

Abstract
EphA2 is associated with tumor growth and distant metastasis in numerous human tumors. Considering the controversial effects of EphA2 in different tumors and the lack of reports in salivary adenoid cystic carcinoma (SACC), we evaluated the effects of EphA2 inhibition by short hairpin RNA on SACC through in vivo and in vitro researches for the first time. Real-time reverse transcriptase-PCR and western blot analysis were conducted to verify the interference effect on SACC cells. Using Cell Counting Kit-8, wound healing, Transwell and Matrigel adhesion assays, we confirm that inhibition of EphA2 promotes the migration, invasion and adhesion ability of SACC cells. In vivo research, we prove that silencing of EphA2 significantly accelerates tumor growth and lung metastasis ability by establishing xenograft models in mice, including subcutaneous inoculation and tail vein injection. In addition, immunostaining of EphA2, E-cadherin and Slug from 40 specimens and in vitro simulation of perineural invasion (PNI) assay imply that suppression of EphA2 partially contribute to epithelial-mesenchymal transition and enhancement of PNI in SACC. In conclusion, all the data suggest that EphA2 may act as a tumor suppressor in SACC progression.
Keywords: Salivary adenoid cystic carcinoma, EphA2, RNA interference, tumor progression, perineural invasion
Introduction
Adenoid cystic carcinoma (ACC) is a highly malignant epithelial tumor that arises from the secretory cells of salivary glands. ACC comprises approximately 1% of head and neck tumors and 10% of all salivary neoplasms [1]. This malignant tumor possesses unique characteristics, such as remarkable morphologic heterogeneity, slow growth, indolent clinical course, multiple local recurrence, tendency for PNI, predilection for lethal distant metastasis to lungs, and poor long-term prognosis [2-5]. Considerable effort has been expended to improve the management of ACC patients, but the overall 5-, 10-, and 15-year survival rates are 73%, 45%, and 35%, respectively [6]. Therefore, investigation of the molecular mechanisms underlying the development and progression of ACC and identification of novel therapeutic targets are urgently needed.
Eph receptors belong to a large family of receptor tyrosine kinases and consist of EphA and EphB subgroups [7]. During embryonic development, the interaction of Eph receptors and ephrin ligands plays an important role in axonal guidance, spine morphogenesis, embryonic patterning, and angiogenesis [8,9]. Emerging evidences have also recently implicated Eph family proteins in the formation and progression in different types of cancers, such as breast carcinoma, melanoma, oral squamous cell carcinoma, and prostate carcinoma [10-12]. However, the functional roles of Eph receptors remain controversial and unclear. Moreover, relatively little is known about the correlation of EphA2 with ACC.
Considering the clinical characteristics of PNI and lung metastasis in ACC and the role of Eph receptors in both neural and vascular development [9], we hypothesized that Eph/ephrin may be involved in the development and progression of ACC. In our previous study, we determined the presence of EphA2/ephrinA1 mRNA and protein in ACC [13]. In the current study, we further investigated the in vitro and in vivo effects of a short hairpin RNA (shRNA) targeting EphA2 on highly metastatic salivary adenoid cystic carcinoma cells (SACC-LM cell line). Results demonstrated that downregulation of EphA2 expression increased the in vitro migration, invasion ability, and PNI of SACC-LM. In vivo assay showed that EphA2-RNAi accelerated tumor growth in xenograft models and promoted lung metastasis ability. These results indicate that EphA2 may act as a tumor suppressor in SACC and a potential target for SACC therapy.
Materials and methods
Antibodies
EphA2, E-cadherin, Slug, and Ki-67 antibodies were purchased from Cell Signaling Technology (Boston, USA). S-100 was acquired from ZSGB-BIO (Beijing, China).
Cell culture
The human SACC cell line SACC-LM with great potential of lung metastasis was obtained from the College of Stomatology, Peking University, China. All cells were cultured in RPMI-1640 complete medium (Hyclone, Logan, USA) supplemented with 10% heat-inactivated fetal bovine serum (Gibco, Carlsbad, USA) in a humidified atmosphere with 5% CO2 at 37°C.
Establishment of EphA2-silenced cell line
Four potential different shRNA sequences (shRNA1, 2, 3, 4) targeting EphA2 and negative shRNA sequence (shRNA-mock) were designed and synthesized by Genepharma (Shanghai, China). These sequences were then inserted into the BamHI/EcoRI restriction sites of pGLV3/H1/GFP+ Puro lentivectors to construct human EphA2 shRNA plasmids. Afterward, 293T cells were co-transduced with lentiviral expression vectors and packaging plasmids according to the manufacturer’s instructions (Genepharma, Shanghai, China). The plasmids used and the sequences of shRNA targeting EphA2 and shRNA-mock are shown in Figure 1A. Viral particles were collected and used to infect SACC-LM cells. The infected cells were identified by selecting and counting with 2 μg/ml puromycin (R&D system, Minneapolis, USA). Transduction with lentivirus-encoded shRNA against EphA2, we obtained five different cellular clones, namely, SACC-EphA2-RNAi (1, 2, 3, 4) and SACC-mock.
Figure 1.
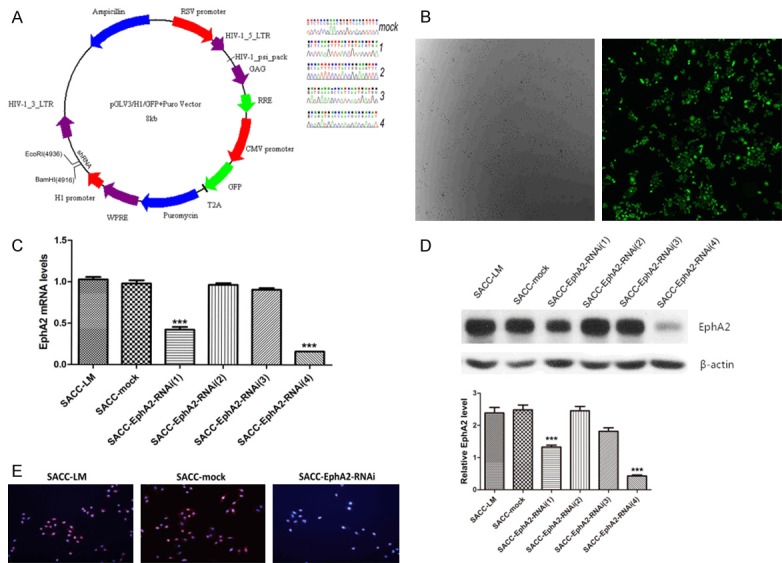
Effects of EphA2-shRNA on the expression of EphA2. A. The shuttle vector we used to construct lentiviral expression vectors and the shRNA sequences inserted, including the shRNA targeting EphA2 and shRNA-mock. B. Stably transduced SACC-LM cells were easily identified by GFP and selected by puromycin. C and D. EphA2 expression in SACC-LM, SACC-mock, SACC-EphA2-RNAi (1, 2, 3, 4) cells was detected by real-time RT-PCR, western blot analysis. Quantity analysis of EphA2 was shown. E. Immunofluorescence assay validated the effect of EphA2-RNAi (4). ***p < 0.001.
Real-time reverse transcriptase (RT)-PCR
Total RNA was extracted from SACC-LM, SACC-mock, and SACC-EphA2-RNAi (1, 2, 3, 4) cells by using TRIzol reagent (Invitrogen, Carlsbad, USA). Gene-specific primers employed in producing cDNA amplification were synthesized as follows: EphA2, forward, 5’-TCACACTAAGAGGGCAGACT-3’; reverse, 5’-GAATGTTTGACACCCTCTGT-3’; β-actin, forward, 5’-CATTAAGGAGAAGCTGTGCT-3’; reverse, 5’-GTTGAAGGTAGTTTCGTGGA-3’ (Sangon Biotech, Shanghai, China). Relative levels of EphA2 were obtained after normalization to an endogenous control β-actin and calculated from the standard curve. All real-time RT-PCR tests were performed in triplicate. The comparative Ct method was used for the final results.
Western blot analysis
The protein expression of EphA2 in SACC-LM, SACC-mock, and SACC-EphA2-RNAi (1, 2, 3, 4) cells was detected by western blot analysis. Seventy-two hours after transduction, proteins were harvested, loaded onto each well, and transferred onto polyvinylidene difluoride membrane (Millipore Co., Billerica, USA). The membrane was blocked and subsequently incubated at 4°C overnight with indicated primary antibodies and at room temperature for 1 h with horseradish peroxide-conjugated secondary antibody (Proteintech, Wuhan, China). The proteins were visualized with ECL plus western blot detection reagents (Beyotime, Haimen, China). Western blot analyses were performed in triplicate under similar conditions.
Cell proliferation assay
Cells were seeded in 96-well culture plates, and cell proliferation was determined after incubation for 24, 48, 72, 96 and 120 h using a Cell Counting Kit-8 (Dojindo, Japan). Growth curves were generated from a colorimetric assay. The absorbance value of each well at 450 nm was measured with a micro-plate reader (Thermo MutliscanMK3; Thermo Fisher Scientific, Waltham, USA).
Migration and invasion assay
Migration assay was performed with wound healing. A wound was made across the subconfluent cells growing in a 24-well plate using a sterilized one-milliliter pipette tip. After washing with PBS to remove the cellular debris, the same position along the wound of each well was photographed at 0 and 24 h. Twenty-four hours later, the rate of wound closure was calculated by the following formula: [1 - (current wound area/initial wound area)] × 100%.
Invasion and PNI assays were performed using Transwell chambers (Corning, Tewksbury, USA) with a polycarbonate membrane (6.5 mm diameter, 8 μm pore size). The membrane was coated with 50 μl of 8 mg/ml reconstructed extracellular matrix (Matrigel; BD Biosciences, USA) prior to use. In the lower chamber, 600 μl of conditioned medium (incubating NIH3T3 cells in serum-free RPMI 1640 medium for 24 h) was employed as chemoattractant. Exponentially growing cells were harvested and seeded into the upper chambers with 2 × 105 cells/well in RPMI 1640 medium supplemented with 1% FBS. Cells were incubated in a humidified atmosphere at 37°C with 5% CO2 for 24 h. For the PNI assay, 25 ng/ml nerve growth factor (NGF; R&D Systems, Minneapolis, USA) was added into the lower chamber to simulate the perineural environment. Invaded cells were fixed, stained, and then counted under a light microscope at a magnification of 400 ×. Five fields of vision were randomly selected and the numbers of the invaded cells were counted. Transwell assays were repeated at least three times under similar conditions.
Adhesion assay
Ninety-six-well plates were coated with Basement Membrane Matrigel (BD Biosciences, USA) according to the manufacturer’s instruction. Cell suspension containing 1 × 104 cells in complete medium were seeded into each well and incubated at 37°C for 45 min. Subsequently, the wells were washed with PBS, and the medium and non-attached cells were removed. MTT (30 μl) was added into each well of 96-well culture plate for 4 h at 37°C, and then 150 μl of DMSO was employed to dissolve the lysate of the cells. Afterward, spectrometric absorbance of each well was measured at the wavelength of 490 nm. The calculation of cell adhesion rate was the same as previously reported [14]. The assays were performed in triplicate independently.
Tumor xenograft model
Fifteen BALB/C nude mice, which were purchased from SLAC Laboratory Animal Center (Hunan, China), were fed under SPF conditions and randomly divided into three groups. Approximately 200 μl of single cell suspension (2 × 106 cells) of SACC-LM, SACC-mock, and SACC-EphA2-RNAi was subcutaneously inoculated into the right axillary fossa of the mice from the three groups. Tumor sizes were monitored every other day and measured weekly. Five weeks after inoculation, mice were sacrificed and tumor tissues were removed and subjected to immunohistochemistry assay. Tumor volume was calculated using the formula V = πabc/6 (a, b, and c are length, width, and height, respectively).
To investigate the effect of EphA2 silencing on SACC-LM metastasis to lung, four-week-old female BALB/C nude mice were randomly divided into four groups of 30 mice each. About 200 μl of cell suspension (SACC-LM, SACC-mock, SACC-EphA2-RNAi; 2 × 106 cells) were injected under aseptic conditions into the tail vein of each mouse, with normal saline as normal control. Five mice randomly selected from each group were sacrificed every week. Fresh bilateral lung tissues were harvested, and then fluorescence images of the lung were captured with the in vivo fluorescence imaging system (Maestro 2, CRI, USA) using the same parameters for the excitation filter range (455 nm, 435-480 nm), emission filter range (490 nm longpass), acquisition setting (500-720 nm in 10 nm steps). Afterward, specimens were weighed, fixed with formalin, and embedded in paraffin for the pathological analysis. Sections with the maximum cross-sectional area were used to calculate the tumor burden. In vivo studies were approved by the ethics committees of School and Hospital of Stomatology, Wuhan University.
Immunohistochemistry and evaluation
Forty cases of clinical specimens of SACC were provided by the Department of Oral Pathology, School and Hospital of Stomatology, Wuhan University. Tumor tissues were fixed in formalin, embedded in paraffin, sectioned to 4 μm, and mounted on slides. Immunohistochemical staining was performed to detect the protein expression of transplanted tumor sections. After deparaffinization and rehydration, antigens on sections were retrieved by boiling in 10 mM sodium citrate buffer (pH 6.0) for 10 min. The activity of endogenous peroxidases was quenched by incubation in 3% H2O2 for 20 min at room temperature. After three washes with PBS, the slides were blocked for 30 min with 100 µl of normal goat serum for 1 h at room temperature. The slides were then incubated with primary antibodies (E-cadherin, Slug, S-100, EphA2, and Ki-67) at 4°C overnight and second antibodies for 1 h at room temperature. Stained slides were visualized using diaminobenzidine as a chromogenic substrate and counterstained with hematoxylin.
E-cadherin and Slug staining were assessed according to a score that added a scale of intensity of staining (magnification × 200) to the proportion of stained cells (magnification × 40) as described in previous research [13]. The integrated optical density (IOD) value of Ki-67 staining was calculated using a semi-automated computerized image analysis system (Image Pro Plus 6.0; Media Cybernetics, Bethesda, USA). For each section, the average IOD score was calculated from triplicate values.
Statistical analysis
Data were treated using one-way ANOVA, two-way ANOVA, Fisher’s exact test, Spearman rank correlation coefficient test, and Student-Newman-Keuls test. GraphPad Prism 5.01 (GraphPad Software, Inc., La Jolla, CA) statistical packages were used to perform statistical data analysis and data were expressed as mean ± standard deviation. p < 0.05 was considered statistically significant.
Results
Effects of EphA2-shRNA on the expression of EphA2
We detected the interference effect of EphA2 shRNA on EphA2 expression in the five stably transduced groups, including SACC-mock, SACC-EphA2-RNAi (1), SACC-EphA2-RNAi (2), SACC-EphA2-RNAi (3), and SACC-EphA2-RNAi (4) cell clones. The plasmids used and the sequences of shRNA targeting EphA2 and shRNA-mock are shown in Figure 1A. The phase contrast and fluorescence images after stable transduction are shown in Figure 1B. Real-time RT-PCR assay showed that EphA2-shRNA (1, 4) significantly inhibited EphA2 mRNA expression in SACC-EphA2-RNAi (1, 4) cells compared with untransduced SACC-LM, and the inhibitory rates were about 58.62% and 84.22%, respectively (p < 0.001, Figure 1C). Western blot analysis and the quantity analysis of EphA2 also confirmed evident decrease in EphA2 protein expression by EphA2-RNAi (1, 4) constructs (p < 0.001, Figure 1D). However, the mock-transduced and SACC-EphA2-RNAi (2, 3) groups showed no obvious difference compared with the SACC-LM cell group, which was consistent with the RT-PCR results (Figure 1D). By contrast, the SACC-mock and SACC-EphA2-RNAi (2, 3) cells displayed no significant differences in EphA2 mRNA level with the inhibitory rates of 2.85%, 3.41%, and 6.98% (p > 0.05, Figure 1C). Results of western blot analysis were consistent with those of RT-PCR (Figure 1D). Therefore, the SACC-EphA2-RNAi (4) was selected as the experimental group with the best interference effect. The effect of EphA2-RNAi (4) was finally validated by immunofluorescence assay, which also verified the above conclusion (Figure 1E).
Effects of EphA2 silencing on tumor cell behaviors
The current study evaluated the effects of EphA2 silencing on SACC-LM cell behaviors, including cell proliferation, migration, invasion, and adhesion ability. Transwell Matrigel assays showed that the number of invaded tumor cells in SACC-EphA2-RNAi group was significantly higher than that in SACC-LM and SACC-mock groups under the stimulation of conditioned medium with or without chemoattractant NGF (p < 0.001, Figure 2A). A significant difference existed in the number of invaded cells between SACC-EphA2-RNAi cells and those stimulated with NGF (p < 0.05, Figure 2A), whereas no significant difference occurred in the parental and mock-transduced cells. These results suggest that silencing of EphA2 remarkably increased the invasive ability of SACC-LM cells. The results also imply a potential role of EphA2 in PNI of ACC because silencing of EphA2 presented much stronger invasive ability at the presence of NGF stimulation, which is usually used as an in vitro PNI model.
Figure 2.
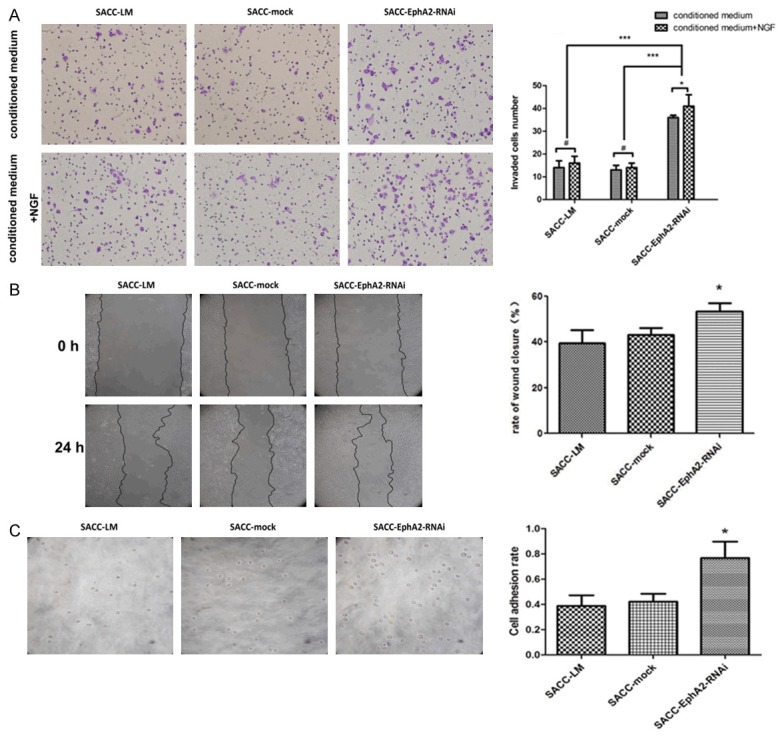
Effects of EphA2 silencing on tumor cell behaviors. A. Invaded cells through the polycarbonate membrane were stained and quantified, and the photographs were taken under a microscope. The up panel represented the invasion assay and bottom panel for the PNI assay. Data in the histogram showed the differences caused by EphA2-RNAi. B. The wound healing assay showed that EphA2 silencing promoted wound closure rate. C. The adhered cells to Matrigel were seen under the microscope, and the adhesion rate was also shown in a histogram. *p < 0.05, ***p < 0.001.
As shown in Figure 2B, cells displayed a significantly higher closure rate of about 53.2% upon EphA2 downregulation than SACC-LM and SACC-mock cell groups (39.5% and 40.2%, respectively), demonstrating an increased migratory ability caused by EphA2 RNAi (p < 0.05). The activity of cell adhesion to extracellular matrix can influence invasion and metastatic spread in tumor progression. After incubation at 37°C for 45 min, cell adhesion rates were 0.421%, 0.362%, and 1.012% in parental, mock-transduced, and EphA2-silenced groups, respectively. An evident increase in the amount of attached cells was observed in RNAi cells (p < 0.05, Figure 2C) compared with the other two groups, indicating the positive role of EphA2 silencing in cell adhesion. However, EphA2 knockdown exhibited no apparent effects on tumor cell proliferation (data not shown).
Downregulation of EphA2 accelerates tumor growth in vivo
We established xenograft models of BALB/C nude mice to assess the effects of EphA2 knockdown on tumorigenicity. All of the 15 mice in the three groups developed detectable tumor nodules 7 days after inoculation. During the observation period, the increase in tumor volume in the EphA2 silencing group was accelerated compared with the parental and mock-transduced groups, whereas no significant difference was observed between the parental and mock-transduced groups. At the end of the observation period (five weeks), silencing of EphA2 resulted in a substantial increase in the volume of transplanted tumors (1622.19 ± 307 mm3) when compared with the other two groups, 582.77 ± 275 mm3 and 588.87 ± 85 mm3 for the parental and mock groups (p <0.001, Figure 3A). Immunostaining of the pathological sections exhibited a remarkable reduction in EphA2 expression level (Figure 3B), which is consistent with our results in vitro. Furthermore, we assessed the expression of Ki-67, a marker reflecting tumor cell proliferation activity, through immunohistochemical staining. EphA2-silencing increased the IOD value of Ki-67 staining (210 ± 13) compared with the parental (107 ± 55) and mock-transduced (115 ± 30) groups (p <0.05, Figure 3C), indicating an increased proliferation ability. These results suggest that downregulation of EphA2 accelerates tumor growth in vivo.
Figure 3.
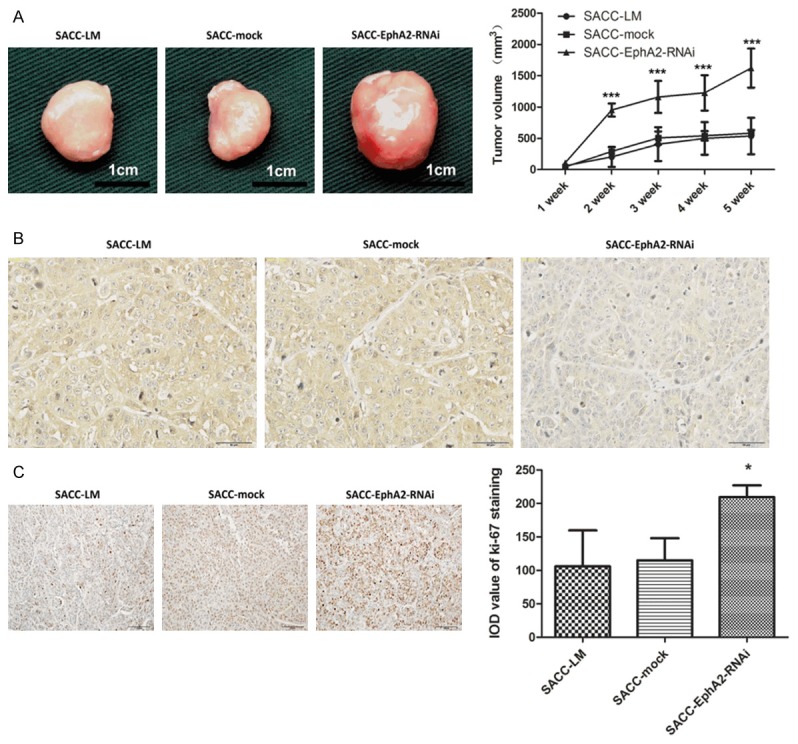
Downregulation of EphA2 accelerates tumor growth in vivo. A. Representative photographs of the transplanted tumors and their growth curves. B and C. Immunohistochemical staining showed a remarkable reduction of EphA2 and Ki-67 expression in the transplanted tumors from the EphA2-RNAi group. In the histogram, IOD value of Ki-67staining represented the proliferation index in transplanted tumors. *p < 0.05, ***p < 0.001.
Suppression of EphA2 promotes tumor metastasis to lung in vivo
Seven days after injection, lung metastatic foci were detected using the in vivo fluorescence imaging system. No significant differences were found among lungs obtained from the three cell groups. Differences were observed on the 14th day and became increasingly significant between lung derived from EphA2-silenced group and the other three groups (Figure 4D). This finding shows that EphA2-RNAi cells resulted in more and larger lung metastatic foci than parental and mock-transduced cells. Six weeks after inoculation, all the remaining mice were sacrificed, and their bilateral lung tissues were then removed and weighed (Figure 4Ai). Sections with maximum cross-sectional area are shown in Figure 4Aii, according to which the tumor burden was calculated. Substantial differences were observed between the EphA2-silenced group (48.6 ± 1.22%) and SACC-LM (27.9 ± 1.01%) and SACC-mock (28.8 ± 2.31%) groups (p < 0.01, Figure 4B). Measurement of lung weight showed that transduction with EphA2-shRNA presented a significant increase at 0.632 ± 0.062 g compared with 0.374 ± 0.057 g and 0.425 ± 0.077 g for SACC-LM and SACC-mock cells (p < 0.05, Figure 4C). These data strongly demonstrate the acceleration of lung metastasis by EphA2-RNAi.
Figure 4.

Suppression of EphA2 promotes tumor metastasis to lung in vivo. Ai. Six weeks after inoculation, photographs of the bilateral lung tissues derived from mice injected with SACC-LM, SACC-mock, SACC-EphA2-RNAi cells and normal saline (N.S) were taken. Aii. Representative images showing hematoxylin-eosin (HE) staining of sections with the maximum cross-sectional area. B. Tumor burden of lungs from SACC-LM, SACC-mock, SACC-EphA2-RNAi groups was calculated. C. Lung weight of each group. D. Lung metastatic foci were detected by the in vivo fluorescence imaging system and photographed at indicated time points (1W, 2W, 3W, 4W, 5W, 6W). In each visual field, from left to right are lungs obtained from N.S, SACC-mock, SACC-EphA2-RNAi groups, respectively. *p < 0.05, **p < 0.01.
Correlation between epithelial-mesenchymal transition (EMT), PNI and EphA2 in SACC
Immunohistochemical analysis was conducted to examine the expression of EphA2, Slug, and E-cadherin in 40 ACC specimens. As shown in Figure 5Ai, the positive staining of EphA2 and E-cadherin was mainly localized into the cytoplasm and membranes of tumor cells, whereas that of Slug was mainly into the nuclei. Spearman rank correlation coefficient test suggested that significant negative correlation existed between EphA2 and Slug expression (p < 0.001, Figure 5Aii), but no remarkable correlation between EphA2 and E-cadherin (p > 0.05, Figure 5Aii). S-100 staining showed that PNI occurred in 32 of 40 SACC specimens. Fisher’s exact test suggested that the expression of Slug was significantly correlated with PNI in all the 40 cases, whereas no significant correlation was found between E-cadherin expression and PNI (p < 0.01, Table 1). In 32 PNI-positive specimens, 11 cases were detected with a relatively lower expression of E-cadherin in the leader cells at the invading front into nerve than that of the tumor cells around (Figure 5B). Similar to E-cadherin, downregulation of EphA2 also occurred at the invading front in 10 cases from the 11 specimens (Figure 5B), suggesting that downregulation of E-cadherin was almost simultaneous with EphA2 suppression at the invading front into nerve. In vitro study, EphA2-RNAi caused downregulation of E-cadherin, overexpression of Slug (Figure 5Ci, 5Cii), and an accelerated invasive ability toward NGF (Figure 2A). Taken together, our in vitro and in vivo studies suggest that suppression of EphA2 partially contributes to EMT and enhancement of PNI in SACC.
Figure 5.
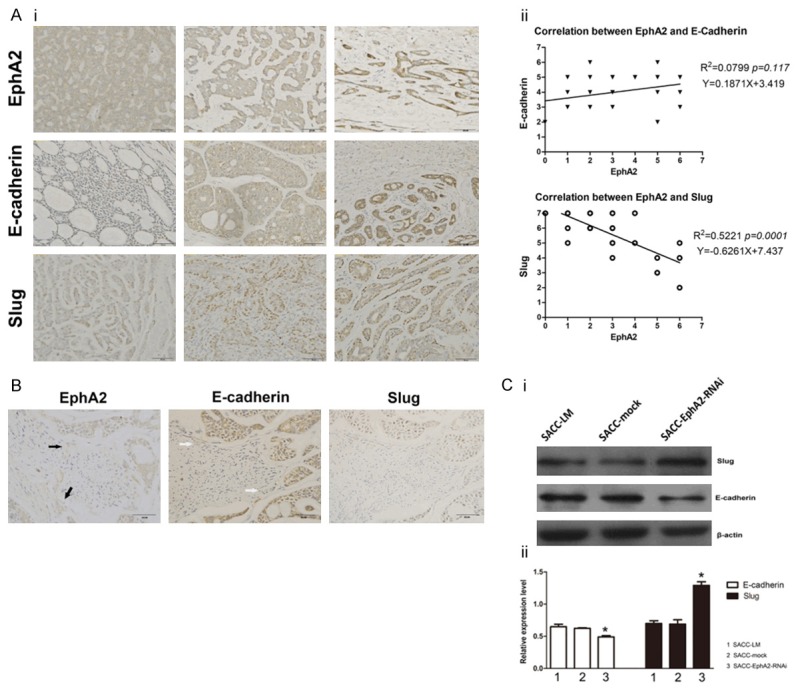
Correlation between EMT, PNI and EphA2 in SACC. Ai. Immunostaining of EphA2, Slug, and E-cadherin at different levels of 40 SACC specimens. Aii. Significant negative correlation existed between EphA2 and Slug expression (p < 0.001), but no remarkable correlation between EphA2 and E-cadherin was found (p > 0.05). B. Immunohistochemical staining showed downregulation of EphA2 (black arrowhead) and E-cadherin (white arrowhead) in the leader cells at the invading front into nerve, but there was no difference in Slug expression. Ci. Detection of Slug and E-cadherin protein expression in SACC-LM, SACC-mock, SACC-EphA2-RNAi cells by western blot analysis. Cii. Quantity analysis showed the band IOD ratio of Slug, E-cadherin and β-actin. *p < 0.05.
Table 1.
Correlation between Slug, E-cadherin expression and PNI in 40 SACC specimens
| Variables | PNI | p | |
|---|---|---|---|
|
| |||
| Yes | No | ||
| Slug | |||
| 0-3 | 2 | 3 | 0.007** |
| 4-5 | 8 | 4 | |
| 6-7 | 22 | 1 | |
| E-cadherin | |||
| 0-3 | 1 | 1 | 0.173 |
| 4-5 | 12 | 5 | |
| 6-7 | 19 | 2 | |
| Total | 32 | 8 | |
p < 0.01.
Discussion
The interaction of Eph receptors and ephrin ligands has been linked to cancer progression, metastatic spread and patient survival in many studies [10]. Over the last few years, increasing evidence has implicated Eph receptors in tumor progression [15-18], but little is known about their role in SACC. This study is the first to report the effects of EphA2 knockdown on the growth and metastasis of SACC through in vitro and in vivo experiments, indicating a tumor suppression activity of EphA2 in SACC tumor progression.
To investigate the role of EphA2 in SACC, we stably transduced SACC-LM cells with EphA2 shRNA-expressing vectors. After isolation of stable clones, EphA2 suppression led to higher migration, invasion, and adhesion rates of SACC-LM cells in vitro. Moreover, EphA2 ablation caused higher tumorigenesis rate, with larger tumor volume and higher IOD value of Ki-67 staining of the tumor section. Consistent with the results in vitro, we also demonstrated that silenced expression of EphA2 resulted in more lung metastatic foci and higher tumor burden in the distant lung metastasis assay. Although EphA2 knockdown actively promoted tumor growth in vivo, no evident effects were found on tumor cell proliferation in vitro. This disparity may be due to the different conditions of tumor microenvironment. Guo et al. [17] reported that tumors arising in EphA2 knockout are more likely to develop fully invasive phenotype compared with tumors from wild-type mice. Previous studies also demonstrated that epigenetic silencing of EphA1 expression is correlated with poor survival in colorectal cancer [19]. Overexpression of EphB3 enhances cell-cell contacts and suppresses tumor growth in HT-29 human colon cancer cells, implying the tumor suppressive activity of Eph receptors in human cancer progression [20]. Another report also reported the growth-inhibiting effects of EphA2 activation after ephrinA1 stimulation in prostatic epithelial and endothelial cells [21]. The present results agree well with these reports and suggest that EphA2 may act as a tumor suppressor gene in local invasiveness and distant metastasis of SACC. In fact, our previous study showed that EphA2/ephrinA1 mRNA expression and protein production are upregulated in ACC of salivary gland. This finding may partially explain why most patients with ACC experienced a relatively long survival time. However, EphA2 overexpression also plays tumor-promoting roles in conferring a malignant phenotype, enhancing tumorigenic potential and increasing tumor growth, angiogenesis, and/or metastasis in several other cancers [22-25]. Therefore, further studies are necessary to address the differential roles of Eph receptors in different types of tumors in the future.
Eph receptor activation induced by ephrin ligands inhibits migration and invasion of many types of cancers both in vitro and in vivo, showing that EphA2 forward signaling suppresses tumor progression [26]. This result indicates that EphA2 activation induced by ephrin may be harmful to tumor development. In this study, we disturbed the ligand-induced EphA2 forward signaling by sudden silencing of EphA2 expression. The disturbance inhibited the suppressive signals to some extent, thus conversely promoting migration and invasion of tumor cells. This phenomenon is reported in several previous reports [20,27].
Both EMT and MET (mesenchymal-epithelial transition) play an important role in tumor progression. Miao et al. [28] found that stimulation of EphB3 receptor with soluble ephrinB1-Fc promotes cell rounding, inhibits cell migration, and impairs cell-matrix adhesion. Further study suggested that EphB3-ephrinB interaction promotes MET through inactivation of the CrkL-Rac1 signaling pathway [20]. Noren [29] has recently reported that EphB4 activation induced by ephrin-B2 suppresses tumorigenicity in breast cancer by activating the Abl-Crk pathway, and disruption of EphB4 signalling induces several properties characteristic of transformed cells in non-transformed mammary epithelial cells. In the present study, epigenetic suppression of EphA2 resulted in increase in Slug and decrease in E-cadherin in SACC-LM cells. Immunohistochemical study further demonstrated that EphA2 expression was negatively correlated with Slug expression in 40 SACC tissue specimens. Although no correlation was found between EphA2 and E-cadherin expression, EphA2 downregulation was typically accompanied by a decrease in E-cadherin expression at the invasive front of PNI-occurred area. In addition, the EphA2-knockdown SACC-LM cells presented active migratory ability under the NGF chemotaxis. A previous study confirmed that ephrinA1-Fc stimulation increases the tyrosine phosphorylation of EphA2 and leads to suppression of Arf6 GTPase activity and inhibition of the internalization of E-cadherin from cell-cell contact areas, thus stabilizing the apical-basal polarity [30,31]. Similarly, our results proved that inhibition of EphA2 tyrosine phosphorylation (caused by EphA2 RNAi) attenuated E-cadherin-based cell junctions, thereby facilitating cell metastasis. These results partially suggest that EphA2 may contribute in tumor progression through regulating EMT and PNI. Whether EphA2 signaling promotes MET or EMT and suppresses tumor progression through the Abl-Crk-Rho-GTPases or some other pathways need further investigation.
In conclusion, the results in this study suggest that EphA2 may act as a tumor suppressor in SACC growth, metastasis, and PNI, but the underlying mechanisms still necessitate further clarification. Understanding the role and mechanism of EphA2 in tumor progression is helpful in designing molecular drugs for SACC therapy.
Acknowledgements
This study was supported by Natural Science Foundation of China (No. 81202143, 81372879).
Disclosure of conflict of interest
None.
References
- 1.Matsuba HM, Spector GJ, Thawley SE, Simpson JR, Mauney M, Pikul FJ. Adenoid cystic salivary gland carcinoma: A histopathologic review of treatment failure patterns. Cancer. 1986;57:519–524. doi: 10.1002/1097-0142(19860201)57:3<519::aid-cncr2820570319>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 2.Barrett AW, Speight PM. Perineural invasion in adenoid cystic carcinoma of the salivary glands: a valid prognostic indicator? Oral Oncol. 2009;45:936–940. doi: 10.1016/j.oraloncology.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 3.Mitani Y, Liu B, Rao PH, Borra VJ, Zafereo M, Weber RS, Kies M, Lozano G, Futreal PA, Caulin C, El-Naggar AK. Novel MYBL1 Gene Rearrangements with Recurrent MYBL1-NFIB Fusions in Salivary Adenoid Cystic Carcinomas Lacking t(6;9) Translocations. Clin Cancer Res. 2016;22:725–733. doi: 10.1158/1078-0432.CCR-15-2867-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sung MW, Kim KH, Kim JW, Min YG, Seong WJ, Roh JL, Lee SJ, Kwon TK, Park SW. Clinicopathologic predictors and impact of distant metastasis from adenoid cystic carcinoma of the head and neck. Arch Otolaryngol Head Neck Surg. 2003;129:1193–1197. doi: 10.1001/archotol.129.11.1193. [DOI] [PubMed] [Google Scholar]
- 5.van der Wal JE, Becking AG, Snow GB, van der Waal I. Distant metastases of adenoid cystic carcinoma of the salivary glands and the value of diagnostic examinations during followup. Head Neck. 2002;24:779–783. doi: 10.1002/hed.10126. [DOI] [PubMed] [Google Scholar]
- 6.Spiro RH, Huvos AG. Stage means more than grade in adenoid cystic carcinoma. Am J Surg. 1992;164:623–628. doi: 10.1016/s0002-9610(05)80721-4. [DOI] [PubMed] [Google Scholar]
- 7.Pasquale EB. Eph receptor signalling casts a wide net on cell behaviour. Nat Rev Mol Cell Biol. 2005;6:462–475. doi: 10.1038/nrm1662. [DOI] [PubMed] [Google Scholar]
- 8.Adams RH, Eichmann A. Axon guidance molecules in vascular patterning. Cold Spring Harb Perspect Biol. 2010;2:a001875. doi: 10.1101/cshperspect.a001875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gelfand MV, Hong S, Gu C. Guidance from above: common cues direct distinct signaling outcomes in vascular and neural patterning. Trends Cell Biol. 2009;19:99–110. doi: 10.1016/j.tcb.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pasquale EB. Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat Rev Cancer. 2010;10:165–180. doi: 10.1038/nrc2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wykosky J, Debinski W. The EphA2 receptor and ephrinA1 ligand in solid tumors: function and therapeutic targeting. Mol Cancer Res. 2008;6:1795–1806. doi: 10.1158/1541-7786.MCR-08-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhuang G, Brantley-Sieders DM, Vaught D, Yu J, Xie L, Wells S, Jackson D, Muraoka-Cook R, Arteaga C, Chen J. Elevation of Receptor Tyrosine Kinase EphA2 Mediates Resistance to Trastuzumab Therapy. Cancer Res. 2009;70:299–308. doi: 10.1158/0008-5472.CAN-09-1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shao Z, Zhu F, Song K, Zhang H, Liu K, Shang Z. EphA2/ephrinA1 mRNA expression and protein production in adenoid cystic carcinoma of salivary gland. J Oral Maxillofac Surg. 2013;71:869–878. doi: 10.1016/j.joms.2012.10.026. [DOI] [PubMed] [Google Scholar]
- 14.Shi H, Wang J, Dong F, Wang X, Li H, Hou Y. The effect of proteoglycans inhibited by RNA interference on metastatic characters of human salivary adenoid cystic carcinoma. BMC Cancer. 2009;9:456. doi: 10.1186/1471-2407-9-456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Batlle E, Bacani J, Begthel H, Jonkheer S, Gregorieff A, van de Born M, Malats N, Sancho E, Boon E, Pawson T, Gallinger S, Pals S, Clevers H. EphB receptor activity suppresses colorectal cancer progression. Nature. 2005;435:1126–1130. doi: 10.1038/nature03626. [DOI] [PubMed] [Google Scholar]
- 16.Brantley-Sieders DM, Fang WB, Hicks DJ, Zhuang G, Shyr Y, Chen J. Impaired tumor microenvironment in EphA2-deficient mice inhibits tumor angiogenesis and metastatic progression. FASEB J. 2005;19:1884–1886. doi: 10.1096/fj.05-4038fje. [DOI] [PubMed] [Google Scholar]
- 17.Guo H, Miao H, Gerber L, Singh J, Denning MF, Gilliam AC, Wang B. Disruption of EphA2 receptor tyrosine kinase leads to increased susceptibility to carcinogenesis in mouse skin. Cancer Res. 2006;66:7050–7058. doi: 10.1158/0008-5472.CAN-06-0004. [DOI] [PubMed] [Google Scholar]
- 18.Huang J, Xiao D, Li G, Ma J, Chen P, Yuan W, Hou F, Ge J, Zhong M, Tang Y, Xia X, Chen Z. EphA2 promotes epithelial-mesenchymal transition through the Wnt/beta-catenin pathway in gastric cancer cells. Oncogene. 2014;33:2737–2747. doi: 10.1038/onc.2013.238. [DOI] [PubMed] [Google Scholar]
- 19.Herath NI, Doecke J, Spanevello MD, Leggett BA, Boyd AW. Epigenetic silencing of EphA1 expression in colorectal cancer is correlated with poor survival. Br J Cancer. 2009;100:1095–1102. doi: 10.1038/sj.bjc.6604970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chiu ST, Chang KJ, Ting CH, Shen HC, Li H, Hsieh FJ. Over-expression of EphB3 enhances cell-cell contacts and suppresses tumor growth in HT-29 human colon cancer cells. Carcinogenesis. 2009;30:1475–1486. doi: 10.1093/carcin/bgp133. [DOI] [PubMed] [Google Scholar]
- 21.Miao H, Wei BR, Peehl DM, Li Q, Alexandrou T, Schelling JR, Rhim JS, Sedor JR, Burnett E, Wang B. Activation of EphA receptor tyrosine kinase inhibits the Ras/MAPK pathway. Nat Cell Biol. 2001;3:527–530. doi: 10.1038/35074604. [DOI] [PubMed] [Google Scholar]
- 22.Lu M, Miller KD, Gokmen-Polar Y, Jeng MH, Kinch MS. EphA2 overexpression decreases estrogen dependence and tamoxifen sensitivity. Cancer Res. 2003;63:3425–3429. [PubMed] [Google Scholar]
- 23.Parri M, Taddei ML, Bianchini F, Calorini L, Chiarugi P. EphA2 reexpression prompts invasion of melanoma cells shifting from mesenchymal to amoeboid-like motility style. Cancer Res. 2009;69:2072–2081. doi: 10.1158/0008-5472.CAN-08-1845. [DOI] [PubMed] [Google Scholar]
- 24.Udayakumar D, Zhang G, Ji Z, Njauw CN, Mroz P, Tsao H. EphA2 is a critical oncogene in melanoma. Oncogene. 2011;30:4921–4929. doi: 10.1038/onc.2011.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zelinski DP, Zantek ND, Stewart JC, Irizarry AR, Kinch MS. EphA2 overexpression causes tumorigenesis of mammary epithelial cells. Cancer Res. 2001;61:2301–2306. [PubMed] [Google Scholar]
- 26.Noblitt LW, Bangari DS, Shukla S, Knapp DW, Mohammed S, Kinch MS, Mittal SK. Decreased tumorigenic potential of EphA2-overexpressing breast cancer cells following treatment with adenoviral vectors that express EphrinA1. Cancer Gene Ther. 2004;11:757–766. doi: 10.1038/sj.cgt.7700761. [DOI] [PubMed] [Google Scholar]
- 27.Dopeso H, Mateo-Lozano S, Mazzolini R, Rodrigues P, Lagares-Tena L, Ceron J, Romero J, Esteves M, Landolfi S, Hernandez-Losa J, Castano J, Wilson AJ, Ramon y Cajal S, Mariadason JM, Schwartz S Jr, Arango D. The receptor tyrosine kinase EPHB4 has tumor suppressor activities in intestinal tumorigenesis. Cancer Res. 2009;69:7430–7438. doi: 10.1158/0008-5472.CAN-09-0706. [DOI] [PubMed] [Google Scholar]
- 28.Miao H, Strebhardt K, Pasquale EB, Shen TL, Guan JL, Wang B. Inhibition of integrin-mediated cell adhesion but not directional cell migration requires catalytic activity of EphB3 receptor tyrosine kinase. Role of Rho family small GTPases. J Biol Chem. 2005;280:923–932. doi: 10.1074/jbc.M411383200. [DOI] [PubMed] [Google Scholar]
- 29.Noren NK, Foos G, Hauser CA, Pasquale EB. The EphB4 receptor suppresses breast cancer cell tumorigenicity through an Abl-Crk pathway. Nat Cell Biol. 2006;8:815–825. doi: 10.1038/ncb1438. [DOI] [PubMed] [Google Scholar]
- 30.Miura K, Nam JM, Kojima C, Mochizuki N, Sabe H. EphA2 engages Git1 to suppress Arf6 activity modulating epithelial cell-cell contacts. Mol Biol Cell. 2009;20:1949–1959. doi: 10.1091/mbc.E08-06-0549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zantek ND, Azimi M, Fedor-Chaiken M, Wang B, Brackenbury R, Kinch MS. E-cadherin regulates the function of the EphA2 receptor tyrosine kinase. Cell Growth Differ. 1999;10:629–638. [PubMed] [Google Scholar]