

Abstract
Methyl jasmonate (MJ) is a botanical hormone that serves as a signal transduction intermediate and regulates cell death in stressed plants. MJ induces cell cycle arrest, apoptosis and non-apoptotic cell death selectively in cancer cells. However, the underlying mechanism of MJ-induced apoptosis remains unclear. In this study, we examined the molecular mechanism through which MJ induces apoptosis in human non-small cell lung cancer (NSCLC). We found that MJ triggered apoptosis via the DDIT3-TNFRSF10B-CASP axis. MJ treatment significantly decreased the expression of CFLAR (CASP8 and FADD-like apoptosis regulator, an inhibitor of CASP8) in NSCLC cells, and ectopic expression of CFLAR partly protected cells from MJ-induced apoptosis. MJ also induced pro-apoptotic autophagy in NSCLC cells. Importantly, inhibition of ROS suppressed both MJ-induced apoptosis and autophagy. Taken together, MJ induces apoptosis and pro-apoptotic autophagy in NSCLC cells through the ROS pathway. Thus, MJ and its derivative treatment may serve as a novel chemotherapeutic strategy for cancer therapy.
Keywords: Methyl jasmonate, apoptosis, TNFRSF10B, DDIT3, autophagy, ROS
Introduction
MJ, a hydrophobic stress hormone, belongs to the jasmonate family that is ubiquitously produced in plants as a defensive component against insects, pathogens and other abiotic damages [1-3]. During the last decade, MJ has been shown to have selective anti-cancer effects [4-6]. Previous studies have shown that MJ arrests the cell cycle at different phases (G0/G1, G0/G1-S, S, S-G2/M and G2/M) and suppresses proliferation in several types of cancer possibly via down-regulation of Myc and up-regulation of p21 protein expression [7,8]. When bound to HK (hexokinase), a rate-limiting enzyme of glycolysis initiation, MJ disrupts the interaction between HK and VDAC (voltage-dependent anion channel) [9]. As HK is associated with mitochondria through VDAC, its over-expression will cause a high rate of glycolysis, which in turn helps cancer cells to survive and grow under aerobic conditions, known as the ‘Warburg effect’ [10]. Thus, the potential role of MJ to inhibit human cancer cell proliferation is indicated. It has also been reported that MJ induces p53-dependent apoptotic and p53-independent non-apoptotic cell death in cancer [8,11]. This may be caused by ROS induction by MJ [12]. In addition, MJ acts as an inhibitor of the NF-κB pathway and the pro-inflammatory 5-LOX-pathway to fight against inflammation in cancer progression [7,13]. Due to its multifaceted toxic effects, particularly against cancer cells, MJ is a promising novel anti-cancer drug that could be used in cancer therapy. However, the molecular mechanisms of MJ-induced apoptosis are still not fully understood. The discovery that the combination treatment of MJ and cisplatin, GA or X-ray irradiation significantly inhibits the survival of cancer cells characterizes the biological behavior of MJ in cancer cell death and may help improve strategies for cancer therapy [14,15].
Generally, there are two apoptotic signaling pathways in mammalian cells: the intrinsic mitochondrial pathway and the extrinsic death receptor pathway [16]. The extrinsic pathway can be induced through TRAIL (tumor necrosis factor-related apoptosis-inducing ligand) [17]. TNFRSF10B (death receptor 5) is a receptor of TRAIL located on the cell membrane. After cells receive the death signal, TNFRSF10B will be trimerized. Then, FADD (Fas-associated death domain) will be recruited, which subsequently forms the DISC complex (death-inducing signaling complex) with pro-CASP8 and CASP10 to initiate the caspase cascade, resulting in the activation of downstream apoptotic effectors [18-21]. CFLAR acts as a suppressor to the extrinsic pathway of apoptosis by preventing the activation of CASP8 [22,23].
MJ was shown to induce non-apoptotic cell death in p53-mutant cancer cells, and its role in necrosis has been clarified [11,24]. However, few studies have explored whether MJ induces autophagy. Macroautophagy (hereafter referred to as autophagy) is a highly conserved process by which unnecessary or damaged cytoplasmic materials are captured and degraded. After induction, the isolation membrane elongates, the autophagosome forms and then its outer membrane fuses with the lysosome (to form an autolysosome), finally leading to content degradation [25]. Autophagy is an indispensable pathway for substrate recycling and cellular homeostasis, indicating a pro-survival or a pro-apoptotic role, depending on cellular conditions [26-29]. The role autophagy plays in cancer cell death is still controversial.
Our study revealed that MJ induces apoptosis via the DDIT3-TNFRSF10B-CASP pathway, while simultaneously inducing pro-apoptotic autophagy in human NSCLC cells. MJ induces pro-apoptotic autophagy in human NSCLC cells, which suggests that combination treatment with MJ and pharmacological autophagy inducers may be an attractive therapeutic strategy for human NSCLC. In addition, our results indicate that MJ induces apoptosis and autophagy via triggering the ROS pathway.
Materials and methods
Cell lines and cell culture
All human NSCLC cell lines used in this study were obtained from the American Type Culture Collection (Manassas, VA). The stable cell line expressing EGFP-MAP1LC3B was constructed in our lab. The A549/CTRL, A549/CFLAR and H157/CTRL, H157/CFLAR cell lines were previously established by infection with lentivirus [30]. The cells were cultured in RMPI-1640 medium (Sigma-Aldrich, R6504) containing 5% fetal bovine serum (SAFC Global, 12003C) at 37°C in humidified atmosphere with 5% CO2 and 95% air. Cells were treated with MJ (Sigma-Aldrich, 392707), which was dissolved in ethanol at a concentration of 200 mM and stored at -20°C, LY294002 (Sigma-Aldrich, L9908) and E64D (Sigma-Aldrich, E8640) which were dissolved in DMSO. The control cells were treated with ethanol and/or DMSO. NAC was purchased from Sigma-Aldrich.
Cell survival assay
Human NSCLC cells were seeded in 96-well cell culture plates and treated with the indicated concentrations of MJ for 12, 24, or 48 h. After the treatment medium was discarded, the cells were fixed with 100 μl cold trichloroacetic acid (TCA) at 4°C for 1 h. Then, the plates were washed with deionized water five times and air dried. Subsequently, each well was incubated with 50 μl 0.4% sulforhodamine B (SRB) solution (dissolved in 1% acetic acid) for 5 min at room temperature. The plates were then washed five times with 1% acetic acid to remove all SRB. The bound SRB was dissolved with 100 μl 10 mM Tris base buffer (pH 10.5) and measured by absorbency using a microtiter plate reader at 495 nm. Absorbency was considered to be positively correlated with cell survival.
Western blot analysis
We have previously described the preparation of whole-cell protein lysates and the western blot protocol used in the present study [21]. The TNFRSF10B and ACTB antibodies were purchased from Sigma-Aldrich (St. Louis, MO, USA). The CFLAR, CASP8, PARP1 and MAP1LC3B antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). The CASP3 antibody was purchased from Imgenex (San Diego, CA, USA). The DDIT3 antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
FACS analysis
Apoptosis was measured by FACS analysis. Based on the Annexin-V-FITC/PI apoptosis detection kit protocol (Bio-BOX Biotech, China), 2×105 cells were harvested for each sample and washed with cold PBS. A total of 5 μl Annexin-V-FITC and 5 μl PI were added to the cells in 500 μl binding buffer for staining. Then, the cells were used for FACS analysis.
siRNA transfection
siRNAs targeting sequences of TNFRSF10B and DDIT3 were synthesized by GenePharma (Shanghai, China). The transfection was performed according to the X-tremeGENE transfection reagent protocol (Roche Molecular Biochemicals, Mannheim, Germany). Cells were cultured in 6-well plates and transfected with the indicated siRNAs. Then, cells were treated with the indicated concentrations of MJ for 24 h and subsequently harvested for Western blotting and FACS analysis. DDIT3 #1 and DDIT3 #2 siRNAs target the sequences 5’-GCC TGG TAT GAG GAC CTG C-3’ and 5’-AAG AAC CAG CAG AGG UCA CAA-3’, respectively. TNFRSF10B #1 and TNFRSF10B #2 siRNAs target the sequences 5’-AAG ACC CTT GTG CTC GTT GTC-3’ and 5’-AAG TTG CAG CCG TAG TCT TGA-3’, respectively.
Fluorescence microscopy
U87-MG-EGFP-MAP1LC3B cells were seeded in 24-well plates. After the indicated treatment, images were detected by fluorescence microscopy (Nikon TS100). Five images were randomly selected for counting the average number of EGFP-MAP1LC3B puncta per cell.
Statistical analysis
The data of EGFP-MAP1LC3B puncta are expressed as the mean ± S.D., and the differences between the groups were evaluated by Student’s t-test. In all statistical analyses, the results were considered to be statistically significant when the P-value was less than 0.05. The same method was used for the results of the FACS analysis.
Results
MJ inhibits cell proliferation in human NSCLC cells
To determine whether MJ inhibits proliferation of human NSCLC cells, we treated four human NSCLC cell lines, A549, Calu-1, H157 and H1792, with different concentrations of MJ (0.4 mM, 0.8 mM and 1.6 mM) for the indicated times (12 h, 24 h and 48 h) and measured cell proliferation by cell survival assay. We found that MJ significantly suppressed proliferation of all four cell lines in a dose-and time-dependent manner. Compared with control cells, 1.6 mM MJ resulted in up to 80% inhibition of cell proliferation at 48 h post-MJ treatment in four NSCLC cell lines (Figure 1A).
Figure 1.
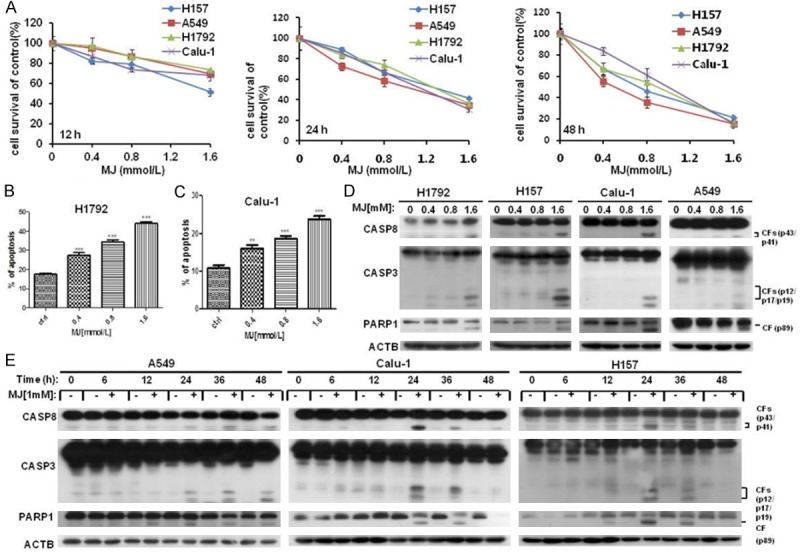
MJ inhibits cell proliferation in human NSCLC cells. (A) Four human NSCLC cell lines were incubated in 96-well cell culture plates and treated with 0.4 mM, 0.8 mM and 1.6 mM MJ for 12, 24, or 48 h. Then, cells were fixed, and cell proliferation was estimated by cell survival analysis. (B-E) MJ induces apoptosis in human NSCLC cell lines. (B and C) H1792 (B) and Calu-1 (C) cells were cultured in 6-well cell culture plates and treated with 0.4 mM, 0.8 mM and 1.6 mM MJ for 24 h. FACS analysis was then performed after staining the cells with Annexin V-FITC and PI. Columns show the percentage of apoptotic cells with different concentrations of MJ treatment. (D) Four human NSCLC cell lines were treated with the indicated concentrations of MJ. Then, full cell lysates were collected for each cell line, and the levels of CASP8, CASP3 and PARP1 were measured by western blot analysis. (E) A549, Calu-1 and H157 cells were treated with 1 mM MJ for 0, 6, 12, 24, 36, or 48 h. Then, full cell lysates were collected, and the levels of CASP8, CASP3 and PARP1 were measured by western blot analysis. Columns: mean values of triplicate treatments; bars: ± SD. The significant differences between the two treatments were analyzed by two-sided unpaired Student’s t-tests (**P < 0.05; ***P < 0.01; ****P < 0.001).
To explore the mechanism of MJ-induced cell survival inhibition in human NSCLC cells, FACS analysis was performed to examine whether MJ induced apoptosis in Calu-1 and H1792 cell lines after treatment at concentrations of 0, 0.4 mM, 0.8 mM and 1.6 mM for 24 hours. The results showed that apoptosis was dose-dependently induced after MJ treatment. Compared with control cells, 1.6 mM MJ resulted in up to approximately 50% apoptosis at 24 h post-MJ treatment (Figure 1B, 1C). To further justify this conclusion at the molecular level, the effect of MJ on the induction of apoptosis was determined by western blot analysis with MJ treatment for the indicated times and concentrations in A549, Calu-1, H157 and H1792 cell lines. The results showed that MJ dramatically triggered cleavage and activation of apoptosis-related proteins including CASP8, CASP3 and PARP1 (poly ADP-ribose polymerase 1, a substrate of CASP3) in both a dose- and time-dependent manner (Figure 1D, 1E). The evidence from both the FACS analysis and western blotting indicates an apoptosis-inducing role of MJ in human NSCLC cells.
MJ induces apoptosis via TNFRSF10B up-regulation in human NSCLC cells
The death receptor TNFRSF10B was also up-regulated after MJ exposure in human NSCLC cells. The dose-dependent western blotting results indicated that MJ up-regulated TNFRSF10B expression in all four NSCLC cell lines (Figure 2A). The time-course dependent western blotting results of 1 mM MJ treatment from 0 to 48 h showed that MJ increased TNFRSF10B protein levels at 6 h, and the effect peaked at 24 h after exposure in these cell lines (Figure 2B). To further determine whether MJ induces apoptosis via up-regulation of TNFRSF10B, we transfected Calu-1 and H1792 cells with siRNAs to inhibit the expression of TNFRSF10B and then measured apoptotic-related proteins following MJ exposure. Not only did the level of TNFRSF10B decrease compared with control cells, but the cleavage of CASP8, CASP3 and PARP1 were also suppressed (Figure 2C). The dramatic decrease in MJ-induced apoptosis in Calu-1 and H1792 cell lines after down-regulation of TNFRSF10B expression observed in the FACS analysis also served as evidence. Compared with control cells, MJ-induced apoptosis decreased up to 70% in TNFRSF10B down-regulated cells (Figure 2D). In conclusion, MJ up-regulates TNFRSF10B expression and activates the apoptotic cascade in human NSCLC cells.
Figure 2.
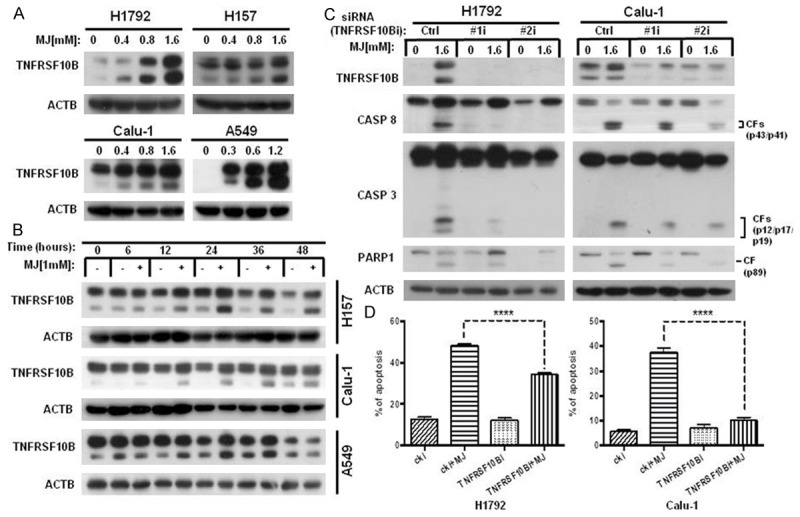
MJ induces apoptosis by up-regulating TNFRSF10B. A. H1792 and H157 cells were treated with 0.4 mM, 0.8 mM and 1.6 mM MJ for 24 h. Then, full cell lysates were collected, and the level of TNFRSF10B was measured by western blot analysis. B. A549, H157 and Calu-1 cells were treated with 1 mM MJ for 0, 6, 12, 24, 36, or 48 h. Then, full cell lysates were collected for each cell line, and the level of TNFRSF10B was measured by western blot analysis. C. H1792 and Calu-1 cells were transfected with TNFRSF10B siRNAs (#1 and #2). Then, cells were treated with 1.6 mM MJ for 24 h. Full cell lysates were collected for each cell line, and levels of TNFRSF10B, CASP8, CASP3 and PARP1 were measured by western blot analysis. D. H1792 and Calu-1 cells were transfected with TNFRSF10B siRNAs (#1 and #2). Then, cells were treated with 1.6 mM MJ for 24 h and subsequently stained with Annexin-V-FITC and PI for FACS analysis. Columns show the percentages of early and late apoptotic cells. Columns: mean values of triplicate treatments; bars: ± SD. The significant differences between the two treatments were analyzed by two-sided unpaired Student’s t-tests (**P < 0.05; ***P < 0.01; ****P < 0.001).
MJ up-regulates TNFRSF10B via DDIT3 induction
Because our earlier research suggested that DDIT3 (DNA-damage-inducible transcript 3) regulated the expression of TNFRSF10B in cancer cells [22,31], we hypothesized that MJ regulates DDIT3 and, thereby, up-regulates TNFSRF10B. To test this hypothesis, the expression levels of DDIT3 and TNFRSF10B were measured by western blot in four human NSCLC cell lines (A549, Calu-1, H157 and H1792) following MJ treatment at the indicated concentrations for 24 h. The protein level of DDIT3 increased along with TNFRSF10B in MJ-treated cells compared with control cells (Figure 3A). The time-dependent western blot showed similar results (Figure 3B). Furthermore, DDIT3 expression was inhibited in Calu-1 and H1792 cell lines with DDIT3 siRNAs. After transfection, we treated cells with 1.6 mM MJ for 24 h and then detected DDIT3, TNFRSF10B, CASP8, CASP3 and PARP1 at the molecular level. Knocking down DDIT3 clearly inhibited TNFSRF10B expression and activated CASP8, CASP3 and PARP1 in both cell lines (Figure 3C), indicating an antagonistic role of DDIT3 gene down-regulation in MJ-induced apoptosis. The dramatic decrease in MJ-induced apoptosis in Calu-1 and H1792 cell lines after down-regulation of DDIT3 expression observed in the FACS analysis also provided evidence. Compared with control cells, MJ-induced apoptosis decreased up to 60% in DDIT3 down-regulated cells (Figure 3D). These data suggest that MJ-induced DDIT3 up-regulation contributes to TNFRSF10B up-regulation and activates apoptosis cascades in NSCLC cells.
Figure 3.
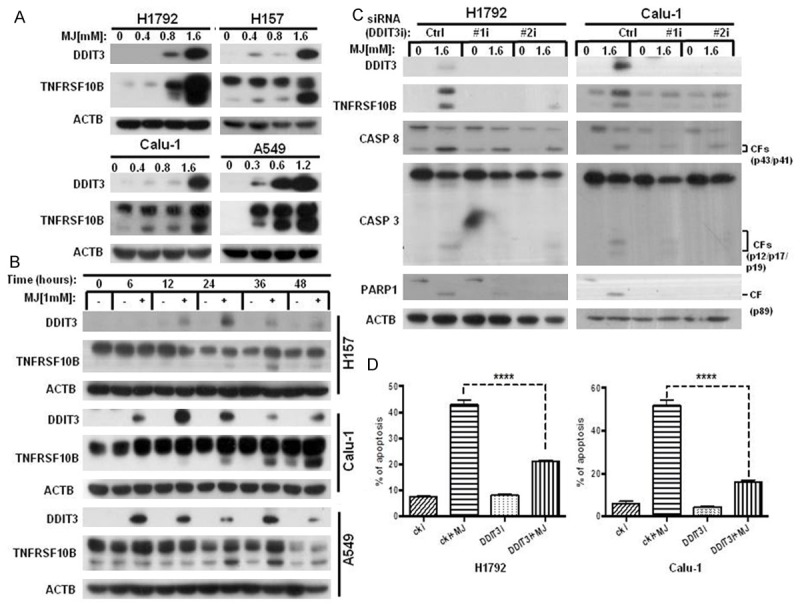
MJ up-regulates TNFRSF10B via DDIT3 induction. A. The four human NSCLC cell lines indicated were treated with 0.4 mM, 0.8 mM and 1.6 mM MJ for 24 h. Then, full cell lysates were collected for each cell line. DDIT3 and TNFRSF10B levels were measured by western blot analysis. B. H157 and Calu-1 cells were treated with 1 mM MJ for 0, 6, 12, 24, 36, or 48 h. Then, full cell lysates were collected for each cell line. DDIT3 and TNFRSF10B levels were measured by western blot analysis. C. H1792 and Calu-1 cells were transfected with DDIT3 siRNAs (#1 and #2). Then, cells were treated with 1.6 mM MJ for 24 h. Full cell lysates were collected for each cell line, and the levels of DDIT3, TNFRSF10B, CASP8, CASP3 and PARP1 were measured by western blot analysis. D. H1792 and Calu-1 cells were transfected with DDIT3 siRNAs (#1 and #2). Then, cells were treated with 1.6 mM MJ for 24 h and subsequently stained with Annexin-V-FITC and PI for FACS analysis. Columns show the percentages of early and late apoptotic cells. Columns: mean values of triplicate treatments; bars: ± SD. The significant differences between the two treatments were analyzed by two-sided unpaired Student’s t-tests (**P < 0.05; ***P < 0.01; ****P < 0.001).
CFLAR down-regulation is triggered in MJ-induced apoptosis
It is known that elevated expression of CFLAR blocks CASP8 activation and confers cancer cell resistance to death receptor-mediated apoptosis [23]. CFLAR is regarded as a promising target for enhancing cell sensitivity to apoptosis-inducing drugs. Thus, we further investigated the effect of MJ on CFLAR in human NSCLC cells. The data showed that CFLAR protein levels were clearly down-regulated by MJ treatment in both a dose- and time-dependent manner, which resulted in the promotion of apoptosis in human NSCLC cells (Figure 4A, 4B). To further determine whether the decrease in CFLAR was important in MJ-induced apoptosis, we examined the effects of MJ in A549/CFLAR cells, which exhibit ectopic CFLAR expression. Compared with A549/CTRL cells, we discovered that CFLAR over-expression reduced the cleavage of CASP8, CASP3 and PARP1 and partly rescued MJ-induced apoptosis. Similar results were obtained in H157/CTRL and H157/CFLAR cells (Figure 4C). The dramatic decrease of apoptosis in A549/CFLAR and H157/CFLAR cells after 1.6 mM MJ exposure for 24 h observed in the FACS analysis also provided evidence. Compared with control cells, MJ-induced apoptosis decreased up to 60% in CFLAR over-expressing cell lines (Figure 4D). Therefore, our data suggest that down-regulation of CFLAR is triggered in MJ-induced apoptosis.
Figure 4.
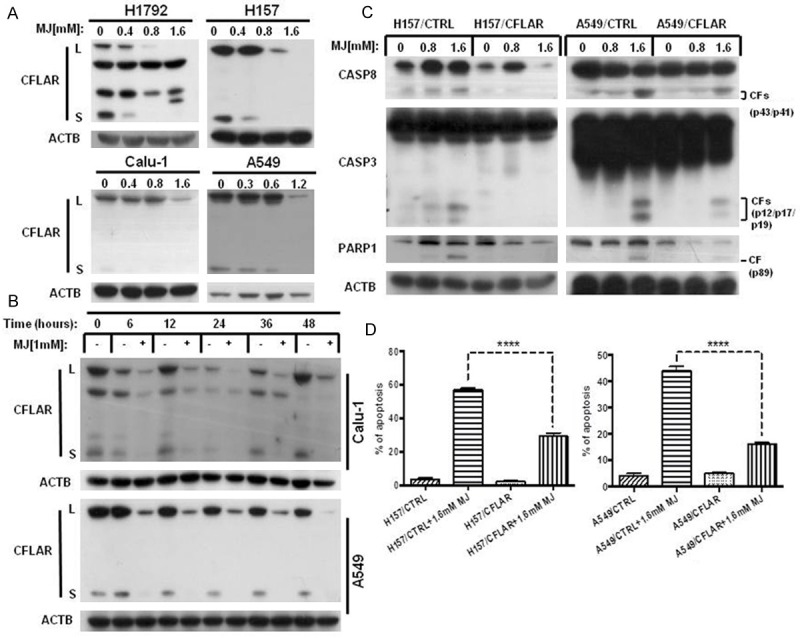
CFLAR down-regulation is triggered in MJ-induced apoptosis. A. The four human NSCLC cell lines indicated were treated with 0.4 mM, 0.8 mM and 1.6 mM MJ for 24 h. Then, full cell lysates were collected for each cell line, and the level of CFLAR was measured by western blot analysis. B. A549 and Calu-1 cells were treated with 1 mM MJ for 0, 6, 12, 24, 36, or 48 h. Then, full cell lysates were collected for each cell line, and the level of CFLAR was measured by western blot analysis. C. H157/CTRL, H157/CFLAR, A549/CTRL, and A549/CFLAR cells were treated with 0.8 mM and 1.6 mM MJ for 24 h. Full cell lysates were collected for each cell line, and the levels of CASP8, CASP3 and PARP1 were measured by western blot analysis. D. H157/CTRL, H157/CFLAR, A549/CTRL, and A549/CFLAR cells were treated with 1.6 mM MJ for 24 h and subsequently stained with Annexin-V-FITC and PI for FACS analysis. Columns show the percentages of early and late apoptotic cells. Columns: mean values of triplicate treatments; bars: ± SD. The significant differences between the two treatments were analyzed by two-sided unpaired Student’s t-tests (**P < 0.05; ***P < 0.01; ****P < 0.001).
MJ-induced autophagy plays a pro-apoptotic role
As MJ has been reported to induce non-apoptotic cell death in cancer cells, we speculated that MJ induced autophagy in human NSCLC cells. To monitor autophagosome formation, we used a U87-MG-EGFP-MAP1LC3B cell line stably expressing EGFP-MAP1LC3B, and treated cells with MJ at the indicated concentrations for 24 h and quantified the EGFP-MAP1LC3B fluorescence. The results showed that the average number of fluorescent EGFP-MAP1LC3B puncta per cell increased in a dose-dependent manner (Figure 5A). We also treated U87-MG-EGFP-MAP1LC3B cells with 1 mM MJ at the indicated times and found that the average number of fluorescent puncta per cell increased in a time-dependent manner (Figure 5B). Then, we treated A549, H157, Calu-1 and H1792 cell lines with MJ at the indicated concentrations for the indicated times and measured the expression of MAP1LC3BI/II by western blot analysis. We found that the protein levels of MAP1LC3B-II were increased following MJ treatment in both a dose- and time-dependent manner, while MAP1LC3B-I level was decreased (Figure 5C, 5D).
Figure 5.
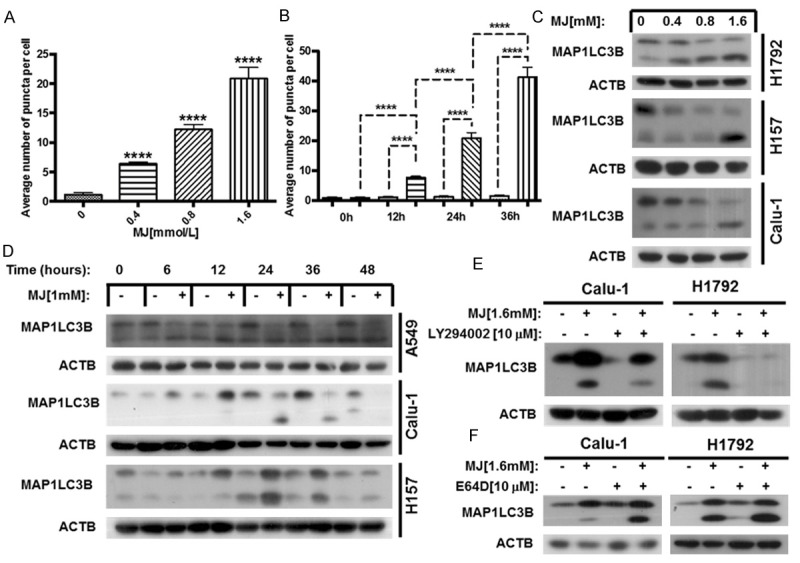
MJ induces autophagy in human NSCLC cells. A. The constructed U87-MG-EGFP-MAP1LC3B cell line was treated with MJ at the indicated concentrations for 24 h, and then fluorescent EGFP-MAP1LC3B puncta were quantified. B. The U87-MG-EGFP-MAP1LC3B cell line was treated with 1.6 mM MJ for 0, 12, 24, and 36 h, then fluorescent EGFP-MAP1LC3B puncta were quantified. C. Calu-1, H157 and H1792 cells were treated with 0.4 mM, 0.8 mM and 1.6 mM MJ for 24 h. Then, full cell lysates were collected for each cell line, and the level of MAP1LC3B was measured by western blot analysis. D. A549, Calu-1 and H157 cells were treated with 1 mM MJ for 0, 6, 12, 24, 36, or 48 h. Then, full cell lysates were collected for each cell line, the level of MAP1LC3B was measured by western blot analysis. E and F. H1792 and Calu-1 cells were treated with 10 mM LY294002 or E64D for 1 h before exposure to 1.6 mM MJ for 24 h. Then, full cell lysates were collected for each cell line, and the level of MAP1LC3B was measured by western blot analysis. The significant differences between the two treatments were analyzed by two-sided unpaired Student’s t-tests (**P < 0.05; ***P < 0.01; ****P < 0.001).
The dynamic generation and conversion of autophagosomes affects the level of MAP1LC3B-II. Therefore, up-regulation of MAP1LC3B-II may be caused by autophagy induction or downstream autophagy inhibition [32]. Accordingly, autophagic flux was monitored using the autophagy initiation inhibitor LY294002 and the lysosome protease inhibitor E64D in Calu-1 and H1792 cell lines. We found that co-treatment with MJ (1.6 mM) and LY294002 (10 μM) reduced the conversion of MAP1LC3B-II. In contrast, co-incubation with MJ (1.6 mM) and E64D (10 μM) increased the protein levels of MAP1LC3B-II (Figure 5E, 5F). These results demonstrate that the up-regulation of MAP1LC3B-II was caused by MJ-induced autophagy.
To explore the relationship between MJ-induced apoptosis and autophagy, we measured the molecular levels of the apoptosis-related proteins CASP8, CASP3 and PARP1 following co-treatment with MJ and the autophagy inhibitor LY294002. We found that the levels of apoptosis-related proteins exhibited the same alterations with MAP1LC3B-II (Figure 6A), suggesting that the inhibition of MJ-induced autophagy actually blocks MJ-induced apoptosis. The dramatic decrease of apoptosis in Calu-1 and H1792 cell lines after co-treatment with MJ and LY294002 observed in the FACS analysis also provided evidence. Compared with control cells, MJ-induced apoptosis decreased up to 40% with LY294002 treatment (Figure 6B). Therefore, MJ-induced autophagy plays a pro-apoptotic role.
Figure 6.
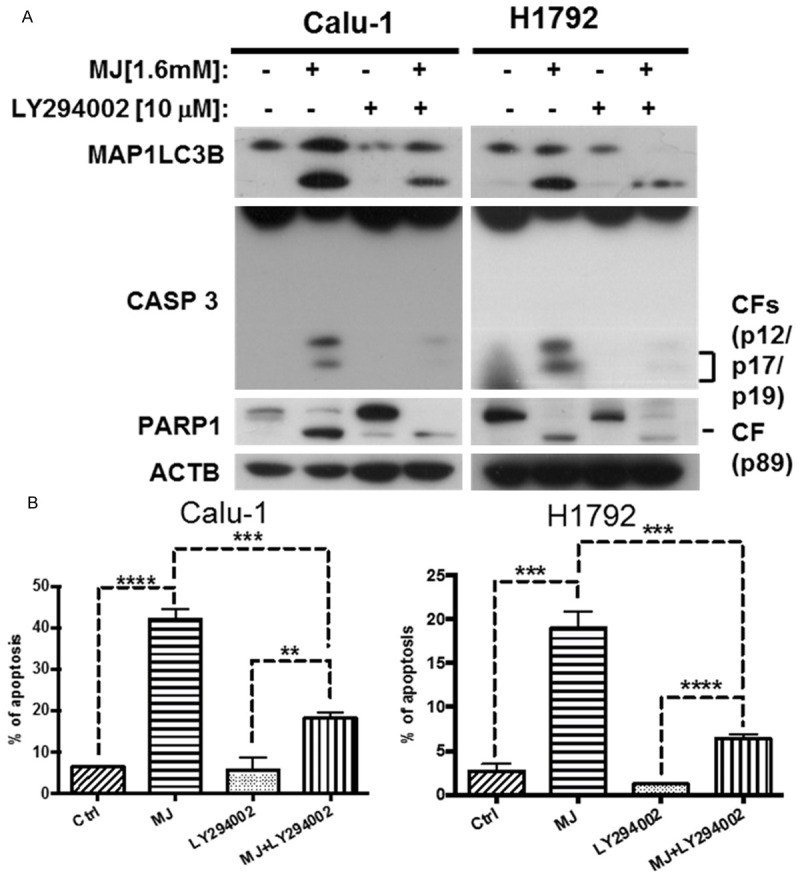
MJ-induced autophagy plays a pro-apoptotic role. A. H1792 and Calu-1 cells were treated with 10 mM LY294002 for 1 h before exposure to 1.6 mM MJ for 24 h. Then, full cell lysates were collected for each cell line, and the levels of MAP1LC3B, CASP8, CASP3 and PARP1 were measured by western blot analysis. B. Calu-1 and H1792 cells were co-treated with 1.6 mM MJ and 10 mM LY294002. Then, cells were stained with Annexin-V-FITC and PI for FACS analysis. Columns show the percentage of early apoptotic cells. Columns: mean values of triplicate treatments; bars: ± SD. The significant differences between the two treatments were analyzed by two-sided unpaired Student’s t-tests (**P < 0.05; ***P < 0.01; ****P < 0.001).
ROS accumulation promotes both MJ-induced apoptosis and autophagy
Excessive ROS promotes cell death [33,34]. Therefore, we further tested whether ROS inhibition affected MJ-induced apoptosis or autophagy. We used 5 mM of the free radical scavenger NAC (antioxidant N-acetylcysteine) to inhibit ROS generation [35]. Cell survival analysis showed that pre-incubation of NAC reduced growth inhibition in MJ-treated Calu-1 and H1792 cells (Figure 7A). To verify whether ROS has an impact on MJ-induced apoptosis and autophagy, we co-treated cells with NAC and MJ (cells were pre-incubated with NAC for 30 min) and we detected the apoptosis-related proteins DDIT3, TNFRSF10B, CASP8, CASP3, and PARP1 as well as MAP1LC3B, a hallmark of autophagy. Both MJ-induced apoptosis and autophagy declined following ROS inhibition (Figure 7B). Thus, we postulated that MJ induces apoptosis and autophagy through ROS accumulation.
Figure 7.
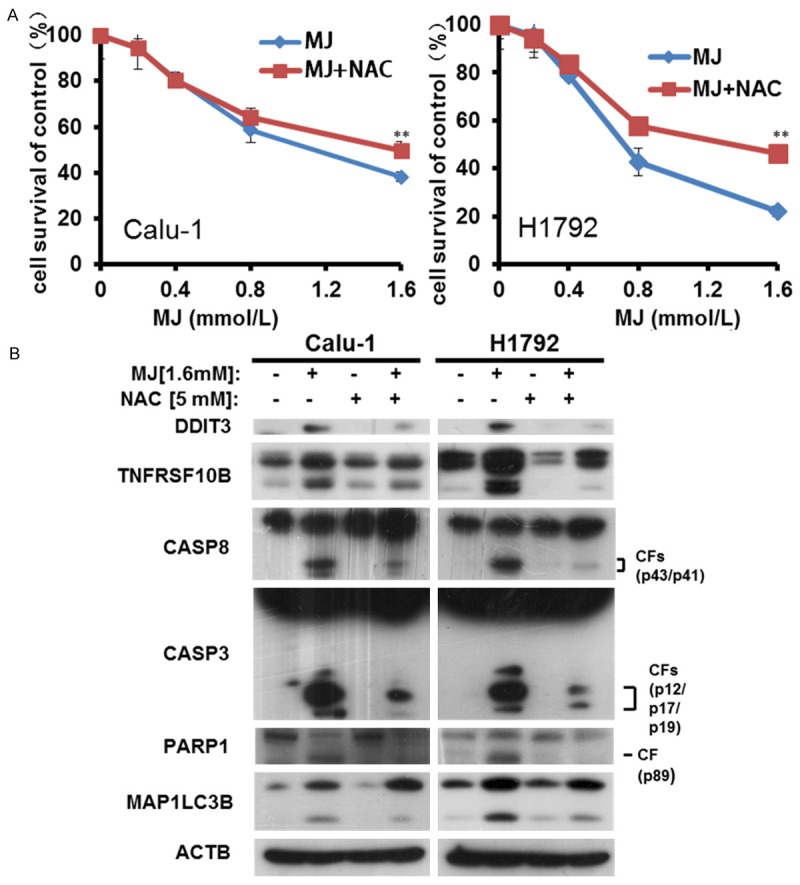
ROS accumulation promotes both MJ-induced apoptosis and autophagy. A. Calu-1 and H1792 cells were treated with 1.6 mM MJ and 5 mM NAC for 24 h. Then, cells were fixed, and cell proliferation was estimated by cell survival assay. B. Calu-1 and H1792 cells were treated with 1.6 mM MJ and 5 mM NAC for 24 h. Full cell lysates were collected for each cell line, and the levels of DDIT3, TNFRSF10B, CASP8, CASP3, PARP1, and MAPLC3B were measured by western blot analysis; bars: ± SD. The significant differences between the two treatments were analyzed by two-sided unpaired Student’s t-tests (**P < 0.05).
Discussion
MJ, a plant stress hormone, is reported to selectively eliminate cancer cells with promising efficiency as a result of its multiple cytotoxic effects and in combination with radio/chemical treatment methods [36]. There have been many efforts to decipher the molecular mechanism by which MJ induces cancer cell death, and yet the mechanism remains unclear. The main purposes of our research were: 1) to measure the feasibility of MJ as a natural anti-cancer drug for human cancer therapy; 2) to discover the mechanisms of MJ-induced apoptosis in human NSCLC cells; 3) to study the relationship between MJ-induced apoptosis and autophagy after demonstrating MJ-induced autophagy in human NSCLC cell lines for the first time.
In our study, cell survival in human NSCLC cell lines decreased after MJ treatment in both a dose- and time-dependent manner, indicating that MJ had toxic effects on human NSCLC cells. It has been reported that MJ induces p53-dependent apoptosis [7]. Our FACS analysis demonstrated that up to 40% of H1792 and Calu-1 cells entered early or late phase apoptosis upon exposure to 1.6 mM MJ. Cleavage and activation of apoptosis-related proteins such as CASP8, CASP3 and PARP1 were increased in both a dose- and time-dependent manner following MJ treatment in A549, H1792, H157 and Calu-1 cell lines. This indicates that MJ also induces p53-independent apoptosis (p53 is mutant in the H1792 cell line). However, the mechanism underlying MJ-induced apoptosis was unclear.
Our group and others have tested TNFRSF10B as a pivotal part of extrinsic apoptosis induced by certain agents [18,21]. We then showed that TNFRSF10B was up-regulated by MJ and that TNFRSF10B gene silencing resulted in subdued cleavage of CASPs, attenuating MJ-induced apoptosis. In addition, DDIT3 is known to regulate TNFRSF10B [22]. We then hypothesized that the mechanistic basis of the up-regulation of TNFRSF10B was the induction of DDIT3 by MJ. Up-regulation of DDIT3 was found to be accompanied by increased TNFRSF10B in human NSCLC cells. Moreover, knocking down DDIT3 abolished the increase in TNFRSF10B and rescued MJ-induced apoptosis, indicating that the MJ-induced up-regulation of TNFRSF10B is DDIT3-dependent. Considering that CFLAR is an important inhibitor of CASP8 and plays an essential role in the regulation of TRAIL-mediated extrinsic apoptosis [22,23], we also determined whether MJ affected CFLAR. After exposure to MJ, CFLAR was decreased, and over-expressing CFLAR weakened MJ-induced apoptosis. Therefore, we conclude that MJ induces apoptosis in human NSCLC cells via the DDIT3-TNFRSF10B-CASPs axis. Our results indicate that MJ would be a promising compound to induce apoptosis in human cancer cell lines.
Autophagy is a catabolic process that has a pivotal role in degrading and recycling damaged or harmful components to maintain cellular homeostasis [37]. By quantifying the aggregation of MJ-induced fluorescent puncta in U87-MG-EGFP-MAP1LC3B cells, we found that MJ induced autophagy in both a dose- and time-dependent manner in cancer cells. The results explain the observation of non-apoptotic cell death in a previous study following MJ exposure [7,11]. Similarly, our western blot analysis demonstrated that MJ stimulated MAP1LC3B-II aggregation in human NSCLC cells. To determine whether this phenomenon was caused by induction of up-stream autophagy or inhibition of down-stream autophagy [32], we tested the level of autophagic flux by co-incubation with MJ and the autophagy inhibitor LY294002. The results demonstrated that MJ induces autophagy in human NSCLC cells.
Given that MJ induces both apoptosis and autophagy in human NSCLC cells, we then explored the relationship between MJ-induced apoptosis and autophagy. Recent research has presented conflicting views of the pro-survival or pro-apoptotic role of autophagy in apoptosis, and this has been tested under different conditions [29,36]. However, our results showed that the level of MAP1LC3BII was positively correlated with the level of TNFRSF10B and the cleavage of CASP8, CASP3 and PARP1, which indicates that MJ-induced autophagy promotes apoptosis. When cells were treated with LY294002 (which prevents the formation of the autophagosome, thus inhibiting autophagy from the beginning), apoptosis was significantly reduced.
The role of autophagy in cell survival is context-specific. The results of our experiment prompted us to hypothesize that MJ-induced autophagy promotes cell death. There are many research studies aimed at determining whether autophagy promotes apoptosis or whether defective autophagy leads to tumor promotion, including through the p62-NRF2 pathway, the NF-κB pathway or other pathways [29,38,39]. While it has been reported that MJ induces ROS in cancer cells [12], we take this a step further, demonstrating that ROS also induces autophagy.
Our future studies will focus on the time sequence of apoptosis and autophagy or on linking the mechanisms between apoptosis and autophagy to explain why MJ-induced autophagy promotes apoptosis. Because autophagy appears to play a pro-apoptotic role in cell survival after MJ treatment, we suggest that combining MJ and autophagy accelerants in cancer therapy may have synergetic effects.
In summary, our study provides evidence for the underlying mechanism of MJ-induced apoptosis and autophagy in human NSCLC cells. MJ triggers ROS and then induces apoptosis by triggering the DDIT3-TNFRSF10B-CASPs axis as well as by down-regulating CFLAR. Simultaneously, MJ induces autophagy via ROS induction. Moreover, MJ-induced autophagy plays a pro-apoptotic role. Our findings provide evidence that MJ is a promising natural anti-cancer drug and predict that combination treatment with MJ and pharmacological autophagy promoters will be an effective strategy for cancer therapy.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (31371402, 81472686, 31571422, and 31171332).
Disclosure of conflict of interest
None.
References
- 1.Farmer EE, Ryan CA. Interplant communication: airborne methyl jasmonate induces synthesis of proteinase inhibitors in plant leaves. Proc Natl Acad Sci U S A. 1990;87:7713–7716. doi: 10.1073/pnas.87.19.7713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kessler A, Halitschke R, Baldwin IT. Silencing the jasmonate cascade: induced plant defenses and insect populations. Science. 2004;305:665–668. doi: 10.1126/science.1096931. [DOI] [PubMed] [Google Scholar]
- 3.Vijayan P, Shockey J, Lévesque CA, Cook RJ. A role for jasmonate in pathogen defense of Arabidopsis. Proc Natl Acad Sci U S A. 1998;95:7209–7214. doi: 10.1073/pnas.95.12.7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohen S, Flescher E. Methyl jasmonate: a plant stress hormone as an anti-cancer drug. Phytochemistry. 2009;70:1600–1609. doi: 10.1016/j.phytochem.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 5.Flescher E. Jasmonates-a new family of anti-cancer agents. Anticancer Drugs. 2005;16:911–916. doi: 10.1097/01.cad.0000176501.63680.80. [DOI] [PubMed] [Google Scholar]
- 6.Raviv Z, Cohen S, Reischer-Pelech D. The anti-cancer activities of jasmonates. Cancer Chemother Pharmacol. 2013;71:275–285. doi: 10.1007/s00280-012-2039-z. [DOI] [PubMed] [Google Scholar]
- 7.Cesari IM, Carvalho E, Figueiredo Rodrigues M, Mendonça Bdos S, Amôedo ND, Rumjanek FD. Methyl jasmonate: putative mechanisms of action on cancer cells cycle, metabolism, and apoptosis. Int J Cell Biol. 2014;2014:572097. doi: 10.1155/2014/572097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tong QS, Jiang GS, Zheng LD, Tang ST, Cai JB, Liu Y, Zeng FQ, Dong JH. Methyl jasmonate downregulates expression of proliferating cell nuclear antigen and induces apoptosis in human neuroblastoma cell lines. Anticancer Drugs. 2008;19:573–581. doi: 10.1097/CAD.0b013e3282fc46b0. [DOI] [PubMed] [Google Scholar]
- 9.Mathupala S, Ko Ya, Pedersen P. Hexokinase II: cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene. 2006;25:4777–4786. doi: 10.1038/sj.onc.1209603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pedersen PL. Warburg, me and Hexokinase 2: Multiple discoveries of key molecular events underlying one of cancers’ most common phenotypes, the “Warburg Effect”, ie, elevated glycolysis in the presence of oxygen. J Bioenerg Biomembr. 2007;39:211–222. doi: 10.1007/s10863-007-9094-x. [DOI] [PubMed] [Google Scholar]
- 11.Fingrut O, Reischer D, Rotem R, Goldin N, Altboum I, Zan-Bar I, Flescher E. Jasmonates induce nonapoptotic death in high-resistance mutant p53-expressing B-lymphoma cells. Br J Pharmacol. 2005;146:800–808. doi: 10.1038/sj.bjp.0706394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maillet A, Pervaiz S. Redox regulation of p53, redox effectors regulated by p53: a subtle balance. Antioxid Redox Signal. 2012;16:1285–1294. doi: 10.1089/ars.2011.4434. [DOI] [PubMed] [Google Scholar]
- 13.Ezekwudo DE, Wang RC, Elegbede JA. Methyl jasmonate induced apoptosis in human prostate carcinoma cells via 5-lipoxygenase dependent pathway. J Exp Ther Oncol. 2006;6:267–277. [PubMed] [Google Scholar]
- 14.Milrot E, Jackman A, Flescher E, Gonen P, Kelson I, Keisari Y, Sherman L. Enhanced killing of cervical cancer cells by combinations of methyl jasmonate with cisplatin, X or alpha radiation. Invest New Drugs. 2013;31:333–344. doi: 10.1007/s10637-012-9870-2. [DOI] [PubMed] [Google Scholar]
- 15.Wang Y, Xiang W, Wang M, Huang T, Xiao X, Wang L, Tao D, Dong L, Zeng F, Jiang G. Methyl jasmonate sensitizes human bladder cancer cells to gambogic acid-induced apoptosis through down-regulation of EZH2 expression by miR-101. Br J Pharmacol. 2014;171:618–635. doi: 10.1111/bph.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 17.Wajant H, Pfizenmaier K, Scheurich P. TNF-related apoptosis inducing ligand (TRAIL) and its receptors in tumor surveillance and cancer therapy. Apoptosis. 2002;7:449–459. doi: 10.1023/a:1020039225764. [DOI] [PubMed] [Google Scholar]
- 18.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 19.Bodmer JL, Holler N, Reynard S, Vinciguerra P, Schneider P, Juo P, Blenis J, Tschopp J. TRAIL receptor-2 signals apoptosis through FADD and caspase-8. Nat Cell Biol. 2000;2:241–243. doi: 10.1038/35008667. [DOI] [PubMed] [Google Scholar]
- 20.Gibson SB, Oyer R, Spalding AC, Anderson SM, Johnson GL. Increased expression of death receptors 4 and 5 synergizes the apoptosis response to combined treatment with etoposide and TRAIL. Mol Cell Biol. 2000;20:205–212. doi: 10.1128/mcb.20.1.205-212.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu X, Yue P, Zhou Z, Khuri FR, Sun SY. Death receptor regulation and celecoxib-induced apoptosis in human lung cancer cells. J Natl Cancer Inst. 2004;96:1769–1780. doi: 10.1093/jnci/djh322. [DOI] [PubMed] [Google Scholar]
- 22.Su L, Liu G, Hao X, Zhong N, Zhong D, Liu X, Singhal S. Death receptor 5 and cellular FLICE-inhibitory protein regulate pemetrexed-induced apoptosis in human lung cancer cells. Eur J Cancer. 2011;47:2471–2478. doi: 10.1016/j.ejca.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 23.Wajant H. Targeting the FLICE Inhibitory Protein (FLIP) in cancer therapy. Mol Interv. 2003;3:124. doi: 10.1124/mi.3.3.124. [DOI] [PubMed] [Google Scholar]
- 24.Kniazhanski T, Jackman A, Heyfets A, Gonen P, Flescher E, Sherman L. Methyl jasmonate induces cell death with mixed characteristics of apoptosis and necrosis in cervical cancer cells. Cancer Lett. 2008;271:34–46. doi: 10.1016/j.canlet.2008.05.031. [DOI] [PubMed] [Google Scholar]
- 25.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen HY, White E. Role of autophagy in cancer prevention. Cancer Prev Res. 2011;4:973–983. doi: 10.1158/1940-6207.CAPR-10-0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eskelinen EL. The dual role of autophagy in cancer. Curr Opin Pharmacol. 2011;11:294–300. doi: 10.1016/j.coph.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 28.Jiang Q, Li F, Shi K, Wu P, An J, Yang Y, Xu C. ATF4 activation by the p38MAPK-eIF4E axis mediates apoptosis and autophagy induced by selenite in Jurkat cells. FEBS Lett. 2013;587:2420–2429. doi: 10.1016/j.febslet.2013.06.011. [DOI] [PubMed] [Google Scholar]
- 29.White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12:401–410. doi: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao X, Liu X, Su L. Parthenolide induce apoptosis via TNFRSF10B and PMAIP1 pathways in human lung cancer cells. J Exp Clin Cancer Res. 2014;33:3. doi: 10.1186/1756-9966-33-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu G, Su L, Hao X, Zhong N, Zhong D, Singhal S, Liu X. Salermide up-regulates death receptor 5 expression through the ATF4-ATF3-CHOP axis and leads to apoptosis in human cancer cells. J Cell Mol Med. 2012;16:1618–1628. doi: 10.1111/j.1582-4934.2011.01401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005;1:84–91. doi: 10.4161/auto.1.2.1697. [DOI] [PubMed] [Google Scholar]
- 33.Jung EM, Lim JH, Lee TJ, Park JW, Choi KS, Kwon TK. Curcumin sensitizes tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis through reactive oxygen species-mediated upregulation of death receptor 5 (DR5) Carcinogenesis. 2005;26:1905–1913. doi: 10.1093/carcin/bgi167. [DOI] [PubMed] [Google Scholar]
- 34.Lee TJ, Um HJ, Park JW, Choi KS, Kwon TK. Withaferin A sensitizes TRAIL-induced apoptosis through reactive oxygen species-mediated up-regulation of death receptor 5 and down-regulation of c-FLIP. Free Rad Biol Med. 2009;46:1639–1649. doi: 10.1016/j.freeradbiomed.2009.03.022. [DOI] [PubMed] [Google Scholar]
- 35.Yang L, Su L, Cao C, Xu L, Zhong D, Xu L, Liu X. The chalcone 2’-hydroxy-4’,5’-dimethoxychalcone activates death receptor 5 pathway and leads to apoptosis in human nonsmall cell lung cancer cells. IUBMB Life. 2013;65:533–543. doi: 10.1002/iub.1161. [DOI] [PubMed] [Google Scholar]
- 36.Rotem R, Heyfets A, Fingrut O, Blickstein D, Shaklai M, Flescher E. Jasmonates: novel anticancer agents acting directly and selectively on human cancer cell mitochondria. Cancer Res. 2005;65:1984–1993. doi: 10.1158/0008-5472.CAN-04-3091. [DOI] [PubMed] [Google Scholar]
- 37.Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo JY, Xia B, White E. Autophagy-mediated tumor promotion. Cell. 2013;155:1216–1219. doi: 10.1016/j.cell.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]