

Abstract
Results of this study showed that the bacterial composition in vagina (V) greatly differed from intrauterine microbiome (I). Microbiomes were present in all intrauterine samples of healthy women (Group H (I)) and patients with endometrial polyps (EP) (including Group EP (I) and Group EP/chronic endometritis (CE) (I)). Indeed, the intrauterine bacteria population in Group EP/CE (I) were more diverse than those in Groups EP (I) and H (I). The result also confirmed the bacterial composition differences between vagina and uterus as well as the intrauterine microbiome alteration in the patients, compared to the healthy. Although bacteria of Proteobacteria, Firmicutes and Actinobacteria, dominated the intrauterine microbiome in all samples, however, proportions of Firmicutes from Group EP/CE (I) and Group EP (I) were much higher than that from Group H (I), in contrast, the proportions of Proteobacteria were far lower than the healthy. At the genus level, compared to Group H (I), it is found that proportions of Lactobacillus, Gardnerella, Bifidobacterium, Streptococcus, and Alteromonas were significantly higher, and that of Pseudomonas were significantly lower in Group EP/CE (I) or Group EP (I). In addition, lower proportions of Enterobacter and Sphingomonas and a higher proportion of Prevotella were also observed in Group EP/CE (I). In conclusion, uterine microbiomes between patients with EP and the healthy are significantly different and all the potentially important variation of uterine microbes may cause EP, but not definitively related to CE. Further experiments should be performed to test these relationships to endometritis occurrence.
Keywords: Endometrial polyps, barcoded sequencing, endometritis
Introduction
Endometrial polyps (EP) is a common gynecologic disease featured as a localized overgrowth of mucosa with a prevalence ranging from 7.8% to 34.9% [1,2]. Clinically, it is often identified in patients with complaints of infertility or abnormal vaginal bleeding, or only observed in routine examination, such as transvaginal ultrasound. However, the etiopathogenesis of EP is not yet fully understood. Some factors such as chronic endometritis (CE), high hormonal influence, imbalance between proliferation and apoptosis, abnormal expression of ovarian steroid hormone receptor, and chromosomal abnormalities are thought to play important roles [3-5]. Previous studies [6,7] indicate a correlation between EP and chronic endometritis (CE), and CE was basically caused by a high load of organisms, including Ureaplasma urealyticum or a wide variety of common bacteria [8]. This suggestion may contribute to an assumption that overgrowth of endometrial tissue is related to continuous stimulation of biological inflammatory factors [6,7]. Many studies have been performed to demonstrate the effect of bacteria on the development of CE [7,9], but no much effect have been devoted to the relationship between bacteria and EP.
Due to the possibility that microorganisms from the endocervical canal can be a contamination of passing through operative instrument, the argument that the high incidence of positive cultures from intrauterine samples is resulted from contamination [10] was raised by who believes that the endometrial cavity is sterile. Based on the traditional microbial research methods, previous studies [8,11,12] have limits to the culturable microbes that only comprise approximately 1% of the total microbiome. Additionally, these studies have failed to assess microbial diversity and community dynamics in the uterine cavity, resulting in a poor understanding of the association between intrauterine microbiota and uterine lesions. Recently, high throughput sequencing techniques based on 16S rRNA gene [13] have facilitated identifying unculturable, low abundance and unclassified microorganisms quickly and accurately. Most importantly, they are also able to analyze the microbial diversity and community dynamics. Here, we used this powerful technique to characterize the intrauterine microbial communities in patients suffering from endometrial polyps combined with or without chronic endometritis, and the intrauterine population difference compared to healthy donors.
Materials and methods
Patients and sample collection
From August 2013 to January 2014, we enrolled 20 patients with endometrial polyps as the study group and 10 healthy women for the healthy controls (group H). Subjects eligible for the study group met the following criteria: 1) regular menstrual cycles, 2) diagnostic hysteroscopy and pathological results showing the presence of EP. Inclusion criteria for patients in the control group: 1) regular menstrual cycles, diagnostic hysteroscopy with endometrial biopsy and laparoscopy as part of their infertility diagnostic work-up prior to IVF, hysteroscopy and subsequent pathological results having shown no abnormality in the uterine cavities and abdominal cavity, 2) the healthy women recruited had male partners who were infertile and diagnosed with defective sperm function, such as asthenozoospermia, oligoasthenozoospermia, severe oligoasthenozoospermia and azoospermia, defined according to guidelines published by the World Health Organization. Exclusion criteria for patients in the control group and study group: 1) other intrauterine lesions such as intrauterine adhesions, submucosal myoma and uterine septum, 2) uterine myoma, endometriosis, ovarian tumor and hydrosalpinx, 3) abnormal sex hormone level, 4) abnormal leucorrhea, vaginitis and pelvic inflammatory disease. The study subjects were further divided into two groups based on pathologic and immunohistochemical results, in which ten patients with both EP and CE were classified as group EP/CE, and the others only with EP, as group EP. As shown in Table 1, clinical characteristics of these three groups are comparable. Meanwhile, samples from vagina were defined as Group V, and those from the uterine cavity as Group I, that is, microbial samples in Group H (V), Group EP/CE (V) and Group EP (V) were respectively collected from vagina, while Group H (I), Group EP/CE (I) and Group EP (I) were collected from the uterus of women in Group H, Group EP/CE and Group EP, respectively. The study was approved by the Medical Ethics Committees at the First Affiliated Hospital of Sun Yat-sen University (No.2014-64), and all women gave their written, informed consents.
Table 1.
Clinical characteristics of women enrolled in the study
| Items | Group H | Group EP/CE | Group EP | P value |
|---|---|---|---|---|
| (n = 10) | (n = 10) | (n = 10) | ||
| Age (years) | 30.90 ± 1.56 | 35.2 ± 1.83 | 34.4 ± 2.44 | 0.289 |
| BMI | 21.04 ± 1.03 | 21.29 ± 0.99 | 20.47 ± 0.67 | 0.811 |
| Gravidity | 1.60 ± 0.45 | 1.50 ± 0.48 | 0.80 ± 0.33 | 0.371 |
| Parity | 0.30 ± 0.15 | 0.90 ± 0.41 | 0.40 ± 0.16 | 0.491 |
| Age of menarche (years) | 13.40 ± 0.45 | 13.50 ± 0.58 | 13.80 ± 0.33 | 0.746 |
| Menstrual duration (days) | 6.00 ± 0.56 | 6.40 ± 0.40 | 5.90 ± 0.41 | 0.663 |
| Menstrual average cycle (days) | 28.70 ± 1.16 | 29.70 ± 0.79 | 27.60 ± 1.12 | 0.370 |
Notes: Mean ± SE are shown. BMI: Body Mass Index.
All 30 participants in Group H, Group EP/CE and Group EP were abstained from intercourse for three or more days and did not receive any antibiotic treatment or other medication, including taking a bath or vaginal douche within at least 3 weeks before sample collection. All samples were obtained in the first week after the participants’ menstrual period. Before vaginal disinfection, vaginal swab samples were collected during a speculum examination from the posterior fornix. Then after vaginal and cervical canal disinfection endometrial swabs and endometrial tissues from the uterine cavity were obtained. In order to minimize the risk of contamination, endometrial swabs with sleeves were inserted under visual control into the uterine cavity, taking care to avoid any contact with the vaginal walls. All the vaginal and endometrial swabs were immediately frozen and stored at -80°C for DNA extraction and 16S rRNA profiling/characterization. The endometrial samples were fixed in formalin and embedded in paraffin for immunohistochemistry.
Instead of classical tissue staining techniques, immunohistochemistry for the transmembrane heparin sulfate proteoglycan syndecan-1 (CD138) was designed to enhance the diagnostic accuracy of CE (Figure S1). The number of immunoreactive cells was identified under a high microscope magnification (400×) by two independent professional observers. The density of immunoreactive cells was determined in 10 nonoverlapping stromal areas and CE was diagnosed when five or more plasma cells presented [14,15].
DNA extraction, bacterial 16S rRNA genes amplification and Miseq sequencing
DNA was extracted from all swabs using TIANamp Swab DNA Kit (TIANGEN, Dusseldorf, Germany). To characterize vaginal (Group V) and intrauterine (Group I) microbial communities in ten healthy people (Group H), ten patients with both EP and CE (Group EP/CE) and ten patients with only EP (Group EP) (Table S1), the V4 region of a broad range of the bacterial and archaeal 16S rRNA genes was amplified [16]with a sample specific, 12-bp barcode sequences on the reverse primer [17] from the extracted DNA using the universal primer set of 515F/806R [18]. Roughly equal amounts of all amplicons were mixed in a single tube, and the amplicon mixture was gel-purified with an E.Z.N.A Gel Extraction Kit (Omega Bio-Tek, USA) and subsequently sequenced using the Illumina Miseq250 (Illumina, Inc. San Diego, California) [13].
DNA sequence data analysis and taxonomy
The paired-end Miseq 250 bp reads sequenced from the 16S rRNA gene (V4 region) PCR products, were firstly quality controlled using in-house perl scripts. Paired-end reads with one or more ambiguous bases were removed, and the retained paired-end reads were trimmed at the 3’ end to eliminate the continuous bases with a quality score < 20. Then the quality paired-end reads were combined using the “make.contigs” command in Mothur (version 1.35.0) [19] to get the full V4 region of 16S rRNA genes. The obtained 16S rRNA gene V4 region sequences were processed with the QIIME software (version 1.8.0) [20]. The sequences were firstly assigned to each sample based on the 12-bp barcoding sequences on the primer 806R, and those sequences with length shorter than 240 bp or longer than 260 bp, or with 8 bp long homopolymer were removed. Potential Chimeric sequences were detected using the software of Uchime [21]. Ultimately, we obtained an average of 9641 ± 777.8 reads per sample with a total of 578495 sequences (Table S1), for thirty intrauterine and thirty vaginal samples. Reads sharing 97% or higher sequence similarity were grouped into operational taxonomic units (OTUs) [20], and taxonomic assignment of representative sequences was performed using the Ribosomal Database Project (RDP) Classifier [22] a minimum confidence threshold of 80%. As a result, over 98% of the OTUs could be assigned to a taxonomic group (phylum), and over 50% could be identified at the genus level. In order to control differences in coverage, OTUs were randomly sub-sampled to 3000 sequences per sample for further analysis. We employed rarefaction curves of observed species (i.e., OTUs) to compare richness and Shannon diversity indices to compare the species diversity (alpha diversity) among different communities. In order to visualize differences in overall bacterial community composition, we employed the principal coordinates analysis (PCoA) based on weighted UniFrac distance. Moreover, analysis of Molecular Variance (AMOVA) or the analysis of similarities (ANOSIM) function based on the weighted UniFrac distance matrix was also conducted to test the differences in beta diversity between or among treatment groups. The sequences were deposited in the European Nucleotide Archive (ENA) database under the accession number of PRJEB9626.
Statistical analyses
Statistical analyses were performed with Sigma Stat statistical software (version 13.0 for windows). The Kolmogorov-Smirnov test was used to test for data normality. The data were presented in the study as means with standard deviations. Differences in the number of taxa detected in EP and healthy women were assessed with the use of the Mann-Whitney U test. The data were compared by the Student t test, the Mann-Whitney test, or the Kruskal-Wallis analysis of variance on ranks, followed by Dunn’s tests to adjust for multiple comparisons as appropriate. P < 0.05 was considered an indication of statistical significance.
Results
Significant differences between vaginal and intrauterine bacterial populations
To clearly characterize the different bacteria population in females’ vagina and uterus, we performed 16S rRNA sequencing, targeting V4 variable region. Our results showed that Firmicutes (65.4%) was the most abundant phylum and Actinobacteria (23.1%) was the second in the vaginal communities; however, compared with these two species in vagina, the mean proportion of them from intrauterine communities were lower, only at 35.1% and 8.9%, respectively (Figure 1A). Actually, the most abundant phylum in uterine cavity (Group I) was Proteobacteria which accounted for 45.3% (Figure 1A). At the genus level (Figure 1B and Table S2), all vaginal bacterial communities were dominated by Lactobacillus (55.1%), followed by Gardnerella (18.5%) and Streptococcus (6.3%); however, intrauterine bacterial communities were dominated by Lactobacillus (26.0%), Enterobacter (16.3) and Pseudomonas (12.6%). In additon, rarefaction curves based on a 97% cluster similarity showed the more number of OTUs in the intrauterine samples than those in the vaginal samples (Figure 1C). Furthermore, we applied UniFrac which is a phylogeny-based metric to evaluate the differences in overall bacterial community composition. A smaller UniFrac distance indicates that two communities are more similar, consisting of lineages sharing a common evolutionary history. As shown in Figure 1D, we observed that clustering was associated with sampling location. We demonstrate that the intrauterine microbiome is significantly different with the vaginal microbiome (AMOVA, P < 0.001, Table 2), indicating that swabs sampled in the uterine cavity were less likely to be contaminated by the vagina than expected. Table S2 shows most of genera in the samples from the vagina and uterine cavities. Meanwhile, some taxa failed to be assigned into any genus/phylum with a confidence level higher than 50%, suggesting they have not been reported before in vagina and uterine samples.
Figure 1.
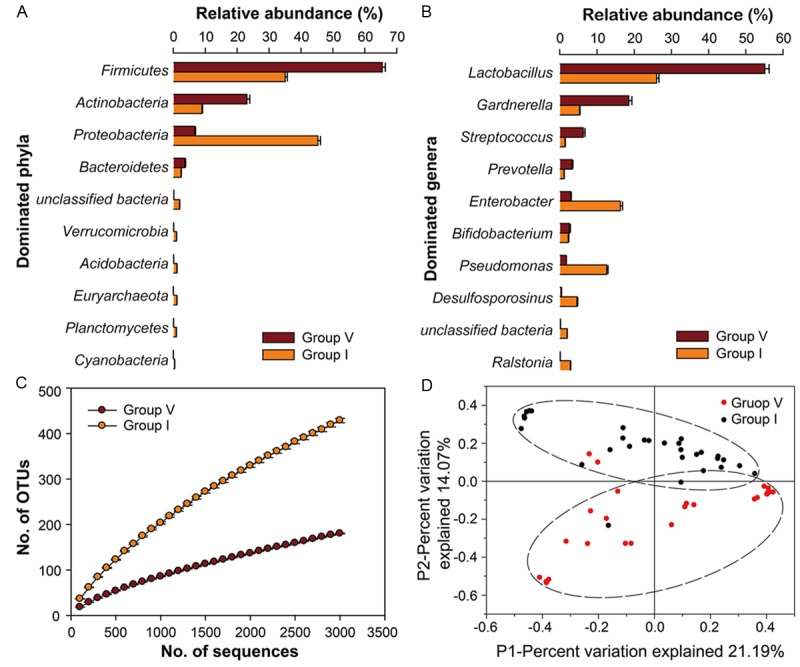
16S rRNA gene analysis revealing taxonomic variations between uterine and vaginal microbiota and high diversity of the uterine microbiota. A. Bar plots showing the relative abundances of the 10 most abundant bacterial phyla in the vagina and uterine samples. B. Bar plots showing the relative abundances of the 10 most abundant bacterial groups at genus level. C. Rarefaction curves for communities sampled from the vagina and uterus (based on 100-3000 sequences per V4 data set; standard error shown). D. Communities clustered using PCoA based on the weighted UniFrac distance matrix. The percentage of variation explained by the plotted principal coordinates is indicated on the axes. Group V, vagina; Group I, uterus.
Table 2.
AMOVA or ANOSIM on the bacterial strains isolated from intrauterine or vagina of healthy women and patients with EP
| Comparison | Source of Variation | d.f. | F stat value/R-value | P value |
|---|---|---|---|---|
| H (V)-H (I)1 | Among populations within regions | 1 | 8.15 | < 0.001* |
| Within populations | 18 | |||
| Total | 19 | |||
| EP/CE (V)-EP/CE (I)1 | Among populations within regions | 1 | 5.17 | < 0.001* |
| Within populations | 18 | |||
| Total | 19 | |||
| EP (V)-EP (I)1 | Among populations within regions | 1 | 3.07 | 0.003* |
| Within populations | 18 | |||
| Total | 19 | |||
| H (I)-EP/CE (I)-EP (I)2 | 0.31 | < 0.001* | ||
| H (I)-EP/CE (I)2 | 0.51 | < 0.001* | ||
| H (I)-EP (I)2 | 0.35 | 0.009 | ||
| EP/CE (I)-EP (I)2 | -0.02 | 0.578 |
AMOVA;
ANOSIM.
In all cases, the probability (P) of having a more extreme variance component thanthe observed value is P < 0.05.
Bacterial species richness and diversity between Groups H (I), EP/CE (I) and EP (I)
To investigate the bacterial species richness and diversity during different intrauterine, we subsequently carried out analysis of alpha diversity of the microbes. Rarefaction curves (Figure 2) show that intrauterine communities varied markedly in their level of bacterial diversity. Surprisingly, we found that the uteruses of healthy women are not sterile and actually harbor diverse kinds of bacteria. Moreover, the uterine cavities from patients with EP and CE harbors more phylotypes and have a significantly higher Shannon diversity index value than those only with or without EP, given our beta diversity analysis (Table S3).
Figure 2.
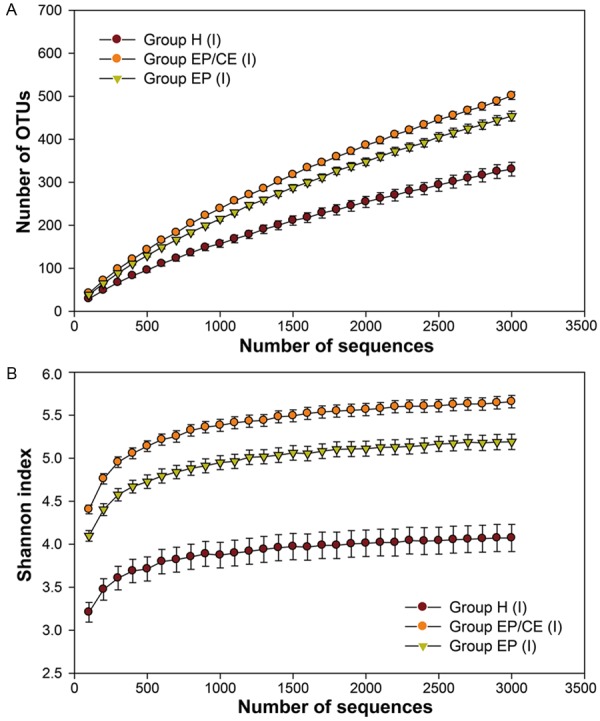
16S rRNA gene analysis indicating lower diversity in Group H (I) and high diversity in the Groups EP/CE (I) and EP (I). A. Rarefaction curves for intrauterine microbiome from Groups H (I), EP/CE (I) and EP (I). B. Shannon index rarefaction curves for intrauterine microbiome from Groups H (I), EP/CE (I) and EP (I). These data were based on 100-3000 sequences per V4 data set. The bars showed the standard errors of observed species and Shannon index of the sample for each group.
Bacterial community composite differences between Groups H (I), EP/CE (I) and EP (I)
We assessed differences in overall bacterial community composition using a phylogeny-based metric, UniFrac. The UniFrac-based principal coordinates analysis (PCoA, Figure 3) showed no apparent clustering among uterine communities. It seems that variation among the uterine communities is not obvious. However, for further analysis about the shared community structure of uterine cavities significantly associated with EP, an ANOSIM (analysis of similarity) test was conducted. The analysis shows that significant differences were present between Groups H (I) and EP/CE (I) (P < 0.001, Table 2) or Groups H (I) and EP (I) (P=0.009 < 0.05, Table 2), indicating that there is a significant relationship between EP and variation of intrauterine bacteria.
Figure 3.
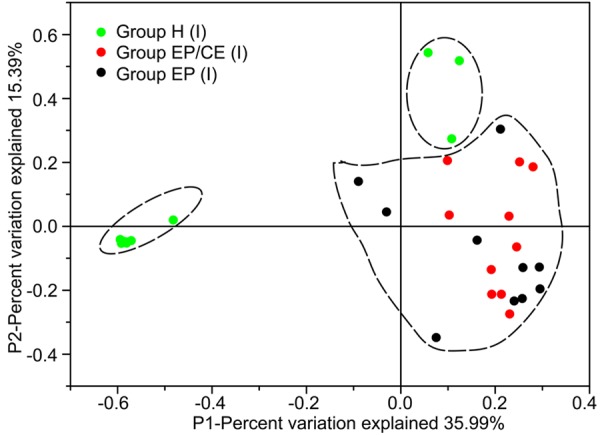
Communities clustered using Principal Component Analysis (PCoA) based on the weighted UniFrac distance matrix. PC1 and PC2 are plotted on x and y axes. Each point is equal to a sample colored by Groups H (I), EP/CE (I) and EP (I). The proportion of variation illustrated by the plotted Principal Component is indicated on the axes. The samples in Group H (I) can be separated distinctly from other samples in Groups EP/CE (I) and EP (I).
Bacterial composition and community structure at the phylum and genus level in Groups H (I), EP/CE (I) and EP (I)
To further clarify the imperative differences present in intrauterine microbiotas of these 30 individuals, we analyzed bacterial composition and community structure at the phylum and genus in Groups H (I), EP/CE (I) and EP (I). Figure 4A shows that these three Groups are all dominated by Proteobacteria (0.729 ± 0.019, 0.343 ± 0.010 and 0.287 ± 0.006), Firmicutes (0.139 ± 0.010, 0.430 ± 0.014 and 0.483 ± 0.014) and Actinobacteria (0.053 ± 0.004, 0.098 ± 0.007 and 0.117 ± 0.008) at the phylum level; however, relative abundance of bacteria-related sequences in different groups may shift significantly. Compared with Group H (I) (Firmicutes: 13.94% and Proteobacteria: 72.90%), the relative abundance of Firmicutes sequences was significantly higher in the uterine communities of Groups EP/CE (I) and EP (I) (43.00% and 48.27%; P < 0.05), whereas that of Proteobacteria sequences was lower markedly (34.30% and 28.73%; P < 0.05).
Figure 4.
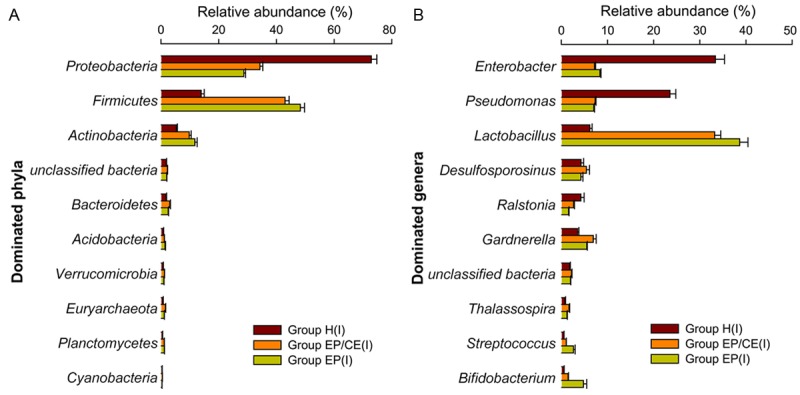
Composition of the top 10 taxa at the (A) phylum and (B) genus level for all intrauterine samples (Groups H (I), EP/CE (I), EP (I)). Relative abundance (percentage) of the 10 most abundant bacterial phyla (A) or genera (B) across 30 intrauterine microbiomes from healthy women (Group H (I)), EP patients with (Group EP/CE (I)) and without CE (Group EP [I]), based on 16S rRNA high throughput sequencing.
At the genus level (Figure 4B), the most abundant OTUs were assigned to Lactobacillus (Groups EP/CE and EP: 33.21% and 38.64%), Enterobacter (7.17% and 8.34%), Pseudomonas (7.32% and 7.02%), Gardnerella (6.91% and 5.50%) and Desulfosporosinus (5.41% and 4.23%) from Groups EP/CE and EP, while the most abundant genus detected in Group H (I) was Enterobacter (33.37%), followed closely by Pseudomonas (23.52%), Lactobacillus (6.17%), Desulfosporosinus (4.25%), Ralstonia (4.21%) and Gardnerella (3.54%). To further test difference in microbiomes between Groups H (I), EP/CE (I) and EP (I), Kruskal-Wallis test was required. Table 3 shows that among the 20 most abundant taxa presented in the communities, Groups EP/CE (I) and EP (I) were composed of more Lactobacillus, Bifidobacterium, Gardnerella, Streptococcus, Alteromonas and decreased Pseudomonas when compared with Group H (I). Meanwhile, the mean proportion of Enterobacter and Sphingomonas was lower and that of Prevotella was higher in Group EP/CE (I) when compared to Group H (I) (P<0.05). Some microorganisms failed to be assigned to a specific genus, but significant differences were present among the three groups - such as unclassified Euryarchaeota (Archaea, P [Group H (I) vs Group EP/CE (I)]: 0.0115; P [Group H (I) vs Group EP (I)]: 0.019) and unclassified Enterobacteriaceae (phylum, P [Group H (I) vs Group EP/CE (I)]: 0.0039; P [Group H (I) vs Group EP (I)]: 0.001).
Table 3.
Differential relative abundance of the 11 most abundant taxa in these uterine communities among healthy women (Group H), patients with EP and CE (Group EP/CE) and patients with unique EP (Group EP)
| Genus | Group H1 (n = 10) | Group EP/CE (n = 10) | Group EP (n = 10) | P value2 | ||
|---|---|---|---|---|---|---|
|
| ||||||
| H:EP/CE | H:EP | EP/CE:EP | ||||
| Lactobacillus | 0.062 ± 0.005 | 0.332 ± 0.013 | 0.386 ± 0.017 | 0.000 | 0.000 | 0.529 |
| Enterobacter | 0.334 ± 0.020 | 0.072 ± 0.002 | 0.083 ± 0.002 | 0.043 | 0.089 | 0.315 |
| Pseudomonas | 0.235 ± 0.013 | 0.073 ± 0.002 | 0.070 ± 0.002 | 0.011 | 0.015 | 0.529 |
| Gardnerella | 0.035 ± 0.003 | 0.069 ± 0.006 | 0.055 ± 0.001 | 0.043 | 0.023 | 0.912 |
| Bifidobacterium | 0.006 ± 0.000 | 0.014 ± 0.001 | 0.048 ± 0.017 | 0.019 | 0.005 | 0.481 |
| Streptococcus | 0.006 ± 0.000 | 0.011 ± 0.000 | 0.026 ± 0.003 | 0.001 | 0.000 | 0.023 |
| Prevotella | 0.008 ± 0.002 | 0.013 ± 0.002 | 0.013 ± 0.001 | 0.029 | 0.105 | 0.912 |
| unclassifid Enterobacteriaceae | 0.016 ± 0.000 | 0.010 ± 0.000 | 0.009 ± 0.000 | 0.004 | 0.001 | 0.190 |
| Alteromonas | 0.004 ± 0.001 | 0.014 ± 0.001 | 0.011 ± 0.001 | 0.003 | 0.019 | 0.739 |
| Euryarchaeota | 0.005 ± 0.001 | 0.010 ± 0.000 | 0.009 ± 0.001 | 0.011 | 0.019 | 0.912 |
| Sphingomonas | 0.008 ± 0.001 | 0.006 ± 0.000 | 0.004 ± 0.000 | 0.043 | 0.143 | 0.280 |
The average relative abundance of genus in the three groups were shown.
The P value was determined by Kruskal-Wallis test of the relative abundance of a given genus between two groups.
Discussion
The results of our study using sequence-based methods show that uterine microbiome is highly personalized and occurs universally through the comparisons between patients with EP and healthy women. The Proteobacteria, Firmicutes and Actinobacteria phyla are the predominate bacteria in the uterus. We suggest that there may be a correlation between the intrauterine microbiota and intrauterine lesions regardless of whether the vaginal microbiota changed.
However, there are some limitations to our study. The first limitation is that our results may not eliminate contamination from vagina and cervical canal. This is a feasibility limitation because it would necessitate transcervical sampling. In our study, samples in the uterine cavity were less likely to be contaminated by the vagina and endocervical canal contamination for several reasons. First, before collecting the intrauterine samples, we sterilized the vagina and endocervical canal in accordance with surgical demands after exposing the cervix using a vaginal speculum and finishing vaginal sampling. Second, when performing endometrial sampling using a 3-mm Novak curette connected to a 20-ml syringe, extreme care was taken to avoid any contact between the curette and vaginal walls. Third, our results revealed that the composition of microbial communities found in the uterine cavity were quite different from microbial populations found in the vagina. Indeed, the vaginal and intrauterine microbiome are composed of the Proteobacteria, Firmicutes and Actinobacteria phyla. However, the dominant bacteria species in the uterine are the Proteobacteria (45.3%) and Firmicutes (35.1%) phyla, while the vaginal microbiome is mainly made of Firmicutes (65.4%) and Actinobacteria (23.1%) phyla. Nevertheless, we found a lower association between vaginal and intrauterine samples, regarding both bacterial composition and diversity (ANOVA, P < 0.001, Table 2). On the other hand, as for cervical contamination, previous studies have shown that cervical and vaginal samples share similar microbial community compositions and that there is a high similarity between the relative quantities of the most abundant bacteria in the cervical and vaginal sites [23-27]. Another limitation to this study is its small sample size, which limits further analysis. However, it is the first study using molecular methods to assess intrauterine microbiome in patients with EP.
In our study, diverse bacterial species were found in the uterine cavity of both healthy women and patients with EP, indicating that the uterine cavity may not be sterile but harbors a rich and unique microbiome. On the contrary, it is believed that the uterine cavity is usually sterile. However, several researchers [28,29] have repeatedly raised the hypothesis based on observational studies that the uterine cavity couldn’t be free of bacteria because of continuous exposure to microorganisms present in the lower genital tract. Unfortunately, most conclusions were speculative since all previous studies detecting the microbiota of the uterine cavity depended on traditional culture techniques, by which most bacteria - especially unknown ones - failed to grow. In a recent study, Stout et al. [30], using morphological techniques, discovered intracellular bacteria in the basal plate of placental specimens without clinical or histologic evidence of chorioamnionitis. Similarly, Aagaard et al. [31], with molecular tools, demonstrated that the placenta harbors a low-abundance but metabolically rich microbiome. These studies raise the possibility that intrauterine colonization may be a possible source of the placenta microbiota. In other words, diverse bacterial communities, including high proportions of fastidious or anaerobic bacterial species, really exist in the uterine cavity. Most importantly, consistent with these studies, the data of Mitchell et al. [32] suggested that the endometrial cavity was not sterile in obtaining a hysterectomy specimen of most women undergoing hysterectomy and low levels of bacteria were detected in the upper genital tract by qPCRs. Regrettably, the use of selective qPCRs weren’t able to capture the entire microbiota.
Surprisingly, we found few differences in the composition and diversity of the microorganisms of the endometrial cavity between EP patients with and without CE. These results are supported by other research based on molecular biology method. Mitchell et al. [32] demonstrated that bacterial presence in the uterine cavity is not related to a significant inflammatory immune response, indicating that the existence of low-level commensal bacteria in the uterus is common and not pathological. However, our results were dissimilar to those found in the study of Ettore Cicinelli et al [8], who cultured diverse bacteria from the endometrial samples of 75% of women with CE. In their study, most of the specimens only tested a single kind of bacterium due to the limitation of culture techniques, and the positive endometrial cultures showed a high correlation to the existence of CE. The reason may be that the majority of commensal intrauterine microbiota can’t be cultivated and intrauterine bacterial colonization is not always benign. Pathological effects of intrauterine bacteria may relate to particularly virulent strains or species, high concentrations of bacteria, or polymicrobial dysbiosis at the endometrial surface.
Moreover, there were significant differences in the EP uterine microbiome compared with healthy women, indicating that alteration of intrauterine flora was found in patients with EP. It suggested that EP may influence the composition of the intrauterine microbiome, or on the contrary, the changes of the intrauterine microbiome may be one of the causes of endometrial polyps, or the results are just coincidence, or they interact as both cause and effect. This potentially critical issue is largely unexplored. Although little is known about the association between alteration of intrauterine flora and EP, previous studies demonstrated that bacteria might have a role in the development of hyperplasia [33-35] either by promoting the proliferation or inhibiting the apoptosis of cells. From our data, we found that an increased abundance of Lactobacillus, Bifidobacterium, Gardnerella, Streptococcus, Alteromonas, Prevotella and unclassified Euryarchaeota (Archaea) as well as a decreased abundance of Enterobacter (genus), Pseudomonas, Enterobacteriaceae (phylum) and Sphingomonas were present in the intrauterine microbiomes of EP patients with or without CE, while a variational abundance of Enterobacter (genus), Sphingomonas and Prevotella were absent in patients with EP and CE. However, there was no research reporting the relationship between some specific species and EP. Rather, the prior studies demonstrated that Lactobacillus and Bifidobacterium might play a vital role in promoting the proliferation and inhibiting the apoptosis of cells [33,36,37], supporting the hypothesis that Lactobacillus and Bifidobacterium might be related to the development of EP. Meanwhile, as the predominant gut microbiota, Escherichia coli had to be proven to inhibit cell proliferation [38,39]. It suggested that a decreased abundance of Enterobacter may play some part in an overgrowth of endometrial tissues, resulting in the formation of EP. Although some function regulated by local microbiomes was not yet clear, it showed considerably that patterns of variability could possibly be attributed to the development of EP.
Surprisingly, we detected Archaea in all samples whether they were from vagina or intrauterine, although that the number of Archaeal sequences was low in comparison to the number of bacterial sequences obtained from the same samples, using the same archaeal primers or bacterial primers (Table S2). To our knowledge, other study fail to find any Archaea in amniotic fluid from healthy or diseased females [40], aside from one single study reporting the occurrence of Archaea by cultivating in the human vaginal microbiome [41]. However, our results were not identical. For example, they isolated methanoarchaea (M. smithii) only from samples suffering from bacterial vaginosis and not healthy ones rather than all samples as we did. These differences might result from differences between experiment methods. Here, we utilized barcoded sequencing targeting V4 of the 16S rRNA gene to identify microbiome in human vagina and uterus, while Belay et al used the traditional culture method. Despite the short-read lengths (~250 bp), this targeted gene region should also provide sufficient resolution for the accurate taxonomic classification of microbial sequences including Archaeal and bacterial ones (Liu et al., 2007). Due to the different sensitivity and detection depth of experimental methods, the differences in results could reflect differences in the ability of certain bacteria present in female reproductive tract. Therefore, further studies are required to determine if this is true.
In general, with the molecular methods and bioinformatic tools, our study indicates that the uteruses of healthy women may harbor a rich and unique microbiome, which is significantly different from that of the vagina. Most importantly, our data reveals that composition and abundance of intrauterine microbiomes vary in patients with EP. This suggests four potential possibilities, 1) changes of microbiota play a role in the development of EP, 2) they do not play a role in the development of EP, 3) they are the result of EP, and not the cause, and 4) there are changes in the microbial communities that are both driving EP and a result of EP.
Conclusions
The uteruses of healthy women were not sterile and actually presented diverse kinds of bacteria. There were significant differences between the bacterial compositions of healthy women and patients with endometrial polyps. Compared to the non-EP uterine microbiome, sequencing experiments revealed that the EP uterine microbiome was composed of increased Lactobacillus, Bifidobacterium, Gardnerella, Streptococcus, Alteromonas and unclassified Euryarchaeota (Archaea) and decreased Pseudomonas and unclassified Enterobacteriaceae (phylum). The results also showed that the EP without CE uterine microbiome harbored decreased Enterobacter and Sphingomonas as well as increased Prevotella. These data suggest that potentially important differences exist between the uterine microbiomes of women with and without EP, but not in the patients with chronic endometritis.
Acknowledgements
We thank the following sources for funding: 1. Science and Technology Planning Project of Guangdong Province, China (2013B021800237), 2. Science and Technology Planning Project of Guangzhou, China (201300000169). In addition, we thank Dr. Tao-tao Yang of Sun Yat-Sen University of Guangzhou, Guangdong, the Republic of China, for his assistance in molecular experiments.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.AAGL practice report: practice guidelines for the diagnosis and management of endometrial polyps. J Minim Invasive Gynecol. 2012;19:3–10. doi: 10.1016/j.jmig.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 2.Salim S, Won H, Nesbitt-Hawes E, Campbell N, Abbott J. Diagnosis and management of endometrial polyps: a critical review of the literature. J Minim Invasive Gynecol. 2011;18:569–581. doi: 10.1016/j.jmig.2011.05.018. [DOI] [PubMed] [Google Scholar]
- 3.Lopes RG, Baracat EC, de Albuquerque Neto LC, Ramos JF, Yatabe S, Depesr DB, Lippi UG. Analysis of estrogen- and progesterone-receptor expression in endometrial polyps. J Minim Invasive Gynecol. 2007;14:300–303. doi: 10.1016/j.jmig.2006.10.022. [DOI] [PubMed] [Google Scholar]
- 4.McGurgan P, Taylor LJ, Duffy SR, O’Donovan PJ. Are endometrial polyps from pre-menopausal women similar to post-menopausal women? An immunohistochemical comparison of endometrial polyps from pre- and postmenopausal women. Maturitas. 2006;54:277–284. doi: 10.1016/j.maturitas.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 5.Vanni R, Dal Cin P, Marras S, Moerman P, Andria M, Valdes E, Deprest J, Van den Berghe H. Endometrial polyp: another benign tumor characterized by 12q13-q15 changes. Cancer Genet Cytogenet. 1993;68:32–33. doi: 10.1016/0165-4608(93)90070-3. [DOI] [PubMed] [Google Scholar]
- 6.Al-Jefout M, Black K, Schulke L, Berbic M, Luscombe G, Tokushige N, Manconi F, Markham R, Fraser IS. Novel finding of high density of activated mast cells in endometrial polyps. Fertil Steril. 2009;92:1104–1106. doi: 10.1016/j.fertnstert.2009.02.016. [DOI] [PubMed] [Google Scholar]
- 7.El-Hamarneh T, Hey-Cunningham AJ, Berbic M, Al-Jefout M, Fraser IS, Black K. Cellular immune environment in endometrial polyps. Fertil Steril. 2013;100:1364–1372. doi: 10.1016/j.fertnstert.2013.06.050. [DOI] [PubMed] [Google Scholar]
- 8.Cicinelli E, De Ziegler D, Nicoletti R, Tinelli R, Saliani N, Resta L, Bellavia M, De Vito D. Poor reliability of vaginal and endocervical cultures for evaluating microbiology of endometrial cavity in women with chronic endometritis. Gynecol Obstet Invest. 2009;68:108–115. doi: 10.1159/000223819. [DOI] [PubMed] [Google Scholar]
- 9.Kitaya K, Yasuo T. Aberrant expression of selectin E, CXCL1, and CXCL13 in chronic endometritis. Mod Pathol. 2010;23:1136–1146. doi: 10.1038/modpathol.2010.98. [DOI] [PubMed] [Google Scholar]
- 10.Espinoza J, Erez O, Romero R. Preconceptional antibiotic treatment to prevent preterm birth in women with a previous preterm delivery. Am J Obstet Gynecol. 2006;194:630–637. doi: 10.1016/j.ajog.2005.11.050. [DOI] [PubMed] [Google Scholar]
- 11.Cicinelli E, Matteo M, Tinelli R, Lepera A, Alfonso R, Indraccolo U, Marrocchella S, Greco P, Resta L. Prevalence of chronic endometritis in repeated unexplained implantation failure and the IVF success rate after antibiotic therapy. Hum Reprod. 2015;30:323–330. doi: 10.1093/humrep/deu292. [DOI] [PubMed] [Google Scholar]
- 12.Cicinelli E, Matteo M, Tinelli R, Pinto V, Marinaccio M, Indraccolo U, De Ziegler D, Resta L. Chronic endometritis due to common bacteria is prevalent in women with recurrent miscarriage as confirmed by improved pregnancy outcome after antibiotic treatment. Reprod Sci. 2014;21:640–647. doi: 10.1177/1933719113508817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol. 2013;79:5112–5120. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Greenwood SM, Moran JJ. Chronic endometritis: morphologic and clinical observations. Obstet Gynecol. 1981;58:176–184. [PubMed] [Google Scholar]
- 15.Kiviat NB, Wolner-Hanssen P, Eschenbach DA, Wasserheit JN, Paavonen JA, Bell TA, Critchlow CW, Stamm WE, Moore DE, Holmes KK. Endometrial histopathology in patients with culture-proved upper genital tract infection and laparoscopically diagnosed acute salpingitis. Am J Surg Pathol. 1990;14:167–175. doi: 10.1097/00000478-199002000-00008. [DOI] [PubMed] [Google Scholar]
- 16.Liu Z, Lozupone C, Hamady M, Bushman FD, Knight R. Short pyrosequencing reads suffice for accurate microbial community analysis. Nucleic Acids Res. 2007;35:e120. doi: 10.1093/nar/gkm541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamady M, Walker JJ, Harris JK, Gold NJ, Knight R. Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat Methods. 2008;5:235–237. doi: 10.1038/nmeth.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bates ST, Berg-Lyons D, Caporaso JG, Walters WA, Knight R, Fierer N. Examining the global distribution of dominant archaeal populations in soil. ISME J. 2011;5:908–917. doi: 10.1038/ismej.2010.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME allows analysis of highthroughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borgdorff H, Tsivtsivadze E, Verhelst R, Marzorati M, Jurriaans S, Ndayisaba GF, Schuren FH, van de Wijgert JH. Lactobacillus-dominated cervicovaginal microbiota associated with reduced HIV/STI prevalence and genital HIV viral load in African women. ISME J. 2014;8:1781–1793. doi: 10.1038/ismej.2014.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith BC, McAndrew T, Chen Z, Harari A, Barris DM, Viswanathan S, Rodriguez AC, Castle P, Herrero R, Schiffman M, Burk RD. The cervical microbiome over 7 years and a comparison of methodologies for its characterization. PLoS One. 2012;7:e40425. doi: 10.1371/journal.pone.0040425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ravel J, Gajer P, Abdo Z, Schneider GM, Koenig SS, McCulle SL, Karlebach S, Gorle R, Russell J, Tacket CO, Brotman RM, Davis CC, Ault K, Peralta L, Forney LJ. Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4680–4687. doi: 10.1073/pnas.1002611107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim TK, Thomas SM, Ho M, Sharma S, Reich CI, Frank JA, Yeater KM, Biggs DR, Nakamura N, Stumpf R, Leigh SR, Tapping RI, Blanke SR, Slauch JM, Gaskins HR, Weisbaum JS, Olsen GJ, Hoyer LL, Wilson BA. Heterogeneity of vaginal microbial communities within individuals. J Clin Microbiol. 2009;47:1181–1189. doi: 10.1128/JCM.00854-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nikolaitchouk N, Andersch B, Falsen E, Strombeck L, Mattsby-Baltzer I. The lower genital tract microbiota in relation to cytokine-, SLPI- and endotoxin levels: application of checkerboard DNA-DNA hybridization (CDH) Apmis. 2008;116:263–277. doi: 10.1111/j.1600-0463.2008.00808.x. [DOI] [PubMed] [Google Scholar]
- 28.Stumpf RM, Wilson BA, Rivera A, Yildirim S, Yeoman CJ, Polk JD, White BA, Leigh SR. The primate vaginal microbiome: comparative context and implications for human health and disease. Am J Phys Anthropol. 2013;152(Suppl 57):119–134. doi: 10.1002/ajpa.22395. [DOI] [PubMed] [Google Scholar]
- 29.White BA, Creedon DJ, Nelson KE, Wilson BA. The vaginal microbiome in health and disease. Trends Endocrinol Metab. 2011;22:389–393. doi: 10.1016/j.tem.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stout MJ, Conlon B, Landeau M, Lee I, Bower C, Zhao Q, Roehl KA, Nelson DM, Macones GA, Mysorekar IU. Identification of intracellular bacteria in the basal plate of the human placenta in term and preterm gestations. Am J Obstet Gynecol. 2013;208:226, e221–227. doi: 10.1016/j.ajog.2013.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aagaard K, Ma J, Antony KM, Ganu R, Petrosino J, Versalovic J. The placenta harbors a unique microbiome. Sci Transl Med. 2014;6:237ra265. doi: 10.1126/scitranslmed.3008599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mitchell CM, Haick A, Nkwopara E, Garcia R, Rendi M, Agnew K, Fredricks DN, Eschenbach D. Colonization of the upper genital tract by vaginal bacterial species in nonpregnant women. Am J Obstet Gynecol. 2015;212:611, e611–619. doi: 10.1016/j.ajog.2014.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ardita CS, Mercante JW, Kwon YM, Luo L, Crawford ME, Powell DN, Jones RM, Neish AS. Epithelial adhesion mediated by pilin SpaC is required for Lactobacillus rhamnosus GG-induced cellular responses. Appl Environ Microbiol. 2014;80:5068–5077. doi: 10.1128/AEM.01039-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwabe RF, Jobin C. The microbiome and cancer. Nat Rev Cancer. 2013;13:800–812. doi: 10.1038/nrc3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoshino Y, Kitazawa T, Ikeda M, Tatsuno K, Yanagimoto S, Okugawa S, Ota Y, Yotsuyanagi H. Clinical features of Bacteroides bacteremia and their association with colorectal carcinoma. Infection. 2012;40:63–67. doi: 10.1007/s15010-011-0159-8. [DOI] [PubMed] [Google Scholar]
- 36.Jones RM, Luo L, Ardita CS, Richardson AN, Kwon YM, Mercante JW, Alam A, Gates CL, Wu H, Swanson PA, Lambeth JD, Denning PW, Neish AS. Symbiotic lactobacilli stimulate gut epithelial proliferation via Nox-mediated generation of reactive oxygen species. EMBO J. 2013;32:3017–3028. doi: 10.1038/emboj.2013.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khailova L, Mount Patrick SK, Arganbright KM, Halpern MD, Kinouchi T, Dvorak B. Bifidobacterium bifidum reduces apoptosis in the intestinal epithelium in necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol. 2010;299:G1118–1127. doi: 10.1152/ajpgi.00131.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ren W, Yu R, Liu G, Li N, Peng Y, Wu M, Yin Y, Li Y, Fatufe AA, Li T. DNA vaccine encoding the major virulence factors of Shiga toxin type 2e (Stx2e)-expressing Escherichia coli induces protection in mice. Vaccine. 2013;31:367–372. doi: 10.1016/j.vaccine.2012.10.107. [DOI] [PubMed] [Google Scholar]
- 39.Nougayrede JP, Homburg S, Taieb F, Boury M, Brzuszkiewicz E, Gottschalk G, Buchrieser C, Hacker J, Dobrindt U, Oswald E. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science. 2006;313:848–851. doi: 10.1126/science.1127059. [DOI] [PubMed] [Google Scholar]
- 40.DiGiulio DB, Romero R, Kusanovic JP, Gomez R, Kim CJ, Seok KS, Gotsch F, Mazaki-Tovi S, Vaisbuch E, Sanders K, Bik EM, Chaiworapongsa T, Oyarzun E, Relman DA. Prevalence and diversity of microbes in the amniotic fluid, the fetal inflammatory response, and pregnancy outcome in women with preterm pre-labor rupture of membranes. Am J Reprod Immunol. 2010;64:38–57. doi: 10.1111/j.1600-0897.2010.00830.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Belay N, Mukhopadhyay B, Conway de Macario E, Galask R, Daniels L. Methanogenic bacteria in human vaginal samples. J Clin Microbiol. 1990;28:1666–1668. doi: 10.1128/jcm.28.7.1666-1668.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.