

Abstract
DEAD-box RNA helicase 3 (DDX3) is a highly conserved family member of DEAD-box protein, which is a cluster of ATP-dependent and the largest family of RNA helicase. DEAD-box family is characterized by the regulation of ATPase and helicase activities, the modulation of RNA metabolism, and the actors of RNA binding proteins or molecular chaperones to interact with other proteins or RNA. For DDX3, it exerts its multifaceted roles in viral manipulation, stress response, hypoxia, radiation response and apoptosis, and is closely related to cancer development and progression. DDX3 has dual roles in different cancer types and can act as either an oncogene or tumor suppressor gene during cancer progression. In the present review, we mainly provide an overview of current knowledge on dual roles of DDX3 in various types of cancer, including breast cancer, lung cancer, colorectal cancer, hepatocellular carcinoma, oral squamous cell carcinoma, Ewing sarcoma, glioblastoma multiforme and gallbladder carcinoma, and illustrate the regulatory mechanisms for leading these two controversial biological effects. Furthermore, we summarize the essential signaling pathways that DDX3 participated, especially the Wnt/β-catenin signaling and EMT related signaling (TGF-β, Notch, Hedgehog pathways), which are crucial to DDX3 mediated cancer metastasis process. Thoroughly exploring the dual roles of DDX3 in cancer development and the essential signaling pathways it involved, it will help us open new perspectives to develop novel promising targets to elevate therapeutic effects and facilitate the “Personalized medicine” or “Precision medicine” to come into clinic.
Keywords: DDX3, cancer, oncogene, tumor suppressor gene, Wnt/β-catenin pathway, EMT related pathway
Introduction
DEAD-box protein is the largest family of RNA helicase, which is able to unwind RNA duplexes and is involved in multiple RNA processing procedures, including mRNA splicing, RNA editing, export, RNA decay, ribosome biogenesis, transcriptional and translational regulation and so on [1,2]. The name of DEAD-box RNA helicase is derived from the conserved amino acid sequence D-E-A-D (Asp-Glu-Ala-Asp) located in the motif II of 12 motifs [3]. The roles of these motifs can be divided into three parts: ATP binding, RNA binding, and link ATP and RNA binding. So DEAD-box family is characterized by the regulation of ATPase and helicase activities, and modulates RNA metabolism in an ATP-dependent manner [4]. Additionally, acting as RNA binding proteins or molecular chaperones, DEAD-box RNA helicase have interaction with other proteins or different forms of RNA, so as to maintain the integrity of the secondary and tertiary structure of RNA and facilitate the transcriptional activation, translational initiation, post-translational modification or miRNA biogenesis processes [5-7]. DEAD-box protein is a widely dispersed family which can be found in almost all organisms, from yeast to human. The genome of the yeast encodes 25 DEAD-box proteins. Besides the counterparts of each 25 proteins, along with 12 additional DEAD-box genes, are found in the human genome [8].
DEAD-box RNA helicase 3 (DDX3) is a highly conserved family member of DEAD-box proteins. The human genome encodes two types of DDX3 genes and two DDX3 homologs, DDX3X and DDX3Y. Based on their locations in chromosome, DDX3X is located on the X-chromosome bands p11.3-11.23 region and escapes from X-inactivation [9,10]. Whereas, DDX3Y is located in the azoospermia factor a (AZFa) region of the Y-chromosome, and is specifically expressed in testis and plays an essential role in spermatogenesis and male fertility [11,12]. DDX3X and DDX3Y share 92% similarity in protein sequence identity, and encodes for a 662- or 661-amino acid polypeptide depending on mRNA alternative splicing [13]. As the specialized role of DDX3Y in male fertility, usually we focus on our study on DDX3X and refer DDX3 to DDX3X.
Being a key RNA binding protein and transcriptional cofactor, DDX3 exerts its multifaceted roles in viral manipulation (especially for HIV, HCV, and HBV), immunology regulation, cancer progression and so on [14-17]. Moreover, DDX3 is closely related to various biological processes, such as stress response, hypoxia, radiation response, apoptosis, and cell cycle regulation [18,19]. For the role of DDX3 in cancer development, it is rather complicated and controversial. DDX3 is a “double-edged sword” gene and can act as either an oncogene or tumor suppressor gene during cancer progression, depending on different cancer types. So in this review, we will illustrate the dual roles of DDX3 in multiple cancer development procedures and explore the essential signaling pathways that DDX3 involved to lead these two conflicting biological effects.
Dual roles of DDX3 in cancer development
Breast cancer
Most of the recent studies demonstrated that DDX3 acts as an oncogenic role in breast cancer biogenesis. The report showed that over-expression of DDX3 in immortalized human breast cancer cell line MCF 10A could promote cell growth, proliferation and neoplastic transformation of epithelial cells. Particularly, DDX3 could repress E-cadherin expression, induced an epithelial-mesenchymal like transformation phenotype and increased the motility and invasive properties of breast cancer cells so as to facilitate metastasis process [20]. Further investigation found that hypoxia inducible factor-1α (HIF-1α) was a transcriptional activator of DDX3 in breast cancer cells. And it has been verified that there were three putative HIF-1 responsive elements located in the promoter region of DDX3 gene. Thus, the expression level of DDX3 can be elevated during hypoxia with the effect of HIF-1 on its promoter, and help tumor cells to survive in this unfavorable condition [21]. Moreover, in invasive breast cancer, the expression of DDX3 was correlated with over-expression of HIF-1α and its downstream genes CAIX, GLUT1, and several hypoxia related genes, including EGFR, HER2, ERα, c-Met and FOXO4 [22]. Those genes worked together under hypoxic conditions to promote tumor cell proliferation and transformation (Figure 1).
Figure 1.
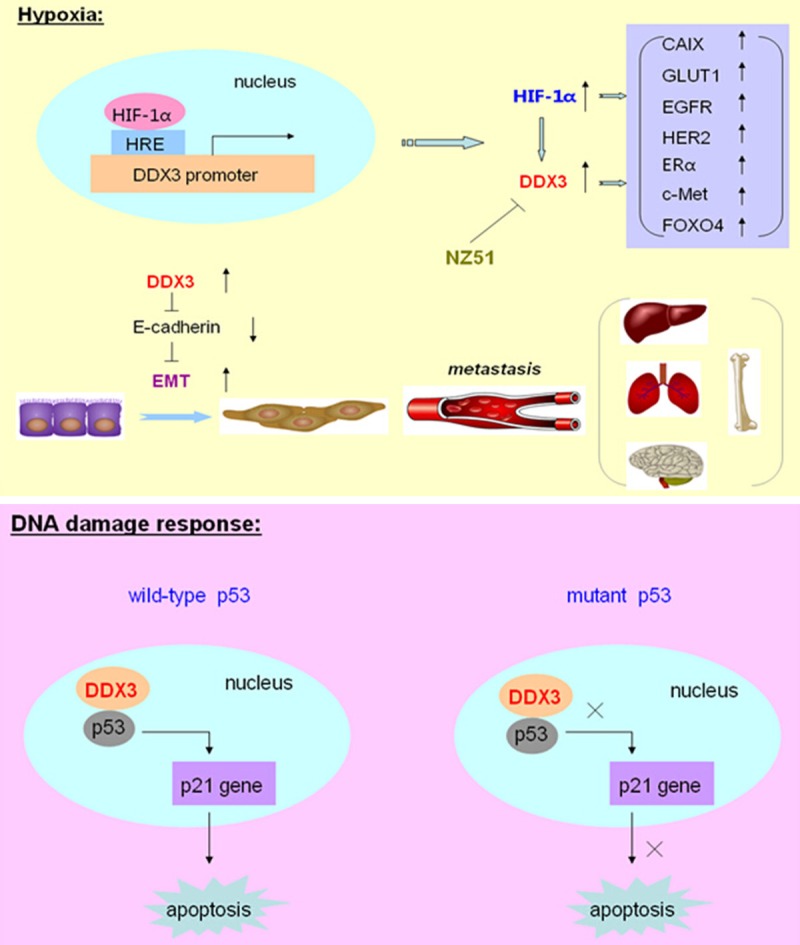
The role of DDX3 in breast cancer. Under hypoxic conditions, hypoxia inducible factor-1α (HIF-1α) could bind to the putative HIF-1 responsive elements (HRE) located in the promoter region of DDX3 gene, and transcriptionally activate the expression of DDX3. Meanwhile, the expression of DDX3 was correlated with over-expression of HIF-1α and its downstream genes CAIX, GLUT1, and several hypoxia related genes, including EGFR, HER2, ERα, c-Met and FOXO4. Moreover, DDX3 could repress E-cadherin expression and induce the Epithelial-mesenchymal transition (EMT) process to facilitate breast cancer metastasis. A novel DDX3 inhibitor, which a ring-expanded nucleoside analogue (REN), named for NZ51, could inhibit the ATP dependent helicase activity of DDX3. During DNA damage response, DDX3 could associate with p53, promote the accumulation of p53 in the nucleus, and activate its downstream target p21 expression, so as to modulate DNA damage induced apoptosis in cells expressed functional wild-type p53. However, in cells expressed non-functional or mutant p53, DDX3 otherwise inhibited the apoptosis process.
In addition, a latest study explored a novel DDX3 inhibitor, which is a ring-expanded nucleoside analogue (REN), named for NZ51. This inhibitor could be incorporated into the ATP binding pocket of DDX3 and therefore inhibited the ATP dependent helicase activity of DDX3. Using NZ51 treatment on MCF-7 and MDA-MB-231 breast cancer cells, it showed the suppression of cell cycle, the inhibition of cell proliferation, and the dramatic anti-cancer properties on cellular motility. Meanwhile, NZ51 could stabilize DDX3 with inactivation of its function and was not affected by hypoxia. So NZ51 had the equal potency in killing breast cancer cells both under hypoxic and normoxic conditions [23]. Furthermore, in breast cancer cell apoptosis process, the role of DDX3 was a “double-edged sword”. During DNA damage response, DDX3 could associate with p53, promote the retention and accumulation of p53 in the nucleus, and activate its downstream target p21 expression, so as to positively modulate DNA damage induced apoptotic signaling and caspase activation in cells which expressed functional wild-type p53. However, in cells which expressed non-functional or mutant p53, DDX3 otherwise inhibited the activity of apoptotic pathway to reduce caspase activation [24] (Figure 1). Overall, these results indicated that DDX3 plays dual roles in regulating apoptosis processes which are closely associated with breast carcinogenesis, radiotherapy and chemotherapy.
Lung cancer
DDX3 acts as diverse roles in lung cancer progression. Some studies showed that DDX3 was over-expressed in lung cancer and associated with lower survival rate and poor prognosis in lung cancer patients. In their studies, Raman et al. designed a novel small molecule inhibitor, RK-33, which could bind to DDX3 and inhibit its helicase activity. Inhibition of DDX3 expression by RK-33 resulted in G1 cell cycle arrest, apoptosis induction, and the promotion of radiotherapy sensitization. Mechanistically, loss of DDX3 functions by RK-33 could disrupt the DDX3-β-catenin regulatory axis and impair key molecules in Wnt signaling pathway. Furthermore, RK-33 could suppress the non homologous end joining (NHEJ) process, which is the major DNA damage repair model in mammalian cells during radiation and DNA damage response [25]. Thus, inhibition of DDX3 by small molecular inhibitors could impede tumor progression and it provided us new insights for developing chemo- or radio-therapy sensitizers.
Whereas, the studies made by another two groups came to quite different conclusions on the role of DDX3 in lung carcinogenesis. In HPV-associated lung cancer, the transcription of DDX3 was regulated by p53, so the expression level of DDX3 was dependent on p53 status. Meanwhile, through increasing Sp1 binding affinity onto the p21 promoter region, DDX3 synergistically enhanced p53-activated p21 transcription and therefore established the p53-DDX3-p21 regulatory axis (Figure 2). Thus, the DDX3 expression level was negatively associated with HPV oncoprotein E6 and was positively related to p21 expression in lung cancer. So the development of HPV-associated lung cancer, appeared to require E6-mediated inactivation of DDX3, degradation of p53, and synergistic suppression of p21 transcription so as to maintain a malignant phenotype and promote cancer progression [26]. In non-small-cell lung cancer, loss of DDX3 by p53 inactivation or mutation could promote tumor cell colony formation and invasiveness capacities. Mechanically, DDX3 loss could decrease Sp1 binding activity to the MDM2 promoter, and the MDM2 transcription process was greatly suppressed. Consequently, Slug expression was elevated due to the suppression of MDM2, and further led to the down-regulation of E-cadherin. Thus, through decreasing MDM2-mediated Slug degradation, loss of DDX3 might result in Slug-suppressed E-cadherin expression [27] (Figure 2). Overall, the studies suggested that DDX3 loss by p53 inactivation via the MDM2/Slug/E-cadherin pathway could facilitate tumor malignancy state and lead to poor clinical outcome for lung cancer patients.
Figure 2.
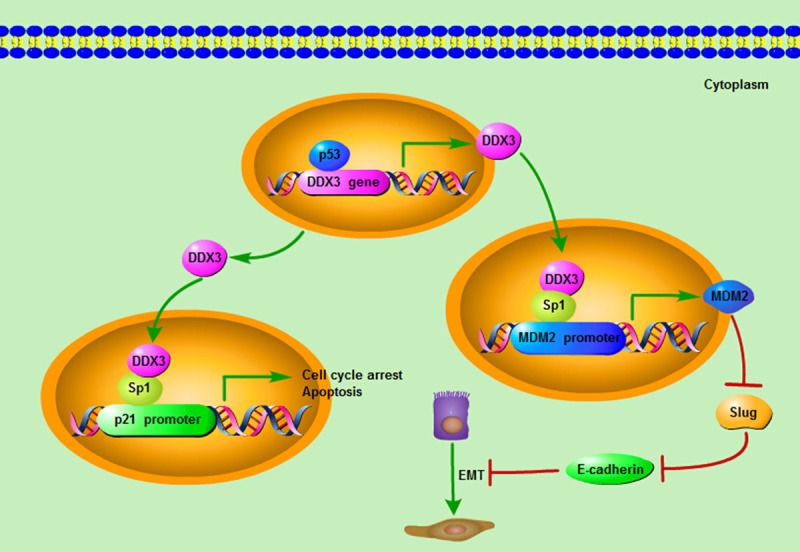
The mechanism of DDX3 acting as a tumor suppressor. The transcription of DDX3 can be regulated by p53, and the expression level of DDX3 is dependent on p53 status. Meanwhile, through interacting and cooperating with Sp1, DDX3 could increase Sp1 binding affinity onto the p21 promoter region and up-regulate the promoter activity of p21 so as to exert its tumor suppressive roles on cell cycle and apoptosis. Moreover, DDX3 could also increase Sp1 binding activity to the MDM2 promoter, and promote the MDM2 transcription process. Consequently, the up-regulation of MDM2 leads to the suppression of Slug, which negatively regulates the expression of E-cadherin. As a result, through increasing MDM2-mediated Slug suppression, DDX3 can act as a tumor suppressor by elevating the E-cadherin expression and impeding the Epithelial-mesenchymal transition (EMT) progression.
Colorectal cancer
The debate on the oncogenic or tumor suppressive role of DDX3 in colorectal cancer is still going on. Some researchers hold the point that DDX3 acted as a tumor suppressive gene and had a significant prognostic predictive power in colorectal cancer. Patients with low DDX3 expression level resulted in poor clinical prognosis and multiple distant metastasis regions. For the molecular mechanisms, down-regulation of DDX3 could lead to the up-regulation of Snail, so as to decrease the expression level of membranous E-cadherin and reduce tumor cell aggregation ability, therefore promoting migration, invasion and metastasis capacities of colon cancer cells [28]. Taken together, these data showed that low DDX3 expression level activated the Snail/E-cadherin pathway, which promoted cancer metastasis process and indicated poor clinical outcome in colorectal cancer patients.
However, other researchers stood at a quite opposite point that DDX3 had an oncogenic role in colorectal cancer. Over-expression of DDX3 was closely correlated with nuclear β-catenin expression, and further activated Wnt signaling pathway. Whereas, down-regulation of DDX3 expression contributed to the reduction of TCF4 reporter activity and the inhibition of mRNA expression levels of TCF4-regulated downstream genes, such as c-MYC, AXIN2, CCND1 and BIRC5A (Figure 3). Moreover, using a small molecule inhibitor, RK-33, which binds to the ATP-binding site of DDX3 and inhibits its helicase activity, could effectively suppress tumor cell growth, proliferation, and induce cell cycle G1 phase arrest. Meanwhile, the study found that in APC wild-type tumors harboring an activating CTNNB1 mutation, RK-33 could exert its highest sensitivity in DDX3 suppression, and therefore had the potential to be a promising treatment strategy for colorectal cancer [29].
Figure 3.
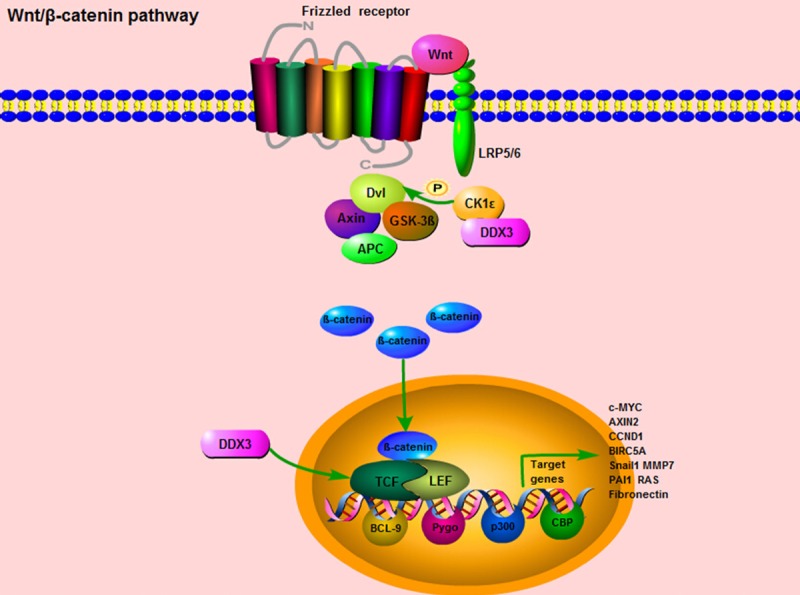
DDX3 is involved in Wnt/β-catenin pathway. When Wnt/β-catenin signaling is activated, the Wnt ligands bind to the Frizzled receptor and the co-receptor lipoprotein receptor related protein 5/6 (LRP5/6) in the membrane. Then, the Dishevelled (Dvl) gathers the cytoplasmic Axin and GSK3β into the membrane, and facilitate the activated β-catenin translocating from cytoplasm to nucleus. The nucleus β-catenin could interact with two major transcription factors, the T-cell factor (TCF) and lymphocyte enhancer factor (LEF), accompanied with several transcription co-activators, BCL9, CBP, p300, Pygo, to regulate multiple genes transcription. These downstream target genes mainly include Snail1, MMP7, PAI1, RAS, and Fibronectin. For the role of DDX3 in Wnt/β-catenin signaling, DDX3 could directly bind to CK1ε, stimulate its kinase activity and further promote the phosphorylation of the scaffold protein Dvl, so as to facilitate β-catenin translocating into the nucleus. Moreover, up-regulation of DDX3 expression contributes to the induction of TCF reporter activity and the elevation of mRNA expression levels of TCF-regulated downstream genes, such as c-MYC, AXIN2, CCND1 and BIRC5A.
Hepatocellular carcinoma
Hepatitis C virus (HCV) is a main leading cause for hepatocellular carcinoma (HCC). HCV infection can lead to different extent of liver damage which includes fibrosis, cirrhosis, and eventually evolving into hepatocellular carcinoma [30-32]. DDX3 has been shown to interact with HCV proteins and regulate HCV replication. HCV core could bind to the C-terminus of DDX3 (amino acids 553-622) and their interaction is mediated by the N-terminus of HCV core (amino acids 1-59). The effect of this interaction between HCV core and DDX3 proposes to be the manipulation of mRNA splicing, transcriptional regulation and translational regulation of HCV, finally affecting HCV replication [33,34]. In hepatitis virus-associated HCC, including HCV positive patients and HBV positive patients, the DDX3 expression level was different. There was a significant lower DDX3 expression level in HBV-positive HCC patients, but not in the HCV-positive ones. Moreover, the expression level of DDX3 was differentially distinguished by the gender, and the tendency of DDX3 down-regulation in HCC was more frequently found in males rather than in females. Knocking down of DDX3 could up-regulate the expression of cyclinD1 and down-regulate the expression of p21, resulting in an entry to S phase to promote cell cycle progression and facilitate tumor cell growth [35]. Together, these findings suggested that DDX3 was deregulated in hepatitis virus-associated HCC and was involved in cell cycle and cell growth control.
Additionally, another report further verified that DDX3 was a candidate tumor suppressor in HCC. A declined expression of DDX3 could be found in HCC, which was accompanied with the reduction of p21 (waf1/cip1) expression. In detail, through an ATPase-dependent but helicase-independent mechanism, DDX3 could transactivate the functions of p21 (waf1/cip1) promoter and transcriptionally regulate its activity. There were four Sp1 binding sites located in the transcription start site of p21 (waf1/cip1) promoter, which were all essential for DDX3 response. Thus, through interacting and cooperating with Sp1, DDX3 could up-regulate the promoter activity of p21 (waf1/cip1) [36] (Figure 2). These findings indicated that DDX3 exerted its tumor suppressive properties on cell cycle and tumor growth mainly through transcriptional regulation of p21 (waf1/cip1) promoter activity.
Oral squamous cell carcinoma
The role of DDX3 in oral squamous cell carcinoma (OSCC) is still conflicting. One study led by a Taiwan group found that DDX3 acted as a tumor suppressor and was a protective factor for OSCC patients. Low expression of DDX3 was significantly associated with life behaviors of OSCC patients, such as smoking, alcohol consumption, betel quid chewing, and also with poor clinical outcomes of OSCC patients, including relapse-free survival (RFS) rate and overall survival (OS) rate. Surprisingly, patients with low/negative DDX3 expression, especially those non-smoker OSCC patients, had relatively worse OS rate compared with smoker patients, and was associated with poor prognosis [37]. All in all, these studies demonstrated that low/negative DDX3 expression was closely correlated with aggressive clinical manifestations and might become a potential survival predictor, particularly for non-smoker OSCC patients.
On the other hand, the research launched by an India group revealed a converse role of DDX3 in OSCC. In their studies, DDX3 was assumed to be an oncogene, which promoted the progression of OSCC. So they developed a new bioactive compound against DDX3, named ketorolac salt. This compound had strong hydrogen bond interactions which were similar to crystallized DDX3 protein, and had less binding free energy than existing synthetic DDX3 inhibitors. Through directly interacting with DDX3, ketorolac salt could inhibit the ATP hydrolysis process of DDX3. Moreover, for in vivo experiment, ketorolac salt could effectively decrease the numbers of neoplastic tongue lesions and reduce the lesion severity in a tongue tumor mouse model [38]. All these data indicated that ketorolac salt might be used as a novel drug candidate to treat DDX3 associated OSCC.
Ewing sarcoma
The latest study showed that DDX3 acted as an oncogenic role in Ewing sarcoma. High expression level of DDX3 could be found in numerous human sarcoma subtypes compared with normal adjacent mesenchymal cells. Knocking down of DDX3 expression by a small molecule inhibitor, RK-33, which was also utilized in lung cancer and colorectal cancer, could efficiently inhibit the oncogenic activities of sarcoma cells and impede Ewing sarcoma progression. Meanwhile, the treatment of RK-33 had preferentially more cyto-toxicity to sarcoma cells, particularly for the chemotherapy-resistant sarcoma stem cells, rather than adjacent non-malignant cells. Moreover, DDX3 inhibition was confirmed to alter the cellular proteome of Ewing sarcoma, especially the proteins which were involved in DNA replication, mRNA translation and proteasome function. For in vivo assay, in human Ewing sarcoma xenograft which expressed high DDX3 level, the tumor growth could be suppressed by RK-33 treatment without overt toxicity [39]. In summary, all these investigations suggested that the development of RK-33 to target DDX3 in Ewing sarcoma could be a promising anti-cancer strategy and more clinical trials should be needed to make it into real clinical use.
Glioblastoma multiforme
In glioblastoma multiforme (GBM), DDX3 was reported to be an oncogene, which facilitated tumor development. In some cases, DDX3 exerted its oncogenic role through impeding the process of death receptor-mediated apoptosis. Meanwhile, there existed another mechanism for DDX3 mediated cancer progression was by elevating the expression levels of transcription factors, such as Snail. Activation of Snail could repress the expressions of several cellular adhesion proteins, and further lead to the malignant phenotypes of cancer cells, including invasion, migration, and metastasis [40-43]. Inhibition of DDX3 expression contributed to the reduced basal level of Snail, which effectively reduced cell proliferation and migration process. In the samples of GBM patients, there was a significantly positive correlation between DDX3 and Snail expression levels [44]. Therefore, these data indicated that DDX3 was required for basal Snail expression, and promoted the expressions of Snail-activated downstream genes, further resulting in tumor progression and development.
Gallbladder carcinoma
DDX3 was found to play an oncogenic role in different pathological subtypes of gallbladder carcinoma, including squamous cell carcinoma, adenocarcinoma, and adenosquamous carcinoma of gallbladder. The study testified that high expression level of DDX3 was significantly correlated with large tumor size, high TNM stage, and lymph node metastasis of gallbladder carcinoma. Using several statistical methods to do univariate Kaplan-Meier analysis and multivariate Cox regression analysis, the results showed that DDX3 expression level, degree of differentiation, tumor size, TNM stage, invasion, lymph node metastasis, and surgical curability were significantly associated with post-operative survival rate in gallbladder patients. And all these risk factors, apart from surgical curability, were essential independent poor-prognostic factors for gallbladder patients [45]. To sums up, this clinical study hinted that high expression of DDX3 was closely related to clinical features, pathological subtypes and biological behaviors of gallbladder carcinoma, thus it provided a promising potential to utilize DDX3 as a novel biomarker for predicting poor prognosis and metastasis in gallbladder carcinoma.
The essential signaling pathways that DDX3 involved
From above studies on the dual roles of DDX3 in multiple cancer development processes, we can summarize several signaling pathways which DDX3 participated in carcinogenesis, including the “HIF-1α-DDX3-E-cadherin” pathway, the “p53-DDX3-p21” pathway, the “DDX3-Rac1-β-catenin” pathway [46], and the “DDX3-MDM2-Slug-E-cadherin” pathway. Due to the different biological functions that DDX3 exerted, especially for tumor invasion, migration and metastasis, the essential signaling pathways DDX3 involved can be illustrated from the following two aspects.
Wnt/β-catenin pathway
Wnt/β-catenin signaling is a conversed indispensable pathway for both embryonic development and adult homeostasis processes [47,48]. It plays key functions in many types of diseases and various cancers [49-51]. And nearly most of the organs and tissues regeneration need the involvement of Wnt/β-catenin signaling, such as brain, spinal cord, eyes, heart, liver, kidney, lungs, gut, skin, hair and bone marrow [52-54]. β-catenin is the key factor and the central mediator of Wnt/β-catenin signaling pathway. Due to its different subcellular locations, in the cytoplasm or in the nucleus, the functions of Wnt/β-catenin signaling can be activated in varying degrees [55-57]. When the cell is quiescent and the Wnt/β-catenin signaling is not activated by stimulus, β-catenin is located in cytoplasm and formed a “destruction complex” with several proteins, including Axin, APC, GSK3β, and CK1α, and further phosphorylated by CK1α and GSK3β at Ser45, 33, 37 and Thr41 [58,59]. Then, the phosphorylated β-catenin will be marked with ubiquitin and go through proteasome-dependent degradation pathway to degrade β-catenin protein [60,61].
On the other hand, when the Wnt/β-catenin signaling is initiated by the stimulus of Wnt ligands, the downstream signal cascades will be activated. Firstly, the Wnt ligands bind to the Frizzled receptor and the co-receptor lipoprotein receptor related protein 5/6 (LRP5/6) in the membrane [62,63]. Second, the Dishevelled (Dvl) gathers the cytoplasmic Axin and GSK3β into the membrane, so as to decompose the “destruction complex” and facilitate the unphosphorylated (activated form) β-catenin translocating from cytoplasm to nucleus. Then, nucleus β-catenin could interact with two major transcription factors, the T-cell factor (TCF) and lymphocyte enhancer factor (LEF), and along with several transcription co-activators, BCL9, CBP, p300, Pygo, to regulate gene transcription and chromatin modification [64-66] (Figure 3). Apart from Wnt ligands, integrin-linked kinase (ILK) and MMP-mediated E-cadherin extracellular domain shedding can also release β-catenin in cytoplasm and activate its functions in nucleus [67,68]. The downstream target genes regulated by Wnt/β-catenin signaling mainly include Snail1, MMP7, PAI1, RAS, Fsp1, and Fibronectin (Figure 3). Most of these genes are involved in the processes of epithelial-mesenchymal transition (EMT), invasion, migration, inflammation, fibrosis and apoptosis [69-73].
For the essential role of DDX3 in the Wnt/β-catenin signaling pathway, the classical study was published on Science in 2013. The researchers explored that DDX3 acted as a regulator of Wnt/β-catenin signaling network through modulating a subunit of casein kinase 1ε (CK1ε). This finding was contrary to the previous notions in this field that CK1 members were “rogue” kinases as their enzymatic activities appeared to be unregulated [74,75]. More specifically, this study verified that DDX3 could directly bind to CK1ε, stimulate its kinase activity and further promote the phosphorylation of the scaffold protein Dishevelled (Dvl), so as to influence Dvl mediated “destruction complex” decomposition, facilitate β-catenin translocating into the nucleus and regulate the activities of Wnt/β-catenin pathway in a Wnt-dependent manner (Figure 3). The regulatory role of DDX3 in Wnt/β-catenin signaling is universal, not only in mammalian cells, but also in the development processes of Xenopus and Caenorhabditis elegans as well [76]. In a word, DDX3 is in command of CK1ε activity, and this newly regulatory relationship between DDX3 and CK1ε will open fresh perspectives for searching promising therapeutic targets in Wnt/β-catenin pathway.
Epithelial-mesenchymal transition (EMT) pathway
Epithelial-mesenchymal transition (EMT) is a key biological process through which an epithelial cell phenotype can be converted into a mesenchymal cell phenotype. During this process, the cells lose epithelial characteristics and acquire mesenchymal features [77-79]. These changes mainly include apical-basolateral polarity losing, cell-cell junctions dissolving, and actin cytoskeleton remodeling [80]. For the changes of molecular hallmarks in EMT process, the epithelial markers are down-regulated, such as E-cadherin, α-catenin, β-catenin, γ-catenin, CK, ZO-1, while the mesenchymal markers are up-regulated, including N-cadherin, Vimentin, α-SMA, fibronectin and so on [81-84]. Under normal physiological conditions, EMT exerts its important functions in embryogenesis, organ development, tissue remodel and wound repairing [85,86]. Moreover, under pathological conditions and carcinogenesis procedures, EMT is an essential necessary step, especially for invasion, migration and metastasis of cancer cells [87,88]. That is because EMT allows cancer cells to leave the primary lesions or environments, cross endothelial barriers, enter blood and lymphatic circulation and migrate to distant locations [89].
Cancer cells can be induced to undergo EMT process by multiple signaling pathways, such as TGF-β signaling, Wnt/β-catenin signaling, Notch, Hedgehog signaling pathways and so on [90-93]. Additionally, there are several key transcription factors which are closely related to these signaling pathways, including zinc-finger proteins Twist, Snail, Slug, ZEB1, SIP1, E47 and other factors Smads, FOXC2 and so on [94-98]. Apart from these transcription factors that involved in EMT and its related signaling, there are a cluster of growth factors, oncogenes, tumor suppressors and miRNAs which are also participated in the control of EMT [99]. More specifically, the growth factors including TGF-β, EGF, HGF, VEGF and FGF2, could effectively activate various signaling pathways such as TGF-β/Smad, MAPK, NF-κB, JAK/STAT3, PI3K/AKT/mTOR, ERK pathways so as to facilitate EMT progression [100-102]. The oncogenes, such as HPV16 E6/E7, PIK3CA, BMI1, AKT2, AEG1 [103-105], and the tumor suppressors, such as p53, RASAL2, SFRPs, Klotho [106,107], along with miR-200 family, miR-155, miR-181, miR-214 and miR-130 [108-110], work together as a complicated network to precisely modulate EMT and the signaling pathways it involved.
Although these signaling pathways may possess different regulatory mechanisms to activate EMT process, they share the common endpoints---E-cadherin, which belongs to adherens junction protein and is a central downstream target being tightly regulated [111,112]. For the role of DDX3 in EMT, E-cadherin is undoubtedly a key target of DDX3 to mediate EMT process. DDX3 could directly repress E-cadherin expression, or elevate the expression level of transcription factor Snail, and further promote the expressions of Snail-activated downstream genes to induce an epithelial-mesenchymal like transformation phenotype and increase the motility and invasive properties of cancer cells [20,44]. On the other hand, loss of DDX3 by p53 inactivation via the MDM2/Slug/E-cadherin pathway might result in Slug-suppressed E-cadherin expression, which could promote EMT process and facilitate tumor invasion, migration, and metastasis [27]. Therefore, E-cadherin is the most important mediator in EMT, and the decreased E-cadherin expression level can be a useful predictor to indicate poor survival in cancer patients.
Conclusion
In this review, we mainly discussed about the recent progress of the DDX3 study in cancer and explored the essential signaling pathways that DDX3 involved in for modulating cancer metastasis. Specifically, DDX3 was reported to be an oncogene in breast cancer, Ewing sarcoma, glioblastoma multiforme and gallbladder carcinoma. In hepatocellular carcinoma, DDX3 was found to act as a tumor suppressive role. Meanwhile, DDX3 led dual roles of both oncogene and tumor suppressor in lung cancer, colorectal cancer and oral squamous cell carcinoma (Table 1). Moreover, for the two major signaling pathways DDX3 participated, Wnt/β-catenin signaling and EMT related signaling (TGF-β, Notch, Hedgehog pathways), were crucial to DDX3 mediated biological functions in cancer development.
Table 1.
Dual roles of DDX3 in cancer development
| Cancer type | Roles | Possible pathways and mechanisms | DDX3 inhibitors | Refs |
|---|---|---|---|---|
| Breast cancer | Oncogene | “HIF-1α-DDX3-E-cadherin-EMT” pathway, promote metastasis | NZ51 (a ring-expanded nucleoside analogue (REN), incorporate into the ATP binding pocket of DDX3) | [20-23] |
| Lung cancer | Oncogene/TSG | Oncogene: “DDX3-Wnt/β-catenin” pathway, affect apoptosis process | RK-33 (a small molecule inhibitor, bind to the ATP-binding site of DDX3) | [25-27] |
| TSG: “p53-DDX3-p21” pathway, affect apoptosis process | ||||
| “p53-DDX3-MDM2-Slug-E-cadherin” pathway, suppress EMT and metastasis | ||||
| Colorectal cancer | Oncogene/TSG | Oncogene: “DDX3-Wnt/β-catenin-TCF4” pathway, affect cell cycle progression | RK-33 (a small molecule inhibitor, bind to the ATP-binding site of DDX3) | [28,29] |
| TSG: DDX3 inhibits Snail expression, and prevents EMT | ||||
| Hepatocellular carcinoma (HCC) | TSG | DDX3 down-regulates cyclinD1, and up-regulates p21, impede cell cycle progression | None | [35,36] |
| “DDX3-Sp1-p21” pathway, affect apoptosis process | ||||
| Oral squamous cell carcinoma (OSCC) | Oncogene/TSG | Oncogene: promote OSCC progression | Ketorolac salt (bioactive compound, inhibit the ATP hydrolysis process of DDX3) | [37,38] |
| TSG: DDX3 is a protective factor and a good survival predictor | ||||
| Ewing sarcoma | Oncogene | Promote Ewing sarcoma progression and the resistance to chemotherapy | RK-33 (a small molecule inhibitor, bind to the ATP-binding site of DDX3) | [39] |
| Glioblastoma multiforme (GBM) | Oncogene | Impede the death receptor mediated apoptosis process, elevate Snail expression, and promote metastasis | To be developed | [44] |
| Gallbladder carcinoma | Oncogene | High DDX3 level is correlated with large tumor size, high TNM stage, lymph node metastasis, and poor prognosis | To be developed | [45] |
Abbreviations: TSG: Tumor Suppressor Gene; EMT: Epithelial-Mesenchymal Transition; TCF: T-Cell Factor; TNM: Tumor, Node, Metastasis.
From the above studies, we can sense that the role of DDX3 in different types of cancer is rather controversial. DDX3 can act as an oncogene in one cancer, but in other type of cancer, it demonstrates a significant tumor suppressor role. Even in same certain type of cancer, the reporters can get to the two totally contradictable conclusions on the role of DDX3. It seems quite confusing about these dual roles of DDX3 in cancer development and progression, and we really want to tell the working mechanisms for this “double-edged sword” gene. Right now, some possible clues might help us to explain these dual roles of DDX3 in cancer. First, as DDX3 has two different phenotypes, unstable and stable fashion, to exert its functions, so in each experiment related to explore cancer cell biological functions, if the experiment system and conditions vary or even change a little bit, the status and activity of DDX3 might be at different degrees, so the conclusions for DDX3 can come to a different direction. Second, DDX3 itself can have mutations in some specific sites and in some types of cancer, so the mutant DDX3 and wild-type DDX3 will exhibit different functions in cancer. Meanwhile, each cancer patients have the heterogeneity for cancer initiation, development and progression, and their cancer samples might have some variations for molecular expressions, which can lead to the dual roles of DDX3 in cancer. Third, DDX3 is closely related to virus infections and replications, particularly for the HIV, HPV, HCV, HBV, so if the cancer cell and cancer patients with different kinds of virus infections, the regulatory mechanisms and roles of DDX3 will not be the same as the cancer patients without virus infections. Virus infections might be a key factor to explain the altered role of DDX3 in different cancers. However, all these possible explanations will need further experiments to confirm and validate.
In summary, systematically studying the dual roles of DDX3 in cancer development and its related essential signaling pathways, it will provide us new insights to explore novel therapeutic targets or small molecule inhibitors aiming at DDX3 and its signaling pathways. Also, these dual roles of DDX3 in cancer will enlighten us to carry out “Personalized medicine” and “Precision medicine” to each patient and each type of cancer to optimize the maximum therapeutic effects and realize the translational medicine from “bench to bedside”.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (30872267, 61572200, 81071290, 81225013, 81430075), China Postdoctoral Science Foundation, the Postdoctoral Science Foundation of Central South University and the China Scholarship Council (CSC). Dr. Luqing Zhao is right now a Postdoctoral Fellow in Department of Dermatology, Xiangya Hospital, Central South University, and thanks all members in Dermatology Lab for their critical comments.
Disclosure of conflict of interest
None.
References
- 1.Linder P, Fuller-Pace F. Happy birthday: 25 years of DEAD-box proteins. Methods Mol Biol. 2015;1259:17–33. doi: 10.1007/978-1-4939-2214-7_2. [DOI] [PubMed] [Google Scholar]
- 2.Andreou AZ, Klostermeier D. Fluorescence methods in the investigation of the DEAD-box helicase mechanism. EXS. 2014;105:161–92. doi: 10.1007/978-3-0348-0856-9_8. [DOI] [PubMed] [Google Scholar]
- 3.Mallam AL, Sidote DJ, Lambowitz AM. Molecular insights into RNA and DNA helicase evolution from the determinants of specificity for a DEAD-box RNA helicase. Elife. 2014;3:e04630. doi: 10.7554/eLife.04630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Linder P, Jankowsky E. From unwinding to clamping - the DEAD box RNA helicase family. Nat Rev Mol Cell Biol. 2011;12:505–16. doi: 10.1038/nrm3154. [DOI] [PubMed] [Google Scholar]
- 5.Jarmoskaite I, Russell R. RNA helicase proteins as chaperones and remodelers. Annu Rev Biochem. 2014;83:697–725. doi: 10.1146/annurev-biochem-060713-035546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bish R, Vogel C. RNA binding protein-mediated post-transcriptional gene regulation in medulloblastoma. Mol Cells. 2014;37:357–64. doi: 10.14348/molcells.2014.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yin J, Park G, Lee JE, Choi EY, Park JY, Kim TH, Park N, Jin X, Jung JE, Shin D, Hong JH, Kim H, Yoo H, Lee SH, Kim YJ, Park JB, Kim JH. DEADbox RNA helicase DDX23 modulates glioma malignancy via elevating miR-21 biogenesis. Brain. 2015;138:2553–70. doi: 10.1093/brain/awv167. [DOI] [PubMed] [Google Scholar]
- 8.Pan C, Potratz JP, Cannon B, Simpson ZB, Ziehr JL, Tijerina P, Russell R. DEAD-box helicase proteins disrupt RNA tertiary structure through helix capture. PLoS Biol. 2014;12:e1001981. doi: 10.1371/journal.pbio.1001981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Epling LB, Grace CR, Lowe BR, Partridge JF, Enemark EJ. Cancer-associated mutants of RNA helicase DDX3X are defective in RNAstimulated ATP hydrolysis. J Mol Biol. 2015;427:1779–96. doi: 10.1016/j.jmb.2015.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ojha J, Secreto CR, Rabe KG, Van Dyke DL, Kortum KM, Slager SL, Shanafelt TD, Fonseca R, Kay NE, Braggio E. Identification of recurrent truncated DDX3X mutations in chronic lymphocytic leukaemia. Br J Haematol. 2015;169:445–8. doi: 10.1111/bjh.13211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramathal C, Angulo B, Sukhwani M, Cui J, Durruthy-Durruthy J, Fang F, Schanes P, Turek PJ, Orwig KE, Reijo Pera R. DDX3Y gene rescue of a Y chromosome AZFa deletion restores germ cell formation and transcriptional programs. Sci Rep. 2015;5:15041. doi: 10.1038/srep15041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vakilian H, Mirzaei M, Sharifi Tabar M, Pooyan P, Habibi Rezaee L, Parker L, Haynes PA, Gourabi H, Baharvand H, Salekdeh GH. DDX3Y, a Male-Specific Region of Y Chromosome Gene, May Modulate Neuronal Differentiation. J Proteome Res. 2015;14:3474–83. doi: 10.1021/acs.jproteome.5b00512. [DOI] [PubMed] [Google Scholar]
- 13.Rauschendorf MA, Zimmer J, Ohnmacht C, Vogt PH. DDX3X, the X homologue of AZFa gene DDX3Y, expresses a complex pattern of transcript variants only in the male germ line. Mol Hum Reprod. 2014;20:1208–22. doi: 10.1093/molehr/gau081. [DOI] [PubMed] [Google Scholar]
- 14.Valiente-Echeverría F, Hermoso MA, Soto-Rifo R. RNA helicase DDX3: at the crossroad of viral replication and antiviral immunity. Rev Med Virol. 2015;25:286–99. doi: 10.1002/rmv.1845. [DOI] [PubMed] [Google Scholar]
- 15.Mahboobi SH, Javanpour AA, Mofrad MR. The interaction of RNA helicase DDX3 with HIV-1 Rev-CRM1-RanGTP complex during the HIV replication cycle. PLoS One. 2015;10:e0112969. doi: 10.1371/journal.pone.0112969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ariumi Y. Multiple functions of DDX3 RNA helicase in gene regulation, tumorigenesis, and viral infection. Front Genet. 2014;5:423. doi: 10.3389/fgene.2014.00423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ko C, Lee S, Windisch MP, Ryu WS. DDX3 DEAD-box RNA helicase is a host factor that restricts hepatitis B virus replication at the transcriptional level. J Virol. 2014;88:13689–98. doi: 10.1128/JVI.02035-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bol GM, Xie M, Raman V. DDX3, a potential target for cancer treatment. Mol Cancer. 2015;14:188. doi: 10.1186/s12943-015-0461-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharma D, Jankowsky E. The Ded1/DDX3 subfamily of DEAD-box RNA helicases. Crit Rev Biochem Mol Biol. 2014;49:343–60. doi: 10.3109/10409238.2014.931339. [DOI] [PubMed] [Google Scholar]
- 20.Botlagunta M, Vesuna F, Mironchik Y, Raman A, Lisok A, Winnard P Jr, Mukadam S, Van Diest P, Chen JH, Farabaugh P, Patel AH, Raman V. Oncogenic role of DDX3 in breast cancer biogenesis. Oncogene. 2008;27:3912–22. doi: 10.1038/onc.2008.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Botlagunta M, Krishnamachary B, Vesuna F, Winnard PT Jr, Bol GM, Patel AH, Raman V. Expression of DDX3 is directly modulated by hypoxia inducible factor-1 alpha in breast epithelial cells. PLoS One. 2011;6:e17563. doi: 10.1371/journal.pone.0017563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bol GM, Raman V, van der Groep P, Vermeulen JF, Patel AH, van der Wall E, van Diest PJ. Expression of the RNA helicase DDX3 and the hypoxia response in breast cancer. PLoS One. 2013;8:e63548. doi: 10.1371/journal.pone.0063548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xie M, Vesuna F, Botlagunta M, Bol GM, Irving A, Bergman Y, Hosmane RS, Kato Y, Winnard PT Jr, Raman V. NZ51, a ring-expanded nucleoside analog, inhibits motility and viability of breast cancer cells by targeting the RNA helicase DDX3. Oncotarget. 2015;6:29901–13. doi: 10.18632/oncotarget.4898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun M, Zhou T, Jonasch E, Jope RS. DDX3 regulates DNA damage-induced apoptosis and p53 stabilization. Biochim Biophys Acta. 2013;1833:1489–97. doi: 10.1016/j.bbamcr.2013.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bol GM, Vesuna F, Xie M, Zeng J, Aziz K, Gandhi N, Levine A, Irving A, Korz D, Tantravedi S, Heerma van Voss MR, Gabrielson K, Bordt EA, Polster BM, Cope L, van der Groep P, Kondaskar A, Rudek MA, Hosmane RS, van der Wall E, van Diest PJ, Tran PT, Raman V. Targeting DDX3 with a small molecule inhibitor for lung cancer therapy. EMBO Mol Med. 2015;7:648–69. doi: 10.15252/emmm.201404368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu DW, Liu WS, Wang J, Chen CY, Cheng YW, Lee H. Reduced p21(WAF1/CIP1) via alteration of p53-DDX3 pathway is associated with poor relapse-free survival in early-stage human papillomavirus-associated lung cancer. Clin Cancer Res. 2011;17:1895–905. doi: 10.1158/1078-0432.CCR-10-2316. [DOI] [PubMed] [Google Scholar]
- 27.Wu DW, Lee MC, Wang J, Chen CY, Cheng YW, Lee H. DDX3 loss by p53 inactivation promotes tumor malignancy via the MDM2/Slug/E-cadherin pathway and poor patient outcome in non-small-cell lung cancer. Oncogene. 2014;33:1515–26. doi: 10.1038/onc.2013.107. [DOI] [PubMed] [Google Scholar]
- 28.Su CY, Lin TC, Lin YF, Chen MH, Lee CH, Wang HY, Lee YC, Liu YP, Chen CL, Hsiao M. DDX3 as a strongest prognosis marker and its downregulation promotes metastasis in colorectal cancer. Oncotarget. 2015;6:18602–12. doi: 10.18632/oncotarget.4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heerma van Voss MR, Vesuna F, Trumpi K, Brilliant J, Berlinicke C, de Leng W, Kranenburg O, Offerhaus GJ, Bürger H, van der Wall E, van Diest PJ, Raman V. Identification of the DEAD box RNA helicase DDX3 as a therapeutic target in colorectal cancer. Oncotarget. 2015;6:28312–26. doi: 10.18632/oncotarget.4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zampino R, Pisaturo MA, Cirillo G, Marrone A, Macera M, Rinaldi L, Stanzione M, Durante-Mangoni E, Gentile I, Sagnelli E, Signoriello G, Miraglia Del Giudice E, Adinolfi LE, Coppola N. Hepatocellular carcinoma in chronic HBV-HCV co-infection is correlated to fibrosis and disease duration. Ann Hepatol. 2015;14:75–82. [PubMed] [Google Scholar]
- 31.Viganò L, Conci S, Cescon M, Fava C, Capelli P, D’Errico A, Torzilli G, Di Tommaso L, Giuliante F, Vecchio FM, Salizzoni M, David E, Pinna AD, Guglielmi A, Capussotti L. Liver resection for hepatocellular carcinoma in patients with metabolic syndrome: A multicenter matched analysis with HCV-related HCC. J Hepatol. 2015;63:93–101. doi: 10.1016/j.jhep.2015.01.024. [DOI] [PubMed] [Google Scholar]
- 32.Arzumanyan A, Reis HM, Feitelson MA. Pathogenic mechanisms in HBV- and HCV-associated hepatocellular carcinoma. Nat Rev Cancer. 2013;13:123–35. doi: 10.1038/nrc3449. [DOI] [PubMed] [Google Scholar]
- 33.Upadya MH, Aweya JJ, Tan YJ. Understanding the interaction of hepatitis C virus with host DEAD-box RNA helicases. World J Gastroenterol. 2014;20:2913–26. doi: 10.3748/wjg.v20.i11.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kang JI, Kwon YC, Ahn BY. Modulation of the type I interferon pathways by culture-adaptive hepatitis C virus core mutants. FEBS Lett. 2012;586:1272–8. doi: 10.1016/j.febslet.2012.03.062. [DOI] [PubMed] [Google Scholar]
- 35.Chang PC, Chi CW, Chau GY, Li FY, Tsai YH, Wu JC, Wu Lee YH. DDX3, a DEAD box RNA helicase, is deregulated in hepatitis virus-associated hepatocellular carcinoma and is involved in cell growth control. Oncogene. 2006;25:1991–2003. doi: 10.1038/sj.onc.1209239. [DOI] [PubMed] [Google Scholar]
- 36.Chao CH, Chen CM, Cheng PL, Shih JW, Tsou AP, Lee YH. DDX3, a DEAD box RNA helicase with tumor growth-suppressive property and transcriptional regulation activity of the p21waf1/cip1 promoter, is a candidate tumor suppressor. Cancer Res. 2006;66:6579–88. doi: 10.1158/0008-5472.CAN-05-2415. [DOI] [PubMed] [Google Scholar]
- 37.Lee CH, Lin SH, Yang SF, Yang SM, Chen MK, Lee H, Ko JL, Chen CJ, Yeh KT. Low/negative expression of DDX3 might predict poor prognosis in non-smoker patients with oral cancer. Oral Dis. 2014;20:76–83. doi: 10.1111/odi.12076. [DOI] [PubMed] [Google Scholar]
- 38.Samal SK, Routray S, Veeramachaneni GK, Dash R, Botlagunta M. Ketorolac salt is a newly discovered DDX3 inhibitor to treat oral cancer. Sci Rep. 2015;5:9982. doi: 10.1038/srep09982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilky BA, Kim C, McCarty G, Montgomery EA, Kammers K, DeVine LR, Cole RN, Raman V, Loeb DM. RNA helicase DDX3: a novel therapeutic target in Ewing sarcoma. Oncogene. 2015 doi: 10.1038/onc.2015.336. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 40.Zheng H, Shen M, Zha YL, Li W, Wei Y, Blanco MA, Ren G, Zhou T, Storz P, Wang HY, Kang Y. PKD1 phosphorylation-dependent degradation of SNAIL by SCF-FBXO11 regulates epithelial-mesenchymal transition and metastasis. Cancer Cell. 2014;26:358–73. doi: 10.1016/j.ccr.2014.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Y, Shi J, Chai K, Ying X, Zhou BP. The Role of Snail in EMT and Tumorigenesis. Curr Cancer Drug Targets. 2013;13:963–72. doi: 10.2174/15680096113136660102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Unternaehrer JJ, Zhao R, Kim K, Cesana M, Powers JT, Ratanasirintrawoot S, Onder T, Shibue T, Weinberg RA, Daley GQ. The epithelial-mesenchymal transition factor SNAIL paradoxically enhances reprogramming. Stem Cell Reports. 2014;3:691–8. doi: 10.1016/j.stemcr.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hsu DS, Wang HJ, Tai SK, Chou CH, Hsieh CH, Chiu PH, Chen NJ, Yang MH. Acetylation of snail modulates the cytokinome of cancer cells to enhance the recruitment of macrophages. Cancer Cell. 2014;26:534–48. doi: 10.1016/j.ccell.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 44.Sun M, Song L, Zhou T, Gillespie GY, Jope RS. The role of DDX3 in regulating Snail. Biochim Biophys Acta. 2011;1813:438–47. doi: 10.1016/j.bbamcr.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miao X, Yang ZL, Xiong L, Zou Q, Yuan Y, Li J, Liang L, Chen M, Chen S. Nectin-2 and DDX3 are biomarkers for metastasis and poor prognosis of squamous cell/adenosquamous carcinomas and adenocarcinoma of gallbladder. Int J Clin Exp Pathol. 2013;6:179–90. [PMC free article] [PubMed] [Google Scholar]
- 46.Chen HH, Yu HI, Cho WC, Tarn WY. DDX3 modulates cell adhesion and motility and cancer cell metastasis via Rac1-mediated signaling pathway. Oncogene. 2015;34:2790–800. doi: 10.1038/onc.2014.190. [DOI] [PubMed] [Google Scholar]
- 47.Lin KY, Kao SH, Lai CM, Chen CT, Wu CY, Hsu HJ, Wang WD. Tumor Suppressor Lzap Suppresses Wnt/β-catenin signaling to Promote Zebrafish Embryonic Ventral Cell Fates via the Suppression of Inhibitory Phosphorylation of GSK3. J Biol Chem. 2015;290:29808–19. doi: 10.1074/jbc.M115.669309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Briggs LE, Burns TA, Lockhart MM, Phelps AL, van den Hoff MJ, Wessels A. Wnt/β-catenin and Sonic Hedgehog pathways interact in the regulation of the development of the dorsal mesenchymal protrusion. Dev Dyn. 2016;245:103–13. doi: 10.1002/dvdy.24339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Su J, Wu S, Wu H, Li L, Guo T. CD44 is functionally crucial for driving lung cancer stem cells metastasis through Wnt/β-catenin-FoxM1-Twist signaling. Mol Carcinog. 2015 doi: 10.1002/mc.22443. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 50.Yang J, Wei D, Wang W, Shen B, Xu S, Cao Y. TRAF4 enhances oral squamous cell carcinoma cell growth, invasion and migration by Wnt-β-catenin signaling pathway. Int J Clin Exp Pathol. 2015;8:11837–46. [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao S, Ma Y, Huang X. Trefoil factor 1 elevates the malignant phenotype of mucinous ovarian cancer cell through Wnt/β-catenin signaling. Int J Clin Exp Pathol. 2015;8:10412–9. [PMC free article] [PubMed] [Google Scholar]
- 52.Wu C, Chen J, Chen C, Wang W, Wen L, Gao K, Chen X, Xiong S, Zhao H, Li S. Wnt/β-catenin coupled with HIF-1α/VEGF signaling pathways involved in galangin neurovascular unit protection from focal cerebral ischemia. Sci Rep. 2015;5:16151. doi: 10.1038/srep16151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ma Q, Yang Y, Feng D, Zheng S, Meng R, Fa P, Zhao C, Liu H, Song R, Tao T, Yang L, Dai J, Wang S, Jiang WG, He J. MAGI3 negatively regulates Wnt/β-catenin signaling and suppresses malignant phenotypes of glioma cells. Oncotarget. 2015;6:35851–65. doi: 10.18632/oncotarget.5323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gao X, Zhang L, Gu G, Wu PH, Jin S, Hu W, Zhan C, Li J, Li Y. The effect of oxLDL on aortic valve calcification via the Wnt/β-catenin signaling pathway: an important molecular mechanism. J Heart Valve Dis. 2015;24:190–6. [PubMed] [Google Scholar]
- 55.Spranger S, Bao R, Gajewski TF. Melanomaintrinsic β-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–5. doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- 56.Azzolin L, Panciera T, Soligo S, Enzo E, Bicciato S, Dupont S, Bresolin S, Frasson C, Basso G, Guzzardo V, Fassina A, Cordenonsi M, Piccolo S. YAP/TAZ incorporation in the β-catenin destruction complex orchestrates the Wnt response. Cell. 2014;158:157–70. doi: 10.1016/j.cell.2014.06.013. [DOI] [PubMed] [Google Scholar]
- 57.Dias C, Feng J, Sun H, Shao NY, Mazei-Robison MS, Damez-Werno D, Scobie K, Bagot R, LaBonté B, Ribeiro E, Liu X, Kennedy P, Vialou V, Ferguson D, Peña C, Calipari ES, Koo JW, Mouzon E, Ghose S, Tamminga C, Neve R, Shen L, Nestler EJ. β-catenin mediates stress resilience through Dicer1/microRNA regulation. Nature. 2014;516:51–5. doi: 10.1038/nature13976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Regmi SC, Park SY, Kim SJ, Banskota S, Shah S, Kim DH, Kim JA. The Anti-Tumor Activity of Succinyl Macrolactin A Is Mediated through the β-Catenin Destruction Complex via the Suppression of Tankyrase and PI3K/Akt. PLoS One. 2015;10:e0141753. doi: 10.1371/journal.pone.0141753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pronobis MI, Rusan NM, Peifer M. A novel GSK3-regulated APC:Axin interaction regulates Wnt signaling by driving a catalytic cycle of efficient βcatenin destruction. Elife. 2015;4:e08022. doi: 10.7554/eLife.08022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Loregger A, Grandl M, Mejías-Luque R, Allgäuer M, Degenhart K, Haselmann V, Oikonomou C, Hatzis P, Janssen KP, Nitsche U, Gradl D, van den Broek O, Destree O, Ulm K, Neumaier M, Kalali B, Jung A, Varela I, Schmid RM, Rad R, Busch DH, Gerhard M. The E3 ligase RNF43 inhibits Wnt signaling downstream of mutated β-catenin by sequestering TCF4 to the nuclear membrane. Sci Signal. 2015;8:ra90. doi: 10.1126/scisignal.aac6757. [DOI] [PubMed] [Google Scholar]
- 61.Shivanna S, Harrold I, Shashar M, Meyer R, Kiang C, Francis J, Zhao Q, Feng H, Edelman ER, Rahimi N, Chitalia VC. The c-Cbl ubiquitin ligase regulates nuclear β-catenin and angiogenesis by its tyrosine phosphorylation mediated through the Wnt signaling pathway. J Biol Chem. 2015;290:12537–46. doi: 10.1074/jbc.M114.616623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee HJ, Bao J, Miller A, Zhang C, Wu J, Baday YC, Guibao C, Li L, Wu D, Zheng J. Structurebased discovery of novel small molecule Wnt signaling inhibitors by targeting the cysteine rich domain of Frizzled. J Biol Chem. 2015;290:30596–606. doi: 10.1074/jbc.M115.673202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ren DN, Chen J, Li Z, Yan H, Yin Y, Wo D, Zhang J, Ao L, Chen B, Ito TK, Chen Y, Liu Z, Li Y, Yang J, Lu X, Peng Y, Pan L, Zhao Y, Liu S, Zhu W. LRP5/6 directly bind to Frizzled and prevent Frizzled-regulated tumour metastasis. Nat Commun. 2015;6:6906. doi: 10.1038/ncomms7906. [DOI] [PubMed] [Google Scholar]
- 64.Barrott JJ, Illum BE, Jin H, Zhu JF, Mosbruger T, Monument MJ, Smith-Fry K, Cable MG, Wang Y, Grossmann AH, Capecchi MR, Jones KB. β-catenin stabilization enhances SS18-SSX2-driven synovial sarcomagenesis and blocks the mesenchymal to epithelial transition. Oncotarget. 2015;6:22758–66. doi: 10.18632/oncotarget.4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lu FI, Sun YH, Wei CY, Thisse C, Thisse B. Tissue-specific derepression of TCF/LEF controls the activity of the Wnt/β-catenin pathway. Nat Commun. 2014;5:5368. doi: 10.1038/ncomms6368. [DOI] [PubMed] [Google Scholar]
- 66.Brown-Clay JD, Shenoy DN, Timofeeva O, Kallakury BV, Nandi AK, Banerjee PP. PBK/TOPK enhances aggressive phenotype in prostate cancer via β-catenin-TCF/LEF-mediated matrix metalloproteinases production and invasion. Oncotarget. 2015;6:15594–609. doi: 10.18632/oncotarget.3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Piao Z, Hong CS, Jung MR, Choi C, Park YK. Thymosin β4 induces invasion and migration of human colorectal cancer cells through the ILK/AKT/β-catenin signaling pathway. Biochem Biophys Res Commun. 2014;452:858–64. doi: 10.1016/j.bbrc.2014.09.012. [DOI] [PubMed] [Google Scholar]
- 68.Bourboulia D, Han H, Jensen-Taubman S, Gavil N, Isaac B, Wei B, Neckers L, Stetler-Stevenson WG. TIMP-2 modulates cancer cell transcriptional profile and enhances E-cadherin/betacatenin complex expression in A549 lung cancer cells. Oncotarget. 2013;4:163–73. doi: 10.18632/oncotarget.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hsu TI, Lin SC, Lu PS, Chang WC, Hung CY, Yeh YM, Su WC, Liao PC, Hung JJ. MMP7-mediated cleavage of nucleolin at Asp255 induces MMP9 expression to promote tumor malignancy. Oncogene. 2015;34:826–37. doi: 10.1038/onc.2014.22. [DOI] [PubMed] [Google Scholar]
- 70.Grande MT, Sánchez-Laorden B, López-Blau C, De Frutos CA, Boutet A, Arévalo M, Rowe RG, Weiss SJ, López-Novoa JM, Nieto MA. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat Med. 2015;21:989–97. doi: 10.1038/nm.3901. [DOI] [PubMed] [Google Scholar]
- 71.Okada T, Sinha S, Esposito I, Schiavon G, López-Lago MA, Su W, Pratilas CA, Abele C, Hernandez JM, Ohara M, Okada M, Viale A, Heguy A, Socci ND, Sapino A, Seshan VE, Long S, Inghirami G, Rosen N, Giancotti FG. The Rho GTPase Rnd1 suppresses mammary tumorigenesis and EMT by restraining Ras-MAPK signalling. Nat Cell Biol. 2015;17:81–94. doi: 10.1038/ncb3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov. 2014;13:928–42. doi: 10.1038/nrd4281. [DOI] [PubMed] [Google Scholar]
- 73.Kenny HA, Chiang CY, White EA, Schryver EM, Habis M, Romero IL, Ladanyi A, Penicka CV, George J, Matlin K, Montag A, Wroblewski K, Yamada SD, Mazar AP, Bowtell D, Lengyel E. Mesothelial cells promote early ovarian cancer metastasis through fibronectin secretion. J Clin Invest. 2014;124:4614–28. doi: 10.1172/JCI74778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cruciat CM. Casein kinase 1 and Wnt/β-catenin signaling. Curr Opin Cell Biol. 2014;31:46–55. doi: 10.1016/j.ceb.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 75.Schittek B, Sinnberg T. Biological functions of casein kinase 1 isoforms and putative roles in tumorigenesis. Mol Cancer. 2014;13:231. doi: 10.1186/1476-4598-13-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cruciat CM, Dolde C, de Groot RE, Ohkawara B, Reinhard C, Korswagen HC, Niehrs C. RNA helicase DDX3 is a regulatory subunit of casein kinase 1 in Wnt-β-catenin signaling. Science. 2013;339:1436–41. doi: 10.1126/science.1231499. [DOI] [PubMed] [Google Scholar]
- 77.Davis FM, Azimi I, Faville RA, Peters AA, Jalink K, Putney JW Jr, Goodhill GJ, Thompson EW, Roberts-Thomson SJ, Monteith GR. Induction of epithelial-mesenchymal transition (EMT) in breast cancer cells is calcium signal dependent. Oncogene. 2014;33:2307–16. doi: 10.1038/onc.2013.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.da Silva SD, Morand GB, Alobaid FA, Hier MP, Mlynarek AM, Alaoui-Jamali MA, Kowalski LP. Epithelial-mesenchymal transition (EMT) markers have prognostic impact in multiple primary oral squamous cell carcinoma. Clin Exp Metastasis. 2015;32:55–63. doi: 10.1007/s10585-014-9690-1. [DOI] [PubMed] [Google Scholar]
- 79.Yoshida T, Ozawa Y, Kimura T, Sato Y, Kuznetsov G, Xu S, Uesugi M, Agoulnik S, Taylor N, Funahashi Y, Matsui J. Eribulin mesilate suppresses experimental metastasis of breast cancer cells by reversing phenotype fromepithelial-mesenchymal transition (EMT) to mesenchymal-epithelial transition (MET) states. Br J Cancer. 2014;110:1497–505. doi: 10.1038/bjc.2014.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ye X, Weinberg RA. Epithelial-Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends Cell Biol. 2015;25:675–86. doi: 10.1016/j.tcb.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yu JM, Sun W, Hua F, Xie J, Lin H, Zhou DD, Hu ZW. BCL6 induces EMT by promoting the ZEB1-mediated transcription repression of E-cadherin in breast cancer cells. Cancer Lett. 2015;365:190–200. doi: 10.1016/j.canlet.2015.05.029. [DOI] [PubMed] [Google Scholar]
- 82.Cheung CT, Bendris N, Paul C, Hamieh A, Anouar Y, Hahne M, Blanchard JM, Lemmers B. Cyclin A2 modulates EMT via β-catenin and phospholipase C pathways. Carcinogenesis. 2015;36:914–24. doi: 10.1093/carcin/bgv069. [DOI] [PubMed] [Google Scholar]
- 83.Mitra A, Satelli A, Xia X, Cutrera J, Mishra L, Li S. Cell-surface Vimentin: A mislocalized protein for isolating csVimentin(+) CD133(-) novel stem-like hepatocellular carcinoma cells expressing EMT markers. Int J Cancer. 2015;137:491–6. doi: 10.1002/ijc.29382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Agajanian M, Runa F, Kelber JA. Identification of a PEAK1/ZEB1 signaling axis during TGFβ/fibronectin-induced EMT in breast cancer. Biochem Biophys Res Commun. 2015;465:606–12. doi: 10.1016/j.bbrc.2015.08.071. [DOI] [PubMed] [Google Scholar]
- 85.Ye X, Tam WL, Shibue T, Kaygusuz Y, Reinhardt F, Ng Eaton E, Weinberg RA. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature. 2015;525:256–60. doi: 10.1038/nature14897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shao DD, Xue W, Krall EB, Bhutkar A, Piccioni F, Wang X, Schinzel AC, Sood S, Rosenbluh J, Kim JW, Zwang Y, Roberts TM, Root DE, Jacks T, Hahn WC. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell. 2014;158:171–84. doi: 10.1016/j.cell.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rokavec M, Öner MG, Li H, Jackstadt R, Jiang L, Lodygin D, Kaller M, Horst D, Ziegler PK, Schwitalla S, Slotta-Huspenina J, Bader FG, Greten FR, Hermeking H. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J Clin Invest. 2014;124:1853–67. doi: 10.1172/JCI73531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ombrato L, Malanchi I. The EMT universe: space between cancer cell dissemination and metastasis initiation. Crit Rev Oncog. 2014;19:349–61. doi: 10.1615/critrevoncog.2014011802. [DOI] [PubMed] [Google Scholar]
- 89.Mitra A, Mishra L, Li S. EMT, CTCs and CSCs in tumor relapse and drug-resistance. Oncotarget. 2015;6:10697–711. doi: 10.18632/oncotarget.4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Derynck R, Muthusamy BP, Saeteurn KY. Signaling pathway cooperation in TGF-β-induced epithelial-mesenchymal transition. Curr Opin Cell Biol. 2014;31:56–66. doi: 10.1016/j.ceb.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Valcourt U, Carthy J, Okita Y, Alcaraz L, Kato M, Thuault S, Bartholin L, Moustakas A. Analysis of Epithelial-Mesenchymal Transition Induced by Transforming Growth Factor β. Methods Mol Biol. 2016;1344:147–81. doi: 10.1007/978-1-4939-2966-5_9. [DOI] [PubMed] [Google Scholar]
- 92.Ghahhari NM, Babashah S. Interplay between microRNAs and WNT/β-catenin signalling pathway regulates epithelial-mesenchymal transitionin cancer. Eur J Cancer. 2015;51:1638–49. doi: 10.1016/j.ejca.2015.04.021. [DOI] [PubMed] [Google Scholar]
- 93.Liu X, Yun F, Shi L, Li ZH, Luo NR, Jia YF. Roles of Signaling Pathways in the Epithelial-Mesenchymal Transition in Cancer. Asian Pac J Cancer Prev. 2015;16:6201–6. doi: 10.7314/apjcp.2015.16.15.6201. [DOI] [PubMed] [Google Scholar]
- 94.Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu CC, LeBleu VS, Kalluri R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527:525–30. doi: 10.1038/nature16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhou Y, Zhang H, Zhuo X, Liu Y, Zhang G, Tan Y. Over-expression of TWIST, an epithelial-mesenchymal transition inducer, predicts poor survival in patients with oral carcinoma. Int J Clin Exp Med. 2015;8:9239–47. [PMC free article] [PubMed] [Google Scholar]
- 96.Cai J, Tian AX, Wang QS, Kong PZ, Du X, Li XQ, Feng YM. FOXF2 suppresses the FOXC2-mediated epithelial-mesenchymal transition and multidrug resistance of basal-like breast cancer. Cancer Lett. 2015;367:129–37. doi: 10.1016/j.canlet.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 97.Zhu G, Li X, Guo B, Ke Q, Dong M, Li F. PAK5-mediated E47 phosphorylation promotes epithelial-mesenchymal transition and metastasis of colon cancer. Oncogene. 2015 doi: 10.1038/onc.2015.259. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 98.Joseph JV, Conroy S, Tomar T, Eggens-Meijer E, Bhat K, Copray S, Walenkamp AM, Boddeke E, Balasubramanyian V, Wagemakers M, den Dunnen WF, Kruyt FA. TGF-β is an inducer of ZEB1-dependent mesenchymal transdifferentiation in glioblastoma that is associated with tumor invasion. Cell Death Dis. 2014;5:e1443. doi: 10.1038/cddis.2014.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–96. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gonzalez DM, Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal. 2014;7:re8. doi: 10.1126/scisignal.2005189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lindsey S, Langhans SA. Crosstalk of Oncogenic Signaling Pathways during Epithelial-Mesenchymal Transition. Front Oncol. 2014;4:358. doi: 10.3389/fonc.2014.00358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wei SC, Fattet L, Tsai JH, Guo Y, Pai VH, Majeski HE, Chen AC, Sah RL, Taylor SS, Engler AJ, Yang J. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat Cell Biol. 2015;17:678–88. doi: 10.1038/ncb3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Katsuno Y, Lamouille S, Derynck R. TGF-β signaling and epithelial-mesenchymal transition in cancer progression. Curr Opin Oncol. 2013;25:76–84. doi: 10.1097/CCO.0b013e32835b6371. [DOI] [PubMed] [Google Scholar]
- 104.Bonelli MA, Cavazzoni A, Saccani F, Alfieri RR, Quaini F, La Monica S, Galetti M, Cretella D, Caffarra C, Madeddu D, Frati C, Lagrasta CA, Falco A, Rossetti P, Fumarola C, Tiseo M, Petronini PG, Ardizzoni A. Inhibition of PI3K Pathway Reduces Invasiveness and Epithelial-to-Mesenchymal Transition in Squamous Lung Cancer Cell Lines Harboring PIK3CA Gene Alterations. Mol Cancer Ther. 2015;14:1916–27. doi: 10.1158/1535-7163.MCT-14-0892. [DOI] [PubMed] [Google Scholar]
- 105.Wei XL, Dou XW, Bai JW, Luo XR, Qiu SQ, Xi DD, Huang WH, Du CW, Man K, Zhang GJ. ERα inhibits epithelial-mesenchymal transition by suppressing Bmi1 in breast cancer. Oncotarget. 2015;6:21704–17. doi: 10.18632/oncotarget.3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jiang FZ, He YY, Wang HH, Zhang HL, Zhang J, Yan XF, Wang XJ, Che Q, Ke JQ, Chen Z, Tong H, Zhang YL, Wang FY, Li YR, Wan XP. Mutant p53 induces EZH2 expression and promotes epithelial-mesenchymal transition by disrupting p68-Drosha complex assembly and attenuating miR-26a processing. Oncotarget. 2015;6:44660–74. doi: 10.18632/oncotarget.6350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Huang Y, Zhao M, Xu H, Wang K, Fu Z, Jiang Y, Yao Z. RASAL2 down-regulation in ovarian cancer promotes epithelial-mesenchymal transition and metastasis. Oncotarget. 2014;5:6734–45. doi: 10.18632/oncotarget.2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rhodes LV, Martin EC, Segar HC, Miller DF, Buechlein A, Rusch DB, Nephew KP, Burow ME, Collins-Burow BM. Dual regulation by microRNA-200b-3p and microRNA-200b-5p in the inhibition of epithelial-to-mesenchymaltransition in triple-negative breast cancer. Oncotarget. 2015;6:16638–52. doi: 10.18632/oncotarget.3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Koutsaki M, Spandidos DA, Zaravinos A. Epithelial-mesenchymal transition-associated miRNAs in ovarian carcinoma, with highlight on the miR-200 family: prognostic value and prospective role in ovarian cancer therapeutics. Cancer Lett. 2014;351:173–81. doi: 10.1016/j.canlet.2014.05.022. [DOI] [PubMed] [Google Scholar]
- 110.Liu F, Kong X, Lv L, Gao J. TGF-β1 acts through miR-155 to down-regulate TP53INP1 in promoting epithelial-mesenchymal transition and cancer stem cell phenotypes. Cancer Lett. 2015;359:288–98. doi: 10.1016/j.canlet.2015.01.030. [DOI] [PubMed] [Google Scholar]
- 111.Sun L, Kokura K, Izumi V, Koomen JM, Seto E, Chen J, Fang J. MPP8 and SIRT1 crosstalk in E-cadherin gene silencing and epithelial-mesenchymal transition. EMBO Rep. 2015;16:689–99. doi: 10.15252/embr.201439792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yu H, Shen Y, Hong J, Xia Q, Zhou F, Liu X. The contribution of TGF-β in Epithelial-Mesenchymal Transition (EMT): Down-regulation of Ecadherin via snail. Neoplasma. 2015;62:1–15. doi: 10.4149/neo_2015_002. [DOI] [PubMed] [Google Scholar]