

Abstract
RNA activation (RNAa) is a gene regulation process in which promoter-targeted short double-stranded RNAs (dsRNAs) or microRNAs (miRs) induce target gene expression at the transcriptional level. Here, we investigate the presence of cryptic promoter transcripts within the VEGF promoter. Single-strand sense and antisense noncoding vascular endothelial growth factor (NcVEGF) promoter transcripts are identified, and their respective expression is studied in cells transfected with a VEGF promoter targeted dsRNA, namely, dsVEGF706, in hypoxic cells and in human malignant lung tissues. Interestingly, in dsVEGF706-transfected, as well as in hypoxic cells, NcVEGF expression levels increase coordinately with coding VEGF expression. Ago2 interaction with both sense and antisense NcVEGFs is increased in hypoxic cells, whereas in dsVEGF706-transfected cells, Ago2 and the antisense strand of the dsRNA interact specifically with the sense NcVEGF transcript. Furthermore, both dsVEGF706 and ectopic NcVEGF transcripts are able to activate the VEGF promoter endogenously present or in a reporter construct. Finally, using small interfering RNA targeting Ago2, we show that RNAa plays a role in the maintenance of increased VEGF and NcVEGF expression after hypoxia. Given the central role of VEGF in major human diseases, including cancer, this novel molecular mechanism is poised to reveal promising possibilities for therapeutic interventions.
INTRODUCTION
Small double-stranded RNAs (dsRNAs) have initially been described as gene silencers through a mechanism known as RNA interference (RNAi). However, in few cases small RNAs can regulate positively cognate sequences (1, 2). Li et al. identified a set of 21-bp dsRNA activating molecules corresponding to the promoter region of three genes, E-cadherin, P21, and VEGF, which are able to activate gene expression at the transcriptional level (3). In contrast to RNAi, the authors proposed the term “RNAa” for dsRNA-induced gene activation or simply for RNA activation. Using several small RNA types, including dsRNA, small hairpin RNA (shRNA) and microRNAs (miRs) others showed that RNAa is a conserved mechanism (4). These distinct molecules are therefore also referred to as small activating RNAs (saRNA). Several other RNAa responsive promoters have been identified, including major regulators of cell cycle and fate, organ development, and cancer (for a review, see references 4, 5, and 6). Given the potential of RNAa as molecular tool, different laboratories have used their ability to induce target gene transcription for studying gene function (7–9). P21, E-cadherin, and VEGF were the first described RNAa-responsive genes, and the applicability of saRNA targeting each of these genes in vivo has received attention (3, 7, 10, 11). Importantly, recent RNAa reports evaluate the promising potential of saRNA for therapeutic intervention (7, 10, 12, 13).
RNAa was originally perceived as an artificial “test-tube” process. However, 2 years before the initial RNAa report (3), Kuwabara et al. already identified an endogenous RNA duplex of ∼20 bp, corresponding to a DNA element commonly found in neuron-specific promoters and which was shown to transcriptionally upregulate a set of neuronal genes (2). Today, mounting evidence indicates that endogenous RNA duplex and miR can also act as saRNAs (14–17). Compared to RNAi, which is induced within hours, exists for 5 to 7 days and disappears subsequently when exogenous small interfering RNA (siRNA) is exhausted (18, 19), RNAa requires generally 2 to 3 days to appear, but it can persist for several weeks (19). Recent observations strongly suggest that modifications of chromatin structure and epigenetic changes, such as histone methylation and deacetylation, could account for the slow kinetics (3, 20, 21). Proteins involved in the RNAi process, such as Dicer, which is required when pre-miRs are used as saRNA (22), Argonaute 2 (Ago2), and GW182, have been shown to be needed also for RNAa (3, 16, 23, 24).
Similar to siRNA, saRNA is submitted to a strand selection in which one strand is discarded and the other strand initiates RNAa via complementary base pairing. In most of the RNAa studies, sense or antisense noncoding sequences are considered target sequences for RNAa (16, 24, 25). The lack of general mechanism to explain the RNAa process results from several shortcomings, including the fact that (i) both strands of RNA duplex are potentially playing the role of guide for RNAa activity, (ii) the exact nature of saRNA molecular targets remains to be clarified, and (iii) a limited number of studies exist (for reviews, see references 5 and 26).
To delve into the RNAa mechanism, we explored here the presence of cryptic transcripts within the targeted promoters initially described by the Dahiya laboratory (3, 22), namely, the E-cadherin, P21, CSDC2, and VEGF promoters, and we studied their regulation in parallel with the expression levels of the corresponding coding genes. We focused on the VEGF promoter because noncoding RNAs within the VEGF promoter and vascular endothelial growth factor (VEGF) mRNA were coordinately regulated in both dsVEGF706-transfected cells and cells subjected to hypoxia. Finally, our results led us to evaluate the role of RNAa in the maintenance of VEGF expression after exposure of cells to hypoxia.
To sum up, our results strengthen the direct implication of noncoding RNAs (NcRNAs) and Ago2 in the mechanism of RNAa for the following reasons. First, cryptic promoter transcripts covering the dsRNA target region were identified in all RNAa-sensitive promoters tested. Second, in the case of VEGF, gene expression and NcRNA are both upregulated during RNAa, but these increases are blunted by siRNAs targeting Ago2. Third, in dsVEGF706-transfected cells, augmented interaction of Ago2 with noncoding VEGF (NcVEGF) promoter transcripts is observed. Finally, a direct interaction between biotinylated dsVEGF706 and sense NcRNA is evidenced during RNAa.
MATERIALS AND METHODS
Cells.
Prostate cancer PC-3 cells were maintained in RPMI medium (Gibco) supplemented with 10% (vol/vol) fetal bovine serum (FBS) and penicillin-streptomycin (100 U/ml). HeLa cells were maintained in Dulbecco modified Eagle medium (DMEM) plus GlutaMAX media (catalog no. 61965-026; Gibco) supplemented with 10% (vol/vol) FBS, 1 mM sodium pyruvate (catalog no. 11360-039; Gibco), and penicillin-streptomycin (100 U/ml). Human primary dermal fibroblast (hDF) lines hDF#1 and hDF#2 were isolated of the foreskin of two donors. hDFs were maintained in DMEM (catalog no. 21969-035; Gibco) supplemented with 10% (vol/vol) FBS. Carcinoma-associated fibroblasts (CAFs) were isolated from tumor biopsy specimens from two patients. CAFs were maintained in DMEM supplemented with 10% (vol/vol) FBS and 1% (vol/vol) insulin-transferrin-selenium solution (catalog no. 41400-045; Gibco). hDFs and CAFs were provided by C. Gaggioli (Nice, France). All cell lines were maintained at 37°C in a humidified atmosphere of 20% O2 and 5% CO2. Cells at high density in a normoxic incubator suffer rapidly from unintentional hypoxia (27). Therefore, it is crucial when studying RNAa of the VEGF promoter to keep cells at low density to maintain normoxic culture conditions after transfection.
Transfection.
The day before transfection, PC3 cells were plated in growth medium without antibiotics at a density of 60 to 70%. Transfections of dsRNA (Table 1), siRNA, and plasmids were carried out using Opti-MEM plus GlutaMAX (catalog no. 51985-26; Gibco) and Lipofectamine 2000 reagent (catalog no. 11668-019; Invitrogen) according to the manufacturer's protocol. About 3 days are required for RNAa to appear. Therefore, except when indicated otherwise, all transfection experiments lasted at least for 72 h. dsRNA at 100 nM was used for RNAa induction, and a 50 nM siRNA mix was used for RNAi.
TABLE 1.
dsRNA used in transfection experiments
| dsRNA | Sequence (5′–3′) |
|---|---|
| dsVEGF-706 S | GCA ACU CCA GUC CCA AAU A [dT][dT] |
| dsVEGF-706 AS | UAU UUG GGA CUG GAG UUG C [dT][dT] |
| dsCon-2 S | ACU ACU GAG UGA CAG UAG A[dT][dT] |
| dsCon-2 AS | UCU ACU GUC ACU CAG UAG U[dT][dT] |
The siRNAs included siCT (On-TargetPlus nontargeting siRNA 1; catalog no. D-001810-01-05; Dharmacon) and siAgo2 (On-TargetPlus human AGO2 (catalog no. 27161) siRNA-SMARTpool (catalog no. L-004639-00-0005; Dharmacon), which corresponds to a mixture of four siRNAs targeting the human Ago2 (EIF2C2) mRNA provided as a single reagent.
Hypoxia.
PC3 cells were exposed to two types of hypoxia. In the first one, cells were cultured in growth medium until 70% confluence and placed for 18 h in a humidified atmosphere of either 20% O2 and 5% CO2 (normoxia) or 1% O2, 5% CO2, 94% N2 (hypoxia) and then immediately used for either RNA or protein extraction. In the second one, PC3 cells were first transfected with a set of either control siRNAs or siRNAs targeting Ago2. The next day, cells were subjected to either normoxia or hypoxia for 24 h. Then, each well was replated using a 1:3 dilution and placed in normoxia. After 72 h in normoxia, cells were used for either RNA or protein extraction. Note that when normalized to 36B4, GAPDH (glyceraldehyde-3-phosphate dehydrogenase) shows a tendency to be increased (1.28-fold; data not shown) after the early hypoxia episode.
For normoxia and hypoxia experiments, when hDFs and CAFs reached confluence, media were replaced with their respective media containing 0.5% (vol/vol) FBS for 3 to 4 additional days at 37°C in a humidified atmosphere of 20% O2 and 5% CO2. The cells were then placed for 24 h in a humidified atmosphere of either 20% O2 and 5% CO2 (normoxia) or 1% O2, 5% CO2, and 94% N2 (hypoxia) and used immediately for RNA extraction.
Nucleic acid extraction.
Total cellular RNA was extracted with TRIzol reagent (catalog no. 15596-026; Invitrogen) as described by the manufacturer. Genomic DNA was isolated with lysis buffer (50 mM KCl, 10 mM Tris [pH 8.3], 25 mM MgCl2, 0.1 mg/ml gelatin, 0.45% [vol/vol] NP-40, 0.45% [vol/vol] Tween 20) supplemented with 30 μg of proteinase K (Merck)/ml. Briefly, the cells were incubated in 100 μl of lysis buffer for 3 h at 55° and then for 5 min at 95°C. Phenol-chloroform and ethanol precipitations were used to purify genomic DNA. After extraction, RNAs and genomic DNAs were quantified using either a Spectro Nano (BMG Labtech) and associated softwares (Spectro Nano and Mars data analysis software) or a UV mc2 spectrophotometer (Safas Monaco) and associated software.
DNase I treatment.
Prior to the reverse transcription (RT), RNAs were subjected to DNase I, RNase-free treatment (catalog no. EN0521; Thermo Scientific) according to the manufacturer's instructions. At the end of the treatment, the DNase was inactivated by exposure to 65°C for 10 min in the presence of 5 mM EDTA.
Semiquantitative and quantitative RT-PCR.
Next, 1 μg of total RNA treated with DNase I was reverse transcribed with SuperScript III reverse transcriptase (catalog no. 18080-044; Invitrogen) and either oligo(dT) plus a random primer (catalog no. 18418-012 and 48190-011; Invitrogen) or a gene-specific primer (as indicated below and in Table 2) as described by the manufacturers. One microliter (40 U) of RiboLock RNase inhibitor (catalog no. E00382; Thermo Scientific) was added to the reaction. The resulting cDNAs in 20 μl (RT) were diluted in water to a final volume of 50 μl. Next, 1-μl portions of cDNA samples with specific primers (Table 2) were used for both semiquantitative and quantitative PCR. In semiquantitative PCR, we used RedTaq ReadyMix (catalog no. R2523; Sigma-Aldrich) as described by the manufacturer, and a T300 Thermocycler (Biometra). For VEGF, NcVEGF, Ago2, 36B4, and GAPDH amplification, the numbers of PCR cycles were 32, 35, 32, 30, and 30, respectively. Amplification of GAPDH or 36B4 served as a loading control in the RNAa or hypoxia experiments, respectively. For quantitative PCR, we used MesaFast qPCR Mastermix Plus for the SYBR assay (RT-SY2X-03+WOUF) in either a 7500 real time PCR system or a 7900HT Fast real-time PCR system (Applied Biosystems) according to the instructions of the manufacturer. Amplification of both GAPDH and 36B4 served as controls for normalization, except for normoxic and hypoxic experiments in which only 36B4 was used for normalization. The results were analyzed on either 7500 software v2.0.6 or an ABI 7900HT SDS2.4 Standalone.
TABLE 2.
PCR primers
| Primer | Sequence (5′–3′) |
|---|---|
| VEGF | |
| VEGF-S | CCCACTGAGGAGTCCAACAT |
| VEGF-AS | AAATGCTTTCTCCGCTCTGA |
| qPCR-Cod-VEGFA-S669 | CTTCTGGGCTGTTCTCGCTTCG |
| qPCR-Cod-VEGFA-R758 | CGGCCCGAGCTAGCACTTCTC |
| VEGFPol2 –S199 | GGTCGAGCTTCCCCTTCA |
| VEGFPol2+R164 | AGGGATAAAACCCGGATCAA |
| VEGFCT+S357 | GAGAGAGACGGGGTCAGAGA |
| VEGFCT+R498 | CTGTCTGTCTGTCCGTCAGC |
| NcVEGF | |
| NCVEGF-S906 | TCTCAGCTCCACAAACTTGGTGC |
| qPCR-VEGF-S788 | CCAGATGAGGGCTCCAGATGGC |
| qPCR-VEGF-R588 | GCACACCCCGGCTCTGGCTAA |
| 5′Race Rev1 VEGF706 | CAGCTACATATTTGGGACTGGAGTTGC |
| 5′Race Rev2 VEGF767 | CCACAGTGTGTCCCTCTGACAATGTG |
| 5′Race Rev3 VEGF787 | AATGTGCCATCTGGAGCCCTCATCTG |
| 3′Race Sens1 VEGF787 | CAGATGAGGGCTCCAGATGGCACATT |
| 3′Race Sens2 VEGF767 | CACATTGTCAGAGGGACACACTGTGG |
| 3′Race Sens3 VEGF706 | GCAACTCCAGTCCCAAATATGTAGCTG |
| NcvegfAS RT full-S990 | CAGACTCCACAGTGCATACG |
| NcvegfAS PCRfull-S976 | CATACGTGGGCTCCAACAG |
| NcvegfAS PCRfull-R146 | GACGCTCAGTGAAGCCTGG |
| NcvegfS RT–R502 | CACACGTCCTCACTCTCGAAG |
| NcvegfS RT–R414 | CTATTGGAATCCTGGAGTGAC |
| NcvegfS RT–R322 | CTGAGAGCCGTTCCCTCTTTG |
| NcvegfS RT–R239 | CCGGAAACTCTGTCCAGAGAC |
| Ago2 | |
| Ago2-S2321 | CAGGCCTTCGCACTATCAC |
| Ago2-R2535 | AGAGGTATGGCTTCCTTCAG |
| GAPDH | |
| GAPDH-S | TCCCATCACCATCTTCCA |
| GAPDH-AS | CATCACGCCACAGTTTCC |
| qPCR-GAPDH-S | TGCACCACCAACTGCTTAGC |
| qPCR-GAPDH-AS | GGCATGGACTGTGGTCATGAG |
| GapdhPol2CHIPS | TACTAGCGGTTTTACGGGCG |
| GapdhPol2CHIPAS | TCGAACAGGAGGAGCAGAGAGCGA |
| 36B4 | |
| qPCR-36B4-S | CAGATTGGCTACCCAACTGTT |
| qPCR-36B4-AS | GGCCAGGACTCGTTTGTACC |
5′ and 3′ RACE.
Rapid amplification of cDNA ends (RACE) experiments were performed with GeneRacer SuperScript TA (catalog no. L1502-01; Invitrogen) as described by the manufacturer. For the 5′ RACE of the sense NcVEGF, we used the qPCR-VEGF-R588 (Table 2) primer for the gene-specific RT and then successively in a PCR and a nested PCR, the primers 5′Race Rev1 VEGF706 (first PCR), 5′Race Rev2 VEGF767 (nested PCR1), and/or 5′Race Rev3 VEGF787 (nested PCR1′ or -2) with the GeneRacer 5′ primers of the kit (Table 2). For the 5′ RACE of the antisense NcVEGF, we used NCVEGF-S906 (Table 2) primer for the gene-specific reverse transcription and then successively in the PCR and the nested PCR, the primers 3′Race Sens1 VEGF787 (first PCR), 3′Race Sens2 VEGF767 (nested PCR1), and/or 3′Race Sens3 VEGF706 (nested PCR1′ or -2). For the 3′ RACE, we used the GeneRacer 3′ primers of the kit for RT, PCR, and nested PCR with the same gene-specific primers as in the 5′ RACE, depending on the NcRNA orientation (as explained above and in Table 2). The advantage of doing these different nested PCRs (1, 1′, and 2) was to directly visualize specific RACE products, which should have molecular masses that are comparable (1 and 1′) or the same (1′ and 2) (data not shown). Specific RACE products were then cloned using TOPO TA FOR SEQ and Top10 One-Shot bacteria in pCR4-TOPO vectors, as described by the manufacturer. The plasmids were sequenced by Millegen (catalog no. 31670; Labege, France).
Primer walking.
To identify the 3′ end of the sense NcVEGF, we used antisense primers for gene-specific RT (Table 2), followed by a control PCR with primers surrounding the dsVEGF706 targeted region (qPCR-VEGF-S788 to qPCR-VEGF-R588).
Plasmids.
VEGF promoter region containing the NcVEGF sequences were amplified using PCR and PC-3 genomic DNA with NcvegfAS RT full-S990 and either NcvegfAS PCRfull-R146 or VegfPol2CHIPAS (Table 2). PCR products were directly cloned in pCRII-TOPO (Invitrogen). Correct sizes of the cloned NcVEGF amplicons were verified with EcoRI enzyme. PCRII-TOPO-NcVEGF vectors were digested with EcoRI, and the purified NcVEGF cDNAs were subcloned into the EcoRI site of the pcDNA3.1 Zeo(+) (catalog no. V860-20; Invitrogen). In these expression vectors, the NcVEGF expression is driven by the cytomegalovirus (CMV) promoter. Sense and antisense orientations of NcVEGF in these vectors were determined with either a ApaI (S990-Pol2CHIPAS) or a SacII/XhoI (S990-R146) digestion profile. pGL3b Luciferase reported vector in which the VEGF promoter from positions −990 to +163 controlled the expression of the luciferase gene was obtained after subcloning the KpnI/XhoI NcVEGF fragment from the “sense” pcDNA3.1 Zeo-Ncvegf (S990-Pol2CHIPAS) vector into the KpnI/XhoI sites of pGL3-Basic vector (catalog no. E1751; Promega). Plasmids were sequenced by Millegen.
RIP assays.
RNA immunoprecipitation (RIP) assays were performed with 10E8 PC3 cells according to the standard ChIP Millipore procedure (chromatin immunoprecipitation [ChIP] assay kit; Doc, certificate of analysis; reference, 17-295), except for the addition of 10 μl of RiboLock RNase inhibitor (Thermo Scientific, E00382)/ml in immunoprecipitation (IP) buffer and washes. After cross-linking RNA to protein with 1% (vol/vol) formaldehyde, the cells were lysed, and nucleic acid (DNA and RNA) was sheared by sonication. RNA-protein complexes were incubated with anti-EIF2C2/AGO2 mouse monoclonal antibody (MBL, catalog no. RN003M), or anti-RNA Pol II antibody (catalog no. sc-899; Sigma). Acetyl-histone H3 rabbit polyclonal antibody (catalog no. 06-599; Upstate) was used to check for nonspecific genomic DNA immunoprecipitation. Normal rabbit serum or nonspecific IgG (catalog no. R9133; Sigma) served as negative controls and dilutions of the “input sample” as positive controls. Instead of the 4 h at 65°C after the immunoprecipitation and washes in ChIP, we used TRIzol to elute the captured RNA from the beads (elution buffer [250 μl plus 250 μl] plus 500 μl of TRIzol). After isopropanol precipitation and a 75% (vol/vol) ethanol wash, the captured RNAs were resuspended in 24 μl of sterile deionized water on ice. Samples were then processed similarly to RNA for DNase I treatment (30 μl final, 3 μl of DNase I) and classical or gene-specific RT-PCR (as indicated in the text and figures).
Biotin pulldown.
For isolation of biotinylated dsRNA/RNA duplex, we adapted the protocol of Ørom et al. (28). Briefly, PC-3 cells were transfected with biotinylated dsRNA (Table 3). After 72 h, the cells were fixed in 1% (vol/vol) formaldehyde for 10 min and then scraped into cold phosphate-buffered saline. After centrifugation, the cells were lysed in ChIP lysis buffer (1% [wt/vol] sodium dodecyl sulfate [SDS], 10 mM EDTA, 50 mM, Tris-HCl [pH 8.1]) and briefly sonicated. Lysates were centrifuged for 10 min at 4°C. The supernatant was diluted in 9 volumes of ChIP dilution buffer (0.01% [wt/vol] SDS, 1.1% [vol/vol] Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl [pH 8.1], 167 mM NaCl) supplemented with 10 μl of RiboLock RNase inhibitor (catalog no. E00382; Thermo Scientific)/ml. Then, 100 μl of the mixture was kept at 4°C as an input sample. Streptavidin-agarose beads (catalog no. N-1000-005; Solulink) were then added to the mixture (20 μl/ml), followed by incubation for 1 h at 4°C on a rotator. Mixtures were centrifuged for 3 min at 3,500 rpm at 4°C to separate the beads. The beads were washed three times for 5 min at 4°C in ChIP lysis buffer-ChIP dilution buffer (1 vol/9 vol) and resuspended in 50 μl of the same wash buffer. Next, 200 μl of TRIzol was used to extract the interacting RNA. After extraction, the RNA was resuspended in water, treated with DNase, and used in a classical RT-PCR.
TABLE 3.
dsRNA used in biotin pulldown experiments
| Biotinylated dSRNA | Sequence (5′–3′) |
|---|---|
| dsCT(S+AS)biot | |
| S | (P)-ACUACUGAGUGACAGUAGA[dT][dT]-biotin |
| AS | (P)-UCUACUGUCACUCAGUAGU[dT][dT]-biotin |
| dsVE(S)biot | |
| S | (P)-GCAACUCCAGUCCCAAAUA[dT][dT]-biotin |
| AS | (P)-UAUUUGGGACUGGAGUUGC[dT][dT] |
| dsVE(AS)biot | |
| S | (P)-GCAACUCCAGUCCCAAAUA[dT][dT] |
| AS | (P)-UAUUUGGGACUGGAGUUGC[dT][dT]-biotin |
SDS-PAGE and Western blots.
Total cell lysates were prepared, electrophoresed, and blotted as described previously (29). We used either a 8.5 or 15% polyacrylamide gel for Ago2 or VEGF Western blotting, respectively. The following antibodies were used: anti-EIF2C2/AGO2 (catalog no. RN003M; MBL) and anti-α-tubulin (catalog no. T6199; Sigma) mouse monoclonal antibodies and anti-VEGF (catalog no. AB1442; Chemicon International) rabbit polyclonal antibody. Peroxidase-coupled anti-rabbit (catalog no. PI-1000; Vector Laboratories) and anti-mouse (catalog no. PI-2000; Vector Laboratories) secondary antibodies were used at a 1:10,000 dilution. Images were captured with Fusion Solo.4.WL (Vilber Lourmat, France) and quantified with the associated Fusion-Capt advance software (Vilber Lourmat).
Patients and tissue samples.
Totals of 20 μg of RNA of either healthy or malignant lung tissues from six patients who underwent surgery for their tumor at the Pasteur Hospital (Department of Thoracic Surgery, University Hospital, Nice, France) was obtained from the Biobank tissue unit of the Pasteur Hospital. The patients were informed, and their consent was obtained. The study was approved by the hospital's ethics committee. The main clinical data are summarized in Table 4.
TABLE 4.
RNAs isolated from healthy and malignant lung tissues of patients
| Patient biobank no. | Sex | Age (yr) | Smoker |
|---|---|---|---|
| LB13-072 | M | 73 | Yes |
| LB13-642 | M | 70 | Yes |
| LB14-322 | M | 83 | Yes |
| LB14-407 | M | 53 | Yes |
| LB11-152 | F | 86 | No |
| LB13-452 | F | 71 | Yes |
Statistical analysis.
Data are expressed as means ± the standard deviations (SD). Statistical analyses were performed by using Student t tests. A P value of <0.05 was considered significant. Note that some experiments were not included in the analysis because of confounding artifacts generated during the experimental procedure. Indeed, exaggerated confluence generating hypoxia and medium acidification at the 72-h time point after transfection led to confounding effects rendering observation of RNAa impossible, since in this case VEGF (and NcVEGF) expression would also be upregulated in control cells and would make the detection of RNAa activation impossible.
RESULTS
NcRNAs are transcribed within target promoters.
Since the VEGF promoter is responsive to RNAa, we hypothesized the presence of noncoding transcripts, which run through the promoter. To demonstrate the presence of such cryptic transcripts, primers within 1 kb of proximal VEGF promoter and surrounding the target sequence of the dsRNA (dsVEGF706) were designed (Fig. 1A). These primers were named sense or antisense according to the orientation of the gene. After a standard RT [oligo(dT) plus random primers], these sense and antisense primers were used to amplify both sense and antisense transcripts (Fig. 1A). Using this strategy, we were able to detect, at the expected size and only after standard RT, cryptic transcripts within the VEGF promoter in various human cell lines (Fig. 1B). To facilitate the reading, cryptic transcripts were designated after the name of the gene, namely, NcVEGF. NcRNAs were also detected in other RNAa sensitive promoters such as E-Cadherin, CSDC2, and P21 (data not shown).
FIG 1.
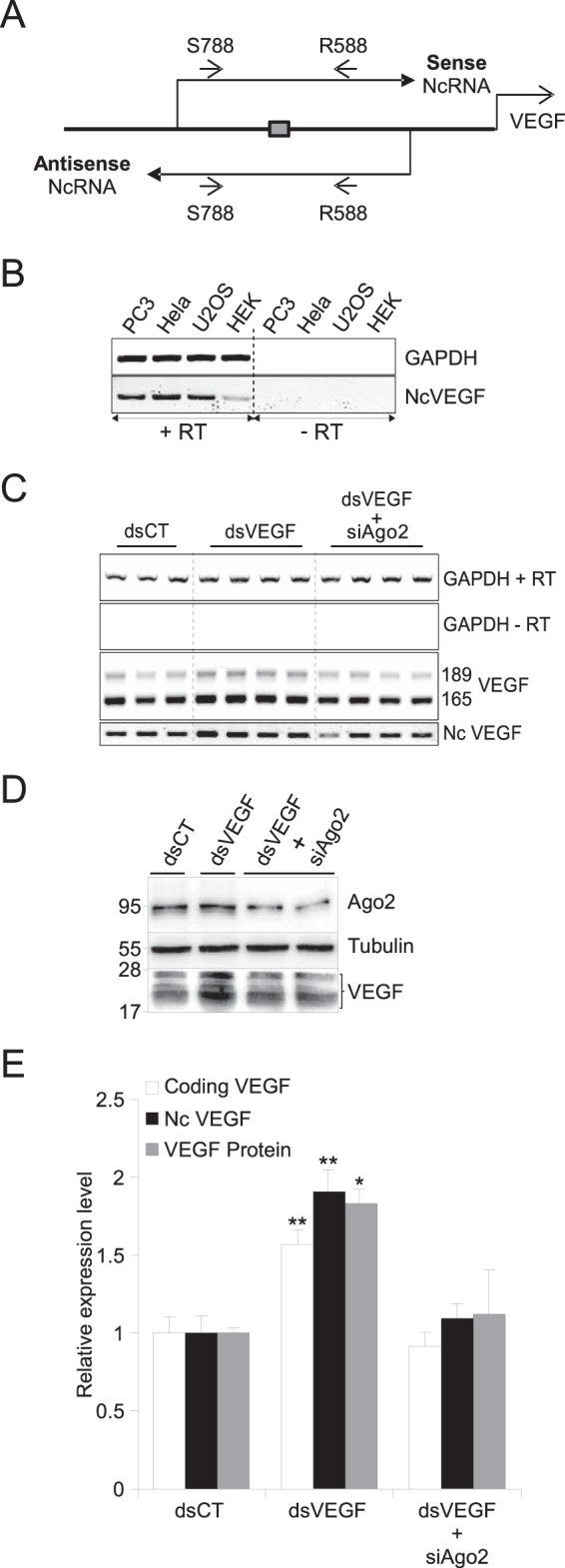
dsVEGF706 induces both VEGF and NcVEGF expression in an Ago2-dependent manner. (A) Schema of the strategy to identify sense and antisense NcVEGF RNA covering the dsVEGF706 target region (box). Small arrows represent sense qPCR-VEGF-S788 (S788) and antisense qPCR-VEGF-R588 (R588) primers listed in Table 2. (B) RNAs from PC-3, HeLa, U2OS, and HEK-293 cells were used in standard RT-PCR experiments with (+RT) or without (−RT) reverse transcriptase, followed by a PCR with the S788 and R588 primers. GAPDH amplification was used as a positive control. (C) Representative RT-PCR with RNA from PC-3 cells transfected with either control dsRNA (dsCT) or dsVEGF706 (dsVEGF) for 72 h without or with a set of siRNA targeting Ago2 (siAgo2). Transfections were processed in triplicate and quadruplicate. Both VEGF and NcVEGF expression levels were assessed by standard RT-PCR with (+RT) or without (−RT) reverse transcriptase. GAPDH served as a loading control. (D) Representative Western blot of PC-3 transfection experiments as described in panel C with anti-Ago2 and anti-VEGF antibodies. Tubulin was used as a loading control. (E) Histogram representing both qPCR and Western blot analyses of the transfection experiments described for panels C and D, respectively. The qPCR VEGF primers were designed to amplify only one fragment from either VEGF189 and VEGF165 cDNA and are not the same as the primers used in panel C (Table 2). 36B4 and GAPDH were used for normalization in qPCR experiments. Values are means ± the SD of three independent experiments, with each sample run in triplicate or quadruplicate. *, P ≤ 0.05; **, P ≤ 0.005.
dsVEGF706 induces both VEGF and NcVEGF expression in an Ago2-dependent manner.
RNAa has been reported to be an Argonaute 2 (Ago2)-dependent process. However, to the best of our knowledge, the effect of Ago2 depletion on VEGF promoter activation by dsRNA has not been reported. Therefore, to monitor the expression of both coding and noncoding VEGF RNA during the RNAa process, PC-3 cells were transfected either with control dsRNAs (dsCT), which is not complementary to the VEGF promoter, or with dsVEGF706 (Table 1), which is complementary to the VEGF promoter from positions −706 to −687 relative to the +1 position of transcription as previously described by Li et al. in the first RNAa study (3). As expected, compared to cells transfected with dsCT, both VEGF189 and VEGF165 were increased in dsVEGF706-transfected cells (Fig. 1C), and qPCR analyses showed a 1.57-fold induction (P = 0.007) (Fig. 1E). Interestingly, with qPCR we found that NcVEGF expression is induced 1.91-fold (P = 0.0025) in dsVEGF706-transfected cells (Fig. 1E). Next, with a set of RNAi targeting Ago2 mRNA, we reduced the expression of the Ago2 protein (Fig. 1D and E). Our results confirm the requirement of Ago2 during RNAa of the VEGF promoter since the depletion of Ago2 abolished the increase in VEGF mRNAs (0.92-fold induction compared to dsCT-transfected cells) (Fig. 1C and E). Induction of NcVEGF is also blunted in the presence of Ago2 siRNAa (1.09-fold induction compared to dsCT-transfected cells). Further, transfection with dsVEGF706 leads to a 1.83-fold increase in VEGF A isoforms (detected at 17 to 25 kDa) expression. However, VEGF induction was blunted when siRNA targeting Ago2 was added (Fig. 1D and E). In summary, our results indicate that small dsRNA targeting the VEGF promoter induced in a coordinated manner and at comparable levels both NcVEGF and VEGF expression. The generation of both coding and NcVEGF is dependent on Ago2. However, a joint increase in cryptic transcripts covering targeted regions does not seem to be a general feature of RNAa since we failed to observe it with other RNAa sensitive promoters, including CSDC2 and E-Cadherin (data not shown).
Hypoxia induces both VEGF and NcVEGF expression.
Next we sought to determine whether cryptic transcriptional activity inside the VEGF promoter was modified in hypoxia, which is known to induce VEGF expression. qPCR analyses showed that VEGF and NcVEGF expression were increased 5.47-fold (P = 7.0 × 10−7) and 5.01-fold (P = 6.0 × 10−9), respectively, in hypoxic PC-3 cells (Fig. 2A). Thus, similarly to dsVEGF706, hypoxia induces both NcVEGF and VEGF expression in a coordinated manner and at comparable levels.
FIG 2.

The VEGF promoter region encodes both sense and antisense NcVEGF transcripts. PC-3 cells were exposed to either normoxia or hypoxia for 18 h. (A) cDNAs were used to perform qPCR, and the results are shown as means ± the SD of three independent experiments, with each sample run in quadruplicate. 36B4 was used to normalize qPCR experiments. **, P ≤ 0.005. (B) Schema of the experimental design and results of 3′ and 5′ RACE PCRs. Briefly, a set of sense primers (S1, S2, and S3) as close as possible to the dsVEGF706 target region was used to identify either the 5′ end of antisense NcVEGF in a 5′ RACE reaction or the 3′ end of sense NcVEGF in a 3′ RACE reaction (Table 2). In parallel, a set of antisense primers (R1, R2, and R3) was used to identify either the 3′ end of antisense NcVEGF or the 5′ end of sense NcVEGF (Table 2). Both sense and antisense NcVEGFs and their positions relative to the VEGF transcription start site obtained after 3′ and 5′ RACE PCR are indicated. Dark and light boxes show the positions of the dsVEGF706 target site and HIF-responsive element (HRE), respectively. (C) Representative gene-specific (R588 or S906) RT-PCR using RNAs from PC-3 cells exposed to either normoxia or hypoxia for 18 h. The respective expressions of the sense (R588) or antisense (S906) NcVEGF transcripts were measured with NcVEGF primers (S788; R588 or S788; R706 [not shown]) (Table 2). PCR performed without cDNA (C−) or with genomic DNA from PC-3 cells (PC3) were used as a negative and positive controls, respectively. (D) Strand-specific cDNAs from three independent experiments with each sample in quadruplicate were also used in a qPCR. The results are presented as a histogram depicting the relative abundance of the antisense (S906) and sense (R588) NcVEGF in normoxia or hypoxia quantified by qPCR. ** indicates P ≤ 0.005 for sense versus antisense NcVEGF in either normoxia or hypoxia. a, b, and c all correspond to P ≤ 0.005 for sense (a), antisense (b), and sense plus antisense (c) NcVEGFs in normoxia versus hypoxia. (E) PC-3 cells were exposed to either dsCT or dsVEGF706 for 72 h. A histogram depicts the relative abundance, in both dsCT- and dsVEGF706-transfected cells, of the antisense (S906) and sense (R588) NcVEGF quantified using qPCR. ** indicates P ≤ 0.005 for sense versus antisense NcVEGF in either dsCT- or dsVEGF706-transfected cells. a corresponds to P ≤ 0.05 and b and c both indicate P ≤ 0.005 for sense (a), for antisense (b), and for sense plus antisense (c) NcVEGF in dsCT- versus dsVEGF706-transfected cells. The results are means ± the SD of three independent experiments with each sample run in quadruplicate.
The VEGF promoter region encodes both sense and antisense NcVEGF transcripts.
To address whether NcRNAs, encoded within the VEGF promoter, reflect expression of a sense and/or an antisense transcript compared to the strand coding for VEGF, we took advantage of the higher expression of NcVEGF in hypoxic cells to perform 5′ and 3′ RACE experiments. Compared to a +1 set at the transcription start position of coding VEGF, we found three 5′-capped antisense transcripts which start at positions −145, −222, and −419, respectively (Fig. 2B) and one 5′ uncapped (either a genuine uncapped NcVEGF or a specific cap-degraded NcVEGF) antisense transcript starting at position −339 (Fig. 2B; also see Materials and Methods). The 3′ ends of these antisense transcripts were located at position −877 or −1013. Interestingly, we found only one 5′-capped sense NcVEGF transcript, which starts only 61 bp downstream of the HIF responsive element [HRE(−975-969)] at position −907 and spans to at least position −588 (Fig. 2B). The 3′ RACE reaction of the sense transcript did not allow us to determine the precise position of the 3′ termini, possibly due to the lack of polyadenylation. However, to identify the 3′ end of the sense NcVEGF, we performed primer walking on the VEGF promoter (see Materials and Methods). With this approach, the sense transcript extended to at least position −146 (Fig. 2B).
Hypoxia induces both sense and antisense NcVEGF expression.
Since the expression of NcVEGF is coordinately increased with VEGF during hypoxia (Fig. 2A), we determined whether it was due to an increase in the sense, the antisense, or both transcripts. According to RACE PCRs, the qPCR-VEGF-S906 (S906) and qPCR-VEGF-R588 (R588) primers were both chosen to perform further gene-specific RT. The S906 or R588 primers specifically reverse the antisense or the sense NcVEGF transcripts, respectively. Contrary to classical reverse transcription [oligo(dT) plus random primers] in which both sense and antisense NcVEGFs were reverse transcribed, the S906 or R588 primers allow strand-specific RT of the antisense or the sense NcVEGF transcripts, respectively. Furthermore, strand specificity is maintained in semiquantitative and quantitative qPCR. With these primers, we found that both sense and antisense transcripts are detected at the expected sizes compared to genomic DNA (Fig. 2C). We also observed that sense and antisense NcRNA expression were increased by hypoxia (Fig. 2C and D). Expression of the antisense NcVEGF was induced to a lower extent (5.27-fold; P = 6.12 × 10−7) than the expression of the sense transcript (11.87-fold; P = 8.77 × 10−8) (Fig. 2D). However, the total NcVEGF (S906+R588) induction, measured by qPCR, is 5.88-fold (P = 2.4 × 10−7) in hypoxic compared to normoxic cells (Fig. 2D). Thus, the level of NcVEGF induction using specific reverse transcription (Fig. 2D) is in agreement with our findings with standard reverse transcription [oligo(dT) plus random primers; Fig. 2A]. The reason behind the relatively weak increment produced by the sense NcVEGF to the total increase in NcVEGF is that it represents 10% of the total NcVEGF in normoxic cells, and it reaches only 20% of the total NcVEGF after hypoxia (Fig. 2D). Nonetheless, the level of the sense NcVEGF after hypoxia is not meaningful since it corresponds to 110% of the total NcVEGF in normoxia (Fig. 2D).
dsVEGF706 induces both sense and antisense NcVEGF expression.
We next studied the expression of the sense and antisense NcVEGFs in dsCT versus dsVEGF706-transfected cells using the same approach as for hypoxic cells. As in hypoxic cells, in dsVEGF706-transfected cells both sense and antisense NcVEGFs were increased. However, in contrast to hypoxic cells, antisense NcVEGF augmented more (1.84-fold, P = 3.23 × 10−5) compared to sense NcVEGF, which is only increased by 1.35-fold (P = 0.036) (Fig. 2E). Thus, the total NcVEGF (sense + antisense) increased 1.78-fold (P = 2.6 × 10−5) in dsVEGF706-transfected cells, which is in agreement with our previous findings (Fig. 1E).
Ago2 is recruited on the NcVEGF RNA in hypoxic cells.
To reveal the binding of Ago2 to NcVEGF RNA, we used normoxic and hypoxic PC-3 cells in RIP experiments (see Materials and Methods). After RNA extraction, gene-specific reverse transcription was done using either S906 or R588 oligonucleotides followed by qPCR analysis (Fig. 3A). After histone H3 IP, qPCR amplification was low and almost at the level observed with negative controls (Non Spec), indicating that, as expected, the acetyl-histone H3 antibody used for negative controls in RIP experiments did not specifically immunoprecipitate NcVEGF RNAs and also that genomic DNA contamination was absent (Fig. 3A). Compared to normoxic cells, we observed after Pol2 IP a significantly increased amplification of the sense NcVEGF transcript in hypoxic cells (Fig. 3A) (42 to 68%). This could reflect the robust increase in expression of the sense NcVEGF (RT R588, black bars) in hypoxic cells. Finally, in the latter cells, increased NcVEGF amplification is found after Ago2 IP, indicating that there is augmented Ago2 binding to both sense (5.1 to 29.4%) and antisense (9.45 to 70.75%) NcVEGF transcripts (Fig. 3A). This 6- to 7-fold increase in NcVEGF amplification after Ago2 IP may reflect a specific phenomenon involving both Ago2 and NcVEGF transcripts during hypoxia.
FIG 3.

Ago2 is recruited on the NcVEGF RNA in both hypoxic and dsVEGF706 transfected cells. PC-3 cells exposed either to normoxia/hypoxia (A) or dsCT/dsVEGF706 transfections (B) were used for RIP experiments. (A) RIP assays were performed on PC3 cells exposed to either hypoxia or normoxia using antibodies to Argonaute 2 (Ago2), RNA polymerase 2 (Pol2), acetyl-histone H3 (H3), or rabbit nonspecific serum (Non Spec) to pulldown-associated RNAs from PC3 cells in hypoxia or normoxia. The precipitated RNAs, as well as undiluted RNA input (10% RNA isolated from the supernatant of rabbit nonspecific serum IP), were used in gene-specific RT with either qPCR-VEGF-S906 or qPCR-VEGF-R588 primers, followed by a qPCR with specific primers surrounding the dsVEGF706 target region (S788-R706 [Table 2]). The results are reported as a histogram depicting the percent relative abundance of either the antisense (RT S906) or the sense (RT R588) NcVEGF compared to the total RNA input. (B) RIP assays were performed with the same antibodies, methods, and primers as in panel A, with pulldown-associated RNAs in PC3 cells transfected using either dsCT or dsVEGF706. The results are reported as a histogram depicting the percent relative abundance of either the antisense (RT S906) or sense (RT R588) NcVEGF compared to the total RNA input. Values are means ± the SD of three independent experiments with each sample run in triplicate. *, P ≤ 0.05.
Ago2 is recruited on the sense NcVEGF RNA in dsVEGF706-transfected cells.
Since Ago2 is required for the RNAa phenomenon, we performed the same RIP experiments with this time PC-3 cells transfected either with dsCT or dsVEGF706 (Fig. 3B). Our RIP results were slightly different compared to those obtained from normoxic and hypoxic cells (compare Fig. 3A and B). In brief, we found that (i) after histone H3 IP, only residual amplification comparable to the level observed in negative controls (Non Spec) was evident (Fig. 3B); (ii) after Pol2 IP and RT-PCRs, there was more NcVEGF amplification in cells transfected with the dsVEGF706 than in control cells (Fig. 3B). Indeed, amplification of the antisense NcVEGF was increased 1.5-fold (16.1 to 25.1%), but that of the sense NcVEGF transcript increased 4-fold (from 2.65 to 10.75%), illustrating more binding of Pol2 RNA polymerase to the NcVEGF transcripts. (iii) Finally, compared to control cells (dsCT), a 10-fold-increased amplification of the sense NcVEGF (from 2.1% to 22%) was observed after Ago2 IP (Fig. 3B). The augmented binding of Ago2 specifically to the sense NcVEGF transcript in dsVEGF706-transfected cells represents the largest difference compared to what happens in hypoxic cells in which Ago2 binding is increased, but it is similar for both sense and antisense NcRNAs.
In dsVEGF-transfected cells, dsVEGF706 is preferentially bound to the sense NcVEGF RNA.
To further characterize the binding of the dsVEGF706, we adapted the protocol of Ørom et al. (28). Briefly, PC-3 cells were transfected either with control dsRNA biotinylated on both sense and antisense 3′ ends [dsCT(S+AS)biot] or with a dsVEGF706 biotinylated on the 3′ end of either a sense strand [dsVE(S)biot] or an antisense strand [dsVE(AS)biot] (Table 3). After 72 h, streptavidin-agarose beads were used to pull down NcVEGF or GAPDH transcripts. As in the Ørom report, we chose to normalize to GAPDH (28). Our reasoning is that GAPDH expression is at least 30 times higher than NcVEGF expression and that a nonspecific pulldown would contain as much NcVEGF/GAPDH as the input, and thus no enrichment would be detected. As shown in Fig. 4, GAPDH amplification is almost similar between pulldown (Fig. 4, left panel) and input (Fig. 4, right panel). Concerning NcVEGF, we observed augmented amplification in the input lane, after transfection of dsVE(AS)biot (4.1-fold) compared to dsCT-transfected cells (Fig. 4). These results are in agreement with the increased NcVEGF expression in dsVEGF706-transfected cells (Fig. 1C and E), but this was not seen when using dsVE(S)biot. In any case, we observed, after the streptavidin/biotin pulldown, amplification of NcVEGF from the captured RNA and normalization to GAPDH, a (10.39 ± 3.82)-fold enrichment of NcVEGF in dsVE(S)biot-transfected cells (Fig. 5, pulldown lanes), and a (39.9 ± 4.48)-fold enrichment in dsVE(AS)biot-transfected cells compared to dsCT(S+AS) biot-transfected cells (set to 1 as nonspecific binding control, Fig. 4). We can presume that dsVE(S)biot and dsVE(AS)biot should pull down specifically the antisense NcVEGF RNA or the sense NcVEGF transcripts, respectively. Thus, despite the use of a standard RT-PCR, only antisense or sense NcRNA should be amplified after biotin pulldown. On the contrary, both antisense and sense NcVEGF transcripts are amplified from the input samples. Since the antisense NcVEGF RNA represents about 90% of the total NcVEGF and the sense only represents ca. 10%, the 40-fold enrichment obtained from the antisense biotin-labeled NcVEGF-transfected cells is even more significant.
FIG 4.

dsVEGF706 is preferentially bound to the sense NcVEGF RNA. PC-3 cells were transfected either with control dsRNA biotinylated on the 3′ ends of both sense and antisense dsCT(S+AS)biot or a dsVEGF706 biotinylated on the 3′ ends of either the sense strand [dsVE(S)biot] or the antisense strand [dsVE(AS)biot]. After 72 h, the cells were fixed and treated as described in RIP experiments using streptavidin-agarose beads to pull down NcVEGF or GAPDH transcripts. Both total RNAs (inputs) and streptavidin pulled-down RNAs were used in standard reverse transcription reactions, followed by semiquantitative (lower panel) and quantitative (top panel) PCRs to amplify either NcVEGF (S788/R588 or S788/R706) or GAPDH. Also, 10% of the cell lysates was kept to obtain the RNA used to generate the input lanes, and nonspecific pulldown of GAPDH mRNA was used to normalize these assays. The histogram shows the NcVEGF/GAPDH qPCR ratio after biotin pulldown (left side) and in the input lanes (right side). To quantify the specific pulldown of NcVEGF using biotin-labeled dsVEGF706, the ratio obtained from dsCT(S+AS)biot-transfected cells was arbitrarily set to 1. The values are means ± the SD of three independent experiments with each sample run in duplicate. *, P ≤ 0.05; **, P ≤ 0.005. The lower panel shows the results from a representative semiquantitative PCR after biotin pulldown with either qPCR-VEGF (R588/S788) or GAPDH primers.
FIG 5.
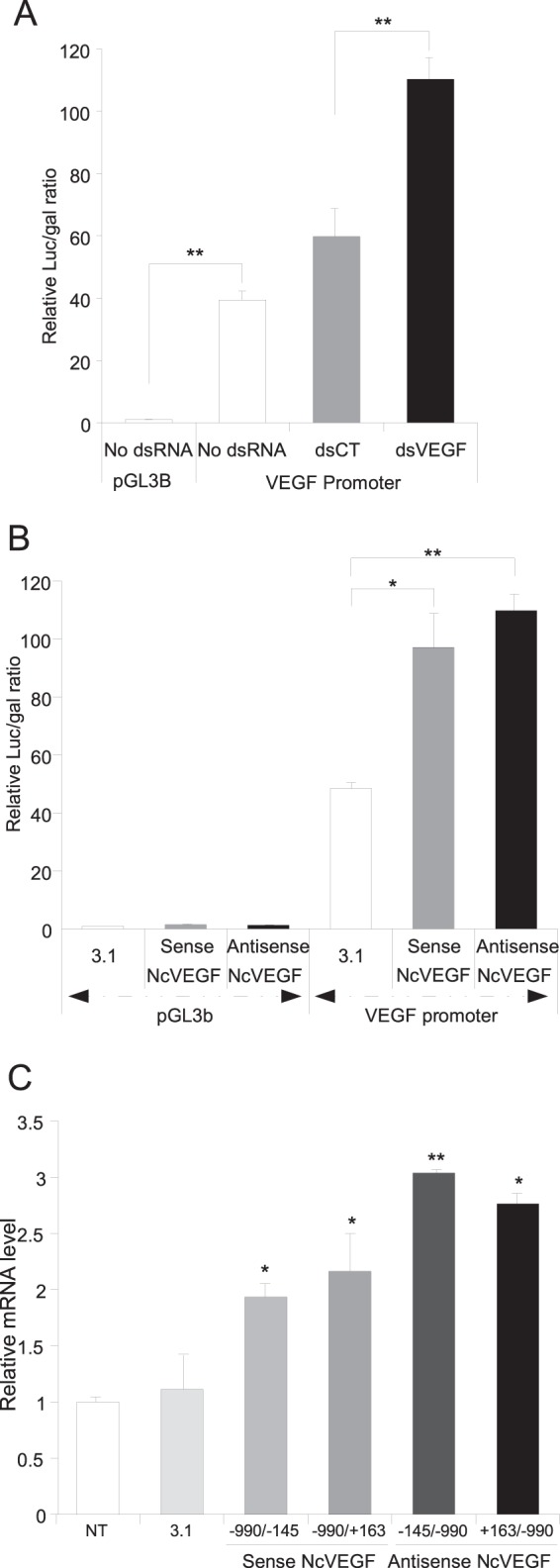
Both dsVEGF706 and NcVEGF positively modify VEGF promoter activity and VEGF expression. (A) PC3 cells were cotransfected, in the absence or presence of dsRNA (control [dsCT] or dsVEGF706 [dsVEGF]), with a β-galactosidase-expressing vector (CMV-βgal) and with either empty (pGL3B) or VEGF promoter (from position −990 to position +145) containing luciferase vector (VEGF promoter). After 72 h, the cells were lysed, and both the luciferase and the β-galactosidase activities were measured. The relative luciferase and β-galactosidase activities are reported on a histogram. Values are means ± the SD of three independent experiments, with each sample run in triplicate. (B) PC3 cells were cotransfected with a β-galactosidase-expressing vector (CMV-βgal), with either empty (pGL3B) or VEGF promoter containing (VEGF promoter) the luciferase vector and with either the expression vector empty (3.1) or encoding either sense (sense NcVEGF) or antisense (antisense NcVEGF) NcVEGF transcripts. After 72 h, the cells were lysed, and both the luciferase and the β-galactosidase activities were measured. The relative luciferase and β-galactosidase activities are reported on a histogram. Values are means ± the SD of three independent experiments, with each sample run in triplicate. (C) PC3 cells transfected for 72 h with an expression vector either empty (3.1) or in which the CMV promoter drives the expression of either sense (sense NcVEGF; −990/−145 or −990/+163) or antisense (antisense NcVEGF; −145/−990 or +168/−990) NcVEGF transcripts. cDNAs were used to analyze VEGF expression with qPCR, and results are reported as the means ± the SD of two independent experiments, with each sample run in triplicate. 36B4 and GAPDH were used for normalization in qPCR experiments. *, P ≤ 0.05; **, P ≤ 0.005.
Both dsVEGF706 and NcVEGF positively modify VEGF promoter activity and VEGF expression.
To delve further into the role of dsVEGF706 and NcVEGF, we constructed a reporter plasmid in which ∼1 kb of the VEGF promoter and a short part of VEGF 5′-noncoding region (from positions −990 to +145) drive the expression of the luciferase gene. The relative luciferase/β-galactosidase ratio of the VEGF promoter construction is ∼40 higher (P = 1.14 × 10−7) than the empty vector (set to 1) (Fig. 5A). The same cotransfections were also realized in the presence of dsRNA control (dsCT) or dsVEGF706 (dsVEGF). The relative activity is increased 1.84-fold in the presence of dsVEGF706 (P = 1.26 × 10−5), indicating that dsVEGF706 is able to positively regulate the activity of an exogenous VEGF promoter.
To evaluate the action of the sense and antisense NcVEGFs, vectors in which the expression of either sense or antisense NcVEGF transcripts (from positions −990 to +145 or positions +145 to −990) are under the control of the CMV promoter (pcDNA3.1 plasmids) were constructed. Compared to cells transfected with empty vector (pcDNA3.1), we found the relative VEGF promoter activity to be increased in the presence of both sense (2.00 ± 0.24-fold; P = 0.011) and antisense (2.26 ± 0.06-fold; P = 0.0008) NcVEGFs, indicating that the NcVEGF transcripts positively regulate the activity of the exogenous VEGF promoter (Fig. 5B).
We subsequently sought to determine whether the NcVEGF transcripts also positively regulate the activity of the endogenous VEGF promoter. The expression of NcVEGF is, as expected, robustly increased in these transfected cells (data not shown). Further, with both semiquantitative (data not shown) and quantitative PCR (Fig. 5C), we found that VEGF expression is augmented ∼1.57-fold in the presence of the sense NcVEGF and ∼2.03-fold in the presence of antisense NcVEGF. To sum up, NcVEGF transcripts are able to positively affect endogenous VEGF promoter activity.
Ago2 activity is required for the maintenance of VEGF expression after hypoxia.
We showed that hypoxia positively regulates NcVEGF (Fig. 2) and that Ago2 binding to NcVEGF transcripts increases during hypoxia (Fig. 3A). We also found that NcVEGF transcripts positively modulate the activity of the VEGF promoter (Fig. 5B and C). Considering these three findings, we explored whether RNAa is involved in the persistence of VEGF expression long after exposure to hypoxia. To this end, PC3 cells were transfected with a set of either control siRNAs (siCT) or siRNAs targeting Ago2 (siAgo2), subjected to either normoxia or hypoxia for 24 h, and then replated and placed in normoxia for 72 h (Fig. 6A; see also Materials and Methods). In siCT-transfected cells, Ago2 (1.31-fold; P = 0.047), VEGF (2.82-fold; P = 7.35 × 10−5), and NcVEGF (10.62-fold; P = 3.09 × 10−5) mRNAs were all significantly increased after this 24-h early hypoxic exposition at the mRNA level (Fig. 6B) compared to the siCT condition in normoxia. This experiment shows also that by 5 days posttransfection, the set of siAgo2 still efficiently reduced the accumulation of both Ago2 RNA and protein by ∼2-fold and in cells maintained either in normoxia or exposed for 24 h to hypoxia (Fig. 6C and D and data not shown). Concerning VEGF, siAgo2 was not able to reduce VEGF mRNA in cells kept in normoxia during the whole experiment (Fig. 6B). In contrast, in cells exposed to hypoxia, siAgo2 transfection blunted the increase in VEGF mRNA from 2.82-fold to 1.73-fold (P = 3.65 × 10−3), and VEGF protein accumulation was accordingly reduced by 1.43-fold (P = 0.016) (Fig. 6B to D). Finally, after 24 h exposure to hypoxia, a major increase in NcVEGF (10.62-fold; P = 3.09 × 10−5) was seen, which was also blunted to 3.34-fold (P = 1.75 × 10−4) in the presence of siAgo2 (Fig. 6B). Together, these results indicate that Ago2 and NcVEGF may through RNAa play a role in the maintenance of VEGF expression long after exposure to hypoxia. In favor of this hypothesis, note that reduced Ago2 RNA in siAgo2-transfected cells can already be seen at the end of 24 h of hypoxia, but it is not yet associated with a reduction in VEGF mRNA (data not shown).
FIG 6.
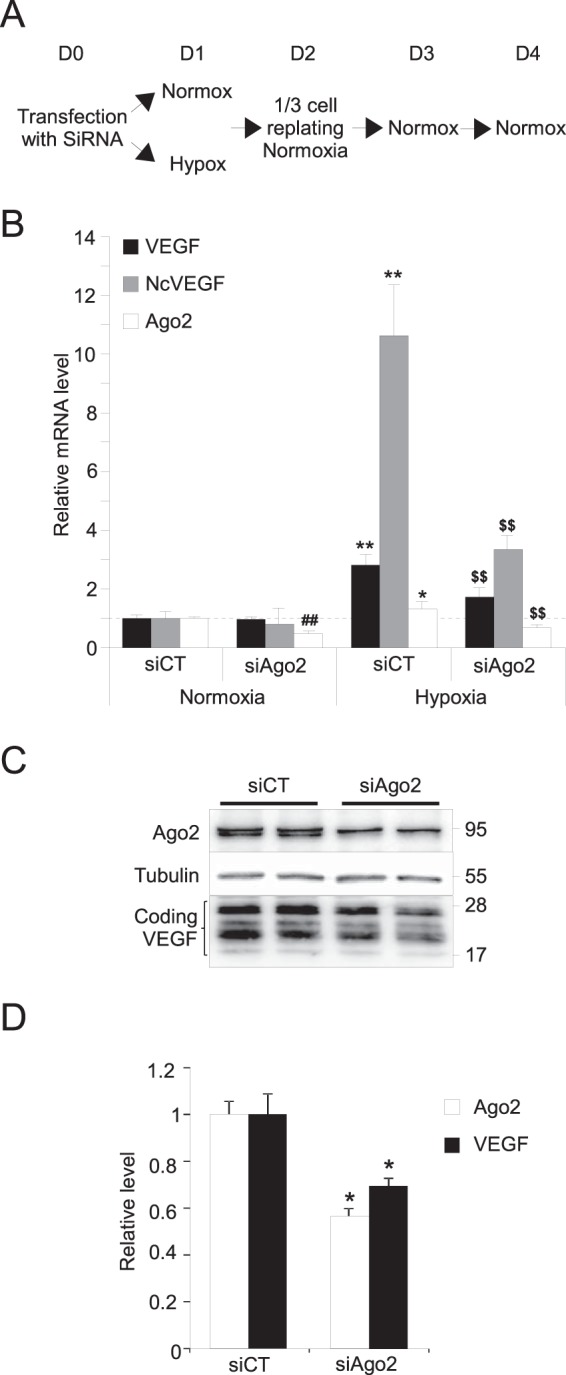
Ago2 activity is required for the maintenance of VEGF expression after hypoxia. (A) Schema of the experimental design to generate the cDNAs and the proteins analyzed in quantitative PCR (B) and Western blotting (C and D). Briefly, PC3 cells transfected at D0 with either control siRNAs (siCT) or siRNAs targeting Ago2 (siAgo2) were subjected to either normoxia or hypoxia for 24 h (D1) and then replated and placed in normoxia during 72 h (D2 to D4) (see Materials and Methods). (B) cDNA were used to perform qPCR. The results are reported on a histogram as the means ± the SD of two independent experiments, with each sample run in quadruplicate. 36B4 was used to normalize qPCR experiments. *, P ≤ 0.05; **, ##, and $$ all indicate P ≤ 0.005. * and ** were used for siCT in normoxia versus hypoxia. For siCT versus siAgo2, we used ## in normoxia and $$ in hypoxia. (C) Representative Western blot of PC3 cell lysates exposed to hypoxia for 24 h and Ago2, VEGF A antibodies. Tubulin was used as a loading control. (D) After normalization and quantification of Western blots, results are reported on a histogram as the means ± the SD of two independent experiments, with each sample run in duplicate. *, P ≤ 0.05.
Sense NcVEGF expression is increased in cancer-associated fibroblasts and in tumors.
A common feature of cancerous tissue is the occurrence of hypoxia associated with increased VEGF expression in both cancer and stromal cells. Therefore, in addition to the expression of VEGF and NcVEGF in tumorigenic PC3 cells, we analyzed their expression in primary cultures of hDFs (normal infant foreskin fibroblasts) and of CAFs (carcinoma-associated fibroblasts) exposed to either normoxia or hypoxia (see Materials and Methods and Fig. 7A and B). VEGF expression was modestly induced by hypoxia in both hDF and CAF cells, although the increase does not reach statistical significance. Compared to hDFs, VEGF expression was slightly higher in CAFs in both normoxia and hypoxia. Of note, the expression of GAPDH was virtually the same under all conditions. Sense and antisense NcVEGFs were detected at the expected sizes and showed a fold expression comparable to that for PC3 cells. In normoxia, the sense NcVEGF was weakly expressed compared to the antisense transcript. After hypoxia, similarly to PC3 cells, a coordinated increase in VEGF and NcVEGF (S+AS) was observed (Fig. 7B). In hypoxia, the antisense expression was moderately decreased, while that of the sense NcVEGF was robustly augmented. Moreover, for CAF cells, a higher level of the sense transcript was seen in both normoxia and hypoxia. Therefore, the hypoxia-related increase in total NcVEGF was more pronounced than in hDF cells (Fig. 7B).
FIG 7.

Sense NcVEGF expression is increased in carcinoma-associated fibroblasts and in tumors. (A) Representative standard and gene-specific (R588 or S906) RT-PCR using RNAs from human primary dermal fibroblasts, hDF 1 and hDF 2, and carcinoma-associated fibroblasts, CAF 1 and CAF 2, exposed for 24 h to either normoxia (N) or hypoxia (H) as described in Materials and Methods. (B) cDNAs from these hDF and CAF fibroblasts were used in qPCR. The results are presented as a histogram depicting VEGF and the relative abundances of both antisense (S906) and sense (R588) NcVEGF in normoxia or hypoxia. The results are reported as the means ± the SD, with each sample run in triplicate. 36B4 was used to normalize qPCR experiments. *, P ≤ 0.05. (C) Representative standard and gene-specific (R588 or S906) RT-PCR using RNAs of either healthy (H) or tumor (T) lung tissues from six patients (P1 to P6 [Table 4]). The respective expressions of the sense (R588), antisense (S906), or both (standard RT) NcVEGF transcripts were revealed with NcVEGF primers (S788; R588 or S788; R706 [data not shown]). Standard RT-PCR with (+RT) or without (−RT) reverse transcriptase for 36B4 and GAPDH (data not shown) amplification were used as positive and negative controls. Experiments were processed in duplicate. (D) cDNAs from these six patients (P1 to P6) were used in qPCR. The results are presented as a histogram depicting the relative abundance of antisense (S906) and sense (R588) NcVEGF in each healthy sample and in corresponding tumor lung tissues. The results are reported as means ± the standard errors of the mean, with each sample run in triplicate. 36B4 and GAPDH were used for normalization in qPCR experiments. *, P ≤ 0.05.
Next, we measured the expression of the NcVEGF transcripts in healthy and malignant lung tissues from six patients using either standard or gene-specific reverse transcription on RNA samples obtained from the Biobank tissue unit of the University Hospital of Nice (Table 3). Interestingly, compared to normal and healthy tissues, increased NcVEGF expression is found in five of six tumor samples when using standard reverse transcription (Fig. 7C, S+AS). In agreement with our previous findings, expression of the sense transcript in healthy tissue was low or even barely detectable (Fig. 7C, S). Importantly, a major increase in the sense transcript is always observed in the tumor sample (Fig. 7C), and it resulted in a 10.74-fold increase (P = 0.027; n = 6) compared to control samples (Fig. 7D), whereas the expression of the antisense does not appear to be correlated with the presence of malignant tissue (Fig. 7C and D). The large increase in sense NcVEGF explained the 5.51-fold increase (P = 0.022; n = 6) in total NcVEGF (S+AS) found in tumor samples (Fig. 7D).
DISCUSSION
The RNAa phenomenon was reported about a decade ago (2, 3). Since then, several studies have illustrated the ability of dsRNA, shRNA, or miR to induce promoter activity of numerous genes both in vitro and in vivo (26). However, only a few reports place it in a physiological context (2), which explains why RNAa is often perceived as an artificial “test-tube” process or only as a tool to specifically affect in vivo gene expression. RNAa is dependent on Ago2 and is in most cases associated with chromatin remodeling due to changes in histone methylation. Mounting evidence suggests that noncoding sequences are the target sequences for RNAa. However, it is still debated whether sense or antisense promoter RNAs within target regions are obligatory partners for RNAa or if dsRNAs are binding directly to genomic DNA driving histone modifications (5, 30). Further, if promoter RNAs are involved, how dsRNAs or miRs affect their expression and stability is not known, and no general mechanism has emerged to the best of our knowledge.
Here, we focused on promoters described for RNA activation by the Dahiya laboratory, namely, P21, CSDC2, E-cadherin, and in particular on VEGF (3, 22). Using various cell lines and primers flanking the dsRNA target region of these RNAa responsive promoters, we revealed in all of them, including P21, cryptic promoter RNAs. As expected using dsRNA, we were able to induce RNAa and increased expression of E-cadherin, CSDC2, and VEGF. In case of E-cadherin and CSDC2, the coding and noncoding transcript levels seem to be unrelated (data not shown), but on the contrary, coordinated changes occurred in the case of the RNAa of the VEGF promoter. One simple explanation could be a general increase in transcription at the VEGF locus, due, for example, to the reported histone H3 modifications induced by RNAa (3, 22), that is not limited to increased mRNA production.
Coordinated upregulation of gene and promoter RNA during RNAa is not limited to VEGF (16). Interestingly, COX-2 mRNA and promoter RNA vary also in a coordinated and potentially coregulated fashion during RNAa, but similarly in stimulated cells, cancer cells, and normal and pathological human tissues (16). Therefore, we investigated the expression of NcVEGF during hypoxia, a well-known stimulus for VEGF expression. As in dsVEGF706-transfected cells, in hypoxic cells coordinated modifications of the coding VEGF and NcVEGF expression also occur. In fact, their upregulation due to hypoxia (between 5- and 6-fold) is higher than the one produced after RNAa (between 1.5- and 2-fold). Further, we observed in hypoxia increased expression of both, the antisense transcript (∼5-fold) and the sense NcVEGF, the latter to a higher extent (∼12-fold). The close proximity between the sole transcription start site of the sense NcVEGF and the HRE (hypoxia response element) on the VEGF promoter (only 61 bp) suggests that, in addition to a general increase in transcription at the VEGF locus, its robust induction during hypoxia is possibly directly linked, at least in part, to the action of hypoxia-inducible factor 1 (HIF-1). However, as in PC3 cells, the sense promoter transcript was scant compared to the antisense VEGF, and it contributed only weakly to the total increase in NcVEGF.
The existence of different antisense NcVEGF species covering the target promoter sequence was suggested by the RACE experiments due to various possibilities of 5′ and 3′ end. For the sense NcVEGF, only one 5′ end was found, but the possible 3′ ends remain to be defined. Various NcVEGF transcripts were indeed detected on Northern blots with both double-stranded DNA and antisense RNA probes. Further, increased levels of these species were revealed in both hypoxic and dsVEGF706-transfected cells (data not shown).
To test whether the presence of Ago2 is required for RNAa activation of the VEGF promoter, we first reduced the amount of both Ago2 mRNA and protein with Ago2 siRNAs. As expected, the upregulation of VEGF induced by dsVEGF706 was blunted in cells cotransfected with siAgo2. Interestingly, in these dsVEGF706-transfected cells, the siAgo2 also diminished the concomitant induction of NcVEGF, suggesting that it was indeed due to a general increase in transcription at the VEGF locus. Changes in Ago2 binding to VEGF promoter DNA were not observed in dsVEGF706-transfected cells or in hypoxic cells (data not shown). On the contrary, an increase in Ago2 binding to NcVEGF transcripts was found in both dsVEGF706-transfected cells and in hypoxic cells. Of interest, augmented Ago2 binding in dsVEGF706-transfected cells is limited to the sense NcVEGF, whereas increased binding to both sense and antisense NcVEGFs is seen in hypoxic cells. Therefore, one can speculate that during RNAa only the antisense strand of dsVEGF706 will, by analogy to an siRNA phenomenon, act as guiding strand affecting Ago2 binding only to the sense NcVEGF. In contrast, binding of Ago2 to both strands of NcVEGF may change during hypoxia after partial dimerization due to augmented expression of sense and antisense transcripts, after a change in their secondary structure or due to increase in miR association.
Concerning RNAa induced with dsVEGF706, we confirmed using strand-specific biotin-labeled dsRNAs that the sense NcVEGF is indeed, despite its low expression level, the preferential target. Therefore, the antisense and sense strands of dsVEGF706 function as guide and passenger strands, respectively. With hypoxic cells, the simplest hypothesis is that increased binding of yet unidentified miRs due to changes in their expression or to improved availability of NcVEGF transcripts may affect the binding of Ago2 to both strands of NcVEGF RNA. However, such miRs or RNA duplexes remain to be identified. Another possibility is that the elevated expression of sense and antisense NcVEGFs during hypoxia results in either an increase in NcVEGF RNA duplexes, or a change in their secondary structure, or a combination of both. Indeed, NcVEGF RNAs can potentially adopt complex secondary structures (data not shown), which may become even more elaborate when considering sense and antisense RNA duplexes. It can be envisioned that after cleavage of such RNA duplexes different saRNAs fully complementary to target sequences are generated during hypoxia and are able to target either sense or antisense NcVEGF. In this context, the 5′-uncapped antisense transcript starting at position −339, which was identified in RACE experiments, may then correspond to a piece of the cleavage product generated during hypoxia; the other piece will then have a size of either 117 or 194 bp. Finally, overexpression of either sense or antisense NcVEGF RNAs induced, in a similar fashion to dsVEGF706, the activity of either exogenous or endogenous VEGF promoters. Such activation may result from a direct effect of the NcVEGF RNA on VEGF promoter, but the generation of small RNA duplexes cannot be excluded.
Altogether, our findings indicate that RNAa is potentially involved in the modulation of VEGF expression during and after hypoxia. Therefore, with either Ago2 or control siRNAs, we explored whether RNAa is involved in the persistence of VEGF expression after exposure to hypoxia. Immediately after hypoxia, VEGF expression remains unchanged despite the presence or absence of Ago2 siRNAs. In contrast, 3 days after the hypoxia exposure, the VEGF expression level is significantly lower in siAgo2-treated cells than in siCT-treated cells. Further, a major and persistent increase in NcVEGF expression is seen in cells exposed to hypoxia and to siCT treatment. This augmented NcVEGF expression after hypoxia is severely reduced in siAgo2-treated cells, indicating that the general increase in transcription at the VEGF locus is blunted when Ago2 levels are reduced. These experiments indicate that Ago2 and augmented NcVEGF may, through RNAa, play a role in the maintenance of VEGF expression long after the fading of HIF1α signaling. RNAa-dependent regulation of VEGF expression after hypoxia could represent an example of the relevance of RNAa in a physiological context. Moreover, one can postulate that, at least in the case of coordinated coding and noncoding promoter RNA expression, an RNAa phenomenon can be initiated and programmed to generate persistence of gene expression through modification of the chromatin structure of the gene locus (Fig. 8).
FIG 8.

Possible model for RNAa in experimental and physiological conditions. The schema depicts how epigenetic modifications and persistent promoter activation are generated in the case of experimental or physiological RNAa activation. Furthermore, when the entire locus is activated, the coordinated increase of noncoding and coding RNA would lead to amplification of the RNAa phenomenon and an increased persistence of the promoter activation (bold boxes and arrows). The broken arrows indicate that NcRNA could possibly affect directly the target promoter activity and/or generate new saRNAs. In the latter case, NcRNA derived saRNA could initiate an RNAa phenomenon.
Using mouse ischemic hindlimb muscles and promoter-targeted lentiviral shRNAs, Turunen et al. demonstrated the possibility to modulate VEGF expression either negatively or positively by RNAa (11). These authors showed that depending on the lentiviral shRNAs used (negative LV-856 or positive LV-451) persistent epigenetic changes occur on the VEGF promoter, including modifications in histone H3K9 methylation and acetylation. In a later report, these shRNAs were successfully applied in an acute therapeutic intervention in a myocardial infarction mice model (31). The identification of such in vivo repressor or inducer of VEGF expression is an obligatory step for future clinical studies.
Interestingly, we found that the expression of VEGF is higher in human carcinoma-associated fibroblasts (CAFs) than in human normal dermal fibroblasts (hDFs) in both normoxia and hypoxia. Furthermore, an augmented level of sense NcVEGF in CAFs is also detected due to higher basal expression in normoxia and a major induction after hypoxia. It may thus reflect a general increase in transcription at the VEGF locus. Whether or not the increased transcription in the VEGF locus is related to RNAa and persistent epigenetic changes due to the previous exposure of CAFs to hypoxia in vivo remains to be established. Importantly, augmented expression of the sense NcVEGF transcript was always detected in malignant lung biopsy specimens compared to healthy tissue from the same patient (n = 6), indicating that hypoxia-related malignant transformation affects the expression of this NcVEGF transcript. Therefore, targeting during tumor progression NcVEGF appropriately with either in vivo repressor or inducer of VEGF expression may represent a potentially interesting therapeutic approach.
To conclude, given the central role of VEGF in major life-threatening diseases such as cancer and cardiovascular diseases, a better understanding of VEGF regulation is poised to reveal promising possibilities for prevention and/or therapeutic interventions.
ACKNOWLEDGMENTS
We thank INSERM and the University Nice Sophia-Antipolis. We are grateful to L. Bel for technical help and to N. Mazure and G. Pages for helpful advice and discussion. We thank C. Gaggioli for the generous gift of primary cultures of hDFs and CAF cells. Finally, we are grateful to the Biobank Tissue Unit, Pasteur Hospital, Nice, France, and we thank E. Selva, V. Tanga, and K. Washetine.
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. 2005. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- 2.Kuwabara T, Hsieh J, Nakashima K, Taira K, Gage FH. 2004. A small modulatory dsRNA specifies the fate of adult neural stem cells. Cell 116:779–793. doi: 10.1016/S0092-8674(04)00248-X. [DOI] [PubMed] [Google Scholar]
- 3.Li LC, Okino ST, Zhao H, Pookot D, Place RF, Urakami S, Enokida H, Dahiya R. 2006. Small dsRNAs induce transcriptional activation in human cells. Proc Natl Acad Sci U S A 103:17337–17342. doi: 10.1073/pnas.0607015103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang V, Qin Y, Wang J, Wang X, Place RF, Lin G, Lue TF, Li LC. 2010. RNAa is conserved in mammalian cells. PLoS One 5:e8848. doi: 10.1371/journal.pone.0008848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Portnoy V, Huang V, Place RF, Li LC. 2011. Small RNA and transcriptional upregulation. Wiley Interdiscip Rev RNA 2:748–760. doi: 10.1002/wrna.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang X, Wang J, Huang V, Place RF, Li LC. 2012. Induction of NANOG expression by targeting promoter sequence with small activating RNA antagonizes retinoic acid-induced differentiation. Biochem J 443:821–828. doi: 10.1042/BJ20111491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Junxia W, Ping G, Yuan H, Lijun Z, Jihong R, Fang L, Min L, Xi W, Ting H, Ke D, Huizhong Z. 2010. Double-strand RNA-guided endogenous E-cadherin up-regulation induces the apoptosis and inhibits proliferation of breast carcinoma cells in vitro and in vivo. Cancer Sci 101:1790–1796. doi: 10.1111/j.1349-7006.2010.01594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mao Q, Li Y, Zheng X, Yang K, Shen H, Qin J, Bai Y, Kong D, Jia X, Xie L. 2008. Up-regulation of E-cadherin by small activating RNA inhibits cell invasion and migration in 5637 human bladder cancer cells. Biochem Biophys Res Commun 375:566–570. doi: 10.1016/j.bbrc.2008.08.059. [DOI] [PubMed] [Google Scholar]
- 9.Wang J, Place RF, Huang V, Wang X, Noonan EJ, Magyar CE, Huang J, Li LC. 2010. Prognostic value and function of KLF4 in prostate cancer: RNAa and vector-mediated overexpression identify KLF4 as an inhibitor of tumor cell growth and migration. Cancer Res 70:10182–10191. doi: 10.1158/0008-5472.CAN-10-2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kang MR, Yang G, Place RF, Charisse K, Epstein-Barash H, Manoharan M, Li LC. 2012. Intravesical delivery of small activating RNA formulated into lipid nanoparticles inhibits orthotopic bladder tumor growth. Cancer Res 72:5069–5079. doi: 10.1158/0008-5472.CAN-12-1871. [DOI] [PubMed] [Google Scholar]
- 11.Turunen MP, Lehtola T, Heinonen SE, Assefa GS, Korpisalo P, Girnary R, Glass CK, Vaisanen S, Yla-Herttuala S. 2009. Efficient regulation of VEGF expression by promoter-targeted lentiviral shRNAs based on epigenetic mechanism: a novel example of epigene therapy. Circ Res 105:604–609. doi: 10.1161/CIRCRESAHA.109.200774. [DOI] [PubMed] [Google Scholar]
- 12.Qin Q, Lin YW, Zheng XY, Chen H, Mao QQ, Yang K, Huang SJ, Zhao ZY. 2012. RNAa-mediated overexpression of WT1 induces apoptosis in HepG2 cells. World J Surg Oncol 10:11. doi: 10.1186/1477-7819-10-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ren S, Kang MR, Wang J, Huang V, Place RF, Sun Y, Li LC. 2013. Targeted induction of endogenous NKX3-1 by small activating RNA inhibits prostate tumor growth. Prostate 73:1591–1601. doi: 10.1002/pros.22709. [DOI] [PubMed] [Google Scholar]
- 14.Crosby ME, Devlin CM, Glazer PM, Calin GA, Ivan M. 2009. Emerging roles of microRNAs in the molecular responses to hypoxia. Curr Pharm Des 15:3861–3866. doi: 10.2174/138161209789649367. [DOI] [PubMed] [Google Scholar]
- 15.Majid S, Dar AA, Saini S, Yamamura S, Hirata H, Tanaka Y, Deng G, Dahiya R. 2010. MicroRNA-205-directed transcriptional activation of tumor suppressor genes in prostate cancer. Cancer 116:5637–5649. doi: 10.1002/cncr.25488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsui M, Chu Y, Zhang H, Gagnon KT, Shaikh S, Kuchimanchi S, Manoharan M, Corey DR, Janowski BA. 2013. Promoter RNA links transcriptional regulation of inflammatory pathway genes. Nucleic Acids Res 41:10086–10109. doi: 10.1093/nar/gkt777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Place RF, Wang J, Noonan EJ, Meyers R, Manoharan M, Charisse K, Duncan R, Huang V, Wang X, Li LC. 2012. Formulation of small activating RNA into lipidoid nanoparticles inhibits xenograft prostate tumor growth by inducing p21 expression. Mol Ther Nucleic Acids 1:e15. doi: 10.1038/mtna.2012.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dykxhoorn DM, Novina CD, Sharp PA. 2003. Killing the messenger: short RNAs that silence gene expression. Nat Rev Mol Cell Biol 4:457–467. doi: 10.1038/nrm1129. [DOI] [PubMed] [Google Scholar]
- 19.Place RF, Noonan EJ, Foldes-Papp Z, Li LC. 2010. Defining features and exploring chemical modifications to manipulate RNAa activity. Curr Pharm Biotechnol 11:518–526. doi: 10.2174/138920110791591463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang V, Place RF, Portnoy V, Wang J, Qi Z, Jia Z, Yu A, Shuman M, Yu J, Li LC. 2012. Upregulation of cyclin B1 by miRNA and its implications in cancer. Nucleic Acids Res 40:1695–1707. doi: 10.1093/nar/gkr934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Janowski BA, Younger ST, Hardy DB, Ram R, Huffman KE, Corey DR. 2007. Activating gene expression in mammalian cells with promoter-targeted duplex RNAs. Nat Chem Biol 3:166–173. doi: 10.1038/nchembio860. [DOI] [PubMed] [Google Scholar]
- 22.Place RF, Li LC, Pookot D, Noonan EJ, Dahiya R. 2008. MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc Natl Acad Sci U S A 105:1608–1613. doi: 10.1073/pnas.0707594105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chu Y, Yue X, Younger ST, Janowski BA, Corey DR. 2010. Involvement of Argonaute proteins in gene silencing and activation by RNAs complementary to a non-coding transcript at the progesterone receptor promoter. Nucleic Acids Res 38:7736–7748. doi: 10.1093/nar/gkq648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwartz JC, Younger ST, Nguyen NB, Hardy DB, Monia BP, Corey DR, Janowski BA. 2008. Antisense transcripts are targets for activating small RNAs. Nat Struct Mol Biol 15:842–848. doi: 10.1038/nsmb.1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matsui M, Sakurai F, Elbashir S, Foster DJ, Manoharan M, Corey DR. 2010. Activation of LDL receptor expression by small RNAs complementary to a noncoding transcript that overlaps the LDLR promoter. Chem Biol 17:1344–1355. doi: 10.1016/j.chembiol.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiao AL, Slack FJ. 2013. RNA-mediated gene activation. Epigenetics 9:27–36. doi: 10.4161/epi.26942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dayan F, Mazure NM, Brahimi-Horn MC, Pouyssegur J. 2008. A dialogue between the hypoxia-inducible factor and the tumor microenvironment. Cancer Microenviron 1:53–68. doi: 10.1007/s12307-008-0006-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ørom UA, Lund AH. 2007. Isolation of microRNA targets using biotinylated synthetic microRNAs. Methods 43:162–165. doi: 10.1016/j.ymeth.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 29.Lendoye E, Sibille B, Rousseau AS, Murdaca J, Grimaldi PA, Lopez P. 2011. PPARβ activation induces rapid changes of both AMPK subunit expression and AMPK activation in mouse skeletal muscle. Mol Endocrinol 25:1487–1498. doi: 10.1210/me.2010-0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu J, Chen Z, Xia D, Wu J, Xu H, Ye ZQ. 2012. Promoter-associated small double-stranded RNA interacts with heterogeneous nuclear ribonucleoprotein A2/B1 to induce transcriptional activation. Biochem J 447:407–416. doi: 10.1042/BJ20120256. [DOI] [PubMed] [Google Scholar]
- 31.Turunen MP, Husso T, Musthafa H, Laidinen S, Dragneva G, Laham-Karam N, Honkanen S, Paakinaho A, Laakkonen JP, Gao E, Vihinen-Ranta M, Liimatainen T, Yla-Herttuala S. 2014. Epigenetic upregulation of endogenous VEGF-A reduces myocardial infarct size in mice. PLoS One 9:e89979. doi: 10.1371/journal.pone.0089979. [DOI] [PMC free article] [PubMed] [Google Scholar]