

Abstract
Mesenchymal stem cells (MSCs), which are capable of differentiating into multiple cell types, are reported to exert multiple effects on tumor development. However, the relationship between MSCs and nasopharyngeal carcinoma (NPC) cells remains unclear. Exosomes are small membrane vesicles that can be released by several cell types, including MSCs. Exosomes, which can carry membrane and cytoplasmic constituents, have been described as participants in a novel mechanism of cell-to-cell communication. In the present study, we investigated the mechanisms underlying the interaction between MSCs and NPC cells. The data showed that MSCs secreted 40-100 nm heterogeneous small vesicles, which were defined as exosomes. Incubation of NPC cells with MSC-derived exosomes resulted in the uptake of exosomes by the cells, which promoted their proliferation, migration and tumorigenesis. After an extended treatment duration, the tumor cells showed morphological changes and significant changes in the expression of epithelial-mesenchymal transition (EMT) markers. Moreover, we found that FGF19 was highly expressed in MSC-exosomes and that exosomes stimulated NPC progression by activating the FGF19-FGFR4-dependent ERK signaling cascade and by modulating the EMT. All of these data indicated that exosomes participate in a novel mechanism by which MSCs influence NPC progression.
Keywords: Mesenchymal stem cells (MSCs), nasopharyngeal carcinoma (NPC), exosomes, metastasis, epithelial-mesenchymal transition (EMT), FGF19/FGFR4 signaling
Introduction
The tumor-associated microenvironment is composed of tumor-promoting and tumor-suppressing cells, soluble molecules and extracellular matrix components, which constitute the tumor stroma [1]. These factors provide pro-malignant signals to cancer cells, which not only support tumor growth but also facilitate the metastatic dissemination of the tumor to distant organs [2]. Many studies have focused on the inhibition of tumor-stroma crosstalk to develop new therapeutic approaches. Recently, mesenchymal stem cells (MSCs) have garnered much attention, as they are important components of tumor stromal cells.
MSCs are multipotent cells that can differentiate into osteoblasts, chondrocytes, adipocytes or myoblasts under the appropriate conditions [3]. Several studies have shown that MSCs can home to primary or metastatic tumor sites and can contribute to the formation of the tumor microenvironment [4,5]. However, the impact of MSCs on tumor progression is still unclear. Some studies have shown that MSCs can support tumor development, whereas other studies have shown that MSCs suppress tumor growth [6]. Many scientists have focused on the mechanism by which MSCs affect tumor progression. Previous studies have shown that MSCs might promote cancer development via the epithelial-mesenchymal transition (EMT), which is characterized as a transition in which cells convert from an epithelial phenotype to a mesenchymal phenotype. The EMT is a critical process during malignant tumor metastasis [7,8].
As paracrine effectors of MSCs, exosomes have become a research focus in recent years. Exosomes are small nanometer-sized membrane vesicles that can be released by a variety of cell types, they can carry membrane and cytoplasmic components of their original cells and can mediate interactions with target cells [9]. Many studies have reported that exosomes might function during the interaction between MSCs and tumor cells [10]. Tao Du et al. found that exosomes derived from MSCs can modulate the tumor microenvironment and promote human renal cancer cell growth and aggressiveness [11,12]. However, whether MSC-derived exosomes impact nasopharyngeal carcinoma (NPC) progression and the mechanisms underlying such an effect remain incompletely understood.
The fibroblast growth factor (FGF) family is composed of 22 structurally related polypeptides that have different biological functions [13]. Most FGFs bind to and activate cell surface FGF receptors to mediate certain biological processes, including cellular functions such as cell growth, differentiation, migration and angiogenesis [14]. Among the FGF family, FGF19 can be secreted into serum and act in an endocrine fashion [15]. Under normal physiological conditions, FGF19 can regulate diverse physiological processes, such as energy metabolism and bile acid homeostasis [16]. However, under disease conditions, the expression of FGF19 may be associated with poor outcomes of breast cancer, prostate cancer and hepatocellular carcinoma and may serve as a therapeutic target in these cancers [10,17,18]. The activity of FGF19 is modulated by the binding and activation of FGFR4; this FGF19-FGFR4 interaction has been proposed to play a role in carcinogenesis. These proteins have been found to be coexpressed in hepatocellular carcinomas, lung squamous cell carcinomas and colon adenocarcinomas [14].
In the present study, we sought to identify the association of MSC-exosomes with NPC cells. Exosomes released by MSCs might influence the progression of NPC, especially in terms of the ability of NPC cells to proliferate, migrate and invade. In addition, we identified the role of FGF19 in the MSC-exosome-mediated signaling network. This study aimed to investigate the impact of MSC-exosomes on NPC development. All of the data implied a novel mechanism by which MSCs influence NPC progression.
Materials and methods
Cell culture
The nasopharyngeal carcinoma cell lines CNE1, CNE2, 5-8F, 6-10B were generously gifted by Sun Yat-sen University Cancer Center and Xiangya Hospital of Central South University. Cells were cultured in RPMI 1640 medium (Gibco BRL, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS) (Gibco) at 37°C in 5% CO2. The immortalized normal nasopharyngeal epithelial cell line NP69 was cultured in Keratinocyte-SFM supplemented with epidermal growth factor (EGF) (Invitrogen, Carlsbad, USA). Human bone marrow MSCs were obtained from three healthy donors and cultured in complete DMEM (Gibco) as previously described [19]. All donors provided consent to participate in the study, which was approved by the Ethics Committee of the Affiliated Hospital of Nantong University.
Immunofluorescence microscopy
MSCs were seeded onto a 24-well plate and fixed with 4% paraformaldehyde. After three rinses in PBS, the MSCs were blocked with 1% normal donkey serum and incubated in a primary anti-vimentin antibody (1:500, Abcam) overnight. Then, the cells were incubated in AlexFluor-conjugated secondary antibodies (Invitrogen Life Technologies, 1:1000). The nuclei were stained with Hoechst and observed under a fluorescence microscope.
Preparation of exosomes from MSCs
After MSCs were cultured to approximately 50% confluence, 4 ml of fresh medium was added. After 48 h, the supernatant was centrifuged at 300 g for 5 minutes, 3000 g for 20 minutes and 6000 g for 1 hour to remove cell debris. The cell-free supernatant was ultracentrifuged at 100,000 g in a 90-Ti swing rotor (Optima L-100XP ultracentrifuge; Beckman Coulter, Fullerton, CA) for 1 h at 4°C, and the resulting pellet was ultracentrifuged a second time. The protein concentration in the exosomes was estimated using a BCA protein assay kit (PIERCE, Rockford, IL, USA). The pellets were stored at -80°C.
Transmission electron microscopy
Exosomes were fixed with 2.5% glutaraldehyde in PBS, washed, and ultracentrifuged. Then, the exosomes were suspended in 100 µl of PBS, and one drop was loaded on a formvar/carbon-coated grid. After staining with 3% aqueous phospho-tungstic acid for 1 minute, the exosomes were observed via transmission electron microscopy (TEM; JEM-1230, JEOL, Tokyo, Japan).
Cellular uptake of MSC-exosomes
Cellular uptake of MSC-exosomes was investigated using the PKH-67 labeling kit [20]. Exosomes were resuspended in 500 µl of FBS and mixed with 500 µl of PHK67 dye diluted in diluent C (1:1 v/v) for 5 min. This mixture was diluted with 8 ml of RPMI 1640 medium and centrifuged at 100,000 g to pellet the PKH-67 labeled exosomes. CNE2 cells that had been cultured to 50% confluency were incubated in the PKH67-labeled exosomes for 3 h. After incubation, the CNE2 cells were fixed in 4% paraformaldehyde for 40 min at room temperature. The cell nuclei were stained with Hoechst. Cellular uptake of MSC-exosomes was observed under a TCS SP-5 confocal microscope (Leica Microsystems, Wetzlar, Germany).
Western blotting
Harvested cells and exosomes were used for immunoblot analysis. The analysis was performed according to previous studies [21]. The primary antibodies used were as follows: anti-CD9, anti-CD63 (1:500, Sangon Biotech, Shanghai), anti-E-cadherin, anti-N-cadherin, anti-vimentin (1:5000, Abcam), anti-FGF19, anti-p-ERK (1:500, Santa Cruz Biotechnology), and anti-p-FGFR4 (1:500, Abcam). β-actin or Flotillin-1 was examined as a loading control.
Transient transfection with siRNAs
We obtained small interfering RNA (siRNA) targeting FGFR4 and silencer negative control siRNA (snc-RNA) from Biomics Biotechnologies Co., Ltd. (Nantong, China). CNE2 cells were plated on a 6-well plate or a 96-well plate overnight and cultured to 30-50% confluence. The siRNAs were transfected into CNE2 cells in vitro using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions.
Transwell migration and invasion assay
Transwell chambers obtained from Millipore were used for the migration assay. The lower chambers were filled with complete RPMI 1640 medium mixed with exosomes extracted from MSCs or with recombinant FGF19 (Sangon Biotech, Shanghai). RPMI 1640 medium without exosomes were used as controls. The cells (5×104) were added to the upper chamber. After incubation for 16 h at 37°C, the medium was removed. The reverse side of the upper chamber was fixed in 100% methanol for 30 minutes and stained with crystal violet, followed by visualization of the migratory cells under a microscope. For the invasion assay, the cells were seeded on a transwell chamber coated with Matrigel (BD Matrigel) for 3 h.
Cell proliferation assay
Cell viability was analyzed using cell counting kit-8 (CCK-8 Kit, Beyotime Institute of Biotechnology). Cells were seeded at a density of 1×104 cells per well and cultured overnight. Then, the medium was replaced with 100 µl of RPMI 1640 medium containing MSC-exosomes or different concentrations of recombinant FGF19 (10, 25, 50, 100, or 200 ng/ml). At various time points, 10 µl of CCK-8 was added to each well and incubated for 1.5 hours. Then, the absorbance of each well was measured at 450 nm using a microplate reader. The untreated cells were used as controls.
CNE2 cells transfected with FGFR4 or control siRNA were seeded on 96-well plates. Then, the cells were incubated in FGF19 or MSC-exosomes at the indicated concentration. At the specified time points, the cell proliferation rate was measured.
Animal studies
Four-week-old female NOD/SCID mice (Laboratory Animal Center of Nantong University, Nantong, China) were housed under specific-pathogen-free conditions. The mice were subcutaneously inoculated with 1×106 CNE2 cells. After three days, the mice were randomly divided into two groups, which were intratumorally injected with 100 µl of either MSC-exosomes or PBS every three days. The size of each tumor was estimated every four days by measuring the longest and shortest tumor diameters. Macroscopic tumor formation was examined at day 20. The animal studies were approved by the Animal Ethics Committee of Nantong University, China.
Immunohistochemistry
Tumor specimens were obtained from tumor-carrying NOD/SCID mice. Then, the specimens were fixed in 4% buffered-formalin solution and embedded in paraffin. Immunohistochemistry was performed on tissue sections using anti-E-cadherin (Abcam 1:500), anti-vimentin (Abcam 1:250) and anti-N-cadherin antibodies (Abcam 1:500) according to previous studies [21].
Statistical analysis
All experiments were independently repeated 3 times, and all data are reported as the means ± standard deviation (SD). Statistical analysis was performed using SPSS17.0 software. Signifi-cant differences between data means were determined using 2-tailed Student’s t tests as appropriate. A p value less than 0.05 was considered statistically significant.
Results
NPC cells took up MSC-derived exosomes
Human bone marrow MSCs exhibited a characteristic morphology of spindle fibroblast-like cells (Figure 1A). They positively expressed vimentin (Figure 1B), which is characteristic of MSCs. CD9 and CD63, which were commonly used as surface markers for exosomes, were detected in exosome preparations obtained from MSC-conditioned medium but were absent from the control RPMI 1640 medium (Figure 1C). Via transmission electron microscopy, we found that the isolated exosomes were heterogeneous small vesicles ranging from 40 to 100 nm in size (Figure 1D).
Figure 1.
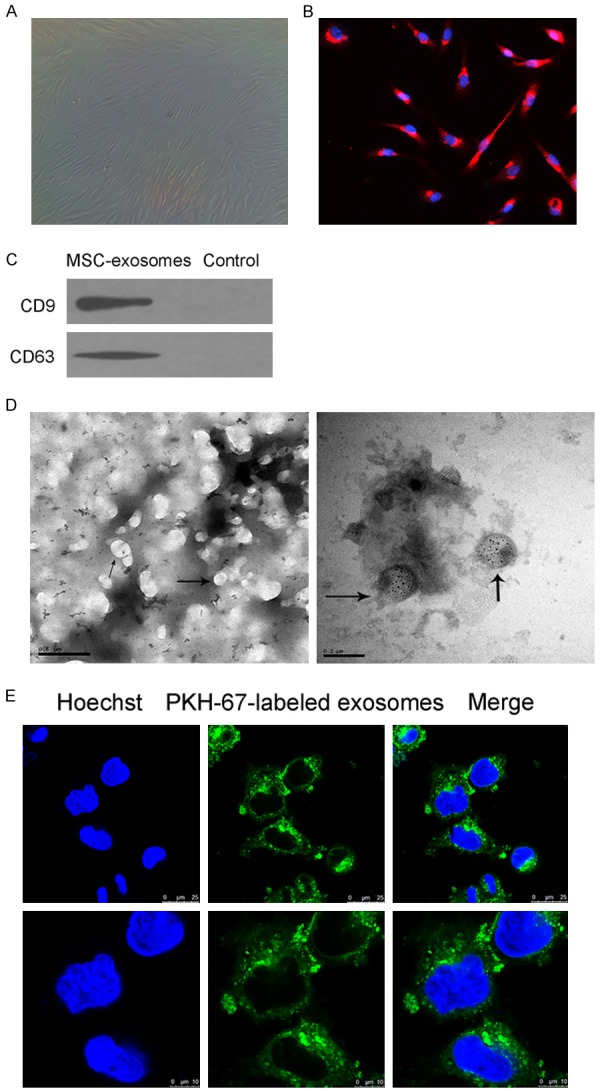
NPC cells took up MSC-exosomes. A. The cell morphology of human bone marrow-derived MSCs was observed under a light microscope. B. The expression of vimentin in MSCs was evaluated using immunofluorescence microscopy. C. Western blotting analysis of CD9 and CD63 expression in lysates from purified MSC-exosomes. D. Exosomes were observed under a transmission electron microscope; some of the exosomes are indicated by arrows. Left: scale bar = 500 nm; right: scale bar = 200 nm. E. CNE2 cells were incubated for 3 h in MSC-exosomes that were labeled with PKH67 (green). The three images at a low magnification presented in the upper portion show CNE2 cells incubated in exosomes. The images in the lower portion show a high magnification.
To confirm that MSC-exosomes could transfer to tumor cells, we assessed the uptake of exosomes by NPC cell line CNE2. Most CNE2 cells exhibited intracellular fluorescence after incubation in MSC-exosomes, and PKH-67-labeled exosomes were localized in the cytoplasm (Figure 1E). This result indicated that exosomes could be taken up by NPC cells through the plasma membrane due to treatment of NPC cells with exosomes.
MSC-exosomes enhanced the growth and migration of NPC cells in vitro
Studies have reported that MSCs can migrate to injury and tumor sites to incorporate into the stroma, where MSCs functionally interact with tumor cells. As paracrine effectors of MSCs, the activities of exosomes on NPC cells are still unclear. As shown in Figure 2A, the addition of MSC-exosomes accelerated the proliferation rate of CNE2, 5-8F and 6-10B cells. However, the proliferation rate of CNE1 cells was not significantly increased by treatment with MSC-exosomes. Moreover, we observed the involvement of MSC-exosomes in NPC cell migration (Figure 2B, 2C). There was no apparent difference in the migration of 6-10B cells, as this cell line exhibits nonmetastatic properties.
Figure 2.
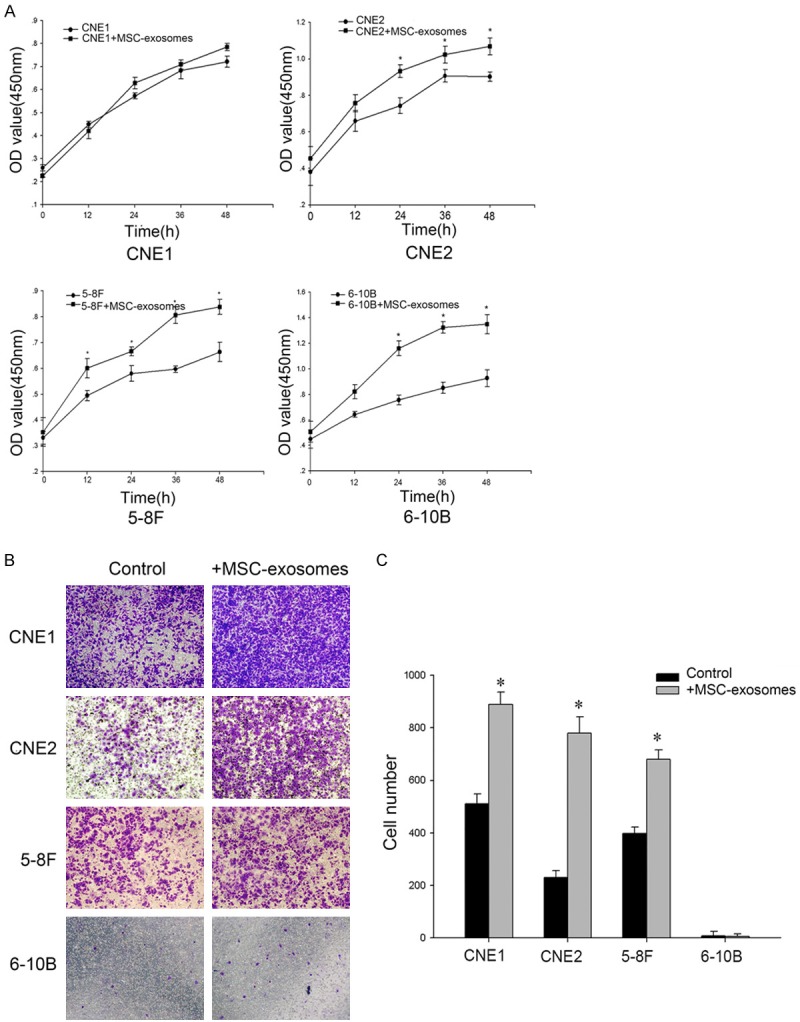
MSC-exosomes enhanced the growth and migration of NPC cells in vitro. A. NPC cells (1×104) were incubated in the presence or absence of exosomes for 12 h, 24 h, 36 h or 48 h. The cell proliferation rates were determined via the CCK8 assay. B. Serum-free cells (5×104) were added to the upper chamber, and conditioned media with or without MSC-exosomes were added to the lower chamber for a transwell migration assay. After 16 h, the cells that had migrated to the bottom of the membrane were stained with crystal violet. Representative images were captured at the same magnification. C. Absolute quantification of the cells that had migrated through the transwell membrane. The data shown are representative of at least three independent experiments. The data were analyzed using Student’s t-test. *p<0.05.
MSC-exosomes promoted CNE2 cell metastasis by stimulating the EMT
We observed effects of MSC-exosomes on NPC cells, but how MSC-exosomes exerted their effects remained unclear. Therefore, we chose CNE2 cells for further studies, as this NPC cell line was non-keratinizing, consistent with the main pathologic subtype of NPC. The occurrence of the EMT, a key step in the process of tumor metastasis, may be associated with poor clinical outcome in cancer patients [22]. We explored whether MSC-exosomes-treated tumor cells were more likely to migrate due to undergoing EMT. Over time, CNE2 cells showed morphological changes, including an elongated morphology, and this change was consistent with the features of the EMT (Figure 3A). Moreover, after an extended exposure duration, significant changes in the expression of EMT markers in CNE2 cells were observed. We found that both N-cadherin and vimentin were upregulated but that E-cadherin was downregulated in a time-dependent manner (Figure 3B, 3C). All of these results suggested that the treatment of tumor cells with MSC-exosomes might induce the EMT.
Figure 3.
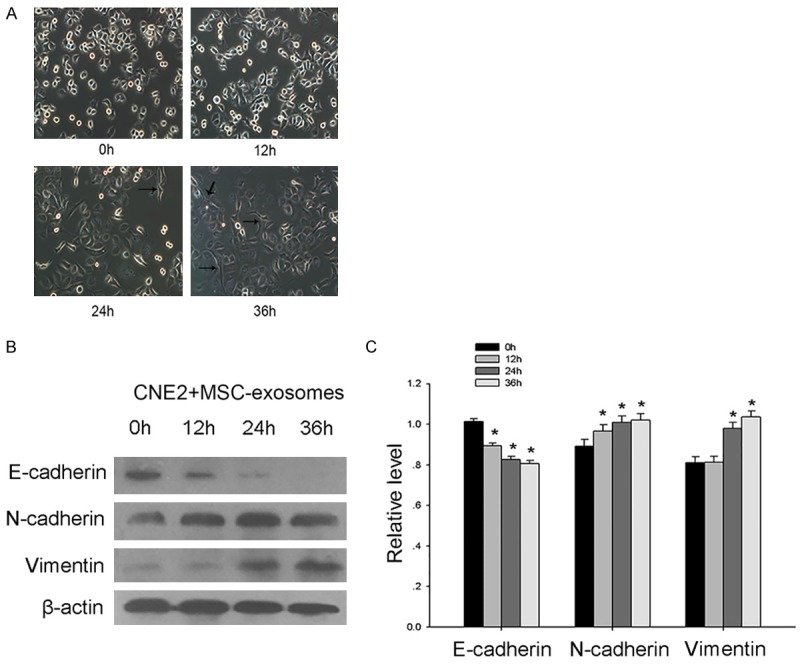
MSC-exosomes promoted the metastasis of CNE2 cells by stimulating the EMT. A. Representative images of the morphology of CNE2 cells after treatment with MSC-exosomes for 0 h, 12 h, 24 h or 36 h. B. Western blot analysis was used to investigate the changes in the expression of EMT markers in CNE2 cells after treatment with exosomes. C. The bar graph demonstrates the expression ratio of the target protein to β-actin based on densitometry. The data shown were representative of at least three independent experiments. The data were analyzed using Student’s t-test. *p<0.05.
MSC-exosomes promoted NPC tumor growth in vivo
To confirm the effects of MSC-exosomes on NPC tumorigenicity, we conducted xenotransplantation experiments in nude mice. CNE2 cells were subcutaneously injected into the mice, and MSC-exosomes were then intratumorally injected every three days. As expected, the MSC-exosomes boosted tumor formation and growth (Figure 4A, 4B). At day 20, the volume of the tumors treated with MSC-exosomes appeared to be larger than control (Figure 4C). Moreover, we performed Western blot and immunohistochemistry analyses to detect the expression of EMT markers in the MSC-exosomes-treated and PBS-treated xenograft tumors. Surprisingly, the expression of N-cadherin and vimentin was upregulated in tumor sections from exosomes-treated mice, and fewer E-cadherin-positive tumor cells were detected after treatment with the exosomes (Figure 4D, 4E). This result validated that MSC-exosomes might induce the EMT in NPC cells.
Figure 4.
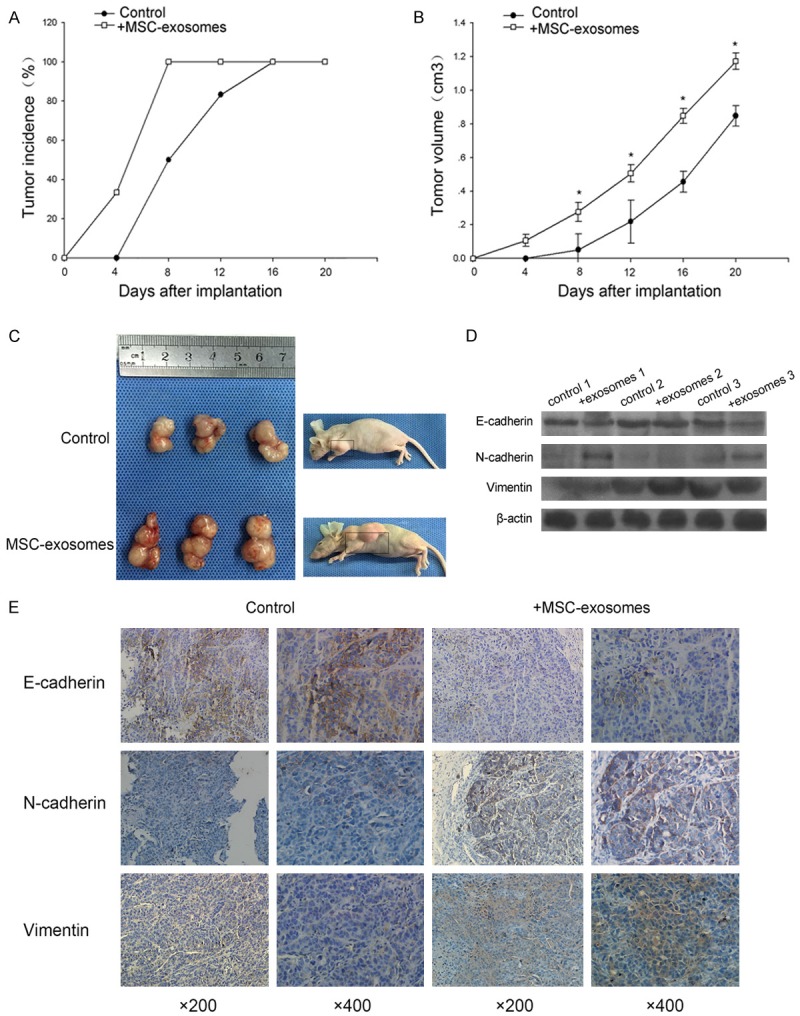
MSC-exosomes promoted NPC tumor growth in vivo. A. The evaluation of tumor incidence in mice treated with MSC-exosomes or with a control solution. B. The tumor volumes were assessed every four days. C. Nude mice were inoculated with CNE2 cells. The figure shows tumors obtained from the sacrificed mice at day 20 after subcutaneous injection of MSC-exosomes or a control solution every 3 days. D. Western blot analysis of E-cadherin, vimentin and N-cadherin expression in the MSC-exosomes-treated and PBS-treated xenograft tumors. E. Immunohistochemical analysis of E-cadherin, vimentin and N-cadherin expression in tumor sections. *p<0.05.
Effects of FGF19-FGFR4 signaling on the proliferation, migration and invasion of CNE2 cells
FGF19 has been found to be highly expressed in a subgroup of primary human tumors [17,23,24]. In these tumors, FGF19 activated FGFR signaling and promoted cell growth, invasion, adhesion and colony formation. To explore the role of FGF19 in NPC, we first stimulated NPC cells with different concentrations of FGF19. The CCK8 assay results showed that recombinant FGF19 significantly promoted cell growth in a dose-dependent manner (Figure 5A). We then evaluated the effect of FGF19 on cell migration. After treatment with 100 ng/mL FGF19, more cells migrated across the membrane than after the control treatment (Figure 5B, 5C). These data showed that FGF19 promoted cell metastasis. As expected, no significant difference in the number of migrating 6-10B cells was observed between treatments.
Figure 5.
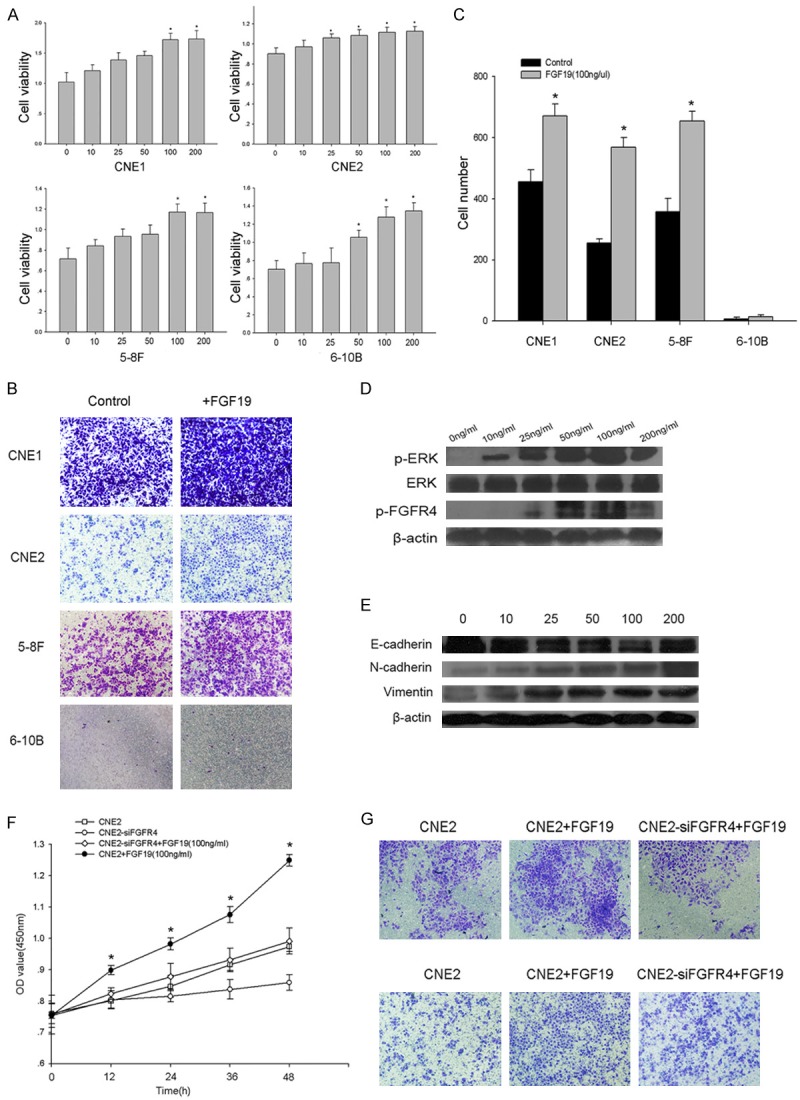
Effects of FGF19-FGFR4 signaling on the proliferation, migration and invasiveness of CNE2 cells. A. NPC cells were treated with different concentrations of FGF19 for 48 h. The cell proliferation rates were determined via a CCK8 assay. B. A transwell migration assay was used to detect cell migration after the cells were incubated in 100 ng/ml FGF19 for 16 h. C. Absolute quantification of the cells that had migrated through the transwell membrane. D. CNE2 cells were stimulated with different concentrations of FGF19 for 15 minutes. Western blots were performed to evaluate the levels of phosphorylated ERK and FGFR4 in CNE2 cells. E. CNE2 cells were stimulated with different concentrations of FGF19 for 48 h. The Western blots showed the EMT marker levels in the cells. F. FGFR4 was stably knocked down using siRNA. A CCK8 assay showed the viability of the cells after transfection with FGFR4 siRNA and treatment with 100 ng/ml FGF19. G. The transwell assays showed cell migration and invasion after transfection with FGFR4 siRNA and treatment with 100 ng/ml of FGF19. Upper: representative images of cell migration. Lower: representative images of cell invasion. The data shown are representative of at least three independent experiments. The data were analyzed using Student’s t-test. *p<0.05.
To explore the molecular mechanism by which FGF19 accelerates NPC progression, we used Western blots to determine the activation of FGFR4, which may be the downstream transducer of FGF19 signaling. As shown in Figure 5D, a significant increase in the tyrosine phosphorylation of ERK and FGFR4 was observed after stimulation with FGF19 for 15 minutes. We then evaluated the levels of EMT markers in cells treated with FGF19 and found a substantial decrease in E-cadherin expression and an increase in N-cadherin and vimentin expression in CNE2 cells (Figure 5E). The above results clearly indicated the involvement of FGF19 in modulating the EMT in NPC cells.
Disruption of the FGF signaling pathway suppressed NPC progression
Studies have reported that FGF19 is a specific FGFR4 activator [14]. To confirm whether the FGF signaling pathway contributes to the development of NPC, we analyzed the effect of silencing FGFR4 on tumor progression after the activation of FGF19. CNE2 cells were transfected with FGFR4 siRNA and treated with 100 ng/ml FGF19. We found that the FGFR4-silenced cells exhibited significantly reduced cell growth, cell migration and invasion (Figure 5F, 5G). The results confirmed that the knockdown of FGFR4 prevented FGF19-activated cell proliferation, migration and invasion. In conclusion, the activation of FGF signaling could promote NPC progression.
Functional role of MSC-exosomes-mediated FGF19-FGFR4 signaling
FGF19 has been suggested to act locally in a paracrine or autocrine fashion, and as exosomes are paracrine effectors, we explored whether the effects of MSC-exosomes were associated with FGF signaling. Not surprisingly, we found a high expression level of FGF19 in MSC-exosomes based on Western blot (Figure 6A). We then examined whether FGF19-FGFR4 signaling played a role in the MSC-exosomes-mediated enhancement of NPC cell survival and tumorigenesis. The results showed that FGFR4 siRNA-treated cells incubated with MSC-exosomes did not exhibit exosomes-induced cell growth, migration and invasion (Figure 6B-F). Moreover, we found that ERK and FGFR4 activation was decreased in association with the knockdown of FGFR4. FGFR4 siRNA also reversed MSC-exosomes-induced ERK phosphorylation and EMT modulation (Figure 6G, 6H).
Figure 6.
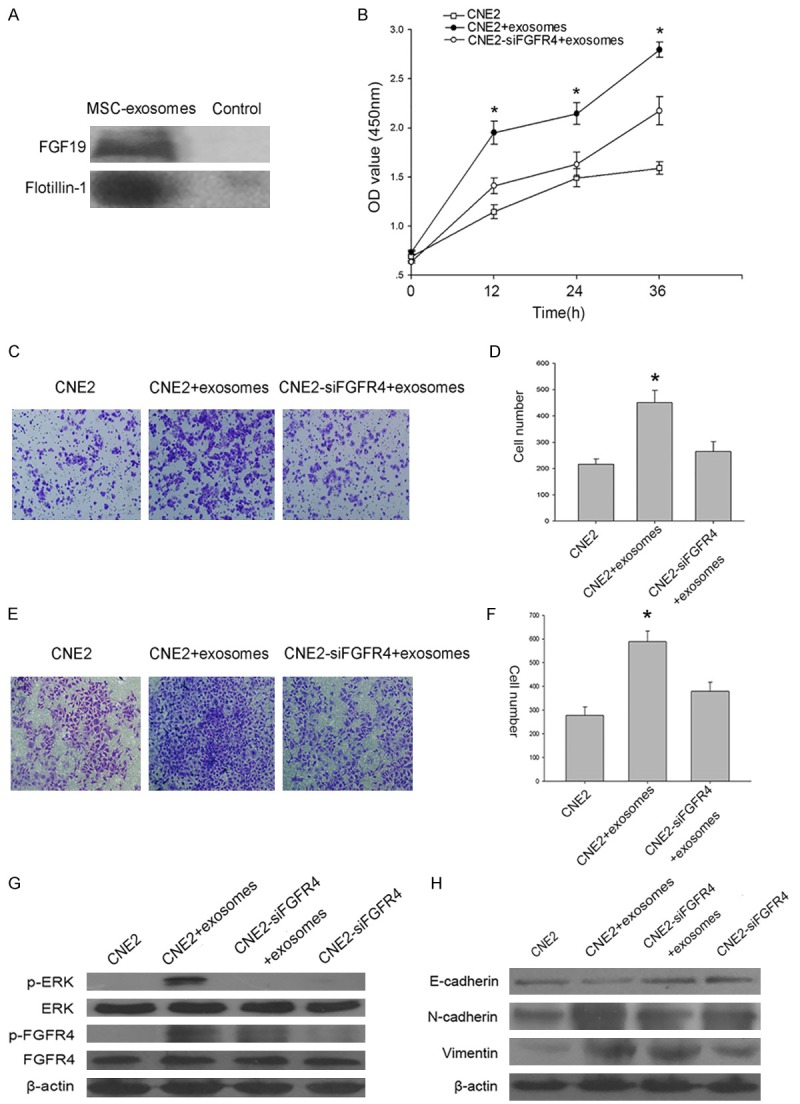
Functional role of MSC-exosomes-mediated FGF19-FGFR4 signaling. A. A Western blot showed the expression of FGF19 in MSC-exosomes. Flotillin-1 was used as a loading control. B. The effect of FGFR4 siRNA transfection on MSC-exosomes-induced cell proliferation in CNE2 cells. C. A transwell assay showed the effect of FGFR4 siRNA transfection on MSC-exosomes-induced cell migration in CNE2 cells. D. Absolute quantification of the migrating cells. E. A transwell assay showed the effect of FGFR4 siRNA transfection on MSC-exosomes-induced cell invasion in CNE2 cells. F. Absolute quantification of the invading cells. G. The levels of phosphorylated ERK and FGFR4 were assessed by Western blot after the treatment of FGFR4-silenced cells with MSC-exosomes. H. Western blot was used to detect the effects of FGFR4 siRNA on MSC-exosomes-mediated EMT modulation. The data shown are representative of at least three independent experiments. The data were analyzed using Student’s t-test. *p<0.05.
Discussion
MSCs have recently gained much attention for their application to tumor therapy, as MSCs can mobilize from bone marrow or other tissues to the tumor microenvironment [25,26]. Although the effects of MSCs on tumor progression remain unclear, it is becoming clear that MSCs can home to tumor sites and that these cells play an indispensable role in tumor development. Many studies have shown that MSCs can either promote or inhibit tumor progression in different tumor models [6,27-29]. However, the molecular mechanisms mediating this particular phenomenon remain to be thoroughly investigated. Studies have reported that secreting paracrine factors is a critical function of MSCs in the tumor microenvironment [30]. Karnoub et al. observed that MSCs integrated into the tumor-associated stroma and acted in a paracrine manner to promote breast cancer cell motility, invasion and metastasis [31]. In the present study, we aimed to explore the interaction between MSCs and NPC cells. We started by isolating BM-derived MSCs, which exhibited common MSC features based on the expression of their surface markers (Figure 1A, 1B).
Exosomes have been described as novel mediators of cell-to-cell communication. Exosomes can affect target recipient cells by inducing intracellular signaling or conferring new materials from the donor cells [32]. The functions of exosomes include genetic material exchange, immune responses, angiogenesis, tumor metastasis and pathogen or oncogene distribution [33]. As paracrine effectors of MSCs, exosomes derived from MSCs might exert effects on tumor development. For example, exosomes from multiple myeloma (MM) patient BM-derived MSCs promoted MM tumor growth, and exosomes derived from MSCs might promote renal cancer cell growth and aggressiveness [12,34]. However, exosomes also showed anti-tumor effects. Bruno S et al. found that exosomes from human MSCs inhibited growth and survival of different tumor cell types, and the same results were observed in SCID mouse models [35]. This discrepancy might be associated with the varying injection time of MSCs or exosomes, the different tumor types and in vivo tumor models examined, and the heterogeneity of MSCs [6]. Whether these isolated exosomes derived from MSCs impact NPC progression remains to be further determined.
In the present study, we isolated exosomes from MSC-conditioned medium using ultracentrifugation and identified the exosomes based on the expression of their surface markers and based on TEM (Figure 1C, 1D). We also found that exosomes could be taken up by CNE2 cells through the plasma membrane after co-incubation (Figure 1E). Further, we found that MSC-exosomes might promote malignant cell proliferation and migration (Figure 2). In vivo, the injection of CNE2 cells followed by isolated MSC-exosomes markedly promoted tumor formation and tumor growth (Figure 4A-C). Our results were consistent with the previously reported studies of renal cancer and MM tumors.
EMT is a process that refers to the loss of epithelial characteristics and the gain of a mesenchymal phenotype. When cells undergo the EMT, they lose cell-cell contacts, exhibit decreased expression of epithelial markers, and gain mesenchymal features such as enhanced motility and invasiveness [36,37]. Zhu et al. found that treating tumor cells with human MSC-conditioned medium (hMSC-CM) induced the EMT, which supported tumor progression. The effective constituents in CM were demonstrated to be exosomes derived from MSCs [38,39]. In our study, after co-culturing CNE2 cells with MSC-exosomes, the morphology of the CNE2 cells was altered (Figure 3A). We also found that this treatment might decrease the expression of the cell adhesion protein E-cadherin and increase the expression of N-cadherin and vimentin in CNE2 cells (Figure 3B). These results were confirmed in nude mice. The expression of EMT markers in tumor sections was altered in exosome-treated mice (Figure 4D, 4E). All of these results suggested that the tumor cells showed a decrease in epithelial characteristics and underwent the EMT.
FGF19, which belongs to the FGF family, plays important roles in cell proliferation, differentiation and motility. In this study, we undertook experiments to investigate the relationship of FGF19 with NPC and found that the application of FGF19 promoted NPC cell proliferation and migration (Figure 5A-C).
Studies have suggested that FGF19 exclusively binds to FGFR4 [40]. Though FGF19-FGFR4 signaling has been demonstrated to be involved in many tumor types, the mechanisms underlying this effect are still under study. In colorectal cancer, inhibition of FGF19 decreased the TAF-induced phosphorylation of FGFR4; this finding suggested that FGF19 plays a role in FGFR4/Wnt activation [41]. In prostate cancer, FGF19 was highly expressed in PCa, and FGF19 signaling activated ERK and p38 MAPK [17]. In our studies, we found that with increasing concentrations of FGF19, ERK and FGFR4 signaling was activated, and the tumor cells underwent the EMT (Figure 5D, 5E).
Roidl A et al. found that an anti-FGFR4 antibody (10F10) alleviated the FGF19-mediated stimulation of the phospho-ERK levels in breast cancer cells [42]. In our study, we found that knockdown of FGFR4 expression inhibited the FGF19-mediated stimulation of cell proliferation, migration and invasion (Figure 5F, 5G). This result might be due to the inhibition of the interaction between FGF19 and FGFR4. All of these results further validated the effects of the FGF19-FGFR4 pathway.
To investigate the mechanisms by which exosomes affect NPC progression, we undertook further studies. Surprisingly, we found that FGF19 was highly expressed in exosomes derived from MSCs (Figure 6A). To confirm whether the impact of exosomes on NPC was associated with FGF19-FGFR4 signaling, we knocked down the expression of FGFR4 in CNE2 cells, and this treatment inhibited cell proliferation, migration and invasion (Figure 6B-F). Moreover, MSC-exosomes-activated FGF19-dependent ERK phosphorylation and EMT induction in NPC cells were neutralized.
In conclusion, we demonstrated that MSC-exosomes accelerate NPC progression in terms of cell proliferation, migration and tumorigenesis through the FGF19-mediated activation of the FGFR4 signaling cascade. Our results support a relationship between MSCs and NPC cells and provide a new understanding of the contribution of MSCs to tumor development.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 81172841 and 81471603), the China Postdoctoral Science Foundation (No. 2013M541708), the Natural Science Foundation of Jiangsu Province (SBK2015022581), the project of “333 Natural Science Foundation” of Jiangsu Grant (No. BRA2013286), the “Top Six Types of Talents” Financial Assistance of Jiangsu Province Grant (No. 6), the project of Jiangsu Provincial Health Department (No. Z201005), the scientific and innovative research project of Nantong (No. BK2014003, HS2014007 201HHS), the innovative research project for Nantong University postgraduate students (No. YKS14009), and the Outstanding Medical Academic Leader Program of Jiangsu Province (No. LJ201136).
Disclosure of conflict of interest
None.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Robinson BD, Sica GL, Liu YF, Rohan TE, Gertler FB, Condeelis JS, Jones JG. Tumor microenvironment of metastasis in human breast carcinoma: a potential prognostic marker linked to hematogenous dissemination. Clin Cancer Res. 2009;15:2433–2441. doi: 10.1158/1078-0432.CCR-08-2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Salem HK, Thiemermann C. Mesenchymal stromal cells: current understanding and clinical status. Stem Cells. 2010;28:585–596. doi: 10.1002/stem.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Droujinine IA, Eckert MA, Zhao W. To grab the stroma by the horns: from biology to cancer therapy with mesenchymal stem cells. Oncotarget. 2013;4:651–664. doi: 10.18632/oncotarget.1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kidd S, Spaeth E, Dembinski JL, Dietrich M, Watson K, Klopp A, Battula VL, Weil M, Andreeff M, Marini FC. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells. 2009;27:2614–2623. doi: 10.1002/stem.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klopp AH, Gupta A, Spaeth E, Andreeff M, Marini F 3rd. Concise review: Dissecting a discrepancy in the literature: do mesenchymal stem cells support or suppress tumor growth? Stem Cells. 2011;29:11–19. doi: 10.1002/stem.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jordan NV, Johnson GL, Abell AN. Tracking the intermediate stages of epithelial-mesenchymal transition in epithelial stem cells and cancer. Cell Cycle. 2011;10:2865–2873. doi: 10.4161/cc.10.17.17188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martin FT, Dwyer RM, Kelly J, Khan S, Murphy JM, Curran C, Miller N, Hennessy E, Dockery P, Barry FP, O’Brien T, Kerin MJ. Potential role of mesenchymal stem cells (MSCs) in the breast tumour microenvironment: stimulation of epithelial to mesenchymal transition (EMT) Breast Cancer Res Treat. 2010;124:317–326. doi: 10.1007/s10549-010-0734-1. [DOI] [PubMed] [Google Scholar]
- 9.Vlassov AV, Magdaleno S, Setterquist R, Conrad R. Exosomes: current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim Biophys Acta. 2012;1820:940–948. doi: 10.1016/j.bbagen.2012.03.017. [DOI] [PubMed] [Google Scholar]
- 10.Buhmeida A, Dallol A, Merdad A, Al-Maghrabi J, Gari MA, Abu-Elmagd MM, Chaudhary AG, Abuzenadah AM, Nedjadi T, Ermiah E, Al-Thubaity F, Al-Qahtani MH. High fibroblast growth factor 19 (FGF19) expression predicts worse prognosis in invasive ductal carcinoma of breast. Tumour Biol. 2014;35:2817–2824. doi: 10.1007/s13277-013-1374-y. [DOI] [PubMed] [Google Scholar]
- 11.Yu B, Zhang X, Li X. Exosomes derived from mesenchymal stem cells. Int J Mol Sci. 2014;15:4142–4157. doi: 10.3390/ijms15034142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Du T, Ju G, Wu S, Cheng Z, Cheng J, Zou X, Zhang G, Miao S, Liu G, Zhu Y. Microvesicles derived from human Wharton’s jelly mesenchymal stem cells promote human renal cancer cell growth and aggressiveness through induction of hepatocyte growth factor. PLoS One. 2014;9:e96836. doi: 10.1371/journal.pone.0096836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ornitz DM, Itoh N. Fibroblast growth factors. Genome Biol. 2001;2:REVIEWS3005. doi: 10.1186/gb-2001-2-3-reviews3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin BC, Desnoyers LR. FGF19 and cancer. Adv Exp Med Biol. 2012;728:183–194. doi: 10.1007/978-1-4614-0887-1_12. [DOI] [PubMed] [Google Scholar]
- 15.Harmer NJ, Pellegrini L, Chirgadze D, Fernandez-Recio J, Blundell TL. The crystal structure of fibroblast growth factor (FGF) 19 reveals novel features of the FGF family and offers a structural basis for its unusual receptor affinity. Biochemistry. 2004;43:629–640. doi: 10.1021/bi035320k. [DOI] [PubMed] [Google Scholar]
- 16.Fu L, John LM, Adams SH, Yu XX, Tomlinson E, Renz M, Williams PM, Soriano R, Corpuz R, Moffat B, Vandlen R, Simmons L, Foster J, Stephan JP, Tsai SP, Stewart TA. Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology. 2004;145:2594–2603. doi: 10.1210/en.2003-1671. [DOI] [PubMed] [Google Scholar]
- 17.Feng S, Dakhova O, Creighton CJ, Ittmann M. Endocrine fibroblast growth factor FGF19 promotes prostate cancer progression. Cancer Res. 2013;73:2551–2562. doi: 10.1158/0008-5472.CAN-12-4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miura S, Mitsuhashi N, Shimizu H, Kimura F, Yoshidome H, Otsuka M, Kato A, Shida T, Okamura D, Miyazaki M. Fibroblast growth factor 19 expression correlates with tumor progression and poorer prognosis of hepatocellular carcinoma. BMC Cancer. 2012;12:56. doi: 10.1186/1471-2407-12-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gu Z, Cao X, Jiang J, Li L, Da Z, Liu H, Cheng C. Upregulation of p16INK4A promotes cellular senescence of bone marrow-derived mesenchymal stem cells from systemic lupus erythematosus patients. Cell Signal. 2012;24:2307–2314. doi: 10.1016/j.cellsig.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 20.Pegtel DM, Cosmopoulos K, Thorley-Lawson DA, van Eijndhoven MA, Hopmans ES, Lindenberg JL, de Gruijl TD, Wurdinger T, Middeldorp JM. Functional delivery of viral miRNAs via exosomes. Proc Natl Acad Sci U S A. 2010;107:6328–6333. doi: 10.1073/pnas.0914843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.You Y, Yao H, You B, Li X, Ni H, Shi S, Shan Y, Cao X. Clinical significance of HAX-1 expression in laryngeal carcinoma. Auris Nasus Larynx. 2015;42:299–304. doi: 10.1016/j.anl.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 22.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 23.Desnoyers LR, Pai R, Ferrando RE, Hotzel K, Le T, Ross J, Carano R, D’Souza A, Qing J, Mohtashemi I, Ashkenazi A, French DM. Targeting FGF19 inhibits tumor growth in colon cancer xenograft and FGF19 transgenic hepatocellular carcinoma models. Oncogene. 2008;27:85–97. doi: 10.1038/sj.onc.1210623. [DOI] [PubMed] [Google Scholar]
- 24.Pai R, Dunlap D, Qing J, Mohtashemi I, Hotzel K, French DM. Inhibition of fibroblast growth factor 19 reduces tumor growth by modulating beta-catenin signaling. Cancer Res. 2008;68:5086–5095. doi: 10.1158/0008-5472.CAN-07-2325. [DOI] [PubMed] [Google Scholar]
- 25.Kidd S, Spaeth E, Watson K, Burks J, Lu H, Klopp A, Andreeff M, Marini FC. Origins of the tumor microenvironment: quantitative assessment of adipose-derived and bone marrow-derived stroma. PLoS One. 2012;7:e30563. doi: 10.1371/journal.pone.0030563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S, Baik GH, Shibata W, Diprete B, Betz KS, Friedman R, Varro A, Tycko B, Wang TC. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. 2011;19:257–272. doi: 10.1016/j.ccr.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Boeck A, Pauwels P, Hensen K, Rummens JL, Westbroek W, Hendrix A, Maynard D, Denys H, Lambein K, Braems G, Gespach C, Bracke M, De Wever O. Bone marrow-derived mesenchymal stem cells promote colorectal cancer progression through paracrine neuregulin 1/HER3 signalling. Gut. 2013;62:550–560. doi: 10.1136/gutjnl-2011-301393. [DOI] [PubMed] [Google Scholar]
- 28.Wang ML, Pan CM, Chiou SH, Chen WH, Chang HY, Lee OK, Hsu HS, Wu CW. Oncostatin m modulates the mesenchymal-epithelial transition of lung adenocarcinoma cells by a mesenchymal stem cell-mediated paracrine effect. Cancer Res. 2012;72:6051–6064. doi: 10.1158/0008-5472.CAN-12-1568. [DOI] [PubMed] [Google Scholar]
- 29.Shinagawa K, Kitadai Y, Tanaka M, Sumida T, Kodama M, Higashi Y, Tanaka S, Yasui W, Chayama K. Mesenchymal stem cells enhance growth and metastasis of colon cancer. Int J Cancer. 2010;127:2323–2333. doi: 10.1002/ijc.25440. [DOI] [PubMed] [Google Scholar]
- 30.Keating A. Mesenchymal stromal cells: new directions. Cell Stem Cell. 2012;10:709–716. doi: 10.1016/j.stem.2012.05.015. [DOI] [PubMed] [Google Scholar]
- 31.Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R, Weinberg RA. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557–563. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- 32.Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9:581–593. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- 33.Zoller M. Tetraspanins: push and pull in suppressing and promoting metastasis. Nat Rev Cancer. 2009;9:40–55. doi: 10.1038/nrc2543. [DOI] [PubMed] [Google Scholar]
- 34.Roccaro AM, Sacco A, Maiso P, Azab AK, Tai YT, Reagan M, Azab F, Flores LM, Campigotto F, Weller E, Anderson KC, Scadden DT, Ghobrial IM. BM mesenchymal stromal cellderived exosomes facilitate multiple myeloma progression. J Clin Invest. 2013;123:1542–1555. doi: 10.1172/JCI66517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bruno S, Collino F, Deregibus MC, Grange C, Tetta C, Camussi G. Microvesicles derived from human bone marrow mesenchymal stem cells inhibit tumor growth. Stem Cells Dev. 2013;22:758–771. doi: 10.1089/scd.2012.0304. [DOI] [PubMed] [Google Scholar]
- 36.Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265–273. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 37.Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA. Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest. 2009;119:1438–1449. doi: 10.1172/JCI38019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu W, Huang L, Li Y, Qian H, Shan X, Yan Y, Mao F, Wu X, Xu WR. Mesenchymal stem cell-secreted soluble signaling molecules potentiate tumor growth. Cell Cycle. 2011;10:3198–3207. doi: 10.4161/cc.10.18.17638. [DOI] [PubMed] [Google Scholar]
- 39.Zhu W, Huang L, Li Y, Zhang X, Gu J, Yan Y, Xu X, Wang M, Qian H, Xu W. Exosomes derived from human bone marrow mesenchymal stem cells promote tumor growth in vivo. Cancer Lett. 2012;315:28–37. doi: 10.1016/j.canlet.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 40.Goetz R, Beenken A, Ibrahimi OA, Kalinina J, Olsen SK, Eliseenkova AV, Xu C, Neubert TA, Zhang F, Linhardt RJ, Yu X, White KE, Inagaki T, Kliewer SA, Yamamoto M, Kurosu H, Ogawa Y, Kuro-o M, Lanske B, Razzaque MS, Mohammadi M. Molecular insights into the klotho-dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol Cell Biol. 2007;27:3417–3428. doi: 10.1128/MCB.02249-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu R, Li J, Xie K, Zhang T, Lei Y, Chen Y, Zhang L, Huang K, Wang K, Wu H, Wu M, Nice EC, Huang C, Wei Y. FGFR4 promotes stromainduced epithelial-to-mesenchymal transition in colorectal cancer. Cancer Res. 2013;73:5926–5935. doi: 10.1158/0008-5472.CAN-12-4718. [DOI] [PubMed] [Google Scholar]
- 42.Roidl A, Berger HJ, Kumar S, Bange J, Knyazev P, Ullrich A. Resistance to chemotherapy is associated with fibroblast growth factor receptor 4 up-regulation. Clin Cancer Res. 2009;15:2058–2066. doi: 10.1158/1078-0432.CCR-08-0890. [DOI] [PubMed] [Google Scholar]