novo steroid synthesis in CRPC, such as STAR, CYP17A, and HSD3β2, has been variably reported [24, 27, 29, 39], alterations in genes mediating the conversion of adrenal androgens to downstream steroids such as the increased expression of AKR1C3 (5–8-fold) and SRD5A1 (2–3-fold) have been more consistently observed (accompanied by a decrease in SRD5A2 (2–9-fold), similar to the shift in these isoforms observed in primary PCa) [24, 27, 29, 39, 40].
Paradoxically, several CRPC studies also show an increased expression of genes involved in DHT catabolism including AKR1C1 (3-fold) and AKR1C2 (2–3-fold) [24, 27] as well as UGT2B15 (3–10-fold) and 17 (34-fold) [27, 29, 40], which would be consistent with a study of CRPC metastases showing an inverted ratio of T to DHT compared to primary PCa (T 0.74 ng/g: DHT 0.25 ng/g in CRCP samples vs T 0.23 ng/g: DHT 2.75 ng/g in primary PCa) [29]. Alternatively, while decreased levels of HSD17B6 have been reported in CRPC tissues compared to untreated primary PCa [10], another study found increased expression of AKR1C2 (6-fold) in association with increased expression of HSD17B10 (3-fold) [24]. The latter observation could suggest that in some cases, steroid flux is directed from androstanedione to androsterone (via AKR1C2), then to 3α-diol (via AKR1C3) and finally back to DHT (via HSD17B10), a testosterone bypass pathway revealed to be active in CRPC by Sharifi et al. [41] (Fig. 2c, blue arrows).
Pre-Receptor Control of DHT Metabolism in Experimental PCa Models
In Vitro Studies of Pre-Receptor Enzymes and DHT Metabolism in PCa Cell Lines
Mechanisms of DHT production and metabolism in PCa have been the subject of extensive investigation in vitro and, to a more limited extent, in vivo. The ability of tumor cells to synthesize DHT from cholesterol de novo has been demonstrated in many (though not all) PCa cell line models, accompanied by expression of transcripts encoding the key steroidogenic genes required for androgen synthesis, including STAR, CYP11A, CYP17A, HSD3B2, and AKR1C3, as well as genes involved in DHT catabolism such as AKR1C2 and SRD5A1 [42–46].
The influence of enzymes involved in pre-receptor control of DHT metabolism in PCa cells has also been demonstrated. Overexpression of AKR1C1 or AKR1C2 in LAPC-4 cells inhibited DHT-stimulated, but not R1881-induced proliferation (consistent with the ability of these enzymes to act on DHT, but not on the non-metabolizable synthetic androgen R1881), resulting in a decrease in secreted DHT in media [26]. In PC-3 cells overexpression of AKR1C2 significantly decreased DHT-dependent AR reporter activity, which was abrogated by increasing DHT levels [8, 26]. DHT treatment was shown to induce AKR1C2 transcript in both DU145 and LNCaP cells, suggesting that a decrease in androgen levels might be countered by a decrease in AKR1C2-mediated catabolism of DHT to preserve DHT levels [8, 33].
Long-term culture of LNCaP cells in androgen-depleted conditions led to a reversal in the ratio of AKR1C2 to HSD17B6, with long-term passages showing lower levels of AKR1C2 and markedly increased levels of HSD17B6 [36]. Although androgen levels were not measured, knockdown of HSD17B6 in the long-term-passaged cells led to a decrease in PSA expression in response to treatment with 3α-diol, consistent with the ability of HSD17B6 to convert this precursor to DHT. Similarly, in a series of PCa cell lines treated with 3α-diol cell, conversion of 3α-diol to DHT led to AR transactivation and stimulation of growth, and was correlated with transcript and protein levels of HSD17B6 [9, 10]. These findings are similar to the increased expression of HSD17B6 observed in primary prostate tissues after ADT [36] and consistent with the hypothesis that alterations in AKR1C2 and HSD17B6 in response to androgen suppression are acting to maintain tissue DHT levels.
UGT2B15 and UGT2B17 have been studied extensively in LNCaP cells where they have been found to be major determinants of the androgen response. Inhibition of their expression by siRNA markedly inhibited glucuronidating activity, resulting in increased DHT levels in cell culture media and an increased proliferative response [15]. Interestingly, these genes are regulated by the AR and subject to DHT-induced down regulation [47], such that their expression is increased in the presence of antiandrogens [48]. This de-repression of UGT2B15 and UGT2B17 expression by AR antagonists, with resultant increased expression and increased glucuronidation capacity was postulated by the authors to be one factor contributing to the anti-tumor activity of these agents. In this regard, the increased expression of these enzymes observed in CRPC specimens is counterintuitive and suggests that alternate explanations of their function in CRPC may be relevant (discussed further below).
In Vivo Studies of Pre-Receptor Enzymes and DHT Metabolism in PCa Xenografts
Alterations in enzymes involved in pre-receptor DHT metabolism in association with changes in intracellular androgen levels have also been reported in PCa xenograft models. In an orthotopic VCaP xenograft model, tumors grown in castrate hosts had levels of intratumoral androgens similar to those in intact mice and demonstrated increased expression of enzymes involved in steroid synthesis (CYP17A, AKR1C3) as well as in prevention of DHT catabolism (HSD17B6) [49]. A study of two AR positive, castration-sensitive LuCaP xenograft models revealed basal differences in intratumoral androgen levels that correlated strongly with their relative expression of genes mediating DHT synthesis vs DHT catabolism. Compared to LuCaP35, LuCaP96 tumors had a lower ratio of intratumoral DHT:T (1:10 vs 1:2), in association with lower expression of genes mediating production and maintenance of DHT (SRD5A1, HSD17B10, HSD17B6) and higher levels of enzymes mediating DHT catabolism (AKR1C2 and UGT2B17) [50]. In a study of castration resistant LuCaP35 and LuCaP23 xenograft tumors that recurred after treatment with abiraterone, levels of DHT in the recurrent tumors were strongly correlated with the expression of genes involved in maintenance of DHT levels, including HSD17B6 and HSD17B10 [51]. These observations are consistent with the hypothesis that genes mediating pre-receptor control of DHT metabolism play an important role in determining intratumoral androgen levels.
Regulation of Enzymes Involved in Pre-Receptor DHT Metabolism in PCa Models
Studies in PCa models have also evaluated the impact of cytokines and growth factors present in the PCa microenvironment on enzymes involved in pre-receptor DHT metabolism. Induction of AKR1C1 expression was induced by insulin-like growth factor 1 (IGF1), interleukin 6 (IL6), and transforming growth factor beta 1 (TGFβ1) in PC-3 cells [52]. In LNCaP cells, expression of UGT2B17 was decreased in response to IL-1α, epidermal growth factor (EGF), and fibroblast growth factor (FGF); expression of UGT2B15 was reduced in response to FGF, while IL4 and IL6 did not affect the expression of either [13, 14, 53, 54]. The decrease in UGT2B17 expression caused by IL-1α, EGF, and FGF was accompanied by a functional decrease in DHT glucuronidation, suggesting the presence of these factors in the prostate tumor microenvironment could lead to higher intratumoral DHT levels. From a therapeutic perspective, calcitriol was found to be a negative regulator of UGT2B15 and UGT2B17 in LNCaPs, resulting in decreased rates of DHT glucuronidation and suggesting that the proposed anti-proliferative properties of calcitriol in PCa cells could be limited by this accompanying decrease in DHT catabolism [55]. The potential chemoprotective agent, sulforaphane (SFN), an isothiocyanate found in cruciferous vegetables, was found to induce expression of multiple AKR1C family members, including AKR1C1, AKR1C2, and AKR1C3 (via activity of the Nrf2 transcription factor) in breast cancer models [56]. Whether the same occurs in PCa cell models and/or whether modulation of cellular androgens plays a role in the proposed chemoprotective activity of SFN in PCa is unknown [57].
Genetic Variation in Genes Involved in Pre-Receptor Control of DHT Metabolism
Genetic variation in key genes involved in DHT synthesis has been convincingly described for HSD3B1 [58] and SRD5A2 [59, 60]. While data on polymorphisms and associations with PCa risk for many of the genes involved in pre-receptor DHT catabolism are limited [61], functional variants in UGT genes and AKR1C2 have been described. UGT genes, in particular, are characterized by common polymorphisms and copy number variants (CNV) that affect gene expression and enzymatic activity and have been associated with PCa risk and outcomes. Although not studied in PCa as extensively as UGT2B15 or UGT2B17, UGT2B28 is another UGT enzyme expressed in prostate with capacity to glucuronidated steroid substrates [17, 19, 62]. Whole gene deletions in UBT2B17 and UGT2B28 occur in Caucasians at 27 and 13.5 %, respectively, with 57 % harboring a deletion in at least one of these genes [63]. A nonsynonymous single nucleotide polymorphism (SNP) in UGT2B15 (D85Y, rs1902923: G>T) increases maximal velocity for DHT and androstanediol 2-fold and occurs in 32 % of Caucasians [64]. CNV in UBT2B17 and UGT2B28 are associated with decreased levels of circulating androgen glucuronides, while CNV in UGT2B17 are also associated with altered levels of intraprostatic and urinary androgen glucuronides [65–67].
Gene deletions in UGT2B17 have been linked with an increased risk of biochemical relapse, while deletion of UGT2B17 and the UGT2B15 SNP D85Y has been linked with increased PCa risk in some (but not all) studies [67–71]. In particular, the UGT2B17 homozygous deletion polymorphism was associated with (1) an increased risk of biochemical recurrence after surgical treatment and significantly lower androgen glucuronides in Caucasian and Asian patients [71]; (2) an increased risk for PCa compared with insertion carriers in Caucasian and Swedish individuals, but not in African American men [72–74]; and (3) in two meta-analyses spanning 17,000 subjects, and was linked to an increased PCa susceptibility [75, 76]. Of note, however, the high activity UGT2B15 D85Y SNP (which would decrease exposure to active androgens) is less prevalent in Asians than Caucasians (18 vs 32 %), while CNV in UGT2B17 (which would increase androgen exposure) is higher in Asians than Caucasians (73 vs 27 %), suggesting that decreases in androgen exposure due to glucuronidating enzymes are not overt mediators of the decreased PCa risk observed in Asian populations [64, 71].
Functional variants in AKR1C2 have also been described, although studies linking these with clinical associations are yet to be reported. Takahashi et al. evaluated the impact of 11 naturally occurring AKR1C2 SNPs on the ability of AKR1C2 to reduce DHT to 3α-diol, demonstrating significant reductions in Vmax for two variants (F46Y and L172Q) [77]. The F46Y variant is of particular interest as the allele frequency of this SNP is the highest in African Americans (15 %), followed by Caucasians (5.9 %), and then Asians (0 %). This parallels the risk for PCa observed in these populations, raising the hypothesis that the decreased catabolism of DHT mediated by the F46Y variant in African American men may influence PCa risk. (The L172Q variant has an allele frequency of 32 % in Caucasians and has not been detected in African American or Asian populations.)
A Non-Classical Role for DHT Metabolizing Enzymes
A significant body of data supports the hypothesis that genes mediating pre-receptor control of DHT metabolism play an important role in determining intratumoral androgen levels in primary and castrate resistant prostate tumors. However, the increased expression of DHT catabolizing enzymes such as AKR1C2, UGT2B15, and UGT2B17 in CRPC (which would theoretically lower ligand levels available for AR activation) are not entirely congruent with this hypothesis. Several potential explanations for these observations exist, including the joint regulation of multiple steroidogenic genes by single transcription factors [78], the potential engagement of metabolism pathways that bypass testosterone synthesis [41], and the possibility that putatively “steroidogenic” enzymes may have cancer-related functions beyond their steroidogenic potential.
The AKR1C family is an important reminder that many steroidogenic enzymes have alternative substrates and have capacity to modify non-steroidal metabolites which can influence disease progression or response to therapy independently of their steroid metabolizing function. For example, AKR1C1 is involved in detoxification of lipid peroxidation products [79] which may influence responses to oxidative stress, and AKR1C3 and AKR1C2 are critical regulators of prostaglandin (PG) synthesis [80]. In particular, AKR1C3 forms PGF2α and 11beta-PGF2α which stimulate the prostaglandin F (FP) receptor and prevent the activation of PPARγ, resulting in a pro-proliferative signal that may stimulate PCa growth independently of an effect on steroidogenesis [81]. Increased expression of AKR1C2 in vitro and its associated increase in levels of prostaglandin F2α has also been associated with resistance to several chemotherapy drugs [82], illustrating another mechanism by which these genes may influence treatment response again independent of androgen signaling.
Alternatively, these proteins may also have functions independent of any enzymatic activity. For example, AKR1C3 has recently been identified as an AR coactivators and thus may play dual roles in promoting ligand synthesis and AR activation [83]. AKR1C3 has also been shown to bind and stabilize the ubiquitin ligase Siah2, inhibiting its degradation and thereby enhancing Siah2-dependent regulation of AR activity in PCa cells [84]. Notably, AKR1C3 may play a role in modulating epigenetic susceptibility in PCa cells independently of an effect on AR. Knockdown of AKR1C3 was accompanied by a significantly reduced expression of a range of histone deacetylases, transcriptional co-regulators, and increased sensitivity towards SAHA, a clinically approved histone deacetylase inhibitor [85]. Looking beyond PCa, UGT2B17 has been identified as a disease accelerator in chronic lymphocytic leukemia [86], and knockdown of UGT2B17 in an endometrial carcinoma cell line resulted in an increase in apoptosis in association with downregulation of the anti-apoptotic protein Mcl-1 and upregulation of the pro-apoptotic target of Mcl-1, Puma [87]. While the mechanism of UGT2B17 involvement in these tumors remains to be elucidated, these reports underscore the potential role of these enzymes in non-steroid metabolizing capacities.
Importantly, it remains to be established whether the increased expression of these genes is truly pathogenic or merely a bystander of altered CRPC signaling. For example, while UGT genes are generally repressed by AR regulated signaling [47, 67], UGT2B17 has been identified as a positively regulated gene target of the constitutively active AR splice variants present in many CRPC tumors [67]. Thus, its presence in CRPC tumors may simply be a reflection of an altered, AR-variant associated transcriptional profile rather than an inherently pathogenic alteration.
Concluding Remarks
In conclusion, there is significant evidence that pre-receptor control of DHT metabolism pathways plays an important role in modulating tumor androgen levels, thereby facilitating continued AR signaling in the progression to castration resistant disease. Whether these pathways will also be prominent mediators of resistance to new agents targeting the AR axis such as abiraterone and enzalutamide is under active investigation [88, 89]. However, while it is tempting to focus on steroid metabolic pathways as drivers of PCa biology, alternative hypotheses remain to be explored, including the capacity of metabolic enzymes to modify non-steroidal substrates with pro or anti-carcinogenic activity and their potential to act in roles independent of their catalytic functions.
Acknowledgments
Support: Pacific Northwest Prostate Cancer Specialize Program of Research Excellence P50 CA97186 (EAM, SP) and Department of Defense Congressionally Directed Medical Research Program W81XWH-12-1-0208 (EAM).
Abbreviations
- PCa
Prostate cancer
- ADT
Androgen deprivation therapy
- CRPC
Castration resistant prostate cancer
- AR
Androgen receptor
- DHT
Dihydrotestosterone
- ER
Estrogen receptor
- IGF1
Insulin-like growth factor 1
- IL6
Interleukin 6
- TGFβ1
Transforming growth factor beta 1
- EGF
Epidermal growth factor
- FGF
Fibroblast growth factor
- CNV
Copy number variant
- SNP
Single nucleotide polymorphism
Compliance with Ethical Standards
Conflict of Interest
The authors declare that they have no competing interest.
References
- 1.Nelson PS, et al. The program of androgen-responsive genes in neoplastic prostate epithelium. Proc Natl Acad Sci U S A. 2002;99:11890–11895. doi: 10.1073/pnas.182376299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nobel S, Abrahmsen L, Oppermann U. Metabolic conversion as a pre-receptor control mechanism for lipophilic hormones. Eur J Biochem / FEBS. 2001;268:4113–4125. doi: 10.1046/j.1432-1327.2001.02359.x. [DOI] [PubMed] [Google Scholar]
- 3.Penning TM, Bauman DR, Jin Y, Rizner TL. Identification of the molecular switch that regulates access of 5alpha-DHT to the androgen receptor. Mol Cell Endocrinol. 2007;265–266:77–82. doi: 10.1016/j.mce.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mostaghel EA, Zhang A, Plymate S. UDP-glucuronosyltransferase enzymes in prostate cancer progression: is only androgen catabolism involved? Eur Urol. 2015 doi: 10.1016/j.eururo.2015.08.025. [DOI] [PubMed] [Google Scholar]
- 5.Mostaghel EA. Steroid hormone synthetic pathways in prostate cancer. Trans Androl Urol. 2013;2:212–227. doi: 10.3978/j.issn.2223-4683.2013.09.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rizner TL, et al. Human type 3 3alpha-hydroxysteroid dehydrogenase (aldo-keto reductase 1C2) and androgen metabolism in prostate cells. Endocrinology. 2003;144:2922–2932. doi: 10.1210/en.2002-0032. [DOI] [PubMed] [Google Scholar]
- 7.Rizner TL, Lin HK, Penning TM. Role of human type 3 3alpha-hydroxysteroid dehydrogenase (AKR1C2) in androgen metabolism of prostate cancer cells. Chem Biol Interact. 2003;143–144:401–409. doi: 10.1016/S0009-2797(02)00179-5. [DOI] [PubMed] [Google Scholar]
- 8.Ji Q, Chang L, VanDenBerg D, Stanczyk FZ, Stolz A. Selective reduction of AKR1C2 in prostate cancer and its role in DHT metabolism. Prostate. 2003;54:275–289. doi: 10.1002/pros.10192. [DOI] [PubMed] [Google Scholar]
- 9.Bauman DR, Steckelbroeck S, Williams MV, Peehl DM, Penning TM. Identification of the major oxidative 3{alpha}-hydroxysteroid dehydrogenase in human prostate that converts 5{alpha}-androstane-3{alpha},17{beta}-diol to 5{alpha}-dihydrotestosterone: a potential therapeutic target for androgen-dependent disease. Mol Endocrinol. 2006;20:444–458. doi: 10.1210/me.2005-0287. [DOI] [PubMed] [Google Scholar]
- 10.Mohler JL, et al. Activation of the androgen receptor by intratumoral bioconversion of androstanediol to dihydrotestosterone in prostate cancer. Cancer Res. 2011;71:1486–1496. doi: 10.1158/0008-5472.CAN-10-1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muthusamy S, et al. Estrogen receptor beta and 17beta-hydroxysteroid dehydrogenase type 6, a growth regulatory pathway that is lost in prostate cancer. Proc Natl Acad Sci U S A. 2011;108:20090–20094. doi: 10.1073/pnas.1117772108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang XF, Luu-The V. Molecular characterization of a first human 3(alpha-- > beta)-hydroxysteroid epimerase. J Biol Chem. 2000;275:29452–29457. doi: 10.1074/jbc.M000562200. [DOI] [PubMed] [Google Scholar]
- 13.Hum DW, et al. Characterization of UDP-glucuronosyltransferases active on steroid hormones. J Steroid Biochem Mol Biol. 1999;69:413–423. doi: 10.1016/S0960-0760(99)00061-8. [DOI] [PubMed] [Google Scholar]
- 14.Guillemette C, et al. Differential regulation of two uridine diphospho-glucuronosyltransferases, UGT2B15 and UGT2B17, in human prostate LNCaP cells. Endocrinology. 1997;138:2998–3005. doi: 10.1210/endo.138.7.5226. [DOI] [PubMed] [Google Scholar]
- 15.Chouinard S, Barbier O, Belanger A. UDP-glucuronosyltransferase 2B15 (UGT2B15) and UGT2B17 enzymes are major determinants of the androgen response in prostate cancer LNCaP cells. J Biol Chem. 2007;282:33466–33474. doi: 10.1074/jbc.M703370200. [DOI] [PubMed] [Google Scholar]
- 16.Chouinard S, Pelletier G, Belanger A, Barbier O. Cellular specific expression of the androgen-conjugating enzymes UGT2B15 and UGT2B17 in the human prostate epithelium. Endocr Res. 2004;30:717–725. doi: 10.1081/ERC-200044014. [DOI] [PubMed] [Google Scholar]
- 17.Turgeon D, Carrier JS, Levesque E, Hum DW, Belanger A. Relative enzymatic activity, protein stability, and tissue distribution of human steroid-metabolizing UGT2B subfamily members. Endocrinology. 2001;142:778–787. doi: 10.1210/endo.142.2.7958. [DOI] [PubMed] [Google Scholar]
- 18.Valentini A, et al. Valproic acid induces neuroendocrine differentiation and UGT2B7 up-regulation in human prostate carcinoma cell line. Drug Metab Dispos Biol Fate Chem. 2007;35:968–972. doi: 10.1124/dmd.107.014662. [DOI] [PubMed] [Google Scholar]
- 19.Levesque E, et al. Isolation and characterization of the UGT2B28 cDNA encoding a novel human steroid conjugating UDP-glucuronosyltransferase. Biochemistry. 2001;40:3869–3881. doi: 10.1021/bi002607y. [DOI] [PubMed] [Google Scholar]
- 20.Koh E, Noda T, Kanaya J, Namiki M. Differential expression of 17beta-hydroxysteroid dehydrogenase isozyme genes in prostate cancer and noncancer tissues. Prostate. 2002;53:154–159. doi: 10.1002/pros.10139. [DOI] [PubMed] [Google Scholar]
- 21.Lin HK, et al. Expression and characterization of recombinant type 2 3 alpha-hydroxysteroid dehydrogenase (HSD) from human prostate: demonstration of bifunctional 3 alpha/17 beta-HSD activity and cellular distribution. Mol Endocrinol. 1997;11:1971–1984. doi: 10.1210/mend.11.13.0026. [DOI] [PubMed] [Google Scholar]
- 22.Lin H-K, Steckelbroeck S, Fung K-M, Jones AN, Penning TM. Characterization of a monoclonal antibody for human aldo-keto reductase AKR1C3 (type 2 3[alpha]-hydroxysteroid dehydrogenase/type 5 17[beta]-hydroxysteroid dehydrogenase); immunohistochemical detection in breast and prostate. Steroids. 2004;69:795–801. doi: 10.1016/j.steroids.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 23.Fung KM, et al. Increased expression of type 2 3alpha-hydroxysteroid dehydrogenase/type 5 17beta-hydroxysteroid dehydrogenase (AKR1C3) and its relationship with androgen receptor in prostate carcinoma. Endocr Relat Cancer. 2006;13:169–180. doi: 10.1677/erc.1.01048. [DOI] [PubMed] [Google Scholar]
- 24.Jernberg E, et al. Characterization of prostate cancer bone metastases according to expression levels of steroidogenic enzymes and androgen receptor splice variants. PLoS One. 2013;8:e77407. doi: 10.1371/journal.pone.0077407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elo JP, et al. Characterization of 17beta-hydroxysteroid dehydrogenase isoenzyme expression in benign and malignant human prostate. Int J Cancer. 1996;66:37–41. doi: 10.1002/(SICI)1097-0215(19960328)66:1<37::AID-IJC7>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 26.Ji Q, et al. Impaired dihydrotestosterone catabolism in human prostate cancer: critical role of AKR1C2 as a pre-receptor regulator of androgen receptor signaling. Cancer Res. 2007;67:1361–1369. doi: 10.1158/0008-5472.CAN-06-1593. [DOI] [PubMed] [Google Scholar]
- 27.Stanbrough M, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66:2815–2825. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- 28.Luo J, Dunn TA, Ewing CM, Walsh PC, Isaacs WB. Decreased gene expression of steroid 5 alpha-reductase 2 in human prostate cancer: implications for finasteride therapy of prostate carcinoma. Prostate. 2003;57:134–139. doi: 10.1002/pros.10284. [DOI] [PubMed] [Google Scholar]
- 29.Montgomery RB, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008;68:4447–4454. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klein H, Bressel M, Kastendieck H, Voigt KD. Androgens, adrenal androgen precursors, and their metabolism in untreated primary tumors and lymph node metastases of human prostatic cancer. Am J Clin Oncol. 1988;11(Suppl 2):S30–S36. doi: 10.1097/00000421-198801102-00008. [DOI] [PubMed] [Google Scholar]
- 31.Titus MA, et al. Steroid 5{alpha}-reductase isozymes I and II in recurrent prostate cancer. Clin Cancer Res. 2005;11:4365–4371. doi: 10.1158/1078-0432.CCR-04-0738. [DOI] [PubMed] [Google Scholar]
- 32.Thomas LN et al (2007) Levels of 5[alpha]-reductase type 1 and type 2 are increased in localized high grade compared to low grade prostate cancer. J Urol In Press, Corrected Proof [DOI] [PubMed]
- 33.Yun H, Xie J, Olumi AF, Ghosh R, Kumar AP (2015) Activation of AKR1C1/ERbeta induces apoptosis by downregulation of c-FLIP in prostate cancer cells: a prospective therapeutic opportunity. Oncotarget [DOI] [PMC free article] [PubMed]
- 34.Khvostova EP, Otpuschennikov AA, Pustylnyak VO, Gulyaeva LF. Gene expression of androgen metabolising enzymes in benign and malignant prostatic tissues. Horm Metab Res. 2015;47:119–124. doi: 10.1055/s-0034-1374631. [DOI] [PubMed] [Google Scholar]
- 35.He XY, et al. Oxidative 3alpha-hydroxysteroid dehydrogenase activity of human type 10 17beta-hydroxysteroid dehydrogenase. J Steroid Biochem Mol Biol. 2003;87:191–198. doi: 10.1016/j.jsbmb.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 36.Ishizaki F, et al. Androgen deprivation promotes intratumoral synthesis of dihydrotestosterone from androgen metabolites in prostate cancer. Sci Rep. 2013;3:1528. doi: 10.1038/srep01528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paquet S, et al. Differential expression of the androgen-conjugating UGT2B15 and UGT2B17 enzymes in prostate tumor cells during cancer progression. J Clin Endocrinol Metab. 2012;97:E428–E432. doi: 10.1210/jc.2011-2064. [DOI] [PubMed] [Google Scholar]
- 38.Nishiyama T, Ikarashi T, Hashimoto Y, Wako K, Takahashi K. The change in the dihydrotestosterone level in the prostate before and after androgen deprivation therapy in connection with prostate cancer aggressiveness using the Gleason score. J Urol. 2007;178:1282–1288. doi: 10.1016/j.juro.2007.05.138. [DOI] [PubMed] [Google Scholar]
- 39.Fankhauser M, et al. Canonical androstenedione reduction is the predominant source of signaling androgens in hormone-refractory prostate cancer. Clin Cancer Res. 2014;20:5547–5557. doi: 10.1158/1078-0432.CCR-13-3483. [DOI] [PubMed] [Google Scholar]
- 40.Pfeiffer MJ, Smit FP, Sedelaar JP, Schalken JA. Steroidogenic enzymes and stem cell markers are upregulated during androgen deprivation in prostate cancer. Mol Med. 2011;17:657–664. doi: 10.2119/molmed.2010.00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chang KH, et al. Dihydrotestosterone synthesis bypasses testosterone to drive castration-resistant prostate cancer. Proc Natl Acad Sci U S A. 2011;108:13728–13733. doi: 10.1073/pnas.1107898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bennett NC, et al. Evidence for steroidogenic potential in human prostate cell lines and tissues. Am J Pathol. 2012;181:1078–1087. doi: 10.1016/j.ajpath.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 43.Jeong CW, et al. Limited expression of cytochrome p450 17alpha-hydroxylase/17,20-lyase in prostate cancer cell lines. Korean J Urol. 2011;52:494–497. doi: 10.4111/kju.2011.52.7.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kumagai J, et al. Intratumoral conversion of adrenal androgen precursors drives androgen receptor-activated cell growth in prostate cancer more potently than de novo steroidogenesis. Prostate. 2013 doi: 10.1002/pros.22655. [DOI] [PubMed] [Google Scholar]
- 45.Dillard PR, Lin MF, Khan SA. Androgen-independent prostate cancer cells acquire the complete steroidogenic potential of synthesizing testosterone from cholesterol. Mol Cell Endocrinol. 2008;295:115–120. doi: 10.1016/j.mce.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Locke JA, Wasan KM, Nelson CC, Guns ES, Leon CG. Androgen-mediated cholesterol metabolism in LNCaP and PC-3 cell lines is regulated through two different isoforms of acyl-coenzyme A:Cholesterol Acyltransferase (ACAT) Prostate. 2008;68:20–33. doi: 10.1002/pros.20674. [DOI] [PubMed] [Google Scholar]
- 47.Bao BY, et al. Androgen receptor mediates the expression of UDP-glucuronosyltransferase 2 B15 and B17 genes. Prostate. 2008;68:839–848. doi: 10.1002/pros.20749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grosse L, et al. Androgen glucuronidation: an unexpected target for androgen deprivation therapy, with prognosis and diagnostic implications. Cancer Res. 2013;73:6963–6971. doi: 10.1158/0008-5472.CAN-13-1462. [DOI] [PubMed] [Google Scholar]
- 49.Knuuttila M, et al. Castration induces up-regulation of intratumoral androgen biosynthesis and androgen receptor expression in an orthotopic VCaP human prostate cancer xenograft model. Am J Pathol. 2014;184:2163–2173. doi: 10.1016/j.ajpath.2014.04.010. [DOI] [PubMed] [Google Scholar]
- 50.Mostaghel EA, et al. Prostate cancer characteristics associated with response to pre-receptor targeting of the androgen axis. PLoS One. 2014;9:e111545. doi: 10.1371/journal.pone.0111545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mostaghel EA, et al. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clin Cancer Res. 2011;17:5913–5925. doi: 10.1158/1078-0432.CCR-11-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tenta R, et al. Microarray analysis of survival pathways in human PC-3 prostate cancer cells. Cancer Genomics Proteomics. 2007;4:309–318. [PubMed] [Google Scholar]
- 53.Belanger A, et al. Characterization and regulation of UDP-glucuronosyltransferases in steroid target tissues. J Steroid Biochem Mol Biol. 1998;65:301–310. doi: 10.1016/S0960-0760(97)00183-0. [DOI] [PubMed] [Google Scholar]
- 54.Levesque E, Beaulieu M, Guillemette C, Hum DW, Belanger A. Effect of interleukins on UGT2B15 and UGT2B17 steroid uridine diphosphate-glucuronosyltransferase expression and activity in the LNCaP cell line. Endocrinology. 1998;139:2375–2381. doi: 10.1210/endo.139.5.6001. [DOI] [PubMed] [Google Scholar]
- 55.Kaeding J, et al. Calcitrol (1alpha,25-dihydroxyvitamin D3) inhibits androgen glucuronidation in prostate cancer cells. Mol Cancer Ther. 2008;7:380–390. doi: 10.1158/1535-7163.MCT-07-0455. [DOI] [PubMed] [Google Scholar]
- 56.Agyeman AS, et al. Transcriptomic and proteomic profiling of KEAP1 disrupted and sulforaphane-treated human breast epithelial cells reveals common expression profiles. Breast Cancer Res Treat. 2012;132:175–187. doi: 10.1007/s10549-011-1536-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Amjad AI, et al. Broccoli-derived sulforaphane and chemoprevention of prostate cancer: from bench to bedside. Curr Pharmacol Rep. 2015;1:382–390. doi: 10.1007/s40495-015-0034-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chang KH, et al. A gain-of-function mutation in DHT synthesis in castration-resistant prostate cancer. Cell. 2013;154:1074–1084. doi: 10.1016/j.cell.2013.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Levesque E, et al. Importance of 5alpha-reductase gene polymorphisms on circulating and intraprostatic androgens in prostate cancer. Clin Cancer Res. 2014;20:576–584. doi: 10.1158/1078-0432.CCR-13-1100. [DOI] [PubMed] [Google Scholar]
- 60.Sissung TM, et al. Genetic variation: effect on prostate cancer. Biochim Biophys Acta. 2014;1846:446–456. doi: 10.1016/j.bbcan.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sun T, et al. The impact of common genetic variations in genes of the sex hormone metabolic pathways on steroid hormone levels and prostate cancer aggressiveness. Cancer Prev Res (Phila) 2011;4:2044–2050. doi: 10.1158/1940-6207.CAPR-11-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Belledant A, et al. The UGT2B28 sex-steroid inactivation pathway is a regulator of steroidogenesis and modifies the risk of prostate cancer progression. Eur Urol. 2015 doi: 10.1016/j.eururo.2015.06.054. [DOI] [PubMed] [Google Scholar]
- 63.Guillemette C, Levesque E, Harvey M, Bellemare J, Menard V. UGT genomic diversity: beyond gene duplication. Drug Metab Rev. 2010;42:24–44. doi: 10.3109/03602530903210682. [DOI] [PubMed] [Google Scholar]
- 64.Lampe JW, Bigler J, Bush AC, Potter JD. Prevalence of polymorphisms in the human UDP-glucuronosyltransferase 2B family: UGT2B4(D458E), UGT2B7(H268Y), and UGT2B15(D85Y) Cancer Epidemiol Biomark Prev a Publ Am Assoc Cancer Res Am Soc Prev Oncol. 2000;9:329–333. [PubMed] [Google Scholar]
- 65.Jakobsson J, et al. Large differences in testosterone excretion in Korean and Swedish men are strongly associated with a UDP-glucuronosyl transferase 2B17 polymorphism. J Clin Endocrinol Metabol. 2006;91:687–693. doi: 10.1210/jc.2005-1643. [DOI] [PubMed] [Google Scholar]
- 66.Olsson M, et al. Correlation between circulatory, local prostatic, and intra-prostatic androgen levels. Prostate. 2011;71:909–914. doi: 10.1002/pros.21307. [DOI] [PubMed] [Google Scholar]
- 67.Gauthier-Landry L, Belanger A, Barbier O. Multiple roles for UDP-glucuronosyltransferase (UGT)2B15 and UGT2B17 enzymes in androgen metabolism and prostate cancer evolution. J Steroid Biochem Mol Biol. 2015;145:187–192. doi: 10.1016/j.jsbmb.2014.05.009. [DOI] [PubMed] [Google Scholar]
- 68.Gallagher CJ, et al. The UGT2B17 gene deletion polymorphism and risk of prostate cancer. A case–control study in Caucasians. Cancer Detect Prev. 2007;31:310–315. doi: 10.1016/j.cdp.2007.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Olsson M, et al. The UGT2B17 gene deletion is not associated with prostate cancer risk. Prostate. 2008;68:571–575. doi: 10.1002/pros.20700. [DOI] [PubMed] [Google Scholar]
- 70.Setlur SR, et al. Genetic variation of genes involved in dihydrotestosterone metabolism and the risk of prostate cancer. Cancer Epidemiol Biomark Prev: Publ Am Assoc Cancer Res Am Soc Prev Oncol. 2010;19:229–239. doi: 10.1158/1055-9965.EPI-09-1018. [DOI] [PubMed] [Google Scholar]
- 71.Nadeau G, et al. Deletions of the androgen-metabolizing UGT2B genes have an effect on circulating steroid levels and biochemical recurrence after radical prostatectomy in localized prostate cancer. J Clin Endocrinol Metab. 2011;96:E1550–E1557. doi: 10.1210/jc.2011-1049. [DOI] [PubMed] [Google Scholar]
- 72.Park J, et al. Deletion polymorphism of UDP-glucuronosyltransferase 2B17 and risk of prostate cancer in African American and Caucasian men. Cancer Epidemiol Biomark Prev a Publ Am Assoc Cancer Res Am Soc Prev Oncol. 2006;15:1473–1478. doi: 10.1158/1055-9965.EPI-06-0141. [DOI] [PubMed] [Google Scholar]
- 73.Park JY, et al. Association between polymorphisms in HSD3B1 and UGT2B17 and prostate cancer risk. Urology. 2007;70:374–379. doi: 10.1016/j.urology.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 74.Karypidis AH, Olsson M, Andersson SO, Rane A, Ekstrom L. Deletion polymorphism of the UGT2B17 gene is associated with increased risk for prostate cancer and correlated to gene expression in the prostate. Pharmacogenomics J. 2008;8:147–151. doi: 10.1038/sj.tpj.6500449. [DOI] [PubMed] [Google Scholar]
- 75.Cai L, Huang W, Chou KC. Prostate cancer with variants in CYP17 and UGT2B17 genes: a meta-analysis. Protein Pept Lett. 2012;19:62–69. doi: 10.2174/092986612798472848. [DOI] [PubMed] [Google Scholar]
- 76.Kpoghomou MA, Soatiana JE, Kalembo FW, Bishwajit G, Sheng W. UGT2B17 polymorphism and risk of prostate cancer: a meta-analysis. ISRN Oncol. 2013;2013:465916. doi: 10.1155/2013/465916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Takahashi RH, Grigliatti TA, Reid RE, Riggs KW. The effect of allelic variation in aldo-keto reductase 1C2 on the in vitro metabolism of dihydrotestosterone. J Pharmacol Exp Ther. 2009;329:1032–1039. doi: 10.1124/jpet.109.150995. [DOI] [PubMed] [Google Scholar]
- 78.Jung KA, et al. Identification of aldo-keto reductases as NRF2-target marker genes in human cells. Toxicol Lett. 2013;218:39–49. doi: 10.1016/j.toxlet.2012.12.026. [DOI] [PubMed] [Google Scholar]
- 79.Burczynski ME, Sridhar GR, Palackal NT, Penning TM. The reactive oxygen species--and Michael acceptor-inducible human aldo-keto reductase AKR1C1 reduces the alpha, beta-unsaturated aldehyde 4-hydroxy-2-nonenal to 1,4-dihydroxy-2-nonene. J Biol Chem. 2001;276:2890–2897. doi: 10.1074/jbc.M006655200. [DOI] [PubMed] [Google Scholar]
- 80.Matsuura K, et al. Identification of a principal mRNA species for human 3alpha-hydroxysteroid dehydrogenase isoform (AKR1C3) that exhibits high prostaglandin D2 11-ketoreductase activity. J Biochem. 1998;124:940–946. doi: 10.1093/oxfordjournals.jbchem.a022211. [DOI] [PubMed] [Google Scholar]
- 81.Penning TM, Byrns MC. Steroid hormone transforming aldo-keto reductases and cancer. Ann N Y Acad Sci. 2009;1155:33–42. doi: 10.1111/j.1749-6632.2009.03700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Huang KH, et al. Overexpression of aldo-keto reductase 1C2 is associated with disease progression in patients with prostatic cancer. Histopathology. 2010;57:384–394. doi: 10.1111/j.1365-2559.2010.03647.x. [DOI] [PubMed] [Google Scholar]
- 83.Yepuru M, et al. Steroidogenic enzyme AKR1C3 is a novel androgen receptor-selective coactivator that promotes prostate cancer growth. Clin Cancer Res. 2013 doi: 10.1158/1078-0432.CCR-13-1151. [DOI] [PubMed] [Google Scholar]
- 84.Fan L, et al. The steroidogenic enzyme AKR1C3 regulates stability of the ubiquitin ligase Siah2 in prostate cancer cells. J Biol Chem. 2015;290:20865–20879. doi: 10.1074/jbc.M115.662155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Doig CL, Battaglia S, Khanim FL, Bunce CM, Campbell MJ. Knockdown of AKR1C3 exposes a potential epigenetic susceptibility in prostate cancer cells. J Steroid Biochem Mol Biol. 2016;155:47–55. doi: 10.1016/j.jsbmb.2015.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gruber M, et al. Overexpression of uridine diphospho glucuronosyltransferase 2B17 in high-risk chronic lymphocytic leukemia. Blood. 2013;121:1175–1183. doi: 10.1182/blood-2012-08-447359. [DOI] [PubMed] [Google Scholar]
- 87.Hirata H, et al. Function of UDP-glucuronosyltransferase 2B17 (UGT2B17) is involved in endometrial cancer. Carcinogenesis. 2010;31:1620–1626. doi: 10.1093/carcin/bgq124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu C, et al. Intracrine androgens and AKR1C3 activation confer resistance to enzalutamide in prostate cancer. Cancer Res. 2015;75:1413–1422. doi: 10.1158/0008-5472.CAN-14-3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tamae D, et al. The DHEA-sulfate depot following P450c17 inhibition supports the case for AKR1C3 inhibition in high risk localized and advanced castration resistant prostate cancer. Chem Biol Interact. 2015;234:332–338. doi: 10.1016/j.cbi.2014.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]