

Abstract
Cancer-associated fibroblasts (CAFs) secrete various pro-tumorigenic cytokines, yet the role of these cytokines in the progression of endometrial cancer remains unclear. We found that CAFs isolated from human endometrial cancer (EC) tissues secreted high levels of interleukin-6 (IL-6), which promotes EC cell proliferation in vitro. Neutralizing IL-6 in CAF-conditioned media reduced (47% inhibition) while IL-6 recombinant protein increased cell proliferation (~2.4 fold) of both EC cell lines and primary cultures. IL-6 receptors (IL-6R and gp130) were expressed only in EC epithelial cells but not in CAF, indicating a one-way paracrine signaling. In the presence of CAF-conditioned media, Janus kinase/signal transducers and activators of transcription (JAK/STAT3) pathway was activated in EC cells. Treatment with JAK and STAT3 specific inhibitors, AD412 and STATTIC, respectively, significantly abrogated CAF-mediated cell proliferation, indicating the role of IL-6 activation in EC cell proliferation. We further showed that one of STAT-3 target genes, c-Myc, was highly induced in EC cells after exposure to CAF-conditioned medium at both mRNA (>105-fold vs. control) and protein level (>2-fold vs. control). EC cell proliferation was dependent on c-Myc expression, as RNAi-mediated c-Myc down-regulation led to a significant 46% reduction in cell viability when compared with scrambled control. Interestingly, CAF-conditioned media failed to promote proliferation in EC cells with reduced c-Myc expression, suggesting that CAF-mediated cell proliferation was also dependent on c-Myc expression. Subcutaneous tumor xenograft model showed that EC cells grew at least 1.4 times larger when co-injected with CAF, when compared to those injected with EC cells alone. Mice injected with EC cells with down-regulated c-Myc expression, however, showed at least 2.5 times smaller tumor compared to those in control group. Notably, there was no increase of tumor size when co-injected with CAFs. Further immunohistochemical staining on human tissues showed positive expression of IL-6 receptors, phosphorylated-STAT3 and c-Myc in human EC tissues with less signals in benign endometrium. Taken together, our data suggests that IL-6 secreted by CAF induces c-Myc expression to promote EC proliferation in vitro and in vivo. IL-6 pathway can be a potential target to disrupt tumor-stroma interaction in endometrial cancer progression.
Keywords: Uterine cancer, epithelial-stroma interaction, cytokine actions, tumor microenvironment, JAK/STAT3, c-Myc activation
Introduction
Endometrial cancer (EC) is the most common gynecological related carcinoma among women worldwide. The latest global EC incidence recorded 319,605 cases in 2012 which accounts for approximately 5% of all new cases for cancer in women [1]. Both EC incidence and death rates in the United States increased 21% since 2008, and increased more than 100% since the last two decades [2]. It is estimated in the recent GLOBOCAN 2012 report, that by 2025, half a million cases of EC would be diagnosed worldwide, emphasizing the exponential rate of this disease (http://globocan.iarc.fr). This increasing trend has been mainly attributed to the increasing rate of obesity among women and the use of tamoxifen as a mainstay treatment in breast cancer who later tend to develop EC [3]. Hysterectomy is the main treatment for patients suffering from EC and this disease is highly curable in early stages [1]. However, resistance to post-surgery treatment and disease recurrence tends to occur in later stages of this disease, rendering significant challenge in further treatment.
Recently, hormone (progesterone) treatment was shown to reduce the progression of EC through the action of the stroma in the EC tumor microenvironment [4]. This finding implies that the tumor microenvironment could be targeted to overcome treatment resistance and to improve the overall disease outcome. The tumor microenvironment, consisting of non-tumorigenic components such as cancer-associated fibroblasts (CAFs), macrophages, natural killer cells as well as lymphocytes, has been shown to sustain the growth and survival of tumors [5,6]. Oncogenic signaling from fibroblast cells surrounding tumor lesion affect the growth of tumor cells, particularly in prostate and pancreatic cancer [6-8]. In epithelial ovarian cancer, CAFs are highly abundant in FIGO stages III-IV compared to earlier stages [9,10], and have been shown to play a role in metastatic disease.
In endometrial cancer, we have previously shown that secretion from CAFs promoted the growth while those from benign fibroblasts inhibited the proliferation of EC cells in vitro. Cytokine profiling performed on these secretions showed high levels of interleukin-6 (IL-6) in CAFs compared to those in benign fibroblasts [11]. High levels of IL-6 are present in the serums of EC patients and are correlated with poor prognostic outcome in these patients [12]. While studies on the role of CAFs are quite abundant in other cancer types, their mechanism in EC is relatively understudied, especially on the tumorigenic role of the secreted IL-6. Hence, we aim to investigate the role of CAFs-secreted IL-6 in the EC progression.
In the current study, we demonstrated that IL-6 secreted by CAFs is responsible for promoting EC cell proliferation. Through IL6-R and gp130, IL-6 activated JAK/STAT3 signaling pathway, leading to induction of proto-oncogene c-Myc expression. Down-regulation of c-Myc expression significantly inhibited EC proliferation, and subsequently abrogated CAF-mediated cell proliferation. The dependence on IL-6 pathway was also observed in vivo in a tumor xenograft model. In addition, we showed that IL-6 downstream molecules including IL-6 receptors, phophorylated-STAT3 and c-Myc are highly expressed in human EC tissues but not in benign endometrial tissues. Our data strongly suggest that IL-6 pathway is activated in EC tumor cells following interaction with CAFs, leading to sustained cell proliferation. Hence, molecules activated in IL-6 pathway may potentially be targeted when designing novel therapeutic options for women with EC.
Materials and methods
Reagents and antibodies
LEAF™ purified anti-human IL-6 antibody, LEAF™ purified rat IgG1, κ Isotype control antibody and recombinant human IL-6 (carrier free) were purchased from Biolegend (CA, USA). STAT3 inhibitor V (STATTIC) and JAK3 inhibitor VII (AD412) were purchased from Santa Cruz Biotechnology (CA, USA), and c-Myc inhibitor, 10058-F4 was purchased from Sigma-Aldrich (MO, USA).
Ethics statement
Fresh EC tissues were obtained for establishment of primary culture from patients undergoing surgery at University of Malaya Medical Center. Endometrium formalin-fixed paraffin blocks for both benign and cancer conditions were obtained for immunohistochemistry work from the Biobank Unit of the University of Malaya. This study was approved by the University of Malaya Medical Center Ethics committee (Ref No. 865.19). Written informed consent was obtained from all participants.
Human endometrial cell lines and primary cultures establishment
Cell lines
Human endometrial cancer cell lines, ECC-1 (CRL-2923) and HEC-1A (HTB-112) and immortalized human normal endometrial fibroblast cell line, T-HESC (CRL-4003) were purchased from American Type Culture Collection (MD, USA) and were cultured in media according to manufacturer’s protocol supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin.
Primary cultures
All cultured primary cells obtained from surgical tissues were subjected to epithelial and stromal cell isolation using human CD326 (EpCAM) magnetic microbeads antibody and human anti-fibroblast magnetic microbeads (Miltenyi Biotech, Cologne, Germany), respectively as described previously [11].
Establishment of ECC-1 cell line with low c-Myc expression
ECC-1 cell and CAF cells (EC11-Fib) were transduced with red fluorescent protein (RFP) and green fluorescent protein (GFP) respectively (Gentarget, CA, USA). Selection was maintained by supplementing the cultures with puromycin with final concentration of 1 µg/ml (Sigma-Aldrich, MO, USA) for a period of 2 weeks. Consequently, the ECC-1 cell line was transfected with short hairpin RNA (shRNA) vector targeting c-Myc. GIPZ MYC shRNA viral particle starter kit was purchased from Dharmacon (CO, USA). Puromycin-resistant clones were selected in the presence of 1 μg/mL puromycin (Sigma-Aldrich).
Preparation of conditioned media from fibroblast cells
Fibroblast cells were seeded and cultured in complete media for 24 hours, before being cultured in media containing 2% FBS for the following 72 hours. Conditioned medium was collected using Amicon ultra centrifugal filters (Merck Milipore, MA, USA) by centrifugation at 5000 × g at 4°C for 1 hour. Protein in the concentrated media was quantified using Bradford assay (Biorad, CA, USA).
Enzyme-linked immunosorbent assay (ELISA)
Biolegend Human IL-6 and ELISA MCP-1/CCL2 MAX™ Deluxe (CA, USA) and Raybiotech #ELH-RANTES, #ELH-VEGF (GA, USA) were used to quantitate levels of these cytokines in conditioned media of CAFs. Briefly, 96 well plates were coated overnight with capture antibody. After blocking the plates with blocking buffer for 2 hours, conditioned media from ten different CAFs and control fibroblasts were added into the plate for another 2 hours, before addition of detection antibody for 1 hour, secondary antibody for 30 minutes and 3,3’5,5’-tetramethylbenzidine (TMB) substrate for 15 minutes. Reactions were terminated with STOP solution, before being analyzed at 450 nm wavelength using spectrophotometer. Assay sensitivity was between 2 to 10 pg/ml.
Methyl thiazolyl tetrazolium (MTT) assay
Proliferation of endometrial cancer cells was assessed by methyl thiazolyl tetrazolium (MTT) test. Briefly, cells were seeded in complete media at 1-3 × 104 cells/well in 96-well plates. At 24 hours post seeding, the cells were treated with either complete media, media with 2% FBS, fibroblast-conditioned media, neutralizing antibodies and/or inhibitors for 72 hours. At the end of treatment, 20 µl of MTT solution (5 mg/ml) was added to each well. Following 4 hours incubation at 37°C, 100 µl of 10% sodium dodecyl sulfate were added to dissolve the formazan crystals by additional 4 hours incubation at 37°C. Absorbance was measured using spectrometer at 575 nm with reference of 650 nm.
Quantitative real time polymerase chain reaction (qRT-PCR)
Total RNA were extracted from cultured cells using TRIzol (Invitrogen, CA, USA) and 1 μg RNA was converted into cDNA using DyNAmo cDNA synthesis kit (Finnzymes, Vantaa, Finland). Primer sequence used to detect IL-6 receptors and IL-6 downstream target genes are listed in Table 1. qRT-PCR was performed using ABI StepOne Plus (Applied Biosystem, CA, USA) in 35 cycles using 5 × HOT FIREPol EvaGreen qPCR Mix (Solis Biodyne, Tartu, Estonia), 10 pmol/µl forward and reverse primer, 10 ng/µl cDNA template and PCR grade H2O.
Table 1.
Primer sequences used for quantitative real time PCR
| Gene | Primers (5’-3’) | Source | |
|---|---|---|---|
| IL-6R | Forward | ATT GCC ATT GTT CTG AGG T | [40] |
| Reverse | TAG TCT GTA TTG CTG ATG TC | ||
| gp130 | Forward | ATT TGT GTG CTG AAG GAG GC | [41] |
| Reverse | AAA GGA CAG GAT GTT GCA GG | ||
| c-Myc | Forward | AAT GAA AAG GCC CCC AAG GTA GTT ATC C | [42] |
| Reverse | GTC GTT TCC GCA ACA AGT CCT CTT C | ||
| TIMP-1 | Forward | CTG TTG TTG CTG TGG CTG ATA | [43] |
| Reverse | CCG TCC ACA AGC AAT GAG T | ||
| PIM1 | Forward | AAC TGG TCT TCC TTT TTG GTT | [44] |
| Reverse | TAC CAT GCC AAC TGT ACA CAC | ||
| SOCS-3 | Forward | AGA CCT TCA GCT CCA AAA GC | [41] |
| Reverse | ACC AGC TTG AGT ACA CAG TCG | ||
| NFkB1 | Forward | AGG AAG AAA ATG GCG GAG TT | [45] |
| Reverse | GCA TAA GCT TCT GGC GTT TC | ||
| NFkB2 | Forward | CGT GAA AGA CCC TCC TGT TC | [46] |
| Reverse | AGA GCG AGA TCC GGA GTT G | ||
Total protein extraction and western blotting
ECC-1 cells were seeded at 1 × 104 cells/well in 6-well plates in complete media. At 24 hours post seeding, the cells were treated with either complete media, media with 2% FBS, fibroblasts-conditioned media, neutralizing antibodies and/or inhibitors for 72 hours. Protein lysates were collected by scraping the cells in cold lysis buffer containing final concentration of 0.1% Triton-X, 0.1% SDS, 50 mM Tris, 150 mM NaCl, 1 × phosphatase and 1 × protease inhibitors. Protein concentration was quantified using Bradford assay (Biorad, CA, USA). Approximately 30 μg of protein were resolved on 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis before being transferred onto polyvinylidene difluoride membrane. Antibodies used were rabbit anti-human phospho-JAK3, JAK3, phospho-STAT3, c-myc, mouse anti-human STAT3 and β-actin (Cell Signaling Technology, MA, USA). Blots were visualized using ECL prime western blotting detection reagent (Amersham, GE Healthcare Lifesciences, Sweden) using gel documentation system (Biospectrum 410, UVP) and Vision Works LS software (CA, USA).
Immunohistochemical analysis
Tumors removed during debulking surgery were fixed overnight in neutral buffered formalin prior to paraffin wax professing and embedding. Tissue sections were cut at 4 µm size. For immunohistochemical analysis, endogenous peroxidase was blocked with 0.3% hydrogen peroxide for 10 minutes. Antigen was retrieved using sodium citrate buffer method by heating at 100°C for 30 minutes. Slides were then incubated with one of the following antibodies for 1 hour, diluted according to the manufacturer protocol: IL-6Rα and gp130 (Santa Cruz Biotechnology, CA, USA), p-STAT3 and c-Myc (Cell Signalling Technology, MA, USA). A labelled streptavidin biotin-system with a horse-radish peroxidase label (DAKO Corp., CA, USA) was used to detect the primary antibodies and visualized by incubation with 3,3’-diaminobenzidine chromogen and hydrogen peroxide substrate (DAKO Corp., CA, USA) for 10 minutes. The slides were then counterstained with hematoxylin and mounted in dibutyl phathalate xylene (Sigma, MO, USA). Images were viewed and captured using Nikon Eclipse 2000 and Nikon ES-Fi1 respectively (Nikon GamBH, Dusseldorf, Germany).
EC tumor xenograft model
Athymic female nude mice (BALB/c, 4 weeks-old) were obtained from Taconic (Cambridge, MA, USA) and were quarantined for 2 weeks. The mice were divided into 7 groups (n=8/group) and were given subcutaneous injection of either ECC-1 cells (5,000 cells) alone or in combination with fibroblast cells (20,000 cells). The groups were Group 1-ECC-1 only; Group 2-EC11-Fib only; Group 3-combination of ECC-1 and EC11-Fib; Group 4-ECC-1 transfected with c-Myc scramble shRNA; Group 5-combination of ECC-1 transfected with c-Myc scramble shRNA and EC11-Fib; Group 6-ECC-1 transfected with c-Myc knockdown shRNA construct; Group 7-combination of ECC-1 transfected with c-Myc knockdown shRNA construct and EC11-Fib. Mice were monitored every other day and tumor size was measured twice weekly using calipers. Tumor volume was calculated according to equation: tumor volume = length (L) × width (W)2/2. Tumors were also imaged once a week using Carestream MS FX-PRO small rodent imager (Molecular Imaging, CT, USA). Animals were maintained in specific pathogen free conditions in an AALAAC-accredited animal housing facility, and were fed with Teklad Global 19% protein extruded rodent diet (Harlan Laboratories, WI, USA). All mice procedures were approved by IACUC committee of University of Malaya (FAR/27/07/2012/IC (R)).
Statistical analysis
Statistical analysis that assessed the differences between means of control and test group was performed using Student’s t-test on GraphPad-Prism (GraphPad Software, CA, USA). A P-value <0.05 was considered to be statistically significant.
Results
CAFs secretes higher levels of inflammatory cytokines compared to control fibroblasts
We previously showed that CAFs secrete high level of inflammatory cytokines compared to the benign counterparts [11]. We further validated this finding in twenty primary fibroblast cultures established from benign and malignant endometrial tissues. Using ELISA, we observed that IL-6 level was at least 10-fold higher in CAFs (897.6 pg/ml) than in benign fibroblasts (87.89 pg/ml) secretion (Figure 1). Other than IL-6, there were also higher levels of MCP-1 (5.6-fold), VEGF (3.3-fold) and RANTES (3-fold) in CAFs secretion than in those from benign fibroblasts (Figure 1).
Figure 1.
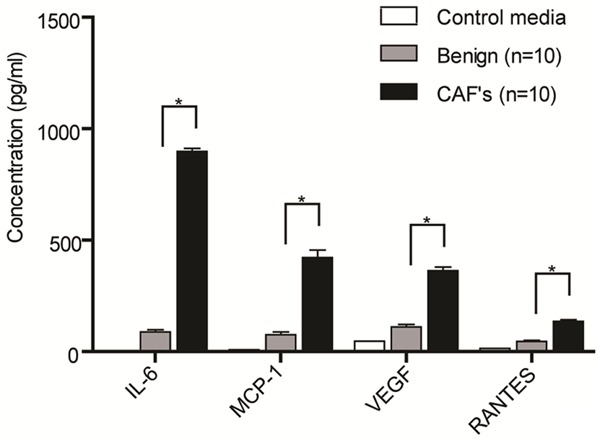
Differential levels of cytokine secreted by cancer and benign-associated fibroblasts. Endometrial cancer and benign-associated fibroblasts were cultured for 72 hours in media containing 2% fetal bovine serum, prior to collection of conditioned media. Cytokines levels were measured using ELISA. Data shown are representative of two independent experiments. Data, average; error bar, S.E.M. *P<0.05.
Activation of IL-6 signaling cascade in EC upon CAFs secretion
To determine if IL-6 mediates EC proliferation, we treated endometrial cancer cell line, ECC-1 and primary endometrial cancer cell, EC6-Ep with IL-6 neutralizing antibody in the presence of CAFs conditioned media. While there were no changes in cell viability in cells treated with isotype control, increasing concentrations of IL-6 neutralizing antibody led to a remarkable inhibitory effect of almost 50% (Figure 2A, 2B). Without CAFs conditioned media, both IL-6 neutralizing antibody and isotype control caused only 5% inhibition in cell proliferation compared to control (non-treated cells) (Figure 2C). Additionally, ECC-1 and EC-6 Ep cells treated with IL-6 recombinant protein without the presence of CAFs conditioned media showed dose-response increase in cell proliferation (Figure 2D). This indicates that IL-6 was present in CAFs secretion and had directly induced EC cell proliferation.
Figure 2.
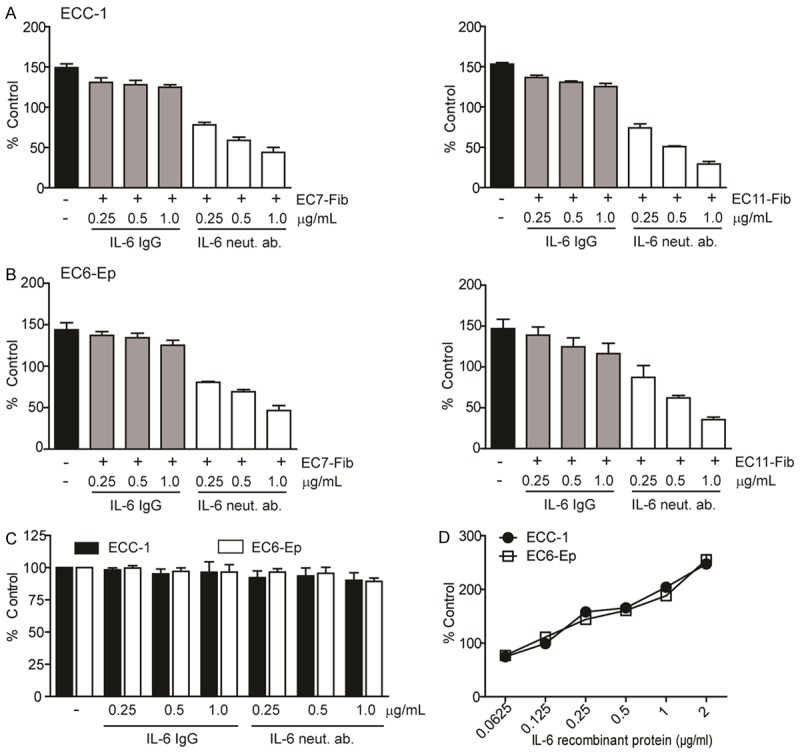
IL-6 secreted by CAFs mediates EC cell proliferation. A, B. ECC-1 cell line and EC6-Ep primary cell were treated with IL-6 neutralizing antibody and its IgG control in the presence of either 1 mg/ml EC7-Fib (left) or EC11-Fib (right) conditioned media. C. ECC-1 and EC6-Ep cells were treated similarly with IL-6 neutralizing antibody and its IgG control but without the presence of CAFs conditioned media. Black bars indicate ECC-1 cells in complete media without any treatment. D. ECC-1 and EC6-Ep cells were treated with IL-6 recombinant protein without the presence of conditioned media. Cell viability was examined using MTT assay and normalized to control media (media containing 2% FBS). Data, average; error bars, S.E.M. Data shown are representative of three independent experiments.
We further showed that IL-6 receptors, IL-6R and gp-130, were expressed only in EC epithelial cells but not in CAFs, at mRNA and protein levels (Figure 3A). Activation of IL-6 receptors by CAF secretion resulted in ~2-fold elevation of both phospho-JAK3 and phospho-STAT3 protein levels in ECC-1 cells (Figure 3B). This activation was abrogated by IL-6 neutralizing antibody, even in the presence of CAFs conditioned media (Figure 3C). Treatment of ECC-1 cells with AD412, a JAK3 selective inhibitor and STATTIC, a STAT3 selective inhibitor in the presence CAFs conditioned media, significantly down-regulated ECC-1 cell proliferation, to about 50% inhibition at the lowest dose of 10 µM (P<0.0001) (Figure 3D). Cells were minimally affected after treatment with vehicle and inhibitors alone. Taken together, our data indicates that IL-6 secreted by CAFs induced paracrine signaling in EC cells, resulting in activation of IL-6 receptor pathway.
Figure 3.
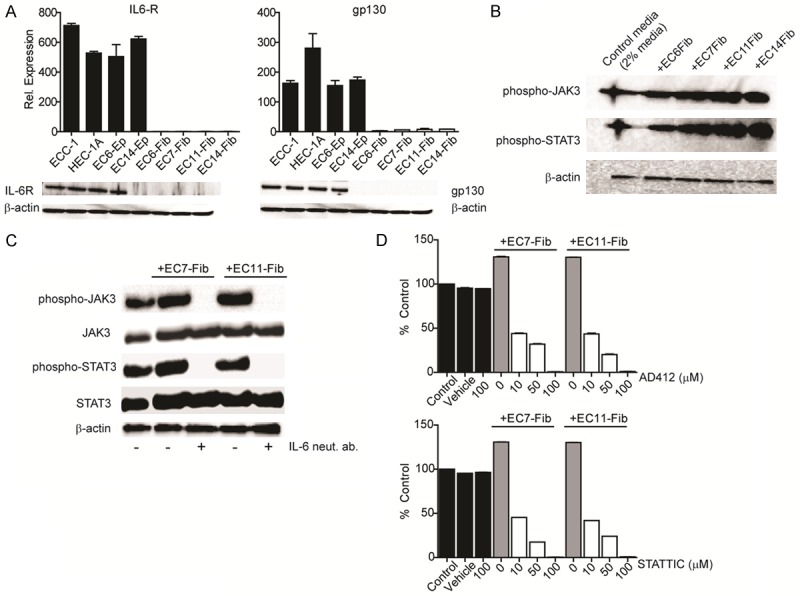
Activation of JAK3/STAT3 signaling is required for CAFs-mediated EC proliferation. A. Expression of IL-6R and gp130 in endometrial epithelial and fibroblast cells were determined using quantitative real-time PCR and Western blot analysis. B. Western blot analysis of phosphorylated-JAK3 and -STAT3 protein expression in ECC-1 cells following treatment with four different CAFs (EC6-Fib, EC7-Fib, EC11-Fib, EC14-Fib). C. Expression of phospho-JAK3 and –STAT3 were determined in ECC-1 cells treated with IL-6 neutralizing antibody in the presence of CAFs conditioned media. D. ECC-1 cells were treated with either JAK3 selective inhibitor (AD 412) or STAT3 selective inhibitor (STATTIC) in the presence of CAFs conditioned media (1 mg/ml) for 72 hours. Cell viability was examined using MTT assay and normalized to control media (media containing 2% FBS). Data, average; error bars, S.E.M. Data shown are representative of two independent experiments.
c-Myc regulates CAFs-mediated EC proliferation
To determine the induction of STAT3 further downstream genes, we measured the expression of six STAT3 target genes that are related to cancer cell proliferation (Figure 4A). Compared to non-treated control, c-Myc was significantly induced in ECC-1 cells, with at least 100-folds at mRNA, and ~2-4 folds at protein levels (Figure 4A, 4B). Similarly, TIMP-1 and SOCS-3 mRNA expressions were higher in the treated ECC-1 cells compared to control. Interestingly, PIM1 was the only gene down-regulated in ECC-1 cells after treatment with different CAFs (<0.5 fold vs. control) (Figure 4A).
Figure 4.
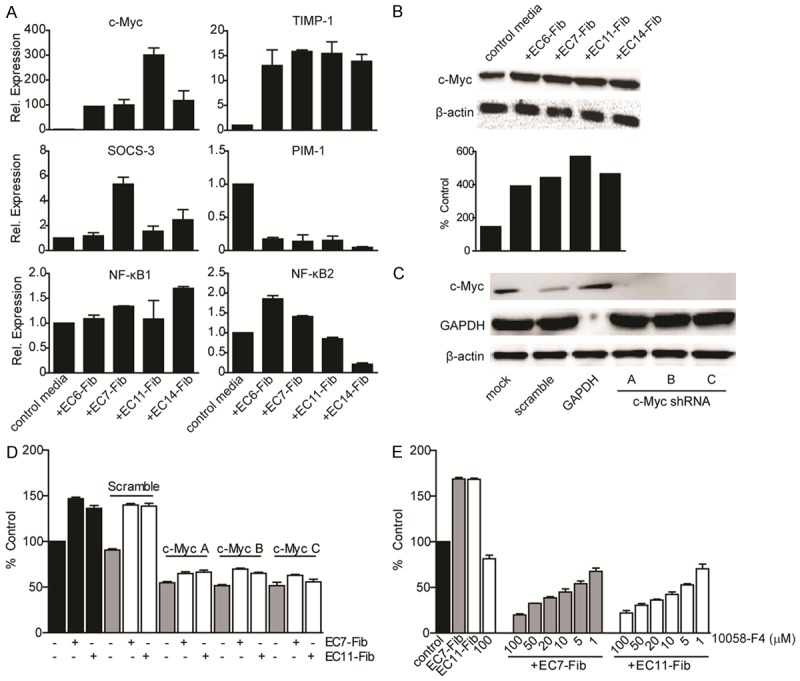
c-Myc regulates CAFs-mediated EC cell proliferation. A. Expression of STAT3 target genes were examined in ECC-1 cells after treated with CAFs for 72 hours using quantitative real-time PCR. Data were normalized to that in untreated ECC-1 cells. B. c-Myc protein expression was analyzed in ECC-1 cell after treated with CAFs. β-actin was used as an endogenous control. C, D. ECC-1 cells were transfected with a mock, shRNA construct targeting with a scrambled c-Myc sequence, shRNA targeting GAPDH and three individual shRNA construct targeting c-Myc. Levels of c-Myc post-knockdown in ECC-1 cells were analysed by Western blotting with β-actin as an endogenous control. The effects of c-Myc shRNA constructs on ECC-1 cell viability with low c-Myc level that were treated with CAF were determined by MTT assay. E. ECC-1 cells treated with c-Myc specific inhibitor 10058-F4 in increasing dose in the presence of CAFs secretion. Cell viability was examined using MTT assay and normalized to control media (media containing 2% FBS). Data, average; error bars, S.E.M. Data shown are representative of two independent experiments.
To determine if CAFs-induced c-Myc mediates EC cell proliferation, we down-regulated its expression in ECC-1 cells using three individual shRNA constructs. Expression of c-Myc protein was found to be downregulated in ECC-1 cells upon transfection with c-Myc specific shRNA constructs (Figure 4C). Cells with reduced c-Myc expression showed a significant decrement in cell proliferation (54% inhibition), when compared to those transfected with c-Myc non-targeting control (~10% inhibition). Treatment of CAFs conditioned media in these cells only marginally increased the proliferation (~11%) (Figure 4D).
We further treated ECC-1 cells with a c-Myc small molecule compound (10058-F4) in the presence of CAFs. This led to a dose-dependent suppression of cell proliferation (Figure 4E). Notably, about 80% inhibition of cell proliferation was observed at 100 µM in the presence of CAF conditioned media. Treatment of 10058-F4 alone (100 µM), however, reduced ECC-1 proliferation by only about 20% (Figure 4E), suggesting that presence of CAFs secretion was necessary for the anti-proliferative action of c-Myc inhibitor. Taken together, we demonstrated that c-Myc was upregulated by IL-6 secreted by CAFs, and c-Myc activation was required for CAF-mediated EC cell proliferation.
CAFs mediated c-Myc induction promotes EC cell growth in vivo
We further investigated the role of c-Myc expression in EC tumor-CAF crosstalk in a subcutaneous human tumor xenograft mouse model. Seven weeks post cell inoculation, ECC-1 tumors were about 736.8 ± 11.1 mm3 while those mice injected with CAFs alone (EC11-Fib cells) alone did not show any signs of cell growth. Mice injected with a combination of ECC-1 and EC11F (1:4 ratio) showed at least 1.4 times greater tumor size (1042.2 ± 27 mm3) compared to those injected with ECC-1 cells alone (P<0.0001). Interestingly, mice injected with ECC-1 with c-Myc knockdown showed at least 2.5 times smaller tumor size (293.9 ± 7 mm3) when compared to those in the scramble group (P<0.0001). Co-injection with CAF failed to induce greater tumor growth in this group (361.9 ± 13.7 mm3) (P=0.013) (Figure 5A). Notably, there was a small but significant difference in growth rate of ECC-1 transfected with scramble shRNA, with and without EC11-Fib (P=0.0118). The tumor size between the groups were also analyzed using in vivo fluorescence imaging, and was closely correlated to the tumor volume measured at the end of experiment (Figure 5C). Our data indicated that c-Myc has a significant role in CAFs-mediated EC cell proliferation in vitro and in vivo.
Figure 5.
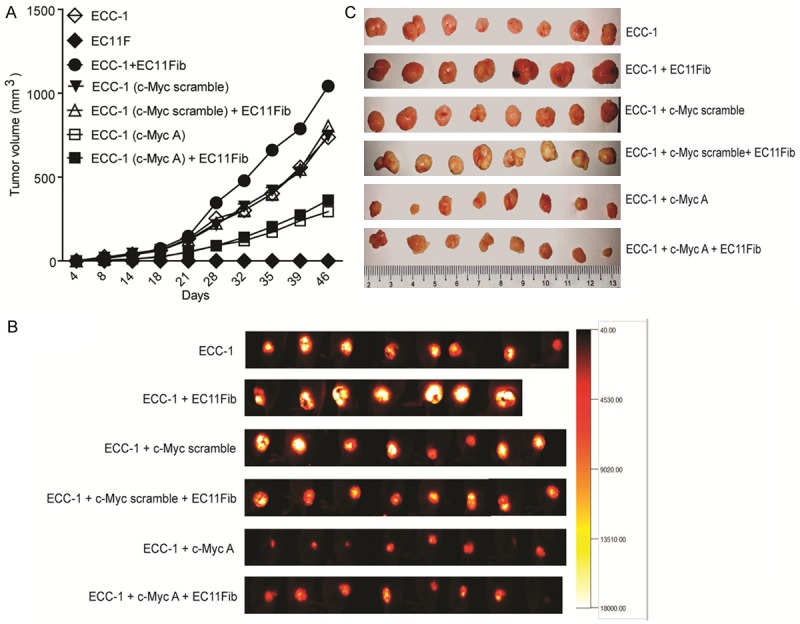
In vivo growth of EC tumor with c-Myc downregulation. (A) Stable clones of ECC-1 cells expressing c-Myc scramble or shRNA A constructs were established prior to subcutaneous injection into the right flank of Balb/c nude mice. These cells were injected with or without EC11Fib cells, in a ratio of 1:4 (tumor:fibroblast). The growth of tumor cells was measured using caliper. (B) At the end of the experiment, the animals were imaged using fluorescence imaging system, followed by tumor excision (C).
Activation of IL-6 signaling markers in cancer tissues
We further validated our finding by analyzing IL-6/STAT3/c-Myc pathway in human endometrial benign and malignant tissues. We analyzed the expression of IL-6R, gp130, phospho-STAT3 and c-Myc in five benign and five cancerous tissues of the human endometrium. Overall, positive staining patterns were observed to be intense and uniform in the glandular linings of the cancer tissues compared to the benign tissues when observed at 10 × magnification. Stronger IL-6R and gp130 staining were seen in the epithelial compartment of the cancerous tissues than in benign tissues. Phospho-STAT3 and c-Myc staining were also evident and predominantly confined to the nucleus in the cancer tissues; although a rather weak and diffuse staining was seen in the cytoplasm of the benign tissues (Figure 6). Our data demonstrated that human endometrial cancer tissues expressed high levels of IL-6R, gp130, phospho-STAT3 and c-Myc proteins which were minimally expressed in benign tissues. This indicates that IL-6/STAT3/c-Myc pathway could be an important player in the pathogenesis of endometrial cancer and could potentially be targeted for future treatments.
Figure 6.
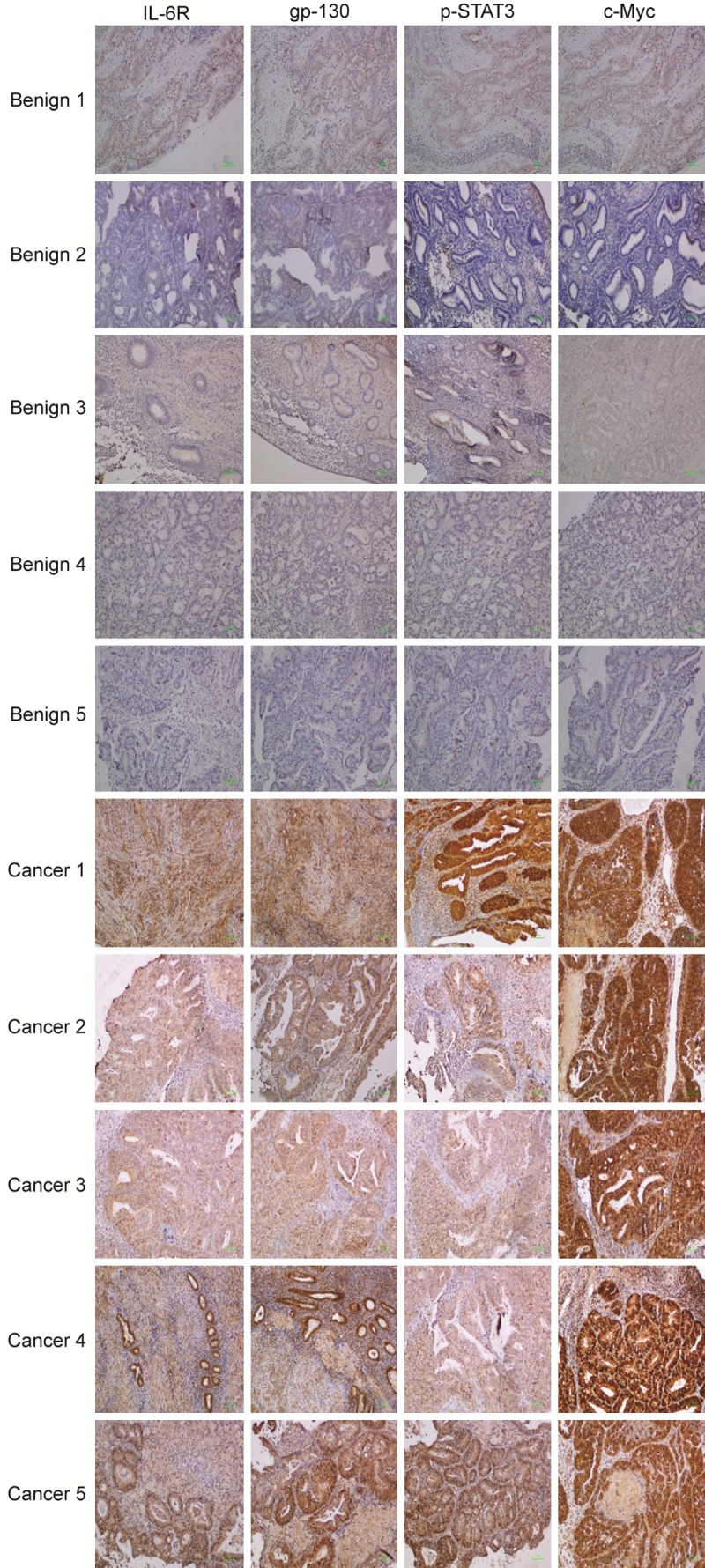
Expression of IL-6 receptors and key downstream signaling molecules in endometrial benign and cancer endometrial tissues. Five formalin-fixed paraffin embedded tissues from endometrial benign and cancer conditions each were stained with antibodies targeting human IL-6R, gp-130, phospho-STAT3 and c-Myc proteins. Detection was performed using light microscope at 10 × magnification. Scale, 100 micron.
Discussion
While CAFs have been implicated as a key player in cancer progression [6,13], its role in EC is relatively understudied. Our study suggests that IL-6 secreted by CAFs could have a role in regulating EC cell proliferation in vitro and in vivo. Elevated levels of IL-6 in the tumor microenvironment led to activation of JAK/STAT3 pathway in tumor cells, which resulted in increased cell proliferation, probably through induction of c-Myc protein. Inhibition of this pathway significantly abrogated the tumor-promoting effects of CAFs. Moreover, IL-6 pathway inhibitors were only effective when CAFs were present, indicating the importance of tumor microenvironment as an element in effective drug screening, which otherwise may be overlooked in conventional screening using only the tumor cells.
It has long been recognized that crosstalk between tumor cells and their microenvironment alters the mechanistic property of tumor cells in proliferation, motility and metastasis [14]. Paracrine signaling regulated by CAFs are shown to contribute to many cancer progression, such as IGF-II, HGF and SDF-1 signaling in lung cancer [15], TGF-β signaling in breast cancer [16] and hedgehog signaling in pancreatic cancer [17]. In pancreatic, prostate, melanoma, multiple myeloma and breast cancers, IL-6 was linked to increased sustainability and survival of tumors [18-20]. In endometrial cancer, however, it was not obvious how the microenvironment affects tumor progression. It was reported that IL-6 levels were elevated in the serum of EC patients [21] and were associated with chemotherapy resistance and poor prognostic outcome [12]. Yet, it is not clear if the presence of IL-6 was due to CAFs residing in the tumor microenvironment. In this study, we demonstrated a role of CAF in promoting EC cell proliferation, and our data showed that this effect was specific to the one-way paracrine signaling by IL-6 secreted by CAFs to activate the receptors expressed only by tumor cells. This in turn translates to CAF’s role as a key player in the tumor microenvironment, modulating the behavior of cancer cells through its secretion of inflammatory cytokines to communicate within the cancer environment.
Aberrant activation of JAK/STAT downstream of IL-6 pathway has been identified as an underlying factor mediating tumor progression and metastasis in various cancers [22], yet its implication and role in EC is unclear. In our study, we found that STAT3 was induced by IL-6 secreted by CAFs, similar to Bromberg’s observation in other cancers [23-25]. Subsequent activation of STAT3 leads to induction of various oncogenic proteins, including c-Myc and was shown to be a pre-requisite to mediate the rapid activation of c-Myc gene [26]. c-Myc is found activated in many human tumors with poor disease prognosis outcome, including EC [27-29]. We observed increased c-Myc expression in EC tissues compared to benign endometrial tissues. Additionally, knockdown of c-Myc in EC cells significantly reduced the proliferation of tumor cells both in vitro and in vivo, despite exposure to CAFs secretion. It is likely that activation of c-Myc via IL-6/STAT3 acts as a “on-switch” in EC cells to progress into a more aggressive stage, as in multiple myeloma [30,31]. Our work may provide one of the first few evidences that c-Myc can be induced via IL-6R/STAT3 pathway following activation by IL-6 from CAF secretion in endometrial cancer. Inhibitors targeting IL-6R downstream molecules were only effective in exerting their anti-tumor effects when in the presence of CAFs secretions. This strongly suggests that IL-6R/STAT3/JAK/c-Myc pathway can be targeted for EC treatment and our study also implies the importance of CAFs in the tumor microenvironment to provide the paracrine stimulants.
IL-6 pathway may not be the only mediator for CAF to affect EC progression. This pathway is shown to communicate with other signaling pathways to achieve significant pro-tumorigenic effects. It was shown that IL-6 in the microenvironment stimulated crosstalk between colorectal cancer cells and immune cells via miRNAs miR-21 and miR-29b, to sustain inflammation and to promote prometastatic behavior [32]. In addition, gastric cancer-derived mesenchymal stem cells was shown to induce chemotaxis of neutrophils, leading to protection against spontaneous apoptosis and activation of cancer cell motility via IL-6-STAT3-ERK1/2 signaling cascade [33]. Moreover, IL-6 was also shown to stimulate receptor activator of NF-κB ligand (RANKL) expression in bone cells, resulting in a direct paracrine-autocrine signaling between osteoblast and cancer cells to enhance the growth of metastatic breast cancers within the bone [34]. Hence, it is possible that IL-6 has further pro-tumorigenic effects in EC when acts in combination with other inflammatory cytokines present in the tumor microenvironment milieu. Further investigations on the crosstalk are crucial to understand the role of CAFs in EC, to design better therapy approaches for this disease.
With increasing role of IL-6 pathway in angiogenesis and cancer progression [35], it may be worthwhile to target this pathway for new treatment for oncology. IL-6 pathway inhibitors which were previously developed mainly for inflammatory conditions are currently tested in oncology settings. For example, tocilizumab, a humanized IL-6R specific monoclonal antibody, was previously approved for rheumatoid arthritis but is now proposed to alleviate cancer cachexia [36,37]. REGN88, a fully humanized IL-6R monoclonal antibody, is currently undergoing phase III clinical trial in rheumatoid arthritis and ankylosing spondilitis with future indication possibility in solid tumor [38]. Additionally, in phase I/II clinical trial in advance or refractory solid tumors, siltuximab or CNTO 328 that neutralizes IL-6, demonstrates promising safety and tolerability outcome [39] as well as a multitude of other indications [38]. Our study suggests IL-6 pathway as a novel target for EC, and hence application of these inhibitors may provide new avenue for treating aggressive EC.
Conclusion
This study shows that IL-6 secreted by cancer-associated fibroblasts in the tumor microenvironment promotes EC proliferation via activation of JAK/STAT3/c-Myc pathway in vitro and in vivo. Targeting IL-6, either through a single agent or by combination with co-activated inflammatory cytokines may be a novel therapeutic approach for women with EC.
Acknowledgements
This study was supported by University of Malaya Research Grant (RG492/13HTM) and HIR-UMCRI/MOHE/MED-12. Tissue collection was undertaken by the University of Malaya’s Faculty of Medicine Biobank Unit and laboratory works were performed at the Translational Core Laboratory, Faculty of Medicine, University of Malaya (RP019b).
Disclosure of conflict of interest
None.
References
- 1.Garg G, Mutch DG. Treatment strategies and prognosis of endometrial cancer, cancer of the uterine endometrium-advances and controversies. InTech. 2012 [Google Scholar]
- 2.Sorosky J. Endometrial cancer. Obstet Gynecol. 2012;120:383–397. doi: 10.1097/AOG.0b013e3182605bf1. [DOI] [PubMed] [Google Scholar]
- 3.Evans T, Sany O, Pearmain P, Ganesan R, Blann A, Sundar S. Differential trends in the rising incidence of endometrial cancer by type: data from a UK population-based registry from 1994 to 2006. Br J Cancer. 2011;104:1505–1510. doi: 10.1038/bjc.2011.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Janzen D, Rosales M, Paik D, Lee D, Smith D, Witte O, Iruela-Arispe M, Memarzadeh S. Progesterone receptor signaling in the microenvironment of endometrial cancer influences its response to hormonal therapy. Cancer Res. 2013;73:4697–4710. doi: 10.1158/0008-5472.CAN-13-0930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Franco OE, Shaw AK, Strand DW, Hayward SW. Cancer associated fibroblasts in cancer pathogenesis. Semin Cell Dev Biol. 2010;21:33–39. doi: 10.1016/j.semcdb.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karagiannis GS, Poutahidis T, Erdman SE, Kirsch R, Riddell RH, Diamandis EP. Cancer-associated fibroblasts drive the progression of metastasis through both paracrine and mechanical pressure on cancer tissue. Mol Cancer Res. 2012;10:1403. doi: 10.1158/1541-7786.MCR-12-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tlsty TD, Coussens LM. Tumor stroma and regulation of cancer development. Ann Rev Pathol. 2006;1:119–150. doi: 10.1146/annurev.pathol.1.110304.100224. [DOI] [PubMed] [Google Scholar]
- 8.Giannoni E, Bianchini F, Masieri L, Serni S, Torre E, Calorini L, Chiarugi P. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res. 2010;70:6945. doi: 10.1158/0008-5472.CAN-10-0785. [DOI] [PubMed] [Google Scholar]
- 9.Schauer I, Sood A, Mok S, Liu J. Cancer-associated fibroblasts and their putative role in potentiating the initiation and development of epithelial ovarian cancer. Neoplasia. 2011;13:393–405. doi: 10.1593/neo.101720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, Tang H, Cai J, Zhang T, Guo J, Feng D, Wang Z. Ovarian cancer-associated fibroblasts contribute to epithelial ovarian carcinoma metastasis by promoting angiogenesis, lymphangiogenesis and tumor cell invasion. Cancer Lett. 2011;303:47–55. doi: 10.1016/j.canlet.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 11.Subramaniam KS, Tham ST, Mohamed Z, Woo YL, Adenan NA, Chung I. Cancer-associated fibroblasts promote proliferation of endometrial cancer cells. PLoS One. 2013;8:e68923. doi: 10.1371/journal.pone.0068923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bellone S, Watts K, Cane S, Palmieri M, Cannon M, Burnett A, Roman J, Pecorelli S, Santin A. High serum levels of interleukin-6 in endometrial carcinoma are associated with uterine serous papillary histology, a highly aggressive and chemotherapy-resistant variant of endometrial cancer. Gynecol Oncol. 2005;98:92–98. doi: 10.1016/j.ygyno.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 13.Cirri P, Chiarugi P. Cancer-associated-fibroblasts and tumour cells: a diabolic liaison driving cancer progression. Cancer Metastas Rev. 2012;31:195–208. doi: 10.1007/s10555-011-9340-x. [DOI] [PubMed] [Google Scholar]
- 14.Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer. 2013;13:759–771. doi: 10.1038/nrc3611. [DOI] [PubMed] [Google Scholar]
- 15.Lin CA, Ho CC, Chen JJ. The roles of cancer-associated fibroblasts in stemness maintenance of lung cancer cells. Cancer Res. 2015;75:3207. [Google Scholar]
- 16.Yu Y, Xiao C, Tan L, Wang Q, Li X, Feng Y. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br J Cancer. 2014;110:724–732. doi: 10.1038/bjc.2013.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hwang R, Moore T, Hattersley M, Scarpitti M, Yang B, Devereaux E, Ramachandran V, Arumugam T, Ji B, Logsdon C, Brown J, Godin R. Inhibition of the hedgehog pathway targets the tumor-associated stroma in pancreatic cancer. Mol Cancer Res. 2012;10:1147–1157. doi: 10.1158/1541-7786.MCR-12-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kroon P, Berry PA, Stower MJ, Rodrigues G, Mann VM, Simms M, Bhasin D, Chettiar S, Li C, Li PK, Maitland NJ, Collins AT. JAK-STAT blockade inhibits tumor initiation and clonogenic recovery of prostate cancer stem-like cells. Cancer Res. 2013;73:5288–98. doi: 10.1158/0008-5472.CAN-13-0874. [DOI] [PubMed] [Google Scholar]
- 19.Oh J, Revel M, Chebath J. A soluble interleukin 6 receptor isolated from conditioned medium of human breast cancer cells is encoded by a differentially spliced mRNA. Cytokine. 1996;8:401–409. doi: 10.1006/cyto.1996.0055. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Yan W, Collins MA, Bednar F, Rakshit S, Zetter BR, Stanger BZ, Chung I, Rhim AD, Magliano MP. Interleukin-6 is required for pancreatic cancer progression by promoting mapk signaling activation and oxidative stress resistance. Cancer Res. 2013;73:6359–6374. doi: 10.1158/0008-5472.CAN-13-1558-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith HO, Stephens ND, Qualls CR, Fligelman T, Wang T, Lin CY, Burton E, Griffith JK, Pollard JW. The clinical significance of inflammatory cytokines in primary cell culture in endometrial carcinoma. Mol Oncol. 2013;7:41–54. doi: 10.1016/j.molonc.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quintás-Cardama A, Verstovsek S. Molecular pathways: Jak/STAT pathway: mutations, inhibitors, and resistance. Clin Cancer Res. 2013;19:1933–1940. doi: 10.1158/1078-0432.CCR-12-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE. Stat3 as an Oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 24.Bromberg J. Stat proteins and oncogenesis. J Clin Invest. 2002;109:1139–1142. doi: 10.1172/JCI15617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ott G, Rosenwald A, Campo E. Understanding MYC-driven aggressive B-cell lymphomas: pathogenesis and classification. Blood. 2013;122:3884–3891. doi: 10.1182/blood-2013-05-498329. [DOI] [PubMed] [Google Scholar]
- 26.Kiuchi N, Nakajima K, Ichiba M, Fukada T, Narimatsu M, Mizuno K, Hibi M, Hirano T. STAT3 is required for the gp130-mediated full activation of the c-myc Gene. J Exp Med. 1999;189:63–73. doi: 10.1084/jem.189.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol. 1999;19:1–11. doi: 10.1128/mcb.19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pelengaris S, Khan M, Evan G. c-MYC: more than just a matter of life and death. Nat Rev Cancer. 2002;2:764–776. doi: 10.1038/nrc904. [DOI] [PubMed] [Google Scholar]
- 29.Geisler J, Geisler H, Manahan K, Miller G, Wiemann M, Zhou Z, Crabtree W. Nuclear and cytoplasmic c-myc staining in endometrial carcinoma and their relationship to survival. Int J Gynecol Cancer. 2004;14:133–137. doi: 10.1111/j.1048-891x.2004.14027.x. [DOI] [PubMed] [Google Scholar]
- 30.Holien T, Våtsveen T, Hella H, Waage A, Sundan A. Addiction to c-MYC in multiple myeloma. Blood. 2012;120:2450–2453. doi: 10.1182/blood-2011-08-371567. [DOI] [PubMed] [Google Scholar]
- 31.Aquino G, Marra L, Cantile M, Chiara AD, Liguori G, Curcio MP, Sabatino R, Pannone G, Pinto A, Botti G, Franco R. MYC chromosomal aberration in differential diagnosis between Burkitt and other aggressive lymphomas. Infect Agent Cancer. 2013;8:1–9. doi: 10.1186/1750-9378-8-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patel S, Gooderham N. IL6 mediates immune and colorectal cancer cell cross-talk via miR-21 and miR-29b. Mol Cancer Res. 2015;13:1502–1508. doi: 10.1158/1541-7786.MCR-15-0147. [DOI] [PubMed] [Google Scholar]
- 33.Zhu Q, Zhang X, Zhang L, Li W, Wu H, Yuan X, Mao F, Wang M, Zhu W, Qian H, Xu W. The IL-6-STAT3 axis mediates a reciprocal crosstalk between cancer-derived mesenchymal stem cells and neutrophils to synergistically prompt gastric cancer progression. Cell Death Dis. 2014;5:e1295. doi: 10.1038/cddis.2014.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng Y, Chow SO, Boernert K, Basel D, Mikuscheva A, Kim S, Fong-Yee C, Trivedi T, Buttgereit F, Sutherland RL, Dunstan CR, Zhou H, Seibel MJ. Direct crosstalk between cancer and osteoblast lineage cells fuels metastatic growth in bone via auto-amplification of il-6 and rankl signaling pathways. J Bone Mineral Res. 2014;29:1938–1949. doi: 10.1002/jbmr.2231. [DOI] [PubMed] [Google Scholar]
- 35.Nagasaki T, Hara M, Nakanishi H, Takahashi H, Sato M, Takeyama H. Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour-stroma interaction. Br J Cancer. 2014;110:469–478. doi: 10.1038/bjc.2013.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ando K, Takahashi F, Motojima S, Nakashima K, Kaneko N, Hoshi K, Takahashi K. Possible role for tocilizumab, an anti-interleukin-6 receptor antibody, in treating cancer cachexia. J. Clin. Oncol. 2013;31:69–72. doi: 10.1200/JCO.2012.44.2020. [DOI] [PubMed] [Google Scholar]
- 37.Berti A, Boccalatte F, Sabbadini MG, Dagna L. Assessment of Tocilizumab in the Treatment of Cancer Cachexia. J. Clin. Oncol. 2013;31:2970. doi: 10.1200/JCO.2012.48.4147. [DOI] [PubMed] [Google Scholar]
- 38.Jones SA, Scheller J, Rose-John S. Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J Clin Invest. 2011;121:3375–3383. doi: 10.1172/JCI57158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen R, Chen B. Siltuximab (CNTO 328): a promising option for human malignancies. Drug Des Devel Ther. 2015;9:3455–3458. doi: 10.2147/DDDT.S86438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiang X, Yang D, Elliott R, Head J. Down-regulation of expression of interleukin-6 and its receptor results in growth inhibition of MCF-7 breast cancer cells. Anticancer Res. 2011;31:2899–2906. [PubMed] [Google Scholar]
- 41.Sriram K, Benkovic SA, Hebert MA, Miller DB, O’Callaghan JP. Induction of gp130-related cytokines and activation of JAK2/STAT3 pathway in astrocytes precedes up-regulation of glial fibrillary acidic protein in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of neurodegeneration. J Biol Chem. 2004;279:19936–19947. doi: 10.1074/jbc.M309304200. [DOI] [PubMed] [Google Scholar]
- 42.Gedman AL, Chen Q, Desmoulin SK, Ge Y, LaFiura K, Haska C, Cherian C, Devidas M, Linda S, Taub J, Matherly L. The impact of NOTCH1, FBW7 and PTEN mutations on prognosis and downstream signaling in pediatric T-cell acute lymphoblastic leukemia: a report from the Children’s Oncology Group. Leukemia. 2009;23:1417–1425. doi: 10.1038/leu.2009.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Castelló R, Estellés A, Vázquez C, Falcó C, España F, Almenar SM, Fuster C, Aznar J. Quantitative real-time reverse transcription-pcr assay for urokinase plasminogen activator, plasminogen activator inhibitor type 1, and tissue metalloproteinase inhibitor type 1 gene expressions in primary breast cancer. Clin Chem. 2002;48:1288–95. [PubMed] [Google Scholar]
- 44.Zemskova M, Sahakian E, Bashkirova S, Lilly M. The PIM1 kinase is a critical component of a survival pathway activated by docetaxel and promotes survival of docetaxel-treated prostate cancer cells. J Biol Chem. 2008;283:20635–20644. doi: 10.1074/jbc.M709479200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim YE, Kang HB, Park JA, Nam KH, Kwon HJ, Lee Y. Upregulation of NF-κB upon differentiation of mouse embryonic stem cells. BMB Rep. 2008;41:705–709. doi: 10.5483/bmbrep.2008.41.10.705. [DOI] [PubMed] [Google Scholar]
- 46.Nowak DE, Tian B, Brasier AR. Two-step cross-linking method for identification of NF-κB gene network by chromatin immunoprecipitation. Biotechniques. 2005;39:715–725. doi: 10.2144/000112014. [DOI] [PubMed] [Google Scholar]