

Abstract
Recovery rates for B-cell Non-Hodgkin’s Lymphoma (NHL) are up to 70% with current standard-of-care treatments including rituximab (chimeric anti-CD20 monoclonal antibody) in combination with chemotherapy (R-CHOP). However, patients who do not respond to first-line treatment or develop resistance have a very poor prognosis. This signifies the need for the development of an optimal treatment approach for relapsed/refractory B-NHL. Novel CD19- chimeric antigen receptor (CAR) T-cell redirected immunotherapy is an attractive option for this subset of patients. Anti-CD19 CAR T-cell therapy has already had remarkable efficacy in various leukemias as well as encouraging outcomes in phase I clinical trials of relapsed/refractory NHL. In going forward with additional clinical trials, complementary treatments that may circumvent potential resistance mechanisms should be used alongside anti-CD19 T-cells in order to prevent relapse with resistant strains of disease. Some such supplementary tactics include conditioning with lymphodepletion agents, sensitizing with kinase inhibitors and Bcl-2 inhibitors, enhancing function with multispecific CAR T-cells and CD40 ligand-expressing CAR T-cells, and safeguarding with lymphoma stem cell-targeted treatments. A therapy regimen involving anti-CD19 CAR T-cells and one or more auxiliary treatments could dramatically improve prognoses for patients with relapsed/refractory B-cell NHL. This approach has the potential to revolutionize B-NHL salvage therapy in much the same way rituximab did for first-line treatments.
Keywords: Non-Hodgkin’s Lymphoma, resistance, apoptosis, immunotherapy, CD19, chimeric antigen receptor, adoptive cell therapy, signal transduction, hematological malignancies
Non-Hodgkin’s Lymphoma (NHL)
Incidence
Non-Hodgkin’s Lymphoma (NHL) is a group of histologically and biologically distinct malignancies that originate from B-cells or T-cells. The exact cause of the disease is poorly understood; irregular incidence patterns may suggest distinct etiologies, yet studies within the past two decades support the notion that there is some common mechanism of development among all lymphomas [1-3]. NHL is most common in developed countries and ranks fifth in cancer mortality in the United States. Incidence rates increase exponentially with age, and are about 50% higher in men than in women in the US. Incidence has increased over the past several decades, with the largest number of increases seen in aggressive lymphoma [4,5]. There is also a greater degree of increase in incidences of the extranodal form than those of the nodal form [6]. The increased number of incidences is likely due to multiple factors, particularly those related to genetics, immune system condition, and lifestyle [7].
Individuals with a family history of NHL show an increased risk of incidence, as well as those with genetic variants that promote B-cell survival and growth [8,9]. Additionally, immune deficiency is known to greatly increase the risk of developing NHL. Immunosuppression is often observed in association with chronic antigenic stimulation due to infection, especially Epstein-Barr virus (EBV). This is thought to be the cause of higher incidence observed among AIDS patients, whose immune systems are weakened and are therefore less likely to successfully respond to oncogenic herpesviruses [10]. Infections with EBV are extremely common; however only in immunocompromised individuals is EBV able to cause B-cell proliferation and eventually lymphoma [11]. There are also other viral infections known to be involved in the pathogenesis of NHL, including human T-cell lymphotrophic virus (HTLV), Kaposi’s sarcoma-associated herpesvirus, herpes zoster (reactivated latent varicella zoster virus), and hepatitis C virus [12-15].
In addition to host factors and immune deficiency due to infection, many incidence factors are associated with lifestyle. While tobacco and alcohol consumption do not show significant and/or consistent associations, diet has been shown to have an effect on NHL incidence [16,17]. Some studies have found that greater consumption of red meat and dietary fat is associated with increased risk, while fish, fruits, and vegetables may be inversely related. Obesity, use of carcinogen-containing hair dyes, and exposure to excess UV radiation are also suggested risk factors [18-21]. However, recreational sun exposure has been shown to potentially lower the risk of NHL development [22]. Additionally, certain occupations show greater incidence, particularly jobs that involve heavy or frequent use of chemicals such as benzene, herbicides, insecticides, fertilizers, and organic solvents [7,23,24].
Classification of NHL and treatment options
In determining the optimal treatment regimen, both the grade and extent of disease must be taken into account. There are approximately 60 subtypes of NHL, which can be classified by their grade. Low-grade (indolent) lymphomas grow slowly and show a high responsiveness to treatment, yet they are incurable when diagnosed at an advanced stage. Intermediate- and high-grade (aggressive) lymphomas are fast-growing and can show long-term remission or cure even at advanced stages. Approximately 85% of NHL cases in the US are of B-cell origin, and of these follicular lymphoma (FL) and diffuse large B-cell lymphoma (DLBCL) account for the greatest number of cases (approximately 20% and 35% of all NHL cases, respectively) [5]. Treatment approaches for these two common subtypes are archetypes for treatments of all B-cell NHL [25].
Follicular lymphoma (FL), an indolent lymphoma, acts as a paradigm for the management of all indolent subtypes of NHL. Treatment options are watchful waiting, single agent alkylators, nucleoside analogues, combination chemotherapy, and immunotherapy with monoclonal antibodies (mAbs), radiolabeled mAbs, or interferon (IFN) [26]. Some patients may be cured with radiation alone, while those with more advanced disease are treated with chemotherapy, immunotherapy, or both. The advent of immunotherapy rituximab was a groundbreaking advancement in treatment of indolent subtypes, and later other forms as well.
Rituximab is directed against the B-cell transmembrane phosphoprotein CD20, which is involved in cell proliferation, activation, differentiation, and apoptosis. CD20 is expressed on more than 90% of B-cell lymphomas, and it is an ideal target for immunotherapy because it does not shed, modulate, or internalize. Rituximab functions by binding to CD20 and inducing apoptosis [27,28]. Though CD20 is expressed on both malignant and normal B-cells, the absence of normal B-cells for the duration of treatment has been proven to be a relatively negligible risk. A momentous clinical trial in 1998 demonstrated a 48% response rate in relapsed low-grade NHL patients over the course of about one year [29]. Since the treatment was first introduced, its application has expanded to nearly all CD20 positive NHL as well as some autoimmune disorders [30]. About 1 in 3 FL cases will transform into an aggressive lymphoma at some stage, dramatically decreasing the probability of 10-year survival [31]. Even after transformation, rituximab is a principal part of treatment.
Diffuse large B-cell lymphoma (DLBCL), an aggressive lymphoma and the most common subtype, acts as a paradigm for the treatment of all aggressive forms of NHL. The standard front-line treatment is R-CHOP, a combination of rituximab and the four chemotherapy drugs cyclophosphamide, doxorubicin, vincristine, and prednisone. Before the introduction of rituximab, CHOP was the optimal treatment regimen for aggressive forms of NHL, providing long-term remission or cure for 30-50% of patients. The later addition of rituximab to CHOP therapy further increased recovery rates to up to 70% [32]. R-CHOP immune-chemotherapy can cure the majority of cases, including those at the most advanced stages, but patients who fail to respond to R-CHOP ultimately die from the disease. Approximately 10-15% of patients showed no response or relapse within the first three months of treatment, a condition also known as primary refractory disease, and 20-25% relapse after exhibiting an initial response [33]. The life expectancy for patients with relapsed or refractory aggressive lymphoma is 3-4 months.
Retreatment with R-CHOP is not effective in such cases, thus other salvage treatments are used as the last line of defense. Salvage agents are combined with high-dose chemotherapy and autologous stem cell transplantation in eligible patients. As with first-line treatment, rituximab may be added to salvage regimens in order to improve outcomes. However, even with rituximab, salvage therapy is successful for relatively few patients [34,35]. Similar salvage approaches and outcomes are found in relapsed or refractory FL and other indolent lymphomas [36]. An optimal treatment for relapsed/refractory NHL has yet to be found, yet a range of novel treatments is being investigated including new applications of antibody-based therapy [37,38].
Antibody-mediated immunotherapy of NHL
Antibody-mediated immunotherapy, which is already central to existing NHL treatments, has the benefit of a high level of specificity and therefore little systematic toxicity. Exploration of cancer therapeutic antibodies began in the 1950s with polyclonal antibodies of murine, rabbit, and rat origins. Large-scale clinical trials became possible in 1975 with the advent of hybridoma technology, which allowed for specifically targeted mAbs to be manufactured in large quantities [39,40]. Within the next decade, new mAbs were engineered that linked mouse or primate antigen binding domains with human constant region domains. While early animal-derived mAbs were problematic in that they elicited a host immune response, struggled to recruit human effector cell functions, and had a short half-life, the new chimeric mAbs had fewer complications [41]. Following the development of chimeric mAbs, humanized mAbs were created which allowed fully human monoclonal antibodies to be generated in the lab [42].
Rituximab, a chimeric mAb, was the first monoclonal antibody to be approved by the FDA for therapeutic use in 1997. Many other mAbs have been introduced since then, including trastuzumab (anti-ERBB2) for treatment of breast cancer, alemtuzumab (anti-CD52) for B-cell chronic lymphocytic leukemia (B-CLL), bevacizumab (anti-VEGF-A) for metastatic colorectal cancer, and ipilimumab (anti-CTLA4) for unresectable or metastatic melanoma [43]. These agents can kill malignant cells either directly via signaling for apoptosis, or indirectly via activated effector mechanisms such as complement-dependent cytotoxicity (CDC) or antibody-dependent cell mediated toxicity (ADCC), among other mechanisms [44]. mAbs can be used either alone or with other agents, both as front-line and salvage treatments.
mAb-based therapies fail when there is proliferation of drug-resistant variants of a malignancy, resulting in relapsed or refractory disease [25]. Some cases have a preexisting resistance to front-line treatments, while others develop resistance after exhibiting an initial response. The mechanisms of both modes of resistance are unknown, but attempts are being made at understanding them. Determining the mechanisms of resistance would be valuable for both the development of new treatment options and the improvement of prognoses [45]. Resistance to rituximab is of particular interest due to its central role in the treatment of both NHL and autoimmune disorders. Some proposed resistance mechanisms are increased metabolism of the mAb, loss or downregulation of CD20, reduced tumor penetration, impaired binding, impaired effector cell recruitment or function, and resistance to effector mechanisms [46]. Novel treatments aim to circumvent these potential means of resistance by employing alternative targets and mechanisms. One such treatment is anti-CD19 chimeric antigen receptor (CAR) therapy, which uses antibody components to target CD19+ B-cells.
Anti-CD19 CAR T-cell therapy
Chimeric antigen receptor (CAR) T-cell immunotherapy: structure and basic design
Chimeric antigen receptor (CAR) T-cell immunotherapy involves the adoptive transfer of patient-derived T-cells engineered to express synthetic antigen-targeted receptors of defined specificity. CARs are able to directly recognize antigens in an MHC-independent. They are designed with a precise combination of components that function in harmony: antigen-recognizing single-chain variable fragments (scFv) derived from mAbs allow for high specificity, intracellular signaling modules trigger that T-cell functions, and spacer domains which provide flexibility and optimize engagement [47]. The antigen-recognizing region triggers T-cell effector functions via the CD3ζ. First generation CARs solely utilize a cytoplasmic CD3ζ region, while subsequent CAR models include additional costimulatory modules for improved T-cell function and persistence. Second generation CAR T-cells with the addition of a CD28 costimulatory domain were shown to be far more effective in a direct comparison with first generation CAR T-cells against B-cell lymphomas [48]. Other costimulatory domains that can be added to CD3ζ to improve efficacy include CD134 and CD137. Third generation CARs include a combination of two or more costimulatory domains that function synergistically for even greater antitumor function [49].
Key characteristics of natural (endogenous) T-cell receptors (TCRs) are considered when designing and testing CARs, including receptor affinity, signal efficiency, and spatial properties. Normal TCRs maintain an optimal level of affinity that elicits an effective response while staying beneath the threshold at which activation-induced cell death (AICD) is triggered. Conversely, optimal CAR affinities, which can vary by target antigen, have yet to be empirically determined. TCRs also have the benefit of forming an immunological synapse with peptide/MHC (pMHC) on target cells, which ensures efficient signal delivery. Because CARs lack this feature, they require higher ligand density for effective recognition. TCR signaling is further supported by the small size of the TCR/pMHC complex, which allows segregation from other cell surface molecules [50]. Accordingly, it is critical to adjust the size and shape of the spacer domain depending on the physical characteristics of the target molecule in order to optimize CAR signaling [51].
CAR therapy can cause prolonged toxicities because the engineered T-cells can persist in the body even after treatment is no longer required. The resulting elevated cytokine levels can lead to cytokine release syndrome, known as cytokine storm in severe cases, in which the immune system creates a positive feedback loop of proinflammatory cytokine production. This is associated with hypotension and systematic inflammatory reaction syndrome, in which the entire body becomes inflamed. These immune reactions are usually resolved with steroids or neutralizing mAbs, but can be fatal in some cases [52]. Neurologic toxic effects have also been reported, including delirium, aphasia, hallucinations, and seizure-like activity [53,54]. There are also complications associated with depletion of healthy cells that also express the target antigen. B-cell aplasia is a typical side effect in the treatment of B-cell malignancies because pan-B-cell markers are usually used as treatment targets. The varying issues that arise in relation to prolonged CAR T-cell activity may be limited by targeting antigens that are largely restricted to tumor cells, such as κ or λ light chains [55]. Other strategies for selective tumor eradication involve the use of two CARs working synchronously, with one acting as the activating receptor and the other serving to either inhibit activation on normal cells or trigger activation on tumor cells [56,57]. Another potential solution is to engineer CAR T-cells to be capable of inducible apoptosis. Trials of the “suicide gene” inducible caspase 9 have shown promising results [58,59].
CAR engineering is still being perfected, but it nonetheless stands as an attractive treatment approach for B-cell malignancies. It can be used as a bridge to stem cell transplantation or potentially as a solo curative therapy for relapsed/refractory diseases. By ensuring the proper connection of active agents to effector T-cells, CAR therapy bypasses several potential points of resistance in relapsed/refractory diseases. It therefore has a greater likelihood of success as a salvage therapy than direct treatment with mAbs. Engineering CARs to target CD19 further increases the likelihood of efficacy in salvage situations because resistance may be specific to CD20-targeted treatments.
Targeting CD19 for therapy of B-cell NHL
Structure and function of CD19 gene and antigen
The human CD19 gene is located on the short arm of chromosome 16 at band p11.2 and contains 15 exons and 18 introns, spanning 7.59 kb of DNA. Transcription is initiated from two probable alternative promoters, as the gene lacks a TATA box. The promoters are presumed to be two regulatory sequences that are highly conserved in mouse and human: a 193 bp sequence that is located directly upstream of the initiation site and a 198 bp sequence that is located 1 kb upstream [60]. The gene produces 8 different mRNA species-7 splice variants and 1 apparently non-coding unspliced form-though only 2 isoforms have been isolated in vivo. [61]. A Southern blot analysis comparison of human and mouse CD19 genes revealed that both were compact single copy genes with 15 exons and identical exon-intron boundaries correlated with the known functional domains of the protein. The gene was highly conserved in all exons and 5’ and 3’ untranslated regions, suggesting that gene expression may be similarly regulated in both species. A 79% homology between mouse and human nucleotide sequences of the CD19 cytoplasmic domains indicates that there must have been significant selective pressure to conserve them, evidence of the intracellular tail’s key role in signal transduction [62,63].
The CD19 gene encodes for a 95 kDa transmembrane glycoprotein of the immunoglobulin superfamily. It is expressed almost continually during B-lymphocyte development, first activated at the late pro-B-cell stage by Paired Box 5 (PAX5) and lost upon terminal plasma cell differentiation [64]. The protein antigen is 556 amino acids in length with an extracellular N-terminus and an intracellular C-terminus. It consists of two extracellular immunoglobulin-like domains, a short hydrophobic transmembrane region, and a roughly 240-amino acid-long cytoplasmic tail. Nadler et al. first identified CD19 as the human B-cell antigen B4, and Schriever et al. later found that it is expressed on almost all B-cells and follicular dendritic cells [65,66]. It functions an essential regulator in both intrinsic and antigen receptor-induced B-cell signal transduction [67].
Physiological functions of CD19
CD19 chiefly operates in a mature B-cell membrane complex comprised of CD21, CD81, and CD225 that modulates B-cell antigen receptor (BCR) signaling. Within the complex, CD19 and the complement receptor CD21 function together to transduce signals when complement C3d-coupled antigens bind to the BCR and to CD21. In the complement receptor system, CD21 has the ability to augment receptor capacity in reaction to decreased antigen concentrations. CD19 serves as the crucial signaling component of the complex due to its long intracellular tail, which transmits signals to downstream components of the signaling machinery [68]. The tetraspanin CD81 links the complex to the actin cytoskeleton and, along with the cytoskeleton, organizes CD19 nanoclusters on the plasma membrane [69]. The function of the fourth protein in this complex, CD225 or Leu-13, is unknown. The complex decreases the threshold for stimulation on the small number of BCRs with which it colligates (approximately 0.03% of the total BCRs), which ensures receptor sensitivity even when antigen concentrations are low. Moreover, the BCRs themselves are low-affinity, which ensures receptor specificity despite the multiplicity of antigens present in the cell’s environment. This system allows BCRs to respond to stimuli in a manner that is both sensitive and specific, which is necessary for proper B-cell proliferation and differentiation [70].
CD19’s function as a B-cell regulator is of crucial importance, as illustrated by observations of CD19 deficiency in mice and CD19 mutations in humans. CD19-/- mice exhibit reduced number of peripheral B-cells, suggesting that the antigen has an important role in B-cell survival. Evidence suggests that CD19 not only propagates BCR-dependent survival signals in mature B-cells, but also promotes the survival of naive recirculating B-cells prior to antigen encounter, indicating that CD19 also functions outside of its BCR-associated complex. CD19-deficient mice also show a drastic reduction in B1, germinal center, and marginal zone B-cells, demonstrating CD19’s significant role in B-cell differentiation [71]. In clinical case studies, mutations of the CD19 gene are associated with severe antibody deficiency and autoimmune disease. The first study to report on CD19 deficiency found homozygous frame shift mutations in the CD19 gene in four patients from two separate families. The mutations resulted in premature stop codons and truncated CD19 proteins that lacked all or part of the cytoplasmic tail, rendering them unstable. Levels of surface CD19 were very low in patients with partial cytoplasmic domains and undetectable in the patient with an absent cytoplasmic domain. All patients had normal numbers of circulating B-cells but a diminished amount of memory B-cells, as well as decreased levels of CD21. Clinical symptoms were increased susceptibility to infection and hypo-gammaglobulinemia, an immune deficiency disease characterized by an abnormally low level of immunoglobulins (Igs), which was caused by defective B-cell antigen responses due to a shortage of the CD19/CD21 complex [72]. In another case study, similar observations were reported in conjunction with two additional CD19 mutations in a single patient. The patient’s CD19 deficiency was suspected to relate to his thrombocytopenia (low platelet count), possibly linking the CD19 mutations to the development of autoimmune disease [73]. Similarly, mutations that cause overexpression of CD19, like those found in systemic sclerosis patients, can also disrupt B-cell regulated autoimmunity and result in autoimmune disorders [74].
CD19-mediated signaling transduction
The cytoplasmic tail of CD19 is responsible for augmenting both basal and BCR-induced Src family kinase activation. Signaling pathways rely on its nine tyrosine residues to activate down-stream protein kinases, resulting in rapid recruitment and activation. The two most essential tyrosines are Y513 and Y482, as tyrosine-to-phenylalanine mutations in these have been found to inhibit phosphorylation of the other residues, however Y391 also takes part in recruiting the adaptor protein Vav. Upon BCR activation, Lyn, or another Src kinase in Lyn’s absence, first binds to Y513 and then phosphorylates Y513, Y482, and Y391 residues on CD19, as well as Vav through processive phosphorylation [67]. Vav activation initiates downstream mitogen-activated protein kinase (MAPK) pathways; phosphorylated Vav activates Rac GTPases, leading to activation of c-Jun N-terminal kinases (JNKs) involved in stress response and phosphatidylinositol 4-phosphate 5-kinases (PIP5Ks) involved in multiple functions via production of PIP2 [74].
In addition to antigen-induced Vav pathways, CD19 also initiates antigen-induced phosphoinositide 3 kinase (PI3K) signaling cascade, a critical and highly conserved pathway is found in all cells of higher eukaryotes. Two key tyrosine residues of CD19, Y513 and Y482, activate PI3K/AKT by binding the tandem SH2 domains of its regulatory p85 subunit. PI3K is an essential signaling module associated with a vast array of cell functions, including cell growth and survival. Activated PI3K uses PIP2 on the plasma membrane to produce second messenger PIP3, which in turn activates the serine/threonine kinase AKT by phosphorylating the threonine 308 and serine 473 residues. AKT/PKB signaling pathway plays a key role by activating the expression of downstream growth factors, cytokines, and other cellular stimuli is involved in a multitude of cell processes including survival, growth, proliferation, metabolism, motility, transcription, protein synthesis, and cell-cycle progression. AKT activation of mTOR is part of a crucial cell cycle regulation pathway that is often aberrant in cancers [75]. PI3K/AKT pathways are largely reliant on CD19 for BCR-mediated activation, though BCR-induced activation of PI3K is also possible in the absence of CD19 due to the complementary functions of the B-cell adaptor [76].
Advantages of targeting CD19 in B-cell malignancies
The CD19 antigen stands as an attractive target for the treatment of B-cell malignancies, particularly in cases of relapsed/refractory B-NHL that may have developed resistance to first-line treatment via loss or down-regulation of CD20. CD19 is a reliable target due to its presence in the vast majority of B-cell malignancies, including more than 95% of NHL cases [77]. Because it is expressed throughout B-cell development, it allows for a wider range of B-cells to be targeted than does CD20. This characteristic can be advantageous even in the treatment of mature B-cell neoplasms due to the likely existence of malignant immature progenitor cells, which have been suggested as a source of relapse in multiple studies [78-82]. Furthermore, effects of CD19+ cell depletion are largely restricted to the lymphoid system because CD19 expression is exclusive to B-cells and follicular dendritic cells. B-cell aplasia as a consequence of treatment is not necessarily associated with life-threatening toxicities. B-cells can normally be regenerated from haematopoietic stem cells following depletion by anti-CD19 CAR therapy, however Ig replacement may be necessary in cases of long-term B-cell deficiency [64].
As an additional benefit of CD19-targeted treatment, the antigen is associated with B-cell lymphomagenesis in at least two ways. First, within the context of its function as a BCR signal amplifier, CD19 plays a role in PAX5-mediated stimulation of neoplastic growth. BCR signaling is constitutively active in multiple NHL subtypes, and it is believed to be capable of driving lymphoma development. Though anti-apoptotic signaling pathways can function in the absence of the BCR by PI3K activation alone, the BCR and its associated components are heavily relied upon for survival signaling [83]. Several crucial BCR signaling components are regulated by the transcription factor PAX5, including CD19, and PAX5’s ability to express these BCR components has been shown to contribute to lymphoma development [84,85] Second, independent of its BCR-associated function, CD19 activates MYC-driven lymphomagenesis. C-MYC, a PAX5 controlled oncogene, plays a major role in promoting neoplastic growth in B-cells and is overexpressed in NHL. CD19 has been found to function in a promoter-independent posttranslational signaling amplification loop with the oncogene to promote B-cell transformation and lymphoma progression. Both the expression and function of c-MYC are enhanced by CD19, so inhibiting this positive feedback loop would greatly reduce its oncogenic capabilities [86-88]. Because these two modes of lymphoma development are related to CD19 expression, eliminating CD19+ cells has the advantage of providing some insurance against relapse.
Clinical trials of anti-CD19 CAR T-cell therapy in B-cell malignancies
The most widely studied and successful applications of CAR therapy have been in CD19-targeted treatment of B-cell leukemia and lymphoma [47]. All clinical trials involve a conditioning chemotherapy regimen prior to infusion, which greatly enhances both T-cell persistence and antitumor efficacy [89]. Antitumor activity of anti-CD19 CAR was first reported in a case of advanced follicular NHL, in which anti-CD19 CAR therapy resulted in dramatic regression. Peripheral blood B-cells were absent for at least 39 weeks after treatment, but no acute toxicities occurred [90]. A similar trial was conducted with 8 patients with advanced, progressive B-cell malignancies, 6 of which obtained remissions. Four patients experienced prolonged B-cell depletion, and 4 of the 8 had elevations of the cytokines IFN-γ and TNF, with correlating acute toxicities [91].
Anti-CD19 CAR therapy used in cases of B-cell leukemia has also shown promising efficacy. In chronic lymphocytic leukemia (CLL), treatment has resulted in partial or complete remissions in a trial of 3 patients with advanced disease, with no persistent toxicities occurring other than B-cell aplasia [92]. Another trial in which complete remission was observed in 1 patient with refractory CLL resulted in tumor lysis syndrome as a notable side effect [93]. Clinical trials of anti-CD19 CAR in relapsed/refractory B-cell acute lymphocytic leukemia (ALL) have shown remarkable response rates. Two trials of 15 and 30 patients both had complete remission rates of roughly 90%, and lasting remissions up to 24 months were observed in the latter. In the larger of the two studies, cytokine-release syndrome was observed in all patients and responding patients also experienced B-cell aplasia [53,54].
A recent study demonstrated efficacy of anti-CD19 CAR in aggressive NHL. Of the 13 evaluable patients with various advanced B-cell malignancies, 8 achieved complete remissions and 4 achieved partial remissions. Seven patients had DLBCL that was refractory to salvage chemotherapy, and 4 of these were cases of complete remission. One patient died due to an unknown cause 16 days post infusion, and some experienced short-lived acute toxicities [94]. Table 1 summarizes the results of various clinical studies using CAR T cell therapy in various hematological malignancies.
Table 1.
Completed trials of anti-CD19 CAR immunotherapy in B-cell hematological malignancies
| Summary of trial | Cell number infused | Results | Toxicities | Ref. |
|---|---|---|---|---|
| Treatment of 2 patients with refractory FL | 108/m2 (dose #1) | 1 patient showed no response, the other was non-evaluable; limited CAR persistence was noted as a barrier to efficacy | Lymphopenia observed in both patients | [134] |
| Lymphodepletion: Flu after dose #1 | 109/m2 (#2, #3) | |||
| Exogenous cytokines: IL-2 | 2×109/m2 (#4, #5) | |||
| Treatment of 6 patients with relapsed or refractory NHL: 1 with SLL, 3 with DLBCL, and 2 with FL/DLBCL | Pt 1, 2: 2×107/m2 | CD28 costimulation greatly enhanced expansion and persistence of CAR T-cells; 2 patients experienced SD | Not reported | [48] |
| Lymphodepletion: None | Pt 3, 4: 1×108/m2 | |||
| Exogenous cytokines: None | Pt 4, 5: 2×108/m2 | |||
| Treatment of 3 patients with relapsed CLL | Pt 1: 1.6×107/kg | 2 patients obtained CRs, 1 obtained PR | Tumor lysis syndrome, lymphopenia, thrombocytopenia, and neutropenia were observed | [92,93] |
| Lymphodepletion: Pt 1: Benda; Pt 2: Benda/Ritux; Pt 3: Pento/Cy | Pt 2: 1.0×107/kg | |||
| Exogenous cytokines: None | Pt 3: 1.5×105/kg | |||
| Treatment of 8 patients with relapsed CLL and 2 patients with relapsed ALL | 1.8×108-3.2×109 | 3 of the 4 evaluable CLL patients who received Cy obtained obtained PRs or SD; the 3 CLL patients who did not receive Cy had no response | Most patients experienced fevers, B-cell aplasia observed in ALL patient | [89] |
| Lymphodepletion: Six of the patients received Cy | ||||
| Exogenous cytokines: None | ||||
| Treatment of 3 patients with relapsed FL, 1 patient with relapsed SMZL, and 4 patients with relapsed CLL | 0.3×107/kg-3.0×107/kg | 5 of the 7 evaluable patients obtained PRs, one obtained CR | Cytokine-associated toxicities including hypotension, fevers, fatigue, renal failure, and obtundation | [90,91] |
| Lymphodepletion: Flu/Cy | ||||
| Exogenous cytokines: IL-2 | ||||
| Treatment of 8 patients with relapsed ALL or CLL who had received prior HSCT | 1.9×107-1.1×108 | 3 patients obtained CRs, 1 obtained PR, 1 experienced SD | No infusion-related toxicities | [135] |
| Lymphodepletion: None | ||||
| Exogenous cytokines: None | ||||
| Treatment of 10 patients with relapsed CLL, DLBCL, and MCL who had received prior HSCT and DLI | 0.4×106/kg-7.8×106/kg | 1 patient obtained CR, 1 obtained PR, 6 experienced SD | Toxicities included transient hypotension and fever | [136] |
| Lymphodepletion: None | ||||
| Exogenous cytokines: None | ||||
| Treatment of 2 children with relapsed and refractory ALL | Pt 1: 1.4×106/kg | Both patients obtained CRs | Cytokine release syndrome and B-cell aplasia were observed in both patients | [95] |
| Lymphodepletion: Pt 1: None; Pt 2: Cy/VP16 | Pt 2: 1.2×107/kg | |||
| Exogenous cytokines: None | ||||
| Treatment of 16 patients with relapsed of refractory ALL | 3.0×106/kg | 88% of patients obtained CRs | 7 patients experienced severe cytokine release syndrome, some patients experienced neurologic toxic effects | [54] |
| Lymphodepletion: Cy | ||||
| Exogenous cytokines: None | ||||
| Treatment of 30 patients with relapsed or refractory ALL | 0.8×106/kg-1.7×107/kg | 90% of patients obtained CRs | 8 patients experienced sever cytokine release syndrome, some patients experienced neurologic toxic effects | [53] |
| Lymphodepletion: 15 patients received Flu/Cy, 12 received a different lymphodepletion regimen | ||||
| Exogenous cytokines: None | ||||
| Treatment of 14 patients with relapsed or refractory B-cell malignancies | Not reported | Cells cultured with IL-7 and IL-15 prior to infusion showed greater persistence and efficacy in vivo than those cultured in IL-2 | None reported | [137] |
| Lymphodepletion: None | ||||
| Exogenous cytokines: None | ||||
| Treatment of 21 patients with relapsed of refractory ALL or NHL | Most patients received 1×106/kg (maximum tolerated dose), four received a second dose at 3×106/kg | 67% of patients obtained CRs | 3 patients experienced sever cytokine release syndrome | [138] |
| Lymphodepletion: Flu/Cy | ||||
| Exogenous cytokines: None | ||||
| Treatment of 15 patients with relapsed of refractory B-cell malignancies: 9 with DLBCL, 2 with indolent lymphomas, and 4 with CLL | 1×106/kg-5×106/kg | 8 of the 13 evaluable patients obtained CRs, 4 obtained PRs, 1 experienced SD | Toxicities in some patients including fever, hypotension, delirium, and other neurologic toxicities; 1 patient died suddenly due to an unknown cause 16 days after cell infusion | [94] |
| Lymphodepletion: Flu/Cy | ||||
| Exogenous cytokines: None | ||||
| Treatment of a patient with refractory multiple myeloma as an addition to high-dose melphalan and HSCT | 5×107 | Durable CR was obtained; enhanced response attributed to anti-CD19 CAR T-cell activity | Hypogammaglobulinemia was observed | [139] |
| Lymphodepletion: Cy | ||||
| Exogenous cytokines: None | ||||
| Treatment of 29 patients with relapsed or refractory NHL: 19 with DLBCL, 8 with FL, and 2 with MCL | 5×108 | Of the 18 evaluable patients, overall response rate was 50% for DLBLC patients and 100% for FL patients | 15 patients experienced cytokine release syndrome, 3 patients experienced neurologic toxic effects | [140] |
| Lymphodepletion: Cy, Benda, EPOCH, radiation/Cy, or Flu/Cy | ||||
| Exogenous cytokines: None | ||||
| Treatment of 7 patients with relapsed or refractory NHL after HDT-ASCT | 7 patients received 5×106/kg, 1 received 1×107/kg | 5 patients obtained CRs | One patient experienced prolonged cytopenias and died of mucormycosis pneumonia 38 days after HDT-ASCT; 4 patients experienced cytokine release syndrome | [141] |
| Lymphodepletion: BEAM | ||||
| Exogenous cytokines: None |
ALL: acute lymphocytic leukemia; ASCT: autologous hematopoietic stem cell transplantation; BEAM: carmustine, etoposide, cytarabine, and melphalan; Benda: bendamustine; Ritux: rituximab; CLL: chronic lymphocytic leukemia; CR: complete response; Cy: Cyclophosphamide; DLBC: diffuse large B-cell lymphoma; DLI: donor lymphocyte infusion; EPOCH: etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin; FL: follicular lymphoma; Flu: fludarabine; HDT: high-dose therapy; MCL: mantle cell lymphoma; NHL: non-Hodgkin’s lymphoma; Pento: pentostatin; PR: partial response; SD: stable disease; SLL: small lymphocytic lymphoma; SMZL, splenic marginal zone lymphoma; VP-16: etoposide.
Speculated resistance mechanisms to CD19 CAR-redirected T cell therapy
Loss or down-regulation of CD19 antigenic target
Targeted immunotherapy exerts selective pressure on tumor cells, resulting in the development of new genetic variants that evade treatment. Potential means of resistance that will be faced in future applications of anti-CD19 CAR therapy in B-cell NHL can be determined by considering resistance mechanisms found in other diseases. One trend found among B-cell ALL trials is relapse with the emergence of CD19-negative lymphoblasts following treatment with anti-CD19 CAR T-cells (Figure 1). A number of trials have reported this observation in a significant minority of B-ALL patients: 1 of 2 patients in a 2013 trial, and 10% of patients in two larger trials of 30 and 20 patients in 2014 and 2015, respectively [53,95,96]. The mechanism by which these escape variants emerge has yet to be determined. This could be the result of selective antigen down-regulation, selection for a null gene mutation, or selection for an abnormal mRNA splice variant. A single mutation in the CD19 or CD81 genes may result in eliminated or greatly decreased CD19 expression, as has been demonstrated by studies of antibody deficiency syndromes. In two aforementioned reports, homozygous frame shift mutations in the CD19 gene led to truncated, unstable protein products in several patients who exhibited low or undetectable levels of surface CD19 [72,73]. In another patient, loss of CD19 expression was caused by a lack of CD81, an associated antigen in the BCR complex, as a result of a splice site mutation in the CD81 gene [97]. The rise of CD19-negative escape variants through alternative mRNA splicing is a novel mechanism that has recently been proposed based on findings of anti-CD19 CAR-relapsed B-ALL specimens. These cells lacked the CD19 antigenic epitope, yet retained intact CD19 gene sequences and normal mRNA levels. Evidence suggested that the observed resistance arose because mRNA isoforms omitting exons 5-6 and/or exon 2 were selectively favored [98]. Though relapse due to selective outgrowth of CD19-negative variants has not yet been reported in applications of anti-CD19 CAR treatment of NHL, this form of resistance will most likely be encountered in future trials.
Figure 1.
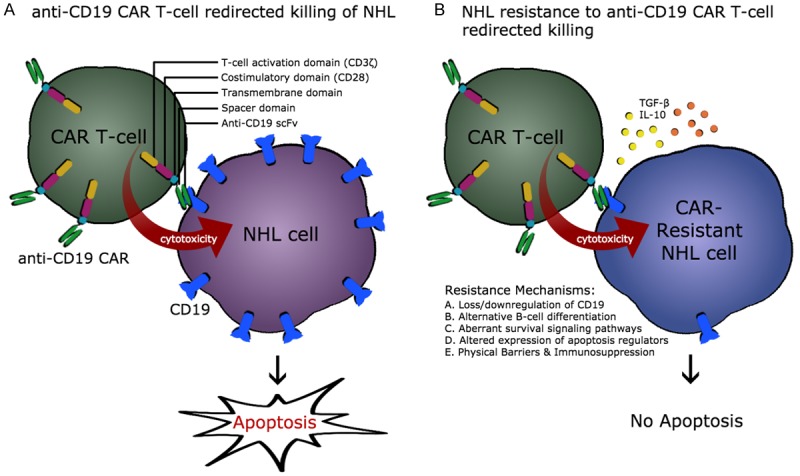
CD19 CAR T cell redirected killing of NHL cells. A. Structure of third generation of anti-CD19 CAR T cells including anti-CD19 single chain Ab fragment, spacer domain, transmembrane domain, T cell costimulatory domain (CD28) and T cell activation domain (CD3z). CD19 redirected CAR T cells induce apoptosis in sensitive NHL cells. B. Various potential mechanisms of the development of resistance by NHL cells to anti-CD19 CAR T cell-mediated apoptosis. These mechanisms may include: i: loos and/or down-regulation of the CD19 target antigen, ii: utilization of alternative B cell differentiation pathways, iii: altered dynamics of cell survival/anti-apoptotic signal transduction pathways, iv: altered expression profile of regulators of apoptosis, and v: physical barriers and immunosuppressive cytokines including TGF-b and IL-10. Please refer to the text for detailed information.
Alternative B-cell differentiation signaling pathways
Resistance may also develop due to selection of alternative differentiation pathways. One recent study reported that a CLL/small lymphocytic lymphoma (SLL) patient with Richter transformation (transformation to an aggressive large cell lymphoma) relapsed after anti-CD19 CAR therapy with two different treatment-resistant clonally related diseases: CD19-negative CLL and overt plasmablastic lymphoma (PBL), an aggressive subtype of NHL. The PBL that emerged was CD19-negative and also lacked most markers of B-cell differentiation, indicating that it was able to evade treatment via divergent lymphoid differentiation. It also had a nonproductive Ig heavy chain reading frame due to a 2 bp insertion mutation that distinguished it from both the original CLL/SLL and the CD19-negative CLL. This suggests that the PBL did not rely on BCR signaling like the original disease, but instead used pathways similar to those found in normal plasma cells to achieve proliferation and survival. Alternative B-cell differentiation of the original clonal tumor was possible because of the existence of residual malignant progenitor cells that survived treatment [82]. Though CLL/SLL affects mature B-cells, this evidence supports the notion that the disease originates in leukemic stem cells [99].
The theory that NHL may likewise have origins in lymphoma stem cells has been supported by several studies. The existence of B-NHL side population cells supports this theory, as these are thought to be lymphoma stem cells. They are isolated by taking advantage of their high expression of ATP-binding cassette (ABC) transporters, which allows them to extrude the nucleic acid dye Hoechst 33342. This is one of the distinctive characteristics that side population cells share with normal stem cells, along with elevated telomerase activity and increased expression of certain transcription factors that regulate pluripotency and self-renewal [78,79]. Further supporting evidence was provided in a study which showed that two biologically and genetically distinct mature B-cell malignancies originate from a common less evolved progenitor cell [80]. Additionally, populations of CD45+/CD19- bone marrow cells that correlate with clinical outcomes in mantle cell lymphoma (MCL) are thought to be lymphoma stem cells [81]. Cancer stem cells pose a serious threat to long-term treatment outcomes, as they are known to be associated with a multi-drug resistance (MDR) phenotype due to the cytotoxic drug-expelling ability of ABC transporters [100]. Given the viable existence of lymphoma stem cells, NHL could be susceptible to anti-CD19 CAR resistance by way of alternative differentiation (Figure 1).
Aberrant cell survival signal transduction pathways
Many human cancers develop as a result of mutations in genes that regulate key cell proliferation and survival signaling pathways, such as the BRAF, NRAS, and TP53 genes. The malignant cell signaling pathways that result from such mutations rely on a reduced number of higher-intensity signals than normal cells. The alternative signaling mechanisms that allow malignant cells to resist normal regulatory immune functions can cause them to possess a related resistance to newly introduced drugs, and vice versa. Such cross-resistance occurs because immune cells and cytotoxic drugs utilize a common apoptotic pathway to eradicate tumor target cells [101]. Therefore, because relapsed/refractory NHL cells have already resisted the apoptotic efforts of the body’s own immune system as well as cytotoxic drugs, they could potentially be preconditioned with cross-resistance to anti-CD19 CAR T-cells even before the initiation of treatment. Several aberrant BCR-activated signal transduction pathways exist in B-cell NHLs, including those involving PI3K/AKT/mTOR signaling, BTK signaling, LYN/SYK signaling, JAK2/STAT signaling, PKCβ signaling, and AAK signaling. Constitutive activation of one or more of these signaling molecules is found in various NHL subtypes, resulting in continually active survival pathways that are apoptosis-resistant [85]. The aberrant signal transduction pathways found in various NHLs could cause resistance to CAR T-cell immunotherapy effector functions, which rely on intact signaling pathways to induce apoptosis in target cells (Figure 1).
Altered expression levels of pro- and anti-apoptotic proteins
Another mechanism that can impede CAR T-cell-induced apoptosis is a modified expression profile of the B-cell lymphoma-2 (Bcl-2) family of apoptosis-regulating proteins, which have been associated with drug resistance in a number of cancers [102]. Though the precise balance of all pro- and anti-apoptotic proteins in a cell ultimately determines the behavior of its apoptosis pathways, it is frequently an overexpression of Bcl-2 family of anti-apoptotic proteins (e.g., Bcl-2, Bcl-xL, Bfl-1/A1, Mcl-1) in tumor cells that is responsible for cancer persistence and drug resistance. Up-regulation of anti-apoptotic proteins is also common in normal immune cells as a preventative measure against AICD, but it can cause uncontrolled growth in tumor cells. These overexpressed Bcl-2 proteins inhibit a cell’s intrinsic apoptosis pathway by blocking Bax and Bak (pro-apoptotic proteins of the Bcl-2 family), thus suppressing Cytochrome c release from mitochondria. Bcl-2 apoptosis inhibitors can contribute to CAR immunotherapy-resistance because CAR T-cells induce apoptosis in target cells through both extrinsic pathways (via death cell receptor or granule-exocytosis signaling pathways and intrinsic apoptotic pathways (via mitochondria). In some cases, simply eliminating the inhibitors that suspend apoptosis signaling in tumor cells can result in activation of the intrinsic pathway, even in the absence of apoptosis-inducing drugs. Hence, removing the block caused by Bcl-2 apoptosis inhibitors may serve not only to increase efficacy of CAR T-cell effector functions by lowering the apoptosis threshold, but also to manage tumor growth by allowing intrinsic apoptosis signaling to resume in malignant B-cells [103,104]. These abnormally expressed anti-apoptotic Bcl-2 family proteins, which were first identified upon discovery that Bcl-2 gene rearrangements in NHL correlated with a poor prognosis, could potentially allow for NHL escape from CAR T-cell immunotherapy [105] (Figure 1).
Physical barriers and immunosuppression
Certain characteristics of lymphoma cells make them inherently more challenging targets for CAR T-cells than leukemia cells. Unlike leukemias, lymphomas are solid tumors that can sometimes physically impede CAR T-cell contact with some target cells. Effector T-cells must be able to infiltrate the tumor parenchyma for maximum efficacy, however their reach is often largely limited. Among other restricting factors, blood vessels in tumor lesions may lack receptors such as ICAM-1, VCAM-1, and P/E-selectins, which are required for T-cell attachment, rolling, and diapedesis. Conversely, T-cells may lack the necessary counterparts, such as the integrins LFA-1 and α4β1 (VLA-4), as well as PSGL-1 and CD43 [106]. CD40/CD40 ligand (CD40L) interaction could also be limited, which would hinder T-cell crossing of endothelial tissues. The activation of CD40 by CD40L in endothelial cells supports T-cell transmigration because it stimulates E-selectin dependent leukocyte attachment and also inhibits endothelial cell migration [107]. Vascular endothelial growth factor (VEGF), which inhibits NF-κB induced endothelial activation, may also contribute to blocked tumor infiltration in NHL. Extracellular matrices can also be obstacles in cases when CAR T-cells are unable to sufficiently degrade them.
CAR T-cells are further obstructed by the immunosuppressive tumor microenvironment in lymphomas. Though much of the tumor microenvironment is comprised of non-malignant cells and the extracellular matrix, it significantly contributes to lymphoma progression. Tumors thwart T-cell proliferation and function by releasing immunosuppressive cytokines like transforming growth factor-β (TGF-β) and interleukin-10 (IL-10). In addition to inhibiting effector cells directly, TGF-β and IL-10 also do so indirectly by driving differentiation of regulatory T cells (Tregs) and M2 macrophages, respectively. Regulatory T-cells suppress T-cell activity by producing a variety of suppressive agents including more TGF-β and IL-10. M2 macrophages, which are anti-inflammatory, pro-angiogenic, and pro-tumorgenic, also produce large amounts of both TGF-β and IL-10. In addition, tumor-produced prostaglandin E2 (PGE2) causes an increase in myeloid-derived suppressor cells (MDSCs) that also contribute to immunosuppression and subsequent resistance to CD19 CAR-redirected T cell therapy [108]. Altogether this network of suppressive components is capable of impeding CAR T-cell persistence and function in NHL, though the degree to which tumors employ microenvironmental cells varies by subtype. Hodgkin’s lymphoma has the greatest ratio of microenvironmental cells to tumor cells, followed by FL, DLBCL, and finally Burkitt’s lymphoma (BL) with the smallest ratio. Other NHL subtypes fall within this range [109] (Figure 1).
Proposed approaches to overcome resistance
Targeting other antigens in addition to CD19
In order to offset potential loss or down-regulation of CD19, multispecific T-cells can be engineered that coexpress two or more CARs targeting different antigens. Multispecfic CAR T-cells require the presence of multiple antigens for maximum activation, yet they have a better chance at maintaining efficacy in the case of antigen-escape than single-target CAR T-cells. Results from a study of CAR T-cell therapy in glioblastroma suggested that this strategy is more effective than using a pooled product of unispecific CAR T-cells that target the same set of antigens [110]. Possible targets to add alongside CD19 include the pan-B-cell markers CD22 and CD20, as well as ROR1, which is expressed in some B-cell malignancies but not in healthy B-cells. Another feasible target to add alongside CD19 is either the κ or λ light Ig chain. Because mature B-cell malignancies are clonally restricted, targeting just one of these light chains would spare those healthy polyclonal B-cells that express only the other light chain [49]. A trial involving κ-directed CARs against relapsed NHL showed efficacy in some patients [52]. Including additional targets that would eliminate EBV-infected cells may provide additional insurance against the recurrence of EBV-related B-NHLs, including BL and DLBCL. Treatment with autologous EBV-specific CAR T-cells targeted against CD30 is currently being investigated for both Hodgkin’s and Non-Hodgkin’s Lymphoma in an ongoing clinical trial (NCT01192464). In implementing a therapeutic approach that utilizes multiple antigen targets, consequences of depletion of healthy cells that also express these antigens must be considered. In order to reduce off-target activation, multispecific CAR T-cells may also be designed to include inhibitory signaling domains that recognize antigens that are only present on normal B-cells [57]. Ideally, a combination of activating and inhibiting CARs could be used to maximize T-cell activation on tumor cells and minimize T-cell activation on normal cells.
Targeting lymphoma stem cells alongside main tumor populations
Targeting lymphoma side population cells can lessen the risk of immune escape by alternative differentiation, as a disease can simply reemerge in a new form unless cancer stem cells are targeted alongside the main population of tumor cells. Treatments that attack lymphoma stem cell populations target one of the following stem cell characteristics: surface markers, signaling cascades, tumor microenvironment, or ABC cassettes. Surface antigens can be targeted with additional CAR T-cells or with other targeted therapies. Further study of lymphoma stem cells is needed in order to determine a unique fingerprint of antigens that exist only on these cells, as has been found in leukemia stem cells [111]. In MCL therapy, multi-specific CAR T-cells that combine activating and inhibiting signals can be used to eliminate the CD45+/CD19- cells that are believed to be MCL stem cells [112]. For signaling cascades, components of pathways that play key roles in stem cell lymphomagenesis like PI3K/AKT and JAK/STAT should be targeted. Kinase inhibitors that could block these pathways in stem cells also serve to regulate the aberrant versions found in mature lymphoma cells, making them particularly advantageous. The tumor microenvironment that protects cancer stem cells from cytotoxic drugs can also be targeted. Treatments can limit the protective microenvironment in a number of ways, including inhibiting stromal cell factors like anti-CXCR4 plerixafor or inhibiting angiogenesis with anti-VEGF sunitnib [113,114]. Additionally, a number of treatments have been developed that inhibit ABC cassettes, the ATP-driven efflux transporters in stem cells that are associated with chemo-resistance [115]. These ATP transporters can be directly targeted with P-glycoprotein (P-gp) efflux pump inhibitors like MRK16, UIC2, and tariquidar, or their expression levels can be downregulated with targeted knockdown of MDR1 and ABCG2 [116-118]. Using one or more of these approaches to diminish NHL stem cells alongside anti-CD19 CAR immunotherapy would aid in preventing stem cell-associated relapse.
Utilization of protein kinase inhibitors
Targeting aberrant signaling molecules with kinase inhibitors can regulate the constitutively active survival pathways that exist in tumor cells. These pathways are responsible for making tumor growth difficult or impossible to manage. Kinase inhibitors are becoming increasingly common in cancer therapies and they can act as sensitizing agents to enhance treatment outcomes of CAR T-cell immunotherapy. Small molecule kinase inhibitors targeting PI3K and BTK have received FDA approval for therapeutic use in NHL. PI3K, central to the PI3K/Akt/mTOR pathway involved in cell cycle regulation, is a promising target because of its key role in lymphoma survival. In 2014, idelalisib (the small molecule PI3Kδ inhibitor) was FDA approved for treatment of relapsed/refractory FL and CLL/SLL. It yields overall response rates of 60-80% in these subtypes with minimal toxicity. For CLL, it is recommended to be used in combination with rituximab. Other inhibitors of PI3K or its leukocyte-specific isoforms PI3Kδ and/or PI3Kγ have shown encouraging outcomes in clinical and preclinical trials. Bruton’s tyrosine kinase (BTK) or also plays a central role in BCR signaling and is essential for the survival of some NHL subtypes. The small molecule BTK inhibitor ibrutinib was FDA approved for treatment of relapsed/refractory cases of MCL in 2013, CLL in 2014, and Waldenstrom Macroglobulinemia (WM) in 2015. Reported overall response rates have been as high as in 58% MCL, 83% in CLL, and 91% in WM [119-121]. Ibrutnib has an impressive safety profile and it has also exhibited efficacy in other NHLs including SLL, DLBCL, and FL [122]. Other kinase inhibitors have also had encouraging results in lymphoma treatment, including the AKT inhibitor perifosine, the mTOR inhibitor temsirolimus, the Src kinase inhibitors dasatinib and fostamatinib, the JAK2 inhibitors pacritinib and ruxolitinib, the PKCβ inhibitors enzastaurin and bryostatin, and the AAK inhibitor alisertib [85]. Kinase inhibitors such as idelalisib, ibrutinib, and others that have had promising results can potentially enhance immunotherapy outcomes in NHL if utilized in combination with an anti-CD19 CAR T-cell treatment regimen.
Modifications of apoptotic machinery
Treatments that regulate expression levels of pro- and anti-apoptotic proteins can act as both sensitizing agents and direct treatments. Inhibitors of pro-survival Bcl-2 proteins can reactivate apoptosis pathways that are deregulated in most tumor cells. The most promising of these agents is ABT-737, a small molecule Bcl-2 inhibitor that is highly effective as an addition to standard treatments in a number of cancers including lymphoma. It binds with high affinity to Bcl-2, Bcl-xL, and Bcl-w, removing their block on the apoptotic pathway and increasing tumor sensitivity to both natural immune cells and cytotoxic agents [104]. The orally active derivative of ABT-737, navitoclax (ABT-263) has shown substantial efficacy in CLL [123]. High levels of Mcl-1, another anti-apoptotic Bcl-2 family protein, are correlated with resistance to ABT-737, so treatment should be combined with an Mcl-1 inhibitor such as RNA silencing Mcl-1 siRNA or the synthetic cytotoxic retinoid N-(4-hydroxyphenyl) retinamide (4-HPR) for maximal efficacy [124,125]. These survival pathway-modulating agents can be highly useful in combination with anti-CD19 CAR immunotherapy, and have already been shown to enhance CAR T-cell function in several NHL cell lines in vitro [103]. Conversely, apoptotic machinery can also be targeted using a different approach; rather than inhibiting anti-apoptotic molecules, pro-apoptotic stimuli may be added. One such agent is recombinant tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), which can activate the apoptosis pathway by binding to TRAIL death receptors DR-4 and DR-5 on tumor cell surface [126]. Recombinant TRAIL is more effective when used together with TRAIL-R2 agonistic antibodies; these treatments can potentially be added to an anti-CD19 CAR regimen [127]. TRAIL may also be directly targeted alongside CD19 in multispecific CAR therapy, as TRAIL-targeted CAR T-cells have shown efficacy in various types of tumor cells [128].
Enhancing CAR T-cell homing abilities and persistence
Due to the fact that NHL can be more difficult to target than other hematological malignancies, CAR T-cells should be modified to possess enhanced tumor infiltrating capabilities. Stimulating CD40 may enhance T-cell transmigration due to its role in endothelial cell migration and leukocyte attachment, but it has also other benefits for T-cell expansion, persistence, and function [107]. A preclinical study of CAR T-cells with the addition of constitutively expressed CD40L showed very promising results. This modification enhanced proliferation of CAR T-cells, increased pro-inflammatory Th1 cytokine secretion, augmented the responsiveness of CD40+ tumor cells via up-regulation of key molecules including Fas death receptor (FasR), induced dendritic cells to secrete proinflammatory IL-12, and resulted in overall increased efficacy against lymphoma in mice [129]. The tyrosine kinase inhibitor sunitnib, which blocks VEGF, has shown efficacy in overcoming the obstructive tumor vasculature [114]. The enzyme heparanase increases the ability of CAR T-cells to degrade the extracellular matrix and infiltrate tumors [130]. The addition of chemokine receptors such as CCR2 or CCR4 may also be used as a method to increase homing abilities of CAR T-cells, as tumor cells often express the correlating chemokine receptors. Additionally, CCR7 may aid CAR T-cells in accessing lymph nodes, which are entered via lymphatics.
In order to overcome the hostile tumor microenvironment and minimize suppression of effector cells, conditioning chemotherapy must be administered prior to CAR T-cell infusion. Lymphodepletion regimens allow for enhanced proliferation and function of CAR T-cells, maximizing treatment efficacy. A recent meta-analysis of anti-CD19 CAR T-cell clinical trials found that patients who received lymphodepletion had a 56% higher response rate than those who did not (88% and 32% respectively). In past trials of CAR T-cells in NHL, a preparative regimen of cyclophosphamide and fludarabin has been used [90,94]. The sensitizing effects of chemotherapeutic drugs may also be substituted with milder immune therapeutics, such as checkpoint blockade antibodies targeting the CTLA-4 and PDL1/PD1 pathways or oncolytic viruses conferring immunostimulatory protein expression. Either of these would be suitable complements to CAR T-cell therapy because they not only enhance CAR T-cell function but also activate natural T-cells to supplement treatment efforts [108]. Surprisingly, the aforementioned meta-analysis also found that IL-2 administration is not favorable to treatment outcomes. This is contrary to the accepted prior belief that IL-2 enhances outcomes by promoting CAR T-cell expansion in vivo [131]. Meta-regression analysis shows that patients who did not receive IL-2 had a significantly higher response rate than those who received IL-2 administrated T-cells. Though IL-2 administration may be beneficial for first generation CAR T-cells that cannot elicit a robust cytokine response alone, it appears to be unfavorable overall for second generation CARs that induce IL-2 secretion upon crosslinking. This may be due to IL-2’s involvement in promoting AICD. The trials that did not add IL-2 instead promoted expansion by stimulating both CD3 and CD28 with mAb-coated magnetic beads, so the enhanced outcomes in these trials could also be caused by CD3/CD28 stimulation [132]. Though IL-2 is not recommended, other pro-inflammatory cytokines such as IL-15, IL-12, and IL-7 may be able to effectively enhance function and proliferation of CAR T-cells [133].
Conclusion
The optimal treatment for relapsed/refractory NHL has yet to be determined, yet anti-CD19 CAR T-cell immunotherapy stands as a highly promising treatment approach for such cases. There have been very few anti-CD19 CAR T-cell clinical trials in NHL reported to date, yet remarkable results in other hematological malignancies and encouraging outcomes in NHL trials thus far have provided more than enough reason to pursue further investigation of this treatment. The experiences gained from other cancers provide valuable indications of the challenges that will likely be faced for CAR T-cells in NHL. In order to ensure maximal efficacy in future clinical trials, multiple supplementary preventative strategies must be employed in order to avoid relapse with anti-CD19 CAR T-cell-resistant strains of disease. With intelligent foresight and application of abundant resistance-preventative measures, anti-CD19 CAR T-cell therapy could soon become the new paradigm for treatment of relapsed/refractory NHL.
Disclosure of conflict of interest
None.
References
- 1.Morton LM, Wang SS, Cozen W, Linet MS, Chatterjee N, Davis S, Severson RK, Colt JS, Vasef MA, Rothman N, Blair A, Bernstein L, Cross AJ, De Roos AJ, Engels EA, Hein DW, Hill DA, Kelemen LE, Lim U, Lynch CF, Schenk M, Wacholder S, Ward MH, Hoar Zahm S, Chanock SJ, Cerhan JR, Hartge P. Etiologic heterogeneity among non-Hodgkin lymphoma subtypes. Blood. 2008;112:5150–5160. doi: 10.1182/blood-2008-01-133587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smedby KE, Foo JN, Skibola CF, Darabi H, Conde L, Hjalgrim H, Kumar V, Chang ET, Rothman N, Cerhan JR, Brooks-Wilson AR, Rehnberg E, Irwan ID, Ryder LP, Brown PN, Bracci PM, Agana L, Riby J, Cozen W, Davis S, Hartge P, Morton LM, Severson RK, Wang SS, Slager SL, Fredericksen ZS, Novak AJ, Kay NE, Habermann TM, Armstrong B, Kricker A, Milliken S, Purdue MP, Vajdic CM, Boyle P, Lan Q, Zahm SH, Zhang Y, Zheng T, Leach S, Spinelli JJ, Smith MT, Chanock SJ, Padyukov L, Alfredsson L, Klareskog L, Glimelius B, Melbye M, Liu ET, Adami HO, Humphreys K, Liu J. GWAS of Follicular lymphoma reveals allelic heterogeneity at 6p21.32 and suggests shared genetic susceptibility with diffuse large B-cell lymphoma. PLoS Genet. 2011;7:e1001378. doi: 10.1371/journal.pgen.1001378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morton LM, Slager SL, Cerhan JR, Wang SS, Vajdic CM, Skibola CF, Bracci PM, de Sanjosé S, Smedby KE, Chiu BC, Zhang Y, Mbulaiteye SM, Monnereau A, Turner JJ, Clavel J, Adami HO, Chang ET, Glimelius B, Hjalgrim H, Melbye M, Crosignani P, di Lollo S, Miligi L, Nanni O, Ramazzotti V, Rodella S, Costantini AS, Stagnaro E, Tumino R, Vindigni C, Vineis P, Becker N, Benavente Y, Boffetta P, Brennan P, Cocco P, Foretova L, Maynadié M, Nieters A, Staines A, Colt JS, Cozen W, Davis S, de Roos AJ, Hartge P, Rothman N, Severson RK, Holly EA, Call TG, Feldman AL, Habermann TM, Liebow M, Blair A, Cantor KP, Kane EV, Lightfoot T, Roman E, Smith A, Brooks-Wilson A, Connors JM, Gascoyne RD, Spinelli JJ, Armstrong BK, Kricker A, Holford TR, Lan Q, Zheng T, Orsi L, Dal Maso L, Franceschi S, La Vecchia C, Negri E, Serraino D, Bernstein L, Levine A, Friedberg JW, Kelly JL, Berndt SI, Birmann BM, Clarke CA, Flowers CR, Foran JM, Kadin ME, Paltiel O, Weisenburger DD, Linet MS, Sampson JN. Etiologic Heterogeneity among non-Hodgkin lymphoma subtypes: the InterLymph Non-Hodgkin Lymphoma Subtypes Project. J Natl Cancer Inst Monogr. 2014;48:130–144. doi: 10.1093/jncimonographs/lgu013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Theodossiou C, Schwarzenberger P. Non-Hodgkin’s Lymphomas. Clin Obstet Gynecol. 2002;43:820–829. doi: 10.1097/00003081-200209000-00029. [DOI] [PubMed] [Google Scholar]
- 5.Howlader N, Noone AM, Krapcho M, Garshell J, Miller D, Altekruse SF, Kosary CL, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA, editors. SEER Cancer Statistics Review, 1975-2011, National Cancer Institute. Bethesda, MD: http://seer.cancer.gov/csr/1975_2011/, based on November 2013 SEER data submission, posted to the SEER web site, April 2014. [Google Scholar]
- 6.Groves FD, Linet MS, Travis LB, Devesa SS. Cancer surveillance series: non-Hodgkin’s lymphoma incidence by histologic subtype in the United States from 1978 through 1995. J Natl Cancer Inst. 2000;92:1240–1251. doi: 10.1093/jnci/92.15.1240. [DOI] [PubMed] [Google Scholar]
- 7.Chiu BC, Hou N. Epidemiology and etiology of non-hodgkin lymphoma. Cancer Treat Res. 2015;165:1–25. doi: 10.1007/978-3-319-13150-4_1. [DOI] [PubMed] [Google Scholar]
- 8.Wang SS, Slager SL, Brennan P, Holly EA, De Sanjose S, Bernstein L, Boffetta P, Cerhan JR, Maynadie M, Spinelli JJ, Chiu BC, Cocco PL, Mensah F, Zhang Y, Nieters A, Dal Maso L, Bracci PM, Costantini AS, Vineis P, Severson RK, Roman E, Cozen W, Weisenburger D, Davis S, Franceschi S, La Vecchia C, Foretova L, Becker N, Staines A, Vornanen M, Zheng T, Hartge P. Family history of hematopoietic malignancies and risk of non-Hodgkin lymphoma (NHL): a pooled analysis of 10,211 cases and 11,905 controls from the International Lymphoma Epidemiology Consortium (InterLymph) Blood. 2007;109:3479–3488. doi: 10.1182/blood-2006-06-031948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skibola CF, Curry JD, Nieters A. Genetic susceptibility to lymphoma. Haematologica. 2007;92:960–969. doi: 10.3324/haematol.11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aboulafia DM, Pantanowitz L, Dezube BJ. AIDS-related non-Hodgkin lymphoma: Still a problem in the era of HAART. AIDS Read. 2004;14:605–617. [PubMed] [Google Scholar]
- 11.Oertel SH, Riess H. Immunosurveillance, immunodeficiency and lymphoproliferations. Recent Results Cancer Res. 2002;159:1–8. doi: 10.1007/978-3-642-56352-2_1. [DOI] [PubMed] [Google Scholar]
- 12.Cleghorn FR, Manns A, Falk R, Hartge P, Hanchard B, Jack N, Williams E, Jaffe E, White F, Bartholomew C, Blattner W. Effect of human T-lymphotropic virus type I infection on non-Hodgkin’s lymphoma incidence. J Natl Cancer Inst. 1995;87:1009–1014. doi: 10.1093/jnci/87.13.1009. [DOI] [PubMed] [Google Scholar]
- 13.Knowles DM, Cesarman E. The Kaposi’s sarcoma-associated herpesvirus (human herpesvirus-8) in Kaposi’s sarcoma, malignant lymphoma, and other diseases. Ann Oncol. 1997;8:123–129. [PubMed] [Google Scholar]
- 14.Engels EA, Chatterjee N, Cerhan JR, Davis S, Cozen W, Severson RK, Whitby D, Colt JS, Hartge P. Hepatitis C virus infection and non-Hodgkin lymphoma: results of the NCI-SEER multi-center case-control study. Int J Cancer. 2004;111:76–80. doi: 10.1002/ijc.20021. [DOI] [PubMed] [Google Scholar]
- 15.Liu YC, Yang YH, Hsiao HH, Yang WC, Liu TC, Chang CS, Yang MY, Lin PM, Hsu JF, Chang PY, Lin SF. Herpes zoster is associated with an increased risk of subsequent lymphoid malignancies - a nationwide population-based matched-control study in Taiwan. BMC Cancer. 2012;12:503. doi: 10.1186/1471-2407-12-503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Stefani E, Fierro L, Barrios E, Ronco A. Tobacco, alcohol, diet and risk of non-Hodgkin’s lymphoma: a case-control study in Uruguay. Leuk Res. 1998;22:445–452. doi: 10.1016/s0145-2126(97)00194-x. [DOI] [PubMed] [Google Scholar]
- 17.Besson H, Brennan P, Becker N, Nieters A, De Sanjose S, Font R, Maynadié M, Foretova L, Cocco PL, Staines A, Vornanen M, Boffetta P. Tobacco smoking, alcohol drinking and non-Hodgkin’s lymphoma: a European multicenter case-control study (Epilymph) Int J Cancer. 2006;119:901–908. doi: 10.1002/ijc.21913. [DOI] [PubMed] [Google Scholar]
- 18.Chiu BC, Cerhan JR, Folsom AR, Sellers TA, Kushi LH, Wallace RB, Zheng W, Potter JD. Diet and risk of non-Hodgkin lymphoma in older women. JAMA. 1996;275:1315–1321. doi: 10.1001/jama.1996.03530410029029. [DOI] [PubMed] [Google Scholar]
- 19.Skibola CF. Obesity, diet and risk of non-Hodgkin lymphoma. Cancer Epidemiol Biomarkers Prev. 2007;16:392–395. doi: 10.1158/1055-9965.EPI-06-1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y, Sanjose SD, Bracci PM, Morton LM, Wang R, Brennan P, Hartge P, Boffetta P, Becker N, Maynadie M, Foretova L, Cocco P, Staines A, Holford T, Holly EA, Nieters A, Benavente Y, Bernstein L, Zahm SH, Zheng T. Personal use of hair dye and the risk of certain subtypes of non-Hodgkin lymphoma. Am J Epidemiol. 2008;167:1321–1331. doi: 10.1093/aje/kwn058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Melbye M, Adami HO, Hjalgrim H, Glimelius B. Ultraviolet light and non-Hodgkin’s lymphoma. Acta Oncol. 1996;35:655–657. doi: 10.3109/02841869609083994. [DOI] [PubMed] [Google Scholar]
- 22.Hartge P, Lim U, Freedman DM, Colt JS, Cerhan JR, Cozen W, Severson RK, Davis S. Ultraviolet radiation, dietary vitamin D, and risk of non-Hodgkin lymphoma (United States) Cancer Causes Control. 2006;17:1045–1052. doi: 10.1007/s10552-006-0040-8. [DOI] [PubMed] [Google Scholar]
- 23.Cocco P, Satta G, Dubois S, Pili C, Pilleri M, Zucca M, ‘t Mannetje AM, Becker N, Benavente Y, de Sanjosé S, Foretova L, Staines A, Maynadié M, Nieters A, Brennan P, Miligi L, Ennas MG, Boffetta P. Lymphoma risk and occupational exposure to pesticides: results of the Epilymph study. Occup Environ Med. 2013;70:91–98. doi: 10.1136/oemed-2012-100845. [DOI] [PubMed] [Google Scholar]
- 24.Espinosa A, Zock JP, Benavente Y, Boffetta P, Becker N, Brennan P, Cocco P, Foretova L, Maynadié M, Staines A, Nieters A, Kogevinas M, de Sanjose S. Occupational exposure to immunologically active agents and risk for lymphoma: the European Epilymph case-control study. Cancer Epidemiol. 2013;37:378–384. doi: 10.1016/j.canep.2013.02.006. [DOI] [PubMed] [Google Scholar]
- 25.Coffey J, Hodgson DC, Gospodarowicz MK. Therapy of non-Hodgkin’s lymphoma. Eur J Nucl Med Mol Imaging. 2003;30:S28–36. doi: 10.1007/s00259-003-1157-6. [DOI] [PubMed] [Google Scholar]
- 26.Jazirehi AR, Bonavida B. Cellular and molecular signal transduction pathways modulated by rituximab (rituxin, anti-CD20 mAb) in non-Hodgkin’s lymphoma: implications in chemosensitization and therapeutic intervention. Oncogene. 2005;24:2121–2143. doi: 10.1038/sj.onc.1208349. [DOI] [PubMed] [Google Scholar]
- 27.Shankland KR, Armitage JO, Hancock BW. Non-Hodgkin lymphoma. Lancet. 2012;380:848–857. doi: 10.1016/S0140-6736(12)60605-9. [DOI] [PubMed] [Google Scholar]
- 28.Bachy E, Salles G. Are we nearing an era of chemotherapy-free management of indolent lymphoma? Clin Cancer Res. 2014;20:5226–5239. doi: 10.1158/1078-0432.CCR-14-0437. [DOI] [PubMed] [Google Scholar]
- 29.McLaughlin P, Grillo-López AJ, Link BK, Levy R, Czuczman MS, Williams ME, Heyman MR, Bence-Bruckler I, White CA, Cabanillas F, Jain V, Ho AD, Lister J, Wey K, Shen D, Dallaire BK. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. J. Clin. Oncol. 1998;16:2825–2833. doi: 10.1200/JCO.1998.16.8.2825. [DOI] [PubMed] [Google Scholar]
- 30.Smith MR. Rituximab (monoclonal anti-CD20 antibody): mechanisms of action and resistance. Oncogene. 2003;22:7359–7368. doi: 10.1038/sj.onc.1206939. [DOI] [PubMed] [Google Scholar]
- 31.Al-Tourah AJ, Gill KK, Chhanabhai M, Hoskins PJ, Klasa RJ, Savage KJ, Sehn LH, Shenkier TN, Gascoyne RD, Connors JM. Populationbased analysis of incidence and outcome of transformed non-Hodgkin’s lymphoma. J. Clin. Oncol. 2008;26:5165–5169. doi: 10.1200/JCO.2008.16.0283. [DOI] [PubMed] [Google Scholar]
- 32.Ferrara F, Ravasio R. Cost-effectiveness analysis of the addition of rituximab to CHOP in young patients with good-prognosis diffuse large-B-cell lymphoma. Clin Drug Investig. 2008;28:55–65. doi: 10.2165/00044011-200828010-00007. [DOI] [PubMed] [Google Scholar]
- 33.Coiffier B, Thieblemont C, Van Den Neste E, Lepeu G, Plantier I, Castaigne S, Lefort S, Marit G, Macro M, Sebban C, Belhadj K, Bordessoule D, Fermé C, Tilly H. Long-term outcome of patients in the LNH-98.5 trial, the first randomized study comparing rituximab-CHOP to standard CHOP chemotherapy in DLBCL patients: a study by the Groupe d’Etudes des Lymphomes del’Adulte. Blood. 2010;116:2040–2045. doi: 10.1182/blood-2010-03-276246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rothe A, Schulz H, Elter T, Engert A, Reiser M. Rituximab monotherapy is effective in patients with poor risk refractory aggressive non-Hodgkin’s lymphoma. Haematologica. 2004;89:875–876. [PubMed] [Google Scholar]
- 35.Tobinai K, Igarashi T, Itoh K, Kobayashi Y, Taniwaki M, Ogura M, Kinoshita T, Hotta T, Aikawa K, Tsushita K, Hiraoka A, Matsuno Y, Nakamura S, Mori S, Ohashi Y. IDEC-C2B8 Japan Study Group. Japanese multicenter phase II and pharmacokinetic study of rituximab in relapsed or refractory patients with aggressive B-cell lymphoma. Ann Oncol. 2004;15:821–830. doi: 10.1093/annonc/mdh176. [DOI] [PubMed] [Google Scholar]
- 36.van Oers MH. Rituximab maintenance therapy: a step forward in follicular lymphoma. Haematologica. 2007;92:826–833. doi: 10.3324/haematol.10894. [DOI] [PubMed] [Google Scholar]
- 37.Gisselbrecht C. Use of rituximab in diffuse large B-cell lymphoma in the salvage setting. Br J Haematol. 2008;143:607–621. doi: 10.1111/j.1365-2141.2008.07383.x. [DOI] [PubMed] [Google Scholar]
- 38.Sehn LH, Gascoyne RD. Diffuse large B-cell lymphoma: optimizing outcome in the context of clinical and biologic heterogeneity. Blood. 2015;125:22–32. doi: 10.1182/blood-2014-05-577189. [DOI] [PubMed] [Google Scholar]
- 39.Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. J Immunol. 1975;174:2453–2455. [PubMed] [Google Scholar]
- 40.Karlitepe A, Ozalp O, Avci CB. New approaches for cancer immunotherapy. Tumour Biol. 2015;36:4075–4078. doi: 10.1007/s13277-015-3491-2. [DOI] [PubMed] [Google Scholar]
- 41.Morrison SL, Johnson MJ, Herzenberg LA, Oi VT. Chimeric human antibody molecules: mouse antigen-binding domains with human constant region domains. Proc Natl Acad Sci U S A. 1984;81:6851–6855. doi: 10.1073/pnas.81.21.6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vaughan TJ, Osbourn JK, Tempest PR. Human antibodies by design. Nat Biotechnol. 1998;16:535–539. doi: 10.1038/nbt0698-535. [DOI] [PubMed] [Google Scholar]
- 43.Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer. 2012;12:278–287. doi: 10.1038/nrc3236. [DOI] [PubMed] [Google Scholar]
- 44.Weiner GJ. Rituximab: mechanism of action. Semin Hematol. 2010;47:115–123. doi: 10.1053/j.seminhematol.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bonavida B. Postulated mechanisms of resistance of B-cell non-Hodgkin lymphoma to rituximab treatment regimens: strategies to overcome resistance. Semin Oncol. 2014;41:667–677. doi: 10.1053/j.seminoncol.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sitaru C, Thiel J. The need for markers and predictors of Rituximab treatment resistance. Exp Dermatol. 2014;23:236–237. doi: 10.1111/exd.12331. [DOI] [PubMed] [Google Scholar]
- 47.Magee MS, Snook A. Challenges to chimeric antigen receptor (CAR)-T cell therapy for cancer. Discov Med. 2014;18:265–271. [PubMed] [Google Scholar]
- 48.Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, Kamble RT, Bollard CM, Gee AP, Mei Z, Liu H, Grilley B, Rooney CM, Heslop HE, Brenner MK, Dotti G. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121:1822–1826. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ramos CA, Heslop HE, Brenner MK. CAR-T Cell Therapy for Lymphoma. Annu Rev Med. 2016;67:165–83. doi: 10.1146/annurev-med-051914-021702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Choudhuri K, Wiseman D, Brown MH, Gould K, van der Merwe PA. T-cell receptor triggering is critically dependent on the dimensions of its peptide-MHC ligand. Nature. 2005;436:578–582. doi: 10.1038/nature03843. [DOI] [PubMed] [Google Scholar]
- 51.Jensen MC, Riddell SR. Designing chimeric antigen receptors to effectively and safely target tumors. Curr Opin Immunol. 2015;33:9–15. doi: 10.1016/j.coi.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ramos CA, Savoldo B, Liu E, Gee AP, Mei Z, Grilley B, Rooney CM, Heslop HE, Brenner MK, Dotti G. Clinical responses in patients infused with T lymphocytes redirected to target κ-light immunoglobulin chain. ASH Annual Meeting and Exposition Abstract. Blood. 2013;122:506. [Google Scholar]
- 53.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, Mahnke YD, Melenhorst JJ, Rheingold SR, Shen A, Teachey DT, Levine BL, June CH, Porter DL, Grupp SA. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M, Qu J, Wasielewska T, He Q, Fink M, Shinglot H, Youssif M, Satter M, Wang Y, Hosey J, Quintanilla H, Halton E, Bernal Y, Bouhassira DC, Arcila ME, Gonen M, Roboz GJ, Maslak P, Douer D, Frattini MG, Giralt S, Sadelain M, Brentjens R. Efficacy and toxicity management of 19-28z CAR cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra25. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vera J, Savoldo B, Vigouroux S, Biagi E, Pule M, Rossig C, Wu J, Heslop HE, Rooney CM, Brenner MK, Dotti G. T lymphocytes redirected against the κ light chain of human immunoglobulin efficiently kill mature B lymphocyte-derived malignant cells. Blood. 2006;108:3890–3897. doi: 10.1182/blood-2006-04-017061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31:71–75. doi: 10.1038/nbt.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Federov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. 2013;5:215ra172. doi: 10.1126/scitranslmed.3006597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, Straathof K, Liu E, Durett AG, Grilley B, Liu H, Cruz CR, Savoldo B, Gee AP, Schindler J, Krance RA, Heslop HE, Spencer DM, Rooney CM, Brenner MK. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365:1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Budde LE, Berger C, Lin Y, Wang J, Lin X, Frayo SE, Brouns SA, Spencer DM, Till BG, Jensen MC, Riddell SR, Press OW. Combining a CD20 chimeric antigen receptor and an inducible caspase 9 suicide switch to improve the efficacy and safety of T cell adoptive immunotherapy for lymphoma. PLoS One. 2013;8:e82742. doi: 10.1371/journal.pone.0082742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moreau T, Bardin F, Imbert J, Chabannon C, Tonnelle C. Restriction of transgene expression to the B-lymphoid progeny of human lentivirally transduced CD34+ cells. Molecular Ther. 2004;10:45–56. doi: 10.1016/j.ymthe.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 61.Thierry-Mieg D, Thierry-Mieg J. AceView: a comprehensive cDNA-supported gene and transcripts annotation - Homo sapiens complex locus CD19, encoding CD19 molecule. Genome Biol. 2006;7:S12.1–14. doi: 10.1186/gb-2006-7-s1-s12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tedder TF, Isaacs CM. Isolation of cDNAs encoding the CD19 antigen of human and mouse B lymphocytes: A new member of the immunoglobulin superfamily. J Immunol. 1989;143:712–717. [PubMed] [Google Scholar]
- 63.Zhou LJ, Ord DC, Omori SA, Tedder TF. Structure of the genes encoding the CD19 antigen of human and mouse B lymphocytes. Immunogenetics. 1992;35:102–111. doi: 10.1007/BF00189519. [DOI] [PubMed] [Google Scholar]
- 64.Weiland J, Elder A, Forster V, Heidenreich O, Koschmieder S, Vormoor J. CD19: A multifunctional immunological target molecule and its implications for B lineage acute lymphoblastic leukemia. Pediatr Blood Cancer. 2015;62:1144–1148. doi: 10.1002/pbc.25462. [DOI] [PubMed] [Google Scholar]
- 65.Nadler LM, Anderson KC, Marti G, Bates M, Park E, Daley JF, Schlossman SF. B4, a human B lymphocyte-associated antigen expressed on normal, mitogen-activated, and malignant B lymphocytes. J Immunol. 1983;131:244–250. [PubMed] [Google Scholar]
- 66.Schriever F, Freedman AS, Freeman G, Messner E, Lee G, Daley J, Nadler LM. Isolated human follicular dendritic cells display a unique antigenic phenotype. J Exp Med. 1989;169:2043–2058. doi: 10.1084/jem.169.6.2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fujimoto M, Fujimoto Y, Poe JC, Jansen PJ, Lowell CA, DeFranco AL, Tedder TF. CD19 regulates Src family protein tyrosine kinase activation in B lymphocytes through processive amplification. Immunity. 2000;13:47–57. doi: 10.1016/s1074-7613(00)00007-8. [DOI] [PubMed] [Google Scholar]
- 68.Fearon DT, Carroll MC. Regulation of B lymphocyte responses to foreign and self-antigens by the CD19/CD21 complex. Annu Rev Immunol. 2000;18:393–422. doi: 10.1146/annurev.immunol.18.1.393. [DOI] [PubMed] [Google Scholar]
- 69.Mattila PK, Feest C, Depoil D, Treanor B, Montaner B, Otipoby KL, Carter R, Justement LB, Bruckbauer A, Batista FD. The actin and tetraspanin networks organize receptor nanoclusters to regulate B cell receptor-mediated signaling. Immunity. 2013;38:461–474. doi: 10.1016/j.immuni.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 70.Carter RH, Fearon DT. CD19: lowering the threshold for antigen receptor stimulation of B lymphocytes. Science. 1992;256:105–107. [PubMed] [Google Scholar]
- 71.Otero DC, Anzelon AN, Rickert RC. CD19 function in early and late B cell development: I. Maintenance of follicular and marginal zone B cells requires CD19 dependent survival signals. J Immunol. 2003;170:73–83. doi: 10.4049/jimmunol.170.1.73. [DOI] [PubMed] [Google Scholar]
- 72.Van Zelm MC, Reisli I, van der Burg M, Castaño D, van Noesel CJ, van Tol MJ, Woellner C, Grimbacher B, Patiño PJ, van Dongen JJ. An antibody-deficiency syndrome due to mutations in the CD19 gene. N Engl J Med. 2006;354:1901–1912. doi: 10.1056/NEJMoa051568. [DOI] [PubMed] [Google Scholar]
- 73.Kanegane H, Agematsu K, Futatani T, Sira MM, Suga K, Sekiguchi T, van Zelm MC, Miyawaki T. Novel mutations in a Japanese patient with CD19 deficiency. Genes Immun. 2007;8:663–670. doi: 10.1038/sj.gene.6364431. [DOI] [PubMed] [Google Scholar]
- 74.Tedder TF, Poe JC, Fujimoto M, Haas KM, Sato S. The CD19-CD21 signal transduction complex of B lymphocytes regulates the balance between health and autoimmune disease: systemic sclerosis as a model system. Curr Dir Autoimmun. 2005;8:55–90. doi: 10.1159/000082087. [DOI] [PubMed] [Google Scholar]
- 75.Manning BD, Cantley LC. AKT/PKB signa-l ing: Navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Reth M, Nitschke L, Hikida M, Kurosaki T. Structure and signaling function of the B-cell antigen receptor and its coreceptors. In: Alt FW, Honjo T, Radbruch A, Reth M, editors. Molecular Biology of B Cells. Second Edition. London: Academic Press; 2015. pp. 151–164. [Google Scholar]
- 77.Ghorashian S, Pule M, Amrolia P. CD19 chimeric antigen receptor T cell therapy for haematological malignancies. Br J Haematol. 2015;169:463–478. doi: 10.1111/bjh.13340. [DOI] [PubMed] [Google Scholar]
- 78.Kim SJ. Lymphoma stem cells: A step toward a new therapeutic target. Korean J Hematol. 2011;46:211–213. doi: 10.5045/kjh.2011.46.4.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee MR, Ju HJ, Kim BS, Ko YH, Kim WS, Kim SJ. Isolation of side population cells in B-cell non-Hodgkin’s lymphomas. Acta Haematol. 2013;129:10–17. doi: 10.1159/000341284. [DOI] [PubMed] [Google Scholar]
- 80.Green MR, Alizadeh AA. Common progenitor cells in mature B-cell malignancies: implications for therapy. Curr Opin Hematol. 2014;21:333–340. doi: 10.1097/MOH.0000000000000049. [DOI] [PubMed] [Google Scholar]
- 81.Kim SM, Lee ST, Ryu KJ, Kim HJ, Kim SH, Yoo KH, Ko YH, Kim WS, Kim SJ. Clinical Relevance of CD45+/CD19- Stem-like Tumor Cells in Patients with Mantle Cell Lymphoma: A Single Center Experience. Blood. 2014;124:1622. [Google Scholar]
- 82.Evans AG, Rothberg PG, Burack WR, Huntington SF, Porter DL, Friedberg JW, Liesveld JL. Evolution to plasmablastic lymphoma evades CD19-directed chimeric antigen receptor T cells. Br J Haematol. 2015;171:205–209. doi: 10.1111/bjh.13562. [DOI] [PubMed] [Google Scholar]
- 83.Srinivasan L, Sasaki Y, Calado DP, Zhang B, Paik JH, DePinho RA, Kutok JL, Kearney JF, Otipoby KL, Rajewsky K. PI3 kinase signals BCR-dependent mature B cell survival. Cell. 2009;139:573–586. doi: 10.1016/j.cell.2009.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cozma D, Yu D, Hodawadekar S, Azvolinsky A, Grande S, Tobias JW, Metzgar MH, Paterson J, Erikson J, Marafioti T, Monroe JG, Atchison ML, Thomas-Tikhonenko A. B cell activator PAX5 promotes lymphomagenesis through stimulation of B cell receptor signaling. J Clin Invest. 2007;117:2602–2610. doi: 10.1172/JCI30842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Arita A, McFarland DC, Myklebust JH, Parekh S, Petersen B, Gabrilove J, Brody JD. Signaling pathways in lymphoma: pathogenesis and therapeutic targets. Future Oncol. 2013;9:1549–1571. doi: 10.2217/fon.13.113. [DOI] [PubMed] [Google Scholar]
- 86.Fellbaum C, Radaszkiewicz T, Ruhri C, Pütz B, Lehmacher W, Höfler H. c-myc mRNA expression in non-Hodgkin’s lymphomas. Virchows Arch B Cell Pathol Incl Mol Pathol. 1992;62:61–68. doi: 10.1007/BF02899666. [DOI] [PubMed] [Google Scholar]
- 87.Chung EY, Psathas JN, Yu D, Li Y, Weiss MJ, Thomas-Tikhonenko A. CD19 is a major B cell receptor-independent activator of MYC-driven B-lymphomagenesis. J Clin Invest. 2012;122:2257–2266. doi: 10.1172/JCI45851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Poe JC, Minard-Colin V, Kountikov EI, Haas KM, Tedder TF. A c-Myc and surface CD19 signaling amplification loop promotes B cell lymphoma development and progression in mice. J Immunol. 2012;189:2318–2325. doi: 10.4049/jimmunol.1201000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brentjens RJ, Rivière I, Park JH, Davila ML, Wang X, Stefanski J, Taylor C, Yeh R, Bartido S, Borquez-Ojeda O, Olszewska M, Bernal Y, Pegram H, Przybylowski M, Hollyman D, Usachenko Y, Pirraglia D, Hosey J, Santos E, Halton E, Maslak P, Scheinberg D, Jurcic J, Heaney M, Heller G, Frattini M, Sadelain M. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, Maric I, Raffeld M, Nathan DA, Lanier BJ, Morgan RA, Rosenberg SA. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, Phan GQ, Hughes MS, Sherry RM, Yang JC, Kammula US, Devillier L, Carpenter R, Nathan DA, Morgan RA, Laurencot C, Rosenberg SA. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor modified T-cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, Yang JC, Phan GQ, Hughes MS, Sherry RM, Raffeld M, Feldman S, Lu L, Li YF, Ngo LT, Goy A, Feldman T, Spaner DE, Wang ML, Chen CC, Kranick SM, Nath A, Nathan DA, Morton KE, Toomey MA, Rosenberg SA. Chemotherapyrefractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J. Clin. Oncol. 2015;33:540–549. doi: 10.1200/JCO.2014.56.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, Milone MC, Levine BL, June CH. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN, Steinberg SM, Stroncek D, Tschernia N, Yuan C, Zhang H, Zhang L, Rosenberg SA, Wayne AS, Mackall CL. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Van Zelm MC, Smet J, Adams B, Mascart F, Schandené L, Janssen F, Ferster A, Kup CC, Levy S, van Dongen JJ, van der Burg M. CD81 gene defect in humans disrupts CD19 complex formation and leads to antibody deficiency. J Clin Invest. 2010;120:1265–1274. doi: 10.1172/JCI39748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sotillo E, Barrett D, Bagashev A, Black K, Lanauze C, Oldridge D, Sussman R, Harrington C, Chung EY, Hofmann TJ, Maude SL, Martinez NM, Raman P, Ruella M, Allman D, Jacoby E, Fry T, Barash Y, Lynch KW, Mackall C, Maris J, Grupp SA, Thomas-Tikhonenko A. Alternative splicing of CD19 mRNA in leukemias escaping CART-19 immunotherapy eliminates the cognate epitope and contributes to treatment failure. Proceedings of the 106th Annual Meeting of the American Association for Cancer Research. Philadelphia, PA: AACR. Cancer Res. 2015;75:3143. [Google Scholar]
- 99.Chiorazzi N, Ferrarini M. Cellular origin(s) of chronic lymphocytic leukemia: cautionary notes and additional considerations and possibilities. Blood. 2011;117:1781–1791. doi: 10.1182/blood-2010-07-155663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Donnenberg VS, Donnenberg AD. Multiple drug resistance in cancer revisited: the cancer stem cell hypothesis. J Clin Pharmacol. 2005;45:872–877. doi: 10.1177/0091270005276905. [DOI] [PubMed] [Google Scholar]
- 101.Jazirehi AR. Functional complementation, molecular targeted strategies, and chemo/immune sensitization in cancer treatment: hurdles and solutions. Current Drug Delivery. 2012;9:1–4. doi: 10.2174/156720112798376069. [DOI] [PubMed] [Google Scholar]
- 102.Kang MH, Reynolds CP. Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res. 2009;15:1126–1132. doi: 10.1158/1078-0432.CCR-08-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Karlsson H, Lindqvist AC, Fransson M, Paul-Wetterberg G, Nilsson B, Essand M, Nilsson K, Frisk P, Jernberg-Wiklund H, Loskog A. Combining CAR T cells and the Bcl-2 family apoptosis inhibitor ABT-737 for treating B-cell malignancy. Cancer Gene Ther. 2013;20:386–393. doi: 10.1038/cgt.2013.35. [DOI] [PubMed] [Google Scholar]
- 104.Karlsson H. CD19-targeting CAR T Cells for Treatment of B Cell Malignancies. From Bench to Bedside. Digital Comprehensive Summaries of Uppsala. Dissertations from the Faculty of Medicine Uppsala, Sweden: Acta Universitatis Upsaliensis. 2014;1031:72. [Google Scholar]
- 105.Yunis JJ, Mayer MG, Arnesen MA, Aeppli DP, Oken MM, Frizzera G. Bcl-2 and other genomic alterations in the prognosis of large-cell lymphoma. N Engl J Med. 1989;320:1047–1054. doi: 10.1056/NEJM198904203201605. [DOI] [PubMed] [Google Scholar]
- 106.Agace WW. Tissue-tropic effector T cells: generation and targeting opportunities. Nat Rev Immunol. 2006;6:682–692. doi: 10.1038/nri1869. [DOI] [PubMed] [Google Scholar]
- 107.Urban D, Thanabalasingam U, Stibenz D, Kaufmann J, Meyborg H, Fleck E, Gräfe M, Stawowy P. CD40/CD40L interaction induces E-selectin dependent leukocyte adhesion to human endothelial cells and inhibits endothelial cell migration. Biochem Biophys Res Commun. 2011;404:448–452. doi: 10.1016/j.bbrc.2010.11.142. [DOI] [PubMed] [Google Scholar]
- 108.Enblad G, Karlsson H, Loskog ASI. CAR T-Cell Therapy: The Role of Physical Barriers and Immunosuppression in Lymphoma. Hum Gene Ther. 2015;26:498–505. doi: 10.1089/hum.2015.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Scott DW, Gascoyne RD. The tumor microenvironment in B cell lymphomas. Nat Rev Cancer. 2014;14:517–534. doi: 10.1038/nrc3774. [DOI] [PubMed] [Google Scholar]
- 110.Hegde M, Corder A, Chow KKH, Mukherjee M, Ashoori A, Kew Y, Zhang YJ, Baskin DS, Merchant F, Brawley VS, Byrd TT, Krebs S, Wu MF, Liu H, Heslop HE, Gottachalk S, Yvon E, Ahmed N. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol Ther. 2013;21:2087–2101. doi: 10.1038/mt.2013.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Guzman ML, Jordan CT. Considerations for targeting malignant stem cells in leukemia. Cancer Control. 2004;11:97–104. doi: 10.1177/107327480401100216. [DOI] [PubMed] [Google Scholar]
- 112.Kim SM, Lee ST, Ryu KJ, Kim HJ, Kim SH, Ko YH, Kim WS, Kim SJ. A subset of CD45+/CD19 - cells in bone marrow may be associated with clinical outcomes of patients with mantle cell lymphoma. Leuk Lymphoma. 2015;56:3052–3057. doi: 10.3109/10428194.2015.1025391. [DOI] [PubMed] [Google Scholar]
- 113.Burger JA, Peled A. CXCR4 antagonists: targeting the microenvironment in leukemia and other cancers. Leukemia. 2008;23:43–52. doi: 10.1038/leu.2008.299. [DOI] [PubMed] [Google Scholar]
- 114.Huang H, Langenkamp E, Georganaki M, Loskog A, Fuchs PF, Dieterich LC, Kreuger J, Dimberg A. VEGF suppresses T-lymphocyte infiltration in the tumor microenvironment through inhibition of NF-κB-induced endothelial activation. FASEB J. 2015;29:227–238. doi: 10.1096/fj.14-250985. [DOI] [PubMed] [Google Scholar]
- 115.Chen K, Huang Y, Chen J. Understanding and targeting cancer stem cells: therapeutic implications and challenges. Acta Pharmacol Sin. 2013;34:732–740. doi: 10.1038/aps.2013.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sims-Mourtada J, Izzo JG, Ajani J, Chao KSC. Sonic hedgehog promotes multiple drug resistance by regulation of drug transport. Oncogene. 2007;26:5674–5679. doi: 10.1038/sj.onc.1210356. [DOI] [PubMed] [Google Scholar]
- 117.Patil Y, Sadhukha T, Ma L, Panyam J. Nanoparticle-mediated simultaneous and targeted delivery of paclitaxel and tariquidar overcomes tumor drug resistance. J Control Release. 2009;136:21–29. doi: 10.1016/j.jconrel.2009.01.021. [DOI] [PubMed] [Google Scholar]
- 118.Ritchie TK, Kwon H, Atkins WM. Conformational analysis of human ATP-binding cassette transporter ABCB1 in lipid nanodiscs and inhibition by the antibodies MRK16 and UIC2. J Biol Chem. 2011;286:39489–39496. doi: 10.1074/jbc.M111.284554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, Grant B, Sharman JP, Coleman M, Wierda WG, Jones JA, Zhao W, Heerema NA, Johnson AJ, Sukbuntherng J, Chang BY, Clow F, Hedrick E, Buggy JJ, James DF, O’Brien S. Targeting BTK with Ibrutinib in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med. 2013;369:32–42. doi: 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, Jurczak W, Advani RH, Romaguera JE, Williams ME, Barrientos JC, Chmielowska E, Radford J, Stilgenbauer S, Dreyling M, Jedrzejczak WW, Johnson P, Spurgeon SE, Li L, Zhang L, Newberry K, Ou Z, Cheng N, Fang B, McGreivy J, Clow F, Buggy JJ, Chang BY, Beaupre DM, Kunkel LA, Blum KA. Targeting BTK with Ibrutinib in Relapsed or Refractory Mantle-Cell Lymphoma. N Engl J Med. 2013;369:507–516. doi: 10.1056/NEJMoa1306220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Treon SP, Tripsas CK, Meid K, Warren D, Varma G, Green R, Argyropoulos KV, Yang G, Cao Y, Xu L, Patterson CJ, Rodig S, Zehnder JL, Aster JC, Harris NL, Kanan S, Ghobrial I, Castillo JJ, Laubach JP, Hunter ZR, Salman Z, Li J, Cheng M, Clow F, Graef T, Palomba ML, Advani RH. Ibrutinib in previously treated Waldenström’s macroglobulinemia. N Engl J Med. 2015;372:1430–1440. doi: 10.1056/NEJMoa1501548. [DOI] [PubMed] [Google Scholar]
- 122.Advani RH, Buggy JJ, Sharman JP, Smith SM, Boyd TE, Grant B, Kolibaba KS, Furman RR, Rodriguez S, Chang BY, Sukbuntherng J, Izumi R, Hamdy A, Hedrick E, Fowler NH. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J. Clin. Oncol. 2013;31:88–94. doi: 10.1200/JCO.2012.42.7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, Carney DA, He SZ, Huang DC, Xiong H, Cui Y, Busman TA, McKeegan EM, Krivoshik AP, Enschede SH, Humerickhouse R. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of aphase I study of navitoclax in patients with relapsed or refractory disease. J. Clin. Oncol. 2012;30:488–496. doi: 10.1200/JCO.2011.34.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kang MH, Wan Z, Kang YH, Sposto R, Reynolds CP. Mechanism of synergy of N-(4-hydroxyphenyl) retinamide and ABT-737 in acute lymphoblastic leukemia cell lines: Mcl-1 inactivation. J Natl Cancer Inst. 2008;100:580–595. doi: 10.1093/jnci/djn076. [DOI] [PubMed] [Google Scholar]
- 125.Keuling AM, Felton KEA, Parker AAM, Akbari M, Andrew SE, Tron VA. RNA Silencing of Mcl-1 Enhances ABT-737-Mediated Apoptosis in Melanoma: Role for a Caspase-8-Dependent Pathway. PLoS One. 2009;4:e6651. doi: 10.1371/journal.pone.0006651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zauli G, Corallini F, Zorzet S, Grill V, Marzari R, Secchiero P. In vivo anti-lymphoma activity of an agonistic human recombinant anti-TRAILR2 minibody. Invest New Drugs. 2012;30:405–407. doi: 10.1007/s10637-010-9519-y. [DOI] [PubMed] [Google Scholar]
- 127.Tuthill MH, Montinaro A, Zinngrebe J, Prieske K, Draber P, Prieske S, Newsom-Davis T, von Karstedt S, Graves J, Walczak H. TRAIL-R2-specific antibodies and recombinant TRAIL can synergise to kill cancer cells. Oncogene. 2015;30:2138–2144. doi: 10.1038/onc.2014.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kobayashi E, Kishi H, Ozawa T, Hamana H, Nakagawa H, Jin A, Lin Z, Muraguchi A. A chimeric antigen receptor for TRAIL-receptor 1 induces apoptosis in various types of tumor cells. Biochem Biophys Res Commun. 2014;453:798–803. doi: 10.1016/j.bbrc.2014.10.024. [DOI] [PubMed] [Google Scholar]
- 129.Curran KJ, Seinstra BA, Nikhamin Y, Yeh R, Usachenko Y, van Leeuwen DG, Purdon T, Pegram HJ, Brentjens RJ. Enhancing antitumor efficacy of chimeric antigen receptor T cells through constitutive CD40L expression. Mol Ther. 2015;23:769–778. doi: 10.1038/mt.2015.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, Ittmann MM, Marchetti D, Dotti G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. 2015;21:524–529. doi: 10.1038/nm.3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhang T, Cao L, Xie J, Shi N, Zhang Z, Luo Z, Yue D, Zhang Z, Wang L, Han W, Xu Z, Chen H, Zhang Y. Efficiency of CD19 chimeric antigen receptor-modified T cells for treatment of B cell malignancies in phase I clinical trials: a metaanalysis. Oncotarget. 2015;6:33961–33971. doi: 10.18632/oncotarget.5582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ramos CA, Savoldo B, Dotti G. CD19-CAR trials. Cancer J. 2015;20:112–118. doi: 10.1097/PPO.0000000000000031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, Ostberg JR, Forman SJ. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 2010;16:1245–1256. doi: 10.1016/j.bbmt.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Cruz CR, Micklethwaite KP, Savoldo B, Ramos CA, Lam S, Ku S, Diouf O, Liu E, Barrett AJ, Ito S, Shpall EJ, Krance RA, Kamble RT, Carrum G, Hosing CM, Gee AP, Mei Z, Grilley BJ, Heslop HE, Rooney CM, Brenner MK, Bollard CM, Dotti G. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: a phase 1 study. Blood. 2013;122:2965–2973. doi: 10.1182/blood-2013-06-506741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kochenderfer JN, Dudley ME, Carpenter RO, Kassim SH, Rose JJ, Telford WG, Hakim FT, Halverson DC, Fowler DH, Hardy NM, Mato AR, Hickstein DD, Gea-Banacloche JC, Pavletic SZ, Sportes C, Maric I, Feldman SA, Hansen BG, Wilder JS, Blacklock-Schuver B, Jena B, Bishop MR, Gress RE, Rosenberg SA. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood. 2013;122:4129–4139. doi: 10.1182/blood-2013-08-519413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Xu Y, Zhang M, Ramos CA, Durett A, Liu E, Dakhova O, Liu H, Creighton CJ, Gee AP, Heslop HE, Rooney CM, Savoldo B, Dotti G. Closely related T-memory stem cells correlate with in vivo expansion of CAR. CD19-T cells and are preserved by IL-7 and IL-15. Blood. 2014;123:3750–9. doi: 10.1182/blood-2014-01-552174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN, Steinberg SM, Stroncek D, Tschernia N, Yuan C, Zhang H, Zhang L, Rosenberg SA, Wayne AS, Mackall CL. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Garfall AL, Maus MV, Hwang WT, Lacey SF, Mahnke YD, Melenhorst JJ, Zheng Z, Vogl DT, Cohen AD, Weiss BM, Dengel K, Kerr ND, Bagg A, Levine BL, June CH, Stadtmauer EA. Chimeric Antigen Receptor T Cells against CD19 for Multiple Myeloma. N Engl J Med. 2015;373:1040–1047. doi: 10.1056/NEJMoa1504542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Schuster SJ, Svoboda J, Nasta S, Porter DL, Mato A, Shah GD, Landsburg DJ, Chong EA, Lacey SF, Melenhorst JJ, Chew A, Hasskarl J, Shah NN, Wasik MA, Marcucci K, Zheng Z, Levine B, June CH. Phase IIa trial of chimeric antigen receptor modified T cells directed against CD19 (CTL019) in patients with relapsed or refractory CD19+ lymphomas. J. Clin. Oncol. 2015;33(suppl) [Google Scholar]
- 141.Sauter CS, Riviere I, Bernal Y, Wang X, Purdon T, Yoo S, Moskowitz CH, Giralt S, Matasar MJ, Curran KJ, Park JH, Sadelain M, Brentjens RJ. Phase I trial of 19-28z chimeric antigen modified T cells (19-28z CAR-T) post-high dose therapy and autologous stem cell transplant (HDT-ASCT) for relapsed and refractory (rel/ref) aggressive B-cell non-Hodgkin lymphoma (B-NHL) J. Clin. Oncol. 2015;33(suppl):abstract 8515. [Google Scholar]