

Abstract
Epidemiological survey identified that the variant patatin-like phospholipase domain-containing protein 3 (PNPLA3) gene at I148M position exerts direct effect in promoting hepatocellular carcinoma (HCC) under extraneous oxidative stress by interaction with obesity. However, the mechanism is still unknown. HepG2 cells were overexpressed by transinfection of PNPLA3 with wild-type 148I (PNPLA3WT) and mutant 148M (PNPLA3I148M), respectively. Variation in metabolic indicators, hepatic steatosis, biological behaviors and signaling molecules related to cancer promotion was measured in hepatocytes using low-dose free fatty acid (FFA) exposure. Effect of PNPLA3I148M on xenograft biology and its interaction with dietary obesity were also evaluated in animal study. Cells overexpresssing PNPLA3I148M in low-dose FFA incubation showed more proliferation, migration, invasion, and less apoptosis (P<0.05). Low-dose FFA specifically activated JAK2/STAT3 phosphorylation of PNPLA3I148M cells via upregulation of interleukin-6. Animal study showed high-fat diet accelerated growth of xenografts derived from PNPLA3I148M cells incubated in low-dose FFA. In low oxidative stress, PNPLA3I148M initiated the hepatocyte malignant transformation through the activation of inflammation-mediated JAK/STAT pathway. Dietary obesity amplified the growth of tumor from PNPLA3I148M cells by interaction with local FFA incubation. Anti-inflammation and weight loss might be potential approaches for preventing HCC in high-risk population carrying PNPLA3 variant.
Keywords: PNPLA3, inflammation, obesity, hepatocellular carcinoma
Introduction
Hepatocellular carcinoma (HCC) is one of the most common malignancies worldwide. It occupies the sixth position in the worldwide incidence of cancer and the third leading cause of death due to all cancers [1]. Following the global increase in the incidence of obesity/diabetes and a simultaneous decrease in traditional hepatic contributors of carcinogenesis (viral infection, excessive alcohol intake, etc.), non-alcoholic fatty liver disease (NAFLD) and its related risk factors have been associated with the occurrence of HCC [2]. Epidemiologic evidence from the U.S demonstrates that NAFLD and progressive nonalcoholic steatohepatitis (NASH) have become the most common etiologies for HCC [3] and the fastest growing indication of liver transplantation in recipients with HCC [4].
Patatin-like phospholipase domain containing 3 (PNPLA3) is a gene that was at first identified using genome-wide association studies in 2008 with validated function on regulation of hepatic fat content [5]. Subsequent studies have confirmed that the missense variant I148M located in PNPLA3 (rs738409 C/G, causes isoleucine-methionine conversation) is comprehensively associated with chronic liver disease including NAFLD [6], alcoholic liver disease [7], hepatitis C virus infection [8], even HCC [9]. The PNPLA3 variant exerts prominent influence on the susceptibility to hepatocarcinogenesis, independent of liver damage and fibrosis [10]. Meanwhile, the effect of PNPLA3 variant is amplified in obese subjects and promotes the development of HCC through its interaction with obesity [11].
Prior mechanistic studies have mainly investigated the pathological role of PNPLA3 and coded adiponutrin (ADPN) on hepatic triglyceride content (HTGC) regulation via various aspects. Increased lysophosphatidic acid-acyltransferase activity [12], decreased activity on triacylglycerol hydrolyzation and remodeling in lipid droplets [13,14] was conferred to the variant PNPLA3 gene. These inner mechanisms might contribute more susceptibility on NAFLD by combination [10]. In case of PNPLA3 variant, positive genomic association with soluble intercellular adhesion molecule-1 as an inflammatory cytokine, and negative correlation with serum adiponectin as an anti-inflammatory cytokine showed potential direct link between ADPN and inflammations [15,16]. Scholars speculated that the carcinogenic effect of PNPLA3 might be due to the long-term, low-grade inflammation triggered by genetic variant [10]. However, experimental evidence about the function of PNPLA3 on processes from simple fat deposition to tumor genesis is still absent and worthy to be elucidated.
Among the inflammatory signaling pathways relevant to the development of tumors, the phosphorylation of Janus Kinase2/Signal transducer and activator of transcription (JAK2/STAT3), as an oncogenic transcription factor activated by interleukin-6 (IL-6), plays a key role in augmenting the pathogenisis of HCC [17,18]. The STAT3 molecule is responsible for the cell proliferation and anti-apoptosis activity of HCC cells [19]. Indeed, IL-6/JAK2/STAT3 is activated in fatty liver-associated inflammation [20], and obesity (accompanied by NASH) is demonstrated as a “bona fide” tumor promoter through this activation [18].
However, whether the PNPLA3 exert its effect in promoting the development of HCC via an inflammation mediator located on up-regulation of IL-6 and subsequent activation of JAK2/STAT3 is still unknown and worthy to be elucidated. Effect of PNPLA3 variant on systemic metabolic disorders and hepatic steatosis as potent confounders in this regard is unclear. As known, low-dose free fatty acids (FFAs) are recognized to promote the development of HCC in previous studies [21]. Overexpression but silence of PNPLA3 gene causes obvious changes in biological behavior and down-stream phenotypes in previous studies [22]. Therefore, overexpression models was constructed in HepG2 cell line (including wild type and variant on PNPLA3 gene, respectively), and a series of experiments were performed to investigate the effects of variant PNPLA3 on carcinogenesis of HCC in the circumstance of fat over accumulation after adjusting the potent metabolic confounders. In addition, interaction of effects of this genetic determinant with dietary obesity on hepatocarcinogenes is also discussed in xenografts from athymic nude mice.
Materials and methods
Study design and flow diagram
Study design and process are presented in Figure 1. Overall, two cellular models with overexpression of PNPLA3WT and PNPLA3I148M gene (cytosine [C]→guanine [G] substitution that changes isoleucine [I] to methionine [M] at codon 148) was conducted, taking cells only trans-infected by vector without target fragment as a control. Cells were incubated in FFA solution with specific concentration (0, 25, 50, and 100 uM) and periods (1, 2, and 3 days) and measured for steatosis, metabolic and liver damage indicators as potent confounders. Genes referred to inflammation signal pathway (IL-6, IL-1β, tumor necrosis factor [TNF-α]) expression and corresponding tumor biological variations (proliferation, migration, and apoptosis) were examined in supernate and cells, if possible. In addition, cells incubated in various FFA concentrations were inoculated into athymic nude mice treated with high-fat diet (HFD) and low-fat diet (LFD), respectively. Tumor was collected after 20 days of growth. Weight and size of xenografted tumor were measured to investigate the gene-environment interaction on tumorigenesis.
Figure 1.
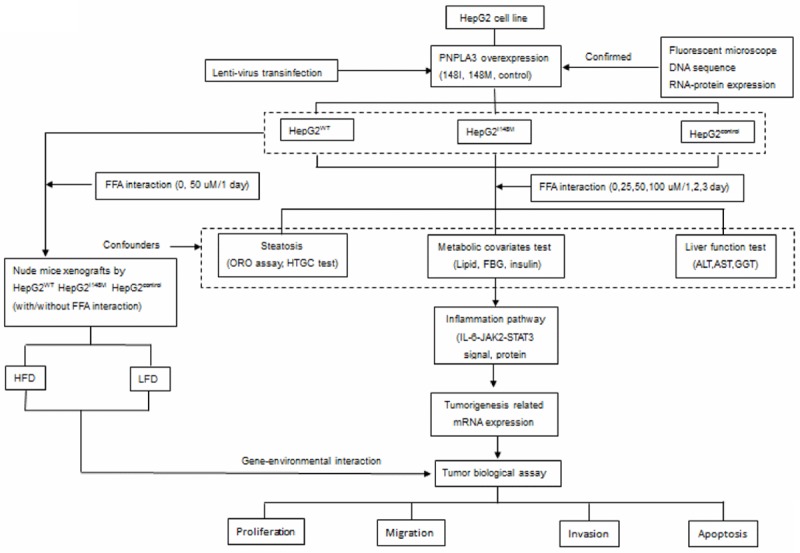
Flow chart of study program. ALT: alanine aminotransferase; AST: Aspartate aminotransferase; FBG: Fasting blood glucose; FFA: Free fatty acid; GGT: Gamma-glutamyl transpeptidase; HFD: High fat diet; HTGC: Hepatic triglyceride content; IL-6: Interleukin-6; JAK2: Janus Activating Kinase 2; LFD: Low fat diet; ORO: Oil red O; STAT3: signal transducer and activator of transcription 3.
Culture of HepG2 cell line
Immortalized HepG2 cell line (obtained from Chinese Typical Culture Collection Center, Shanghai) is sensitive to FFA stimulation [23] and selected for construction of hepatic cellular model of PNPLA3 overexpression. Routinely, HepG2 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Hyclone, Logan, UT), with 10% fetal bovine serum (FBS), 100 U/mL of penicillin, and 0.1 mg/mL of streptomycin. The cell lines were incubated in humidity at 5% CO2 and temperature at 37°C.
Construction of recombinant PNPLA3 148I wild type (PNPLA3WT) and PNPLA 148M mutant (PNPLA3I148M) vectors
Human 148I wild-type or 148M mutant PNPLA3 cDNA (GenBank ID: NM_025225) was synthesized and cloned into the NheI and AscI double enzyme restricted pLenti6.3_MCS_IRES2- EGFP vector containing blasticidin-resistance gene (Invitrogen, Life Technologies, Carlsbad, CA). The I148M substitution corresponding to G allele on rs738409 variant was introduced into the PNPLA3 cDNA sequence. Then recombinant lentiviral plasmids were generated in the supernate of 293T cell line by co-transfection with the constructed pLenti vector, packaging mix (including pLP1, pLP2, pLP/VSVG) and lipofectamine 2000 (ViraPower™; Invitrogen, Life Technologies) according to the manufacturer’s instructions, after 72 h culture in DMEM with 10% FBS. Meanwhile, lentiviral vectors without insertion of target gene clone (namely HepG2control) were also constructed and designed as control. Then, supernates containing lentiviral plasmid were collected and stored at -80°C until transinfection. Recombinant lentiviral vectors were validated using direct sequence after construction.
Cellular model construction by lentiviral vector transduction
HepG2 cells (5×104) were seeded onto 24-well plate (Corning Inc, Corning, NY) and cultured in 5% CO2 at 37°C for 1 day. When grown to 60%-70% confluency on the bottom, HepG2 cells were infected with lentivirus with multiplicity of infection (MOI) by gradient (2.5, 5, 10, 20, and 40) for 3 days. And the viral MOIs (ratios between lentivirus and target cells) were choosen as the optimal transduction index, with the most efficiency (based on the number of cells under fluorescence microscopy) and lowest cytotoxicity.
HepG2 cells were then transinfected at the optimal MOI of lentivirus in 24-well plates (4×104 per well) with common growth medium (DMEM containing 10% of FBS) at 10 mg/mL of polybrene (Sigma, St. Louis, Mo) for 24 h. Following 24 h incubation in 5% CO2 at 37°C, medium with lentivirus was removed and replaced by common medium. The transinfected cells were further cultured in common medium for 6 days and seeded onto six-well plate at a concentration of 1×105 per well. To construct cell line that stably express the PNPLA3 gene, the cells were treated with 4 ug/mL of blasticidin (Invitrogen, Life Technologies) after seeding onto six-well plate for 24 h. At 14 days of culture, cells not transducted by lentiviral vector were killed due to blasticidin. The effect of lentivirus on the expression of PNPLA3 of HepG2 was validated by the percentage of cells with green fluorescent protein (GFP), real time-polymerase chain reaction (RT-PCR), and western blot.
FFA induction of HepG2 cell line
According to prior rapid method on biological model of hepatic steatosis [23], oleate (O3880; Sigma) and palmitate (D9767; Sigma) were choosen at 1:1 ratio to prepare for FFA mixture. In general, the oleate and palmitate powders were dissolved in phosphate-buffered saline (PBS) and methanol, respectively for stock solution at 50 mM. The solution was diluted in DMEM containing 1% bovine serum albumin (BSA) to 1 mM of working solution after filtering through 0.22 um syringe filter (Millipore, Billerica, MA). After seeding onto culture plate for 24 h, working solution was added with pre-set FFA concentration by mixing common medium (without FFA) proportionately. Duration and dose of FFA exposure on HepG2 cells were determined according to pre-designed study protocols.
Qualitative and quantitative measurement of hepatic steatosis with oil red O staining
For detecting hepatic steatosis, HepG2 cells were stained with oil red O (ORO) kit (D027; Jiancheng Biotech, Nanjing) according to the manufacturer’s instructions. The degree of steatosis degree was observed and qualitatively assessed under light microscopes.
Moreover, steatosis was also quantitatively measured using methods referred in a previous study [24]. Specifically, HepG2 cells were stained in ORO dye for 15 min after growing up to 70% of confluency. Later, dye was removed and washed with PBS buffer solution. And isopropyl alcohol was added to extract ORO dye released into hepatocytes and transferred into a new plate. The degree of steatosis was assessed and compared using the value of optical density (OD) examined using ELX800 Universal Microplate Reader (BioTek, Winooski, VT) at a wavelength of 405 nm.
HTGC, as a quantative indicator of steatosis, was also measured using commercial kit (Applygen Technologies Inc, Beijing, China) according to the manufacturer’s instructions.
Biochemical indicator and inflammatory cytokine assay
The supernate of HepG2 cell was collected from culture medium after pre-treatment with FFA on pre-designed concentrations and duration of exposure. According to the manufacturer’s instructions, concentration of metabolic covariates (Triglyceride [TG], total cholesterol [TC], high-density lipoproteincholesterol [HDL-C], insulin), liver enzymes (alanine aminotransferase [ALT], aspartate aminotransferase [AST]) of supernate were determined using corresponding commercial kit (e-Bioscience, San Diego, CA). The IL-6 protein in supernate was also detected using enzyme-linked immunosorbent assay (ELISA) kit from another company (D6050; R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions.
RT-PCR
RNA was isolated from cells using TRIzol reagent (15596-026; Invitrogen, Waltham, MA), and the first-strand cDNA was synthesized using iScript cDNA Synthesis kit (1708891; Bio-Rad, Hercules, CA ). For normalization, the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) gene was set as an internal control. Quantitative RT-PCR was performed using SYBR Green in ABI 7500 system (Applied Biosystems, Carlsbad, CA). The primer sequences for PCR assay on individual target gene are listed in Table S1.
Western blot assay
Cells were washed using pre-cooled FBS twice and incubated in cell lysis buffer (Solabio, Shanghai, China) for 30 min. Lysates were centrifuged at 12000 rpm at 4°C for 15 min and the supernate was extracted as a mixture of total protein and examined using bicinchoninic acid assay kit (Thermo Scientific, Rockford, IL) for concentration test.
Equal protein samples mixed with loading buffer at 3:1 were loaded onto 10% Bis-Tris Gel (Invitrogen) for electrophoresis in sodium dodecyl sulfate running buffer (Invitrogen) at 120 V for 60 min. Protein was transferred to polyvinylidene fluoride (PVDF) membrane (Millipore) at 350 mA for 70 min. Membrane was blocked in Tris-buffered saline and Tween 20 (TBST) buffer (containing 3% of BSA) and shaken mildly for 60 min. PVDF membrane was taken out and incubated with primary anti-body (listed in Table S2) at 4°C overnight with mild shaking. Membrane was incubated in suitable second antibody (HRP-conjugated goat anti-rabbit antibody, Jackson ImmunoResearch, West Grove, USA) for 60 min, after routine clearance with TBST buffer (10 min×3). ECL substrate (Thermo Scientific) was used to develop the blots. Quantitative analysis of protein expression was performed by detecting the gray intensity of target band using the Image Lab software (Bio-Rad). GAPDH protein was selected as a reference in the western blot process for standardization.
Cell viability, proliferation, apoptosis, migration and invasion assay
Series of tumor biological assays were performed according to pre-designed study program and instructions recommended by the manufacturer. Cell viability was analyzed using Trypan blue dye exclusion assay on Vi-CELL™ XR Cell Viability Automatic Analyzer (Beckman Coulter, Brea, CA). Proliferative activity was detected using Cell Counting Kit (CCK-8) kit (Dojindo, Tokyo, Japan) after mixing for 1 h in the cell incubator. Proliferation was quantitatively assigned by the measurement of OD using microplate reader (ELX800; BioTek) at a wavelength of 405 nm. The extent of cell apoptosis stained using Annexin V-fluorescein isothiocyanate/propidium ioidide Apoptosis Kit (eBioscience) and detected on BD FACS Canto flow cytometer after 30 min incubation in dark at room temperature.
Cell migration and invasion were assayed using transwell system (Corning) with a pore size of 8 um. For migration, 5×104 cells at 85% confluency were plated on upper chamber in 100 uL FBS-free DMEM. And the lower compartment was filled with 500 uL DMEM containing 10% FBS with or without 50 uM FFA. After 1 day’s culture at 37°C, cells on the upper surface were wiped out and the migrated cells on the bottom of the membrane were fixed with 95% ethanol and dyed with crystal violet (0.3%) for 15 min. Later, stained cells were washed with PBS thrice and observed under microscope for counting to evaluate the cellular migration. Six foci were randomly selected for each sample, and the cells were counted at 200× field of microscope. The procedure for invasion test was similar to the migration assay. The only difference was that cells were seeded onto the membrane with pre-coated Matrigel (Corning).
Xenografts in nude mice and deoxynucleotidyl transferase dUTP nick end labeling assay for tumor tissue
According to the pre-designed study program, 6-week-old athymic BALB/c nude male mice (provided by Xipuer-BiKai Experimental Animal Company, Shanghai, China) were randomly selected for the experiment. In brief, 100 uL Matrigel containing pre-treated cells were injected into the armpits of each mice (5×106 cells per mouse), after measuring the weight at baseline. Later enrolled mice were categorized into two groups on average and fed with HFD and LFD, respectively, with different kcal (60% to 10%) of fat (D12492 and D12450B; Research Diets Inc, New Brunswick, NJ). The weight and size of xenografted tumor of the mice were measured and recorded every 2 days. Tumor-bearing mice were killed after 20 days of cell injection. Tumors were obtained, measured for weight and volume, formalin-fixed, and paraffin-embedded for the subsequent examinations. This study was approved by the Animal Ethics Committee of the First Affiliated Hospital of Zhejiang University.
Xenograft was fixed in 10% neutral formalin, embedded in paraffin, and sectioned at 6 μm to three slides per tissue for examination. Apoptosis of tumor was detected using Situ Cell Death Detection Kit (Roche, Mannheim, Germany), according to the manufacturer’s instructions. Slides were counterstained with hematoxylin and finally mounted for microscopic examination. Results of apoptosis were analyzed based on the observation under the light microscopy (200×). Specifically, the number of apoptotic cells (with obvious chromatin condensation, nucleic fragmentation presentation) and total cells, respectively, were counted under every five fields (200×) per slide. The apoptotic rate was calculated using the formula: apoptotic rate=(average apoptotic cells/total cells) ×100%.
Statistical analysis
Results were expressed as mean ± standard error. Comparisons of indicators between two subgroups (like comparison of steatosis, mRNA expression between cells with 0 uM and 50 uM of FFA intervention) were done using Student’s t test. While, comparison of values in three or more subgroups (like comparisons of steatosis, metabolic traits, relative mRNA/protein expression, cell proliferation, apoptosis, migration, invasion, tumor size from cells with different PNPLA3 expression at the same extent of FFA exposure) were performed by Student-Newman-Keuls’ (SNK) test in one-way ANOVA.
Statistical analyses were performed using SPSS, version 13.0 (IBM Corporation, Somers, NY). P-values were two-sided. P<0.05 was considered as significant difference, except for specific illustration.
Results
Construction and confirmation of Hep-G2 cellular model with overexpression of PNPLA3WT, PNPLA3I148M, and PNPLA3control
According to the manufacturer’s instruction, cellular models overexpressing PNPLA3WT, PNPLA3I148M, and PNPLA3control were built using transfection of corresponding lenti-virus (Figure 2). Almost all cells were observed to be tagged by GFP attached to lenti-viral vectors under fluorescent microscope, after 14 days of culture on pressure of blasticidin. Conversion on PNPLA3 rs738409 polymorphism was confirmed using direct Sanger’s sequencing of PCR products. Compared to PNPLA3control, PNPLA3WT and PNPLA3I148M had significantly higher expression on PNPLA3 and encoded ADPN (all P<0.05).
Figure 2.
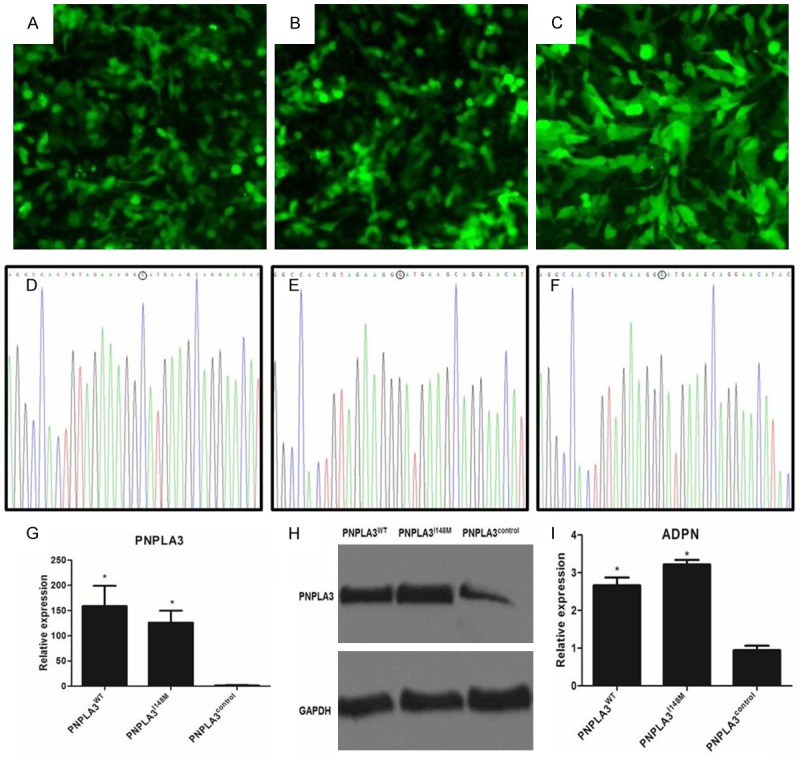
Construction of cellular models with different PNPLA3 overexpression (PNPLA3WT, PNPLA3I148M, and PNPLA3control). A-C. Respectively represented the green fluorescent protein expression under fluorescent microscope for PNPLA3WT, PNPLA3I148M, and PNPLA3control cells (magnification: 200×); D-F. Respectively showed the genotype of PNPLA3WT, PNPLA3I148M, and PNPLA3control detected by direct sequen (base composition in “O” represented the variant genotype in PNPLA3I148M position); G. Showed quantitative analysis of PNPLA3 expression in cells with different PNPLA3 trans-infection; H. Showed results of western blot assay for PNPLA3 expression in PNPLA3WT, PNPLA3I148M, and PNPLA3control cells; I. Showed quantitative analysis of relative expression for PNPLA3 encoded ADPN in PNPLA3WT, PNPLA3I148M, and PNPLA3control cells. Relative ADPN expression in western blot assay was expressed as the ratio of ADPN to GAPDH as internal reference. *Represented significant difference across cells with different PNPLA3 expression in the same extent of FFA exposure (P<0.05); Data was expressed as means ± standard error of three independent experiments. ADPN: adiponutrin.
Variation in metabolic profiles across cellular models in different dose periods of FFA exposure
Indicators of hepatic injury (ALT, AST), lipid (TG, TC, HDL-C) and insulin concentration were measured in culture supernate (Figure 3). Overall, variation in micro-environment in different models was consistent. No significant difference was observed across cellular models on the same intensity of FFA exposure, except for TG concentration (P<0.05) using paired t-test. It is noteworthy that a significant effect of FFA exposure on inter-group supernate variation in TG was mainly attributed to the long-term exposure of high FFA concentration(duration≥4 days, concentration≥100 uM). And the difference in TG between PNPLA3WT and PNPLA3I148M turned to be insignificant (P=0.10, data not shown) when excluding data on above-mentioned FFA circumstance.
Figure 3.

FFA induced variation on metabolic profiles in supernates across cell models with different PNPLA3 expression. Indicators were measured in 0, 25, 50, 100, 200 uM of FFA exposure respectively at 1, 2, 3, 4 days. A. Represented the ALT variation in PNPLA3WT, PNPLA3I148M and PNPLA3control cells; B. Represented the AST variation in PNPLA3WT, PNPLA3I148M and PNPLA3control cells; C. Represented the TG variation in PNPLA3WT, PNPLA3I148M and PNPLA3control cells; D. Represented the TC variation in PNPLA3WT, PNPLA3I148M and PNPLA3control cells; E. Represented the HDL-C variation in PNPLA3WT, PNPLA3I148M and PNPLA3control cells; F. Represented the insulin variation in PNPLA3WT, PNPLA3I148M and PNPLA3control cells. *Represented significant difference across cells with different PNPLA3 expression in the same extent of FFA intervention (P<0.05); Results are expressed as means ± standard error of five independent experiments. ALT: Alanine aminotransferase; AST: Aspartate aminotransferase; FFA: Free fatty acid; HDL-C: High-density lipoproteincholesterol; TC: Total cholesterol; TG: Triglyceride.
Measurement of HTGC and degree of steatosis
Figure 4 shows the doseperiod related variation in HTGC and steatosis of hepatocytes through the exposure of FFA. When exposed to oxidative stress with high FFA concentration (≥100 uM), HepG2 cells overexpressing variant PNPLA3I148M were more susceptible to higher accumulation of HTGC and severe steatosis than PNPLA3WT and PNPLA3control. However, this increasing trend was unstable, and inter-group difference was insignificant when treated with a medium with relatively lower FFA concentration (<100 uM). When cells were exposed to different FFA concentrations for 1 day (Figure 4C), colorimetric quantitative assay revealed no significant difference in the degree of steatosis for PNPLA3WT and PNPLA3I148M on exposure to low-dose FFA (≤50 uM ). The results were confirmed using conventional ORO staining methods, comparing the images under fluorescence microscopy (Figure 4D). These data indicated that FFA has steatogenic effect on hepatocytes, but low-dose FFA did not cause significant differentiation on steatosis of cells in spite of different expression of PNPLA3.
Figure 4.

Dose-period effect of FFA exposure on accumulation of HTGC and steatosis of hepatocytes with different expression of PNPLA3. A. Represented HTGC accumulation of hepatocytes (PNPLA3WT, PNPLA3I148M and PNPLA3control) treated in different dose-period FFA exposure (25 uM, 50 uM, 100 uM, 200 uM in 1 day, 2 days, 3 days and 4 days); B. Showed steatosis degree of hepatocytes (PNPLA3WT, PNPLA3I148M and PNPLA3control) treated in different dose-period FFA exposure (25 uM, 50 uM, 100 uM, 200 uM in 1 day, 2 days, 3 days and 4 days); C. Showed comparison of steatosis degree for hepatocytes (PNPLA3WT, PNPLA3I148M and PNPLA3control) incubated in different FFA concentration for 1 day; D. Showed ORO staining for hepatocytes (PNPLA3WT, PNPLA3I148M and PNPLA3control) in medium with 0/50 uM of FFA incubation under light microscope (magnification: 200×); E. Showed quantitative analysis on steatosis of hepatocytes (PNPLA3WT, PNPLA3I148M and PNPLA3control) by integral optical density. *Represented significant difference across cells with different PNPLA3 expression in the same extent of FFA exposure (P<0.05); Results are expressed as means ± standard error of five independent experiments. FFA: Free fatty acid; HTGC: Hepatic triglyceride content; ORO: Oil red O.
Overexpression of variant PNPLA3 affects the viability, proliferation, apoptosis, and invasiveness of HepG2 cells mediated in low-dose FFA exposure
Effect of overexpression of PNPLA3 on the biological behavior of tumor was evaluated in respective cellular models incubated with FFA for 24 h (Figure 5). The viability was similar for cell models when incubated in low-dose FFA (≤50 uM), but prominently decreased in the medium with higher FFA concentration (≥200 uM). The results of biological assay on the tumorigenesis across the cell models were confirmed and compared to the medium with lower FFA concentration (≤100 uM) for analogous cell viability. Subsequently, the results of the CCK-8 assay found obviously higher proliferation in PNPLA3I148M than PNPLA3WT and control cells when treated with low dose FFA. Notably, proliferation of PNPLA3I148M was activated in FFA medium with concentration <50 uM. The slower proliferation in 100 uM FFA might be contributed for more cell deaths (P=0.07, at borderline significance) and severe steatosis (Figure 4C).
Figure 5.

Variation in tumor-related biological behavior modulated by PNPLA3 expression in low-dose FFA exposure. A. Presented viability for hepatocytes (PNPLA3WT, PNPLA3I148M and PNPLA3control) treated in different FFA exposure (25 uM, 50 uM, 100 uM, 200 uM) for 1 day; B. Presented proliferation for hepatocytes (PNPLA3WT, PNPLA3I148M and PNPLA3control) treated in different FFA exposure (0 uM, 25 uM, 50 uM, and 100 uM) for 1 day; C. Showed hepatocytes (PNPLA3WT, PNPLA3I148M and PNPLA3control) were treated with 0/50 uM of FFA exposure for 1 day, then stained with Annexin V-APC/PI and measured by flow cytometry; D. Showed quantitative comparison of apoptotic rate for hepatocytes (PNPLA3WT, PNPLA3I148M and PNPLA3control) incubated in different FFA exposure (0 uM, 25 uM, 50 uM, and 100 uM) for 1 day; E. Presented cell migration assay for hepatocytes (PNPLA3WT, PNPLA3I148M and PNPLA3control) incubated in different FFA exposure (0 uM, 50 uM) for 1 day; F. Showed quantitative comparison of migration for hepatocytes (PNPLA3WT, PNPLA3I148M and PNPLA3control) incubated in different FFA exposure (0 uM, 50 uM) for 1 day; G. Presented cell invasion assay for hepatocytes (PNPLA3WT, PNPLA3I148M and PNPLA3control) incubated in different FFA exposure (0 uM, 50 uM) for 1 day; H. Showed quantitative comparison of invasion for hepatocytes (PNPLA3WT, PNPLA3I148M and PNPLA3control) incubated in different FFA exposure (0 uM, 50 uM) for 1 day. *Represented significant inter-group difference incubated in the same FFA exposure (P<0.05); #Represented significant difference for the same cells incubated in different FFA exposure (P<0.05); Results are means ± standard error of five independent experiments. FFA: Free fatty acid.
Low-dose FFA exposure caused variation in apoptotic status across cellular models. In case of PNPLA3WT and blank cells, no significant variation in apoptosis was observed due to low-dose FFA exposure. In case of PNPLA3I148M cells, FFA (≤50 uM) stimulation caused less apoptosis in a dose-response manner (R2=-0.894, P<0.001). However, inter-group difference cannot be observed in case of elevated FFA exposure levels (≥100 uM). The result seemed to contradict pro-apoptotic effect of PNPLA3I148M variant detected in patients with NAFLD, based on a previous study [25]. It also suggested the dual effect of PNPLA3 variant on apoptosis through its interaction with different concentrations of FFA. Low-dose FFA exposure might be considered as a carcinogen leading to less apoptosis of PNPLA3I148M cells by activating tumorigenesis pathway accompanied with higher hepatocyte proliferation. When concentration of FFA is elevated, more lipids are stored in PNPLA3I148M hepatocytes and toxicity occurs by inducing more cell damage and apoptosis.
Further, cell migration and invasion assays revealed increasing number of PNPLA3I148M hepatocytes invading the membrane than other cell models incubated in 50 uM FFA. This result indicated that PNPLA3I148M exerts its effect on accelerating cell migration and invasion ability due to low level of oxidative stress.
Taken together, overexpression of PNPLA3 per se did not influence the biological behavior of hepatocytes on tumorigenesis. Overexpression of variant PNPLA3 was shown to promote more progressive development of HCC in low-dose FFA environment. However, this function is not present in hepatocytes overexpressing wild-type PNPLA3 gene.
Overexpression of variant PNPLA3 gene activated the phosphorylation of JAK2/STAT3 signaling pathway via IL-6 up-regulation
Furthermore, the effect of overexpression of variant PNPLA3 on IL-6- dependent JAK/STAT3 signaling activation was investigated in respective cellular models treated with specific FFA concentration for 24 h (Figure 6). Compared to cells in the FFA-free medium, low-dose FFA caused higher expression of PNPLA3 on mRNA level in spite of cells with overexpression of PNPLA3WT or PNPLA3I148M. However, the effect of FFA on the expression of PNPLA3 was not present in regulating the PNPLA3-encoded ANPN protein levels. The ADPN protein levels in respective cells (PNPLA3WT, PNPLA3I148M and PNPLA3control) did not change significantly at 50 uM FFA concentration (P>0.05).
Figure 6.

Variant PNPLA3 overexpression induced activation of JAK2/STAT3 phosphorylation via IL-6 up-regulation. A. Showed PNPLA3 expression of hepatocytes (PNPLA3WT and PNPLA3I148M) treated by 0 uM and 50 uM of FFA for 1 day; B. Showed the quantitative IL-6 mRNA expression in hepatocytes with different PNPLA3 expression (PNPLA3WT, PNPLA3I148M and PNPLA3control) by different FFA intervention (0 uM, 25 Um, 50 Um, and 100 uM) for 1 day; C. Showed concentration of IL-6 protein in hepatocytes with different PNPLA3 expression (PNPLA3WT, PNPLA3I148M and PNPLA3control), supernate of cells were assayed by ELISA kit after treatment by different FFA exposure (0 uM, 25 Um, 50 Um, and 100 uM) for 1 day; D. Presented results of western blot assay for PNPLA3, total JAK2, phosphorylated JAK2, total STATA3, phosphorylated STAT3 at serine 727 and tyrosine 705, and GAPDH protein in hepatocytes with different PNPLA3 expression (PNPLA3WT, PNPLA3I148M, and PNPLA3control) treated by 0 uM and 50 uM of FFA intervention for 1 day; E. Showed quantitative comparison of relative expression for PNPLA3, total JAK2, phosphorylated JAK2, total STATA3, phosphorylated STAT3 protein at serine 727 and tyrosine 705 in hepatocytes with different PNPLA3 expression (PNPLA3WT, PNPLA3I148M, and PNPLA3control) treated by 0 uM and 50 uM of FFA intervention for 1 day, relative expression in western blot assay was evaluated by the ratio of target protein to GAPDH as internal reference. Data are expressed as means ± standard error of three independent experiments. aRepresented STAT3 protein phosphorylated at serine 727; bRepresented STAT3 protein phosphorylated at tyrosine 705; *Represented significant difference in hepatocytes (PNPLA3WT, PNPLA3I148M and PNPLA3control) by the same FFA intervention (P<0.05); #Represented significant difference for the same cells incubated in different FFA exposure (P<0.05). ELISA: Enzyme-linked immunoabsorbent assay; FFA: Free fatty acid; IL-6: Interleukin-6; JAK2: Janus Activating Kinase 2; STAT3: Signal transducer and activator of transcription.
ELISA assay revealed significant difference in IL-6 protein levels across hepatocytes (PNPLA3WT, PNPLA3I148M, and PNPLA3control) when treated with low-dose FFA concentration. The inter-cellular difference in IL-6 protein levels was not obvious (P>0.05) in FFA free medium. But the IL-6 protein level was up-regulated in PNPLA3I148M cells at low-dose FFA concentration (P<0.05). And this trend was not observed in PNPLA3WT and PNPLA3control cells. Consistent with the results of ELISA, overexpression of PNPLA3I148M also resulted in higher extent of elevations in IL-6 elevation than PNPLA3WT and PNPLA3control on mRNA level when treated with the same FFA concentration (P<0.05). Higher expression of IL-6 indicated severe inflammatory status of hepatocytes caused by the overexpression of variant PNPLA3 when exposed to constant external oxidative stress.
IL-6 is a pleiotropic cytokine with crucial function in mediating the cancer-related pathway, especially in activating JAK2/STAT3 signaling [26]. Therefore, total and phosphorylated protein expression of inflammation-related down-stream signaling molecules on JAK2-STAT3 pathway was assayed based on the variations in IL-6 across hepatocytes with lower FFA exposure (≤50 uM, Figure 4) with consistent oxidative stress.
The results of western-blot assay revealed that the overexpression of variant PNPLA3 caused substantial elevation in JAK2 and STAT3 phosphorylation in low dose FFA concentration (50 uM) than cells in medium without FFA exposure (P<0.05). Whereas, overexpression of wild-type PNPLA3 did not show this variation. For total protein of JAK2 and STAT3, the expression levels were unchanged in low-dose FFA incubation in spite of variation in phosphorylation (P>0.05). These results suggested that PNP-LA3I148M might promote the activation of the JAK2/STAT3 pathway related to tumorigenesis by up-regulating the secretion and release of IL-6 from hepatocytes.
It is noteworthy that these trends still remained even after adjusting for slight variation in the degree of steatosis and metabolic profiles in low-dose FFA exposure (data not shown), indicating that the carcinogenicity of variant PNPLA3 is independent of its activity on metabolic derangements.
Effect of modification in PNPLA3 on gene expression related to inflammation and tumorigenesis in low-dose FFA incubation
Biological behavior and malignant transformation of hepatocytes are mediated by gene expression in regulating hallmarks of cancer including cell cycle, proliferation, apoptosis, metastasis, invasion, angiogenesis, oxidative stress, and inflammation [27]. Therefore, the expression of genes related to neoplastic transformation of hepatocytes (PNPLA3WT, PNPLA3I148M, and PNPLA3control) was measured using different FFA concentration (50 uM/0 uM) to detect the mechanism of effect of variant PNPLA3 overexpression on tumorigenesis and progression.
As shown in Figure 7, no significant difference was found across hepatocytes in common medium. However, low-dose FFA mediated profound changes in carcinogenic gene expression by interaction with variant PNPLA3. After incubation in the medium with FFA for 24 h, oncogenes representing progression of cell cycle (Hsp70, cyclin D1), promotion of cell proliferation (TIMP1), invasion (MMP2, MMP9), angiogenesis (HIF-α, VEGF) were highly expressed in PNPLA3I148M cells. While the anti-oncogenes encoding inhibitor of cell cycle progression (P53, MAD1), suppressor of proliferation (TIMP2), and scavenger of free superoxide radicals (SOD1) were significantly down-regulated in PNPLA3I148M-overexpressed hepatocytes. In case of apoptosis, the Bax: Bcl-2 ratio was lower for lower expression of Bax as activator of apoptosis in PNPLA3I148M cells. With regard to pro-inflammatory cytokines, the change in expression of TNF-α and IL-1β did not follow the PNPLA3 variant in low-dose FFA exposure. The elevation in expression of TNF-α and IL-1β in PNPLA3I148M cell was not prominent and could not cannot be distinguished from PNPLA3WT and control cells.
Figure 7.
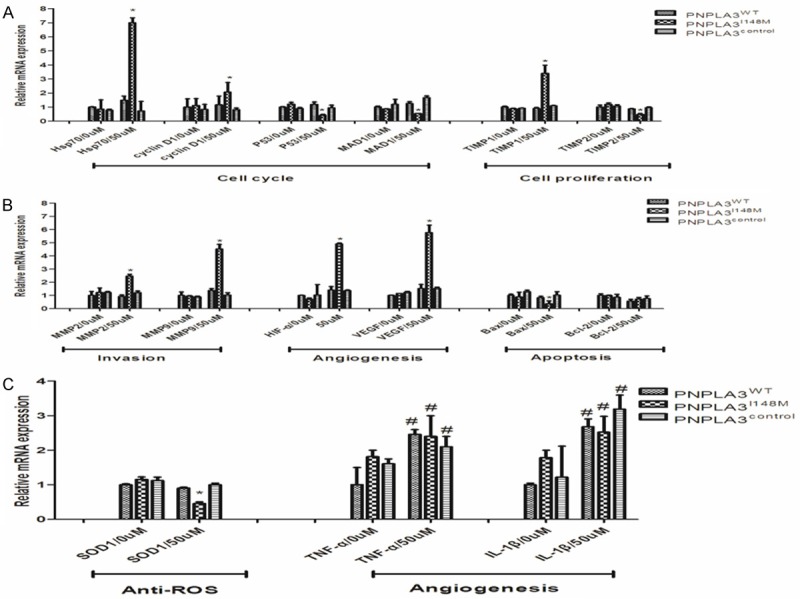
Up-regulation of variant PNPLA3 mediated gene expression involved in the progression of inflammation and tumor carcinogenesis by interaction with FFA exposure. Cells were treated by 0 uM and 50 uM of FFA exposure for 1 day and assayed using RT-PCR analysis. Quantative relative expression in real-time PCR assay was evaluated by the ratio of target mRNA expression to GAPDH as internal reference, gene expression of PNPLA3WT cells in FFA free medium was assigned as control. A. Showed expression of genes related to cell cycle and proliferation in hepatocytes (PNPLA3WT, PNPLA3I148M and PNPLA3control) by different FFA intervention (0 uM and 50 uM); B. Showed expression of genes related to cell invasion, angiogenesis and apoptosis in hepatocytes (PNPLA3WT, PNPLA3I148M and PNPLA3control) with different FFA incubation (0 uM and 50 uM); C. Showed expression of genes related to anti-ROS and pro-inflammatory cytokine in hepatocytes (PNPLA3WT, PNPLA3I148M and PNPLA3control with different FFA incubation (0 uM and 50 uM). Results were expressed as means ± standard error of five independent experiments. *Represented significant difference across cells with different PNPLA3 expression in the same FFA exposure (P<0.05); #Represented significant difference for gene expression between cells in with/without FFA treatment (P<0.05). FFA: Free fatty acid; ROS: Reactive oxygen species.
Taken together, these results indicated that the variant PNPLA3I148M has a comprehensive effect in mediating the expression of genes with regard to inflammation-related transformation of hepatocyte and tumor development in low-dose FFA exposure.
PNPLA3I148M amplified the carcinogenic effect of FFA on BALB/c nude mice xenografts by interaction with dietary obesity in vivo
Effect of overexpression of PNPLA3I148M on biological behavior of hepatocytes was evaluated by subcutaneous inoculation of cells in athymic BALB/c nude mice, as a more complex microenvironment. The tumor from PNPLA3control incubated in FFA-free medium was assigned as a control. Mice were randomly selected for the experiment. No significant inter-group difference in the body weight of mice was observed when enrolled for inoculation (data not shown). HFD induces higher increase in body weight than LFD-treated mice when killed at 20 days (P>0.05). About 1.65 g increase in weight gain was observed in HFD-fed mice than LFD-fed mice (equal to 6.9% of LFD-fed mice, P<0.05; Figure 8A).
Figure 8.

Overexpression of variant PNPLA3 gene caused significant variation in biological behavior of BALB/c nude mice xenograft in low-dose FFA exposure by interaction with dietary obesity. A. Showed body weight of nude mice treated by HFD and LFD respectively for 20 days; B. Presented xenograft of nude mice derived from hepatocytes (PNPLA3WT, PNPLA3I148M and PNPLA3control) categorized by FFA exposure (0 uM, 50 uM) and mice dietary (HFD, LFD); C. Showed tumor weight of xenograft in nude mice derived from hepatocytes (PNPLA3WT, PNPLA3I148M and PNPLA3control) categorized by FFA exposure (0 uM, 50 uM) and mice dietary (HFD, LFD); D. Showed growth of xenograft derived from hepatocytes (PNPLA3WT, PNPLA3I148M and PNPLA3control) categorized by FFA exposure (0 uM, 50 uM) in LFD fed mice; E. Showed growth of xenograft derived from hepatocytes (PNPLA3WT, PNPLA3I148M and PNPLA3control) categorized by FFA exposure (0 uM, 50 uM) in HFD fed mice; F. Presented apoptotic expression under light microscope (magnification: 200×) for xenografts derived from hepatocytes (PNPLA3WT, PNPLA3I148M and PNPLA3control) with different FFA exposure (0 uM, 50 uM) in BALB/c nude mice categorized by dietary (HFD, LFD); G. Showed the quantitative comparison of apoptosis for xenografts derived from hepatocytes (PNPLA3WT, PNPLA3I148M and PNPLA3control) with different FFA exposure (0 uM, 50 uM) in nude mice categorized by different dietary pattern (HFD, LFD). Results were expressed as means ± standard error of five independent experiments. *Represented significant difference on biological behavior derived from hepatocytes (PNPLA3WT, PNPLA3I148M and PNPLA3control) in the same FFA exposure. FFA: Free fatty acid; HFD: High fat diet; LFD: Low fat diet.
In the LFD group, no prominent variation was observed with regard to tumor size (including volume/weight), derived from distinct cells without treatment with FFA. However, tumor from PNPLA3I148M presented higher growth rate in vivo when treated with low-dose FFA (50 uM). In subsequent necropsy, larger tumor generated in mice inoculated with FFA-treated PNPLA3I148M cells than control (2.35 ± 0.61 g vs. 1.48 ± 0.20 g, respectively) indicated higher proliferation and tumorigenicity in variant PNPLA3 overexpression.
Apoptosis of xenograft was measured using terminal deoxynucleotidyl transferase dUTP nick-end labeling assay. As shown in Figure 8, the difference was less (P>0.05) with regard to apoptotic rate of tumors generated from diverse cells (PNPLA3WT, PNPLA3I148M, or PNPLA3control) untreated with low dose FFA. In case of cells in FFA environment, the apoptotic cells significantly decreased (about 45%) in tumors derived from PNPLA3I148M cells than the control, while the apoptosis for xenografts derived from PNPLA3WT and PNPLA3control was unchanged. Less apoptosis in tumors from FFA-incubated cells with overexpressed PNPLA3I148M might result in higher proliferation and larger size of xenografts.
It is of interest to understand the role of HFD on the growth of xenograft and apoptosis. Consistent with prior study [28], consumption of HFD also promoted progression of xenografts. When categorized by the type of cells for inoculation, higher growth of tumors was found in case of PNPLA3I148M cells incubated in low-dose FFA. Compared to the LFD-fed mice, about 34% of increase in tumor volume was observed in the HFD group treated with FFA-incubated PNPLA3I148M cells. With regard to apoptosis, obesity up-regulated the apoptitic level, which is considered as a contributor of compensatory tumor progression in proliferation [29]. Low dose FFA (50 uM) did not cause significant difference in apoptotic rate in case of PNPLA3I148M-induced xenografts than wild-type or blank cells (Figure 8G, P>0.05). Apoptotic resistance of PNPLA3I148M might be covered by the intake of HFD. These results indicated that systemic obesity might amplify the effect of cells with overexpressed PNPLA3I148M xenografts in low-dose FFA exposure with consistent apoptotic status.
Discussion
PNPLA3I148M (rs738409) is a common genetic variation associated with hepatic steatosis and metabolic derragements [6,30]. Recently, accumulative evidences have shown the independent predisposition to PNPLA3I148M polymorphism on the incidence of HCC. PNPLA3I148M variant is indicative of severe fibrosis, poor prognosis in patients with HCC despite the primary etiologies [9,31]. Dietary obesity plays a positive role in amplifying the genetic susceptibility to the occurrence of HCC in patients carrying variant on PNPLA3I148M polymorphism [11]. A previous study has identified that free fatty acids have oncogenic effect and causes malignant transformation in hepatocytes [21]. However, the function of variant PNPLA3 gene on mediation of genesis of HCC and progression in FFA environment is unknown. Otherwise, the effect of systemic metabolic disorders and hepatic steatosis caused by FFA exposure on PNPLA3-HCC association is also worthy to be elucidated. In HepG2 cellular models with similar wild/variant type of PNPLA3 overexpression (CC/GG distinction on rs738409 polymorphism, respectively), it was found that overexpression of PNPLA3I148M significantly deteriorated the biological behavior of hepatocytes through the interaction with low-dose FFA treatment via up-regulation of IL-6 and subsequent phosphorylation of JAK2/STAT3. Dietary obesity specifically promoted the growth of xenograft derived from hepatocytes with overexpression of PNPLA3I148M.
The PNPLA3 gene plays a role in hydrolyzing hepatic triacylglycerols, and I148M substitution inducs a loss of this function by blocking the integration between substrate and catalytic site [13]. Previous studies have shown that the knock-in of PNPLA3I148M variant resulted in differentiating steatosis than wild type in transgenic mice with exogenous oxidative stress (induced by high sucrose diets) [32]. Thus, it was hypothesized that the difference across cells with overexpression of PNPLA3 might be trigged by specific oxidative stress threshold and fatty acid was chosen as a common metabolic-related carcinogens [21] to stimulate the hepatocytes. Consistent with this hypothesis, insignificant difference was found on the severity of steatosis in cells with different overexpression of PNPLA3 (wild type, variant and control) on low-dose FFA treatment (Figure 4). In addition, metabolic indicators in cultural medium extracted from different cells (PNPLA3WT, PNPLA3I148M, and PNPLA3control) were unchanged on low dose FFA exposure (Figure 3). These results indicated that incubation with low-dose FFA caused similar steatosis and metabolic status independent of the expression of PNPLA3.
Effect of FFA exposure on the expression of PNPLA3 is not fully elucidated. Previous study revealed that PNPLA3 mRNA expression was up-regulated in western-type diet-fed mice with no distinction in I148M polymorphism, but the ADPN protein level was not compared [33]. In the present study, it was found that the ADPN protein level remained same in hepatocytes with different overexpression of PNPLA3 (wild type or variant) before/after FFA exposure, in spite of mRNA elevation. The reason for unchanged ADPN level in case of FFA exposure might be due to the overexpression of PNPLA3 per se and low extent of oxidative stress under low-dose FFA exposure.
The effect of PNPLA3I148M variant on malignance transformation of hepatocytes in FFA medium is still unknown. Low-dose fatty acid was previously reported to promote cancer [21,34]. In the present study, overexpressed PNPLA3I148M was found to increase tumor malignancy by interaction with FFA (Figure 5). Low dose FFA incubation caused higher proliferation, migration, and invasion in PNPLA3I148M-overexpressed hepatocytes. In case of PNPLA3WT and control cells, the difference in using FFA was insignificant on biological behavior (P>0.05). Differential effects of fatty acid on apoptosis were reported in a previous study [35]. The results of the present study showed less apoptosis in PNPLA3I148M cells in low-dose FFA medium, but this phenomenon does not occur in the medium with elevated FFA concentration. The reduction in quantitative apoptotic rate might be due to the response to excessive cell proliferation in PNPLA3I148M cells. The results indicate that FFA might cause transformation of hepatocytes at specific threshold. Consistent with a previous study [36], variant PNPLA3 increased the sensitivity of hepatocytes to induce severe tumor progression on exposure to FFA-related carcinogenesis. It is noteworthy that tumorgenesis effect of PNPLA3I148M is independent of FFA-related steatosis, indicating this variant might initiate this process on carcinogenesis before accelerated accumulation of fat, which is consistent with an epidemiological survey [37]. Otherwise, similar PNPLA3 protein levels (for wild/variant type) on FFA exposure suggest promotion of cancer from PNPLA3I148M that might contribut to the polymorphism itself but not the expression.
PNPLA3I148M variant is a risk factor for the occurrence and progression of HCC, independent of steatosis and metabolic derangement, but the inner mechanism is still unknown. Scholars speculated that the association between PNPLA3 variant and progression of HCC might be linked by its direct regulation on inflammation [10]. Corresponding to the correlation between variant PNPLA3 and inflammatory mediator [15], it was found that the variant PNPLA3 up-regulated the IL-6 level in hepatocytes with low-dose FFA exposure. IL-6 is a pleiotropic cytokine and its release further mediated the downstream JAK2/STAT3 phosphorylation. The IL-6/JAK2/STAT3 signaling pathway is demonstrated to build a link between obesity/NAFLD and hepatocarcinogenesis in a prior animal study [18]. Combined with the present data, the malignant transformation in cells with PNPLA3I148M overexpression might be due to its direct pro-inflammatory effect and subsequent activation of oncogenic JAK2/STAT3 pathway on lower extent of FFA exposure. Interestingly, other inflammatory cytokine genes (such as TNF-α, IL-1β) were also slightly elevated, but cannot be differentiated by the expression of PNPLA3 (wild/variant type). The result indicates that the mechanism of inflammation and tumor promotion in the variant PNPLA3 might be closer to the IL-6/JAK/STAT pathway.
Excessive endogenous fatty acid synthesis was considered to be associated with progressive tumor behavior [38]. But the effect of fatty acid on the growth of xenograft in vivo was not determined and this difference might be due to the components of fatty acid [38]. Synergistic effect of local FFA incubation with systemic obesity was also observed in the proliferation of xenograft derived from hepatocytes [21]. The results of the present study showed that the FFA (proportion oleate: palmitate=1:1) had an overall effect in promoting the malignancy of xenograft from cells with PNPLA3I148M overexpression, which is corresponding to the in vitro data. In case of HFD-fed mice, obesity amplified the effect of variant PNPLA3 gene on tumor proliferation with gene-environment interaction, consistent with the results of clinical epidemiological survey [11]. Obesity covered the resistant effect of variant PNPLA3 on apoptosis. These results suggest more complexity in the transformation of hepatocytes, and other signaling pathways might be involved in obese environment.
Taken together, these results showed that the variant PNPLA3 has carcinogenic effect by mediating the cell inflammation with low-dose exogenous oxidative stress. A further animal study also confirmed that the variant PNPLA3 exerted positive effect on tumor growth by interaction with systemic obesity. These results indicate that high-risk patients carrying this variant might be more sensitive to oxidative stress, and more attention should be paid on cancer prevention even without definite metabolic disorder or steatosis. Comprehensive measures including anti-inflammation and weight loss might be an effective therapeutic/preventive approach to ameliorate the effect exerted by high-risk variant on inflammation and subsequent tumor progression.
Acknowledgements
This work was supported by National S&T Major Project (No. 2016ZX10002020), China Postdoctoral Science Foundation Project (Grant No. 2015M570518), and Foundation for Innovative Research Groups of the National Natural Science Foundation of China (Grant No. 81421062). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.McGlynn KA, London WT. The global epidemiology of hepatocellular carcinoma: present and future. Clin Liver Dis. 2011;15:223–243. doi: 10.1016/j.cld.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sanyal A, Poklepovic A, Moyneur E, Barghout V. Population-based risk factors and resource utilization for HCC: US perspective. Curr Med Res Opin. 2010;26:2183–2191. doi: 10.1185/03007995.2010.506375. [DOI] [PubMed] [Google Scholar]
- 4.Wong RJ, Cheung R, Ahmed A. Nonalcoholic steatohepatitis is the most rapidly growing indication for liver transplantation in patients with hepatocellular carcinoma in the US. Hepatology. 2014;59:2188–2195. doi: 10.1002/hep.26986. [DOI] [PubMed] [Google Scholar]
- 5.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC, Hobbs HH. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nature genetics. 2008;40:1461–1465. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sookoian S, Pirola CJ. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology. 2011;53:1883–1894. doi: 10.1002/hep.24283. [DOI] [PubMed] [Google Scholar]
- 7.Salameh H, Raff E, Erwin A, Seth D, Nischalke HD, Falleti E, Burza MA, Leathert J, Romeo S, Molinaro A. PNPLA3 Gene Polymorphism Is Associated With Predisposition to and Severity of Alcoholic Liver Disease. The Am J Gastroenterol. 2015;110:846–56. doi: 10.1038/ajg.2015.137. [DOI] [PubMed] [Google Scholar]
- 8.Valenti L, Rumi M, Galmozzi E, Aghemo A, Del Menico B, De Nicola S, Dongiovanni P, Maggioni M, Fracanzani AL, Rametta R. Patatin-Like phospholipase domain-containing 3 I148M polymorphism, steatosis, and liver damage in chronic hepatitis C. Hepatology. 2011;53:791–799. doi: 10.1002/hep.24123. [DOI] [PubMed] [Google Scholar]
- 9.Trepo E, Nahon P, Bontempi G, Valenti L, Falleti E, Nischalke HD, Hamza S, Corradini SG, Burza MA, Guyot E. Association between the PNPLA3 (rs738409 C>G) variant and hepatocellular carcinoma: Evidence from a metaanalysis of individual participant data. Hepatology. 2014;59:2170–2177. doi: 10.1002/hep.26767. [DOI] [PubMed] [Google Scholar]
- 10.Valenti L, Dongiovanni P, Corradini SG, Burza MA, Romeo S. PNPLA3I148M variant and hepatocellular carcinoma: A common genetic variant for a rare disease. Dig Liver Dis. 2013;45:619–624. doi: 10.1016/j.dld.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 11.Burza MA, Pirazzi C, Maglio C, Sjöholm K, Mancina RM, Svensson PA, Jacobson P, Adiels M, Baroni MG, Borén J. PNPLA3I148M (rs738409) genetic variant is associated with hepatocellular carcinoma in obese individuals. Dig Liver Dis. 2012;44:1037–1041. doi: 10.1016/j.dld.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 12.Kumari M, Schoiswohl G, Chitraju C, Paar M, Cornaciu I, Rangrez AY, Wongsiriroj N, Nagy HM, Ivanova PT, Scott SA. Adiponutrin functions as a nutritionally regulated lysophosphatidic acid acyltransferase. Cell Metabolism. 2012;15:691–702. doi: 10.1016/j.cmet.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang Y, Cohen JC, Hobbs HH. Expression and characterization of a PNPLA3 protein isoform (I148M) associated with nonalcoholic fatty liver disease. J Biol Chem. 2011;286:37085–37093. doi: 10.1074/jbc.M111.290114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruhanen H, Perttilä J, Hölttä-Vuori M, Zhou Y, Yki-Järvinen H, Ikonen E, Käkelä R, Olkkonen VM. PNPLA3 mediates hepatocyte triacylglycerol remodeling. J Lipid Res. 2014;55:739–746. doi: 10.1194/jlr.M046607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paré G, Ridker PM, Rose L, Barbalic M, Dupuis J, Dehghan A, Bis JC, Benjamin EJ, Shiffman D, Parker AN. Genome-wide association analysis of soluble ICAM-1 concentration reveals novel associations at the NFKBIK, PNPLA3, RELA, and SH2B3 loci. PLoS Genet. 2011;7:e1001374. doi: 10.1371/journal.pgen.1001374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Valenti L, Rametta R, Ruscica M, Dongiovanni P, Steffani L, Motta BM, Canavesi E, Fracanzani AL, Mozzi E, Roviaro G. The I148M PNPLA3 polymorphism influences serum adiponectin in patients with fatty liver and healthy controls. BMC Gastroenterology. 2012;12:111. doi: 10.1186/1471-230X-12-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He G, Karin M. NF-κB and STAT3-key players in liver inflammation and cancer. Cell Research. 2011;21:159–168. doi: 10.1038/cr.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, Österreicher CH, Takahashi H, Karin M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140:197–208. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He G, Yu GY, Temkin V, Ogata H, Kuntzen C, Sakurai T, Sieghart W, Peck-Radosavljevic M, Leffert HL, Karin M. Hepatocyte IKKβ/NF-κB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell. 2010;17:286–297. doi: 10.1016/j.ccr.2009.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller AM, Wang H, Bertola A, Park O, Horiguchi N, Hwan Ki S, Yin S, Lafdil F, Gao B. Inflammation-associated interleukin-6/signal transducer and activator of transcription 3 activation ameliorates alcoholic and nonalcoholic fatty liver diseases in interleukin-10-deficient mice. Hepatology. 2011;54:846–856. doi: 10.1002/hep.24517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vinciguerra M, Carrozzino F, Peyrou M, Carlone S, Montesano R, Benelli R, Foti M. Unsaturated fatty acids promote hepatoma proliferation and progression through downregulation of the tumor suppressor PTEN. J Hepatol. 2009;50:1132–1141. doi: 10.1016/j.jhep.2009.01.027. [DOI] [PubMed] [Google Scholar]
- 22.Li JZ, Huang Y, Karaman R, Ivanova PT, Brown HA, Roddy T, Castro-Perez J, Cohen JC, Hobbs HH. Chronic overexpression of PNPLA3I148M in mouse liver causes hepatic steatosis. J Clin Invest. 2012;122:4130. doi: 10.1172/JCI65179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gomez-Lechon MJ, Donato MT, Martínez-Romero A, Jiménez N, Castell JV, O’Connor JE. A human hepatocellular in vitro model to investigate steatosis. Chem Biol Interact. 2007;165:106–116. doi: 10.1016/j.cbi.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 24.Cui W, Chen SL, Hu KQ. Quantification and mechanisms of oleic acid-induced steatosis in HepG2 cells. Am J Transl Res. 2010;2:95. [PMC free article] [PubMed] [Google Scholar]
- 25.Valenti L, Al-Serri A, Daly AK, Galmozzi E, Rametta R, Dongiovanni P, Nobili V, Mozzi E, Roviaro G, Vanni E. Homozygosity for the patatin-like phospholipase-3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology. 2010;51:1209–1217. doi: 10.1002/hep.23622. [DOI] [PubMed] [Google Scholar]
- 26.Hodge DR, Hurt EM, Farrar WL. The role of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer. 2005;41:2502–2512. doi: 10.1016/j.ejca.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 27.Mantovani A. Cancer: inflaming metastasis. Nature. 2009;457:36–37. doi: 10.1038/457036b. [DOI] [PubMed] [Google Scholar]
- 28.Tang FY, Pai MH, Chiang EP. Consumption of high-fat diet induces tumor progression and epithelial-mesenchymal transition of colorectal cancer in a mouse xenograft model. J Nutr Biochem. 2012;23:1302–1313. doi: 10.1016/j.jnutbio.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 29.Khandekar MJ, Cohen P, Spiegelman BM. Molecular mechanisms of cancer development in obesity. Nat Rev Cancer. 2011;11:886–895. doi: 10.1038/nrc3174. [DOI] [PubMed] [Google Scholar]
- 30.Mangge H, Baumgartner B, Zelzer S, Prüller F, Schnedl W, Reininghaus E, Haybaeck J, Lackner C, Stauber R, Aigner E. Patatin-like phospholipase 3 (rs738409) gene polymorphism is associated with increased liver enzymes in obese adolescents and metabolic syndrome in all ages. Aliment Pharmacol Ther. 2015;42:99–105. doi: 10.1111/apt.13232. [DOI] [PubMed] [Google Scholar]
- 31.Singal AG, Manjunath H, Yopp AC, Beg MS, Marrero JA, Gopal P, Waljee AK. The effect of PNPLA3 on fibrosis progression and development of hepatocellular carcinoma: a metaanalysis. Am J Gastroenterol. 2014;109:325–334. doi: 10.1038/ajg.2013.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smagris E, BasuRay S, Li J, Huang Y, Lai KM, Gromada J, Cohen JC, Hobbs HH. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology. 2015;61:108–118. doi: 10.1002/hep.27242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoekstra M, Li Z, Kruijt JK, Van Eck M, Van Berkel TJ, Kuiper J. The expression level of non-alcoholic fatty liver disease-related gene PNPLA3 in hepatocytes is highly influenced by hepatic lipid status. J Hepatol. 2010;52:244–251. doi: 10.1016/j.jhep.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 34.Nath A, Li I, Chan C. Elevated uptake of free fatty acids via CD36 promotes epithelialmesenchymal transition in hepatocellular carcinoma. Cancer Research. 2015;75:5157–5157. [Google Scholar]
- 35.Ricchi M, Odoardi MR, Carulli L, Anzivino C, Ballestri S, Pinetti A, Fantoni LI, Marra F, Bertolotti M, Banni S. Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. J Gastroenterol Hepatol. 2009;24:830–840. doi: 10.1111/j.1440-1746.2008.05733.x. [DOI] [PubMed] [Google Scholar]
- 36.Limagne E, Cottet V, Cotte AK, Hamza S, Hillon P, Latruffe N, Delmas D. Potential role of oxidative DNA damage in the impact of PNPLA3 variant (rs 738409 C>G) in hepatocellular carcinoma risk. Hepatology. 2014;60:1110–1111. doi: 10.1002/hep.27004. [DOI] [PubMed] [Google Scholar]
- 37.Liu YL, Patman G, Leathart J, Piguet AC, Burt A, Dufour JF, Day C, Daly A, Reeves H, Anstee Q. Carriage of the PNPLA3 rs738409 C>G polymorphism confers an increased risk of non-alcoholic fatty liver disease associated hepatocellular carcinoma. J Hepatol. 2014;61:75–81. doi: 10.1016/j.jhep.2014.02.030. [DOI] [PubMed] [Google Scholar]
- 38.Kuhajda FP. Fatty-acid synthase and human cancer: new perspectives on its role in tumor biology. Nutrition. 2000;16:202–208. doi: 10.1016/s0899-9007(99)00266-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.