

Abstract
The dynamic exchange of histones alleviates the nucleosome barrier and simultaneously facilitates various aspects of cellular DNA metabolism, such as DNA repair and transcription. In response to DNA damage, the acetylation of Lys5 in the histone variant H2AX, catalyzed by TIP60, plays a key role in promoting histone exchange; however, the detailed molecular mechanism still is unclear. Here, we show that the TIP60 complex includes poly(ADP-ribose) polymerase 1 (PARP-1). PARP-1 is required for the rapid exchange of H2AX on chromatin at DNA damage sites. It is known that PARP-1 binds dynamically to damaged chromatin and is crucial for the subsequent recruitment of other repair factors, and its auto-poly(ADP-ribosyl)ation is required for the dynamics. We also show that the acetylation of histone H2AX at Lys5 by TIP60, but not the phosphorylation of H2AX, is required for the ADP-ribosylation activity of PARP-1 and its dynamic binding to damaged chromatin. Our results indicate the reciprocal regulation of K5 acetylation of H2AX and PARP-1, which could modulate the chromatin structure to facilitate DNA metabolism at damage sites. This could explain the rather undefined roles of PARP-1 in various DNA damage responses.
INTRODUCTION
Posttranslational modifications of histones are a fundamental process for the chromatin remodeling machinery in DNA metabolism, including transcription, DNA replication, and DNA repair (1, 2). These histone modifications either serve as the binding interface for regulatory factors in chromatin reorganization or function as a platform in a signaling cascade, such as in the DNA damage checkpoint response (1, 3). In addition to histone modifications, the incorporation or eviction of histone variants regulates chromatin dynamics and could directly promote DNA metabolism in the context of chromatin (4–9). Thus, it is important to clarify how the histone variants' dynamics at DNA damage sites and their modifications are coordinated upon commitment to each type of DNA metabolism. Such clarification would promote a better understanding of the significance of histone variants, which potentially play active roles, rather than simply functioning as barriers, during DNA metabolism (10).
Upon DNA damage, Ser139 (S139) of H2AX, a histone H2A variant, is phosphorylated at DNA damage sites, and the phosphorylated H2AX functions as an anchor to retain DNA damage response (DDR) factors on the DNA damage sites (11–14). We previously reported that the acetylation of H2AX at lysine 5 (K5Ac) is required to facilitate histone H2AX exchange at DNA damage sites and also is necessary for the efficient assembly of NBS1 at these sites by promoting its turnover rate (9, 15). K5 acetylation of H2AX is catalyzed by the histone acetyltransferase TIP60 complex, which coordinates the signaling and repair of DNA damage via chromatin reorganization (9, 16–18). Importantly, the phosphorylation of H2AX on S139 is not required to facilitate H2AX exchange or to promote NBS1 turnover at DNA damage sites (9, 15). These findings indicated the distinct role of H2AX acetylation from that of H2AX phosphorylation in the H2AX dynamics upon DNA damage.
Poly(ADP-ribose) polymerase 1 (PARP-1) is responsible for the major cellular poly(ADP-ribose) synthesis following DNA damage (reviewed in reference 19). PARP-1 reportedly is involved in several DNA repair pathways, such as single-strand break repair (SSBR) and homologous recombination (HR) (reviewed in references 19 and 20). However, the precise role of PARP-1 in these processes still is controversial. For instance, in the case of SSBR, most of the in vitro studies have failed to show any acceleration of the repair rate by PARP-1 (reviewed in reference 19). Thus, it was proposed that PARP-1 is involved in accelerating the detection of DNA damage by the DNA repair machineries or promoting the repair process in the context of chromatin, although the underlying mechanism remains largely unknown. In the nucleus, PARP-1 is predominantly associated with chromatin, and its binding properties are highly dynamic (21). Upon DNA damage, PARP-1 accumulates at the damage site and exerts its function by directly (ADP-ribosyl)ating substrates, including the core histones and chromatin-associated proteins, and thereby promotes the dissociation of nucleosomes and the decondensation of chromatin (21; reviewed in reference 22). PARP-1 itself is also the primary protein target for PARP-1-mediated (ADP-ribosyl)ation in vivo. The activity of PARP-1 is rapidly stimulated upon binding to DNA breaks; however, auto(ADP-ribosyl)ation facilitates PARP-1 dissociation from DNA, possibly by charge repulsion (23–26). Thus, the binding of PARP-1 to DNA breaks is transient, and this allows the assembly of other repair proteins at the sites of DNA damage. Previous studies have shown that the ADP ribosylation of the histone chaperone FACT by PARP-1 is necessary for the dissociation of FACT from nucleosomes (27). Thus, these findings suggested that a regulatory relationship exists between TIP60 and PARP-1 in histone H2AX exchange at DNA damage sites.
In this study, we found that PARP-1 is present in the purified H2AX complex but only after the induction of DNA damage. We then performed an inverted fluorescence recovery after photobleaching (iFRAP) analysis in combination with microirradiation to analyze the effect of PARP-1 on the histone H2AX exchange within the microirradiated area. The depletion of PARP-1 in cells inhibited the histone H2AX exchange within the microirradiated area. This result indicated that PARP-1 is involved in the histone H2AX exchange at DNA damage sites in vivo. We next examined the effect of the TIP60 acetyltransferase activity on the (ADP-ribosyl)ation activity of PARP-1 in histone H2AX exchange at DNA damage sites. The depletion of TIP60 or the defective acetylation of H2AX at Lys5 in cells was accompanied by the decreased (ADP-ribosyl)ation activity of PARP-1 after the induction of DNA damage. Moreover, we performed the microirradiation experiment to monitor the accumulation of PARP-1–green fluorescent protein (GFP) at the damaged area and found that the fluorescence recovery of PARP-1–GFP after photobleaching at the microirradiated area in TIPM (TIP60 mutant lacking acetyltransferase activity)- or H2AX K5R-expressing cells is reduced compared to that in TIP60- or H2AX wild-type (WT)-expressing cells. Since the (ADP-ribosyl)ation activity of PARP-1 is required for its dissociation from damaged chromatin, we concluded that the acetylation of H2AX at Lys5 by TIP60 is required for the (ADP-ribosyl)ation activity and the dynamic binding of PARP-1 to chromatin after the induction of DNA damage.
MATERIALS AND METHODS
Plasmids.
FLAG-hemagglutinin (HA)-tagged H2AX (eH2AX; WT and K5R), pCAGGS-tagged H2AX (WT, K5R, and S139A), TIP60 (eTIP60), and TIPM (eTIPM; the acetyltransferase mutant of TIP60) were reported previously (9, 28). For GFP-tagged PARP-1, the PARP-1 coding sequence was cloned into pEGFP-N1 to fuse a GFP tag to the C terminus.
Cell culture and transfection.
HeLa cells (kind gift from Y. Nakatani, Dana-Faber Cancer Institute), murine embryonic fibroblast (MEF) cells, and GM0637 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum. Plasmid transfection was performed, according to the manufacturer's protocol, using the GeneJuice transfection reagent (Novagen). H2AX knockout (KO) MEF cells were a kind gift from H. Honda (Hiroshima University). PARP-1 KO MEF cells were a kind gift from M. Masutani (Nagasaki University). For the analysis using TIP60- or TIPM-expressing cells, we constructed MEF cells or HeLa cells in which shTIP60, which effectively knocks down both human and mouse TIP60, on a retroviral vector was stably expressed (9, 15). TIP60 genes on retroviral plasmid pOZ-eTIP60 or pOZ-eTIPM were stably expressed in shTIP60-expressing cells. The plasmid constructions were reported previously (9, 28). For the knockdown of PARP-1, shPARP-1 (1) (GGACATCGAGGTGGCCTACAGTCT) and shPARP-1 (2) (AAGGCTGGAGAGAGATTCTGTT) were used.
Antibodies.
Immunoblot analyses were performed with anti-TIP60 (kind gift from H. Hosokawa, Kyoto University), anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH) (Abcam), anti-H2AX (Sigma), anti-H2B (Upstate), anti-K5Ac-H2AX (Millipore), anti-PARP-1 (Santa Cruz), and anti-poly-ADP-ribosylation (anti-PAR; Trevigen).
TIP60 and H2AX complex purification.
eTIP60 and eH2AX were purified via affinity chromatography on anti-Flag antibody-conjugated agarose, followed by elution with Flag peptide under native conditions. These affinity-purified proteins were further purified by immunoaffinity chromatography with anti-HA antibody-conjugated agarose and then examined by SDS-PAGE (28, 29). For the 10 to ∼35% glycerol gradient sedimentation, 200 μl of FLAG-purified H2AX complex was used. The detailed procedure was described previously (28).
Microirradiation iFRAP and FRAP.
The iFRAP and FRAP analyses with the UVA microirradiation of GFP-H2AX were performed as previously described (9). For the FRAP analysis of PARP-1–GFP with microirradiation, the 405-nm line of the UV laser (Leica) was used. The FRAP analyses with the UVA microirradiation for PARP-1–GFP dynamics were performed as previously described (9, 15). We used PARP-1 C-terminally tagged with a single GFP. Cells that expressed similar amounts of PARP-1–GFP in eTIP60/M-expressing MEFs were subjected to FRAP analysis. For the quantification of the fluorescence recovery, the measurement was accomplished with ImageJ. The background fluorescence intensity (BG) and the average fluorescence intensity in the UV-microirradiated and bleached region at each time point (It) were measured as described previously (9). For each time point, the relative intensity was calculated as Irel;t = (It − BG)/(I0 − BG), where I0 is the average intensity of the region of interest before bleaching. The percent recovery after each time point in the FRAP analysis (Precovery;t) was calculated and normalized as Precovery;t = 100 × (Irel;t)/(Inorm;t), where Inorm;t is the relative fluorescence intensity, calculated by the same method as that for Irel;t, in the UV-microirradiated but unbleached region. Statistical comparisons were made using the Student t test.
Poly(ADP-ribosyl)ation assay.
N-terminally His-tagged PARP-1 or PARP-1 harboring the E988K mutation was expressed in Escherichia coli cells and purified with Talon beads (Clontech). Further purification was performed using a MonoQ column with an AKTA purifier system, followed by dialysis in buffer containing 100 mM KCl. The poly(ADP-ribosyl)ation assay was performed as follows. The total volume of a 20-μl single reaction mixture contained 6.7 μl of His–PARP-1 WT or E988K protein (7.5 ng/μl), 1 μl of NAD (1 mM), 1 μl of calf thymus DNA (1 mg/ml), and 4 μl of 5× reaction buffer (500 mM Tris-Cl, pH 7.0, 50 mM MgCl2). The mixture was incubated at 37°C for 1 h. The reaction was terminated by adding 5 μl of 5× SDS sample buffer and incubated at 95°C for 3 min. The reaction mixtures subsequently were analyzed by immunoblotting using anti-PAR and anti-PARP-1 antibodies.
In situ proximity ligation assay.
After the induction of irradiation (IR), the cells were fixed in 4% paraformaldehyde for 10 min. Nuclei were permeabilized with 0.5% Triton X-100 in phosphate-buffered saline (PBS) for 5 min and then stained with anti-TIP60 (1/20,000) and anti-PARP-1 (1/20,000) in 1% bovine serum albumin–1× phosphate-buffered saline (PBS) overnight. The in situ PLA was performed using specific antibodies (Duolink; Sigma) and a Duolink in situ PLA kit (Olink Bioscience). After in situ PLA, the cells were fixed with 4% paraformaldehyde for 10 min. The fixed cells were incubated in blocking buffer (Blocking One Histo; 06349-64; Nacalai Tesque) at room temperature (RT) for 30 min and then stained with direct-labeled γ-H2AX (Zenon Alexa Fluor 488-mouse IgG labeling kit [Z25002]; Molecular Probes by Life Technologies). Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (catalog number 10236276001; Roche). Samples were examined by immunofluorescence microscopy using a Keyence BZ-9000 microscope. Red PLA spots were scored in three independent experiments, and >100 cells were analyzed in each experiment.
RESULTS
PARP-1 is present in the purified TIP60 and H2AX complex.
We noticed that PARP-1 was included in TIP60 purified from the nuclear extracts of gamma-irradiated or nonirradiated HeLa cells stably expressing TIP60 tagged with a Flag-HA epitope (N-terminal tag for eTIP60) (15). We confirmed the interaction by the immunoblotting of the Flag-purified eTIP60 complex with an anti-PARP-1 antibody (Fig. 1A).
FIG 1.

Functional interaction of PARP-1 and TIP60 histone acetyltransferase. (A) Immunopurified TIP60 complexes were subjected to immunoblot analysis using anti-PARP-1 and anti-TIP60. Nondamaged cells and damaged cells (2 min after 12 Gy of IR) were used for the analysis. (B) PLA analysis using anti-TIP60 and anti-PARP-1 antibodies. The interaction between TIP60 and PARP-1 is detected as PLA spots (red). Costaining with the direct fluorescently labeled anti-γ-H2AX antibody (green) was performed to monitor the DNA damage response. Bars, 10 μm. (C) The numbers of PLA foci within cells were plotted. cont, control. (D) The immunopurified H2AX complex was subjected to immunoblot analyses using anti-PARP-1, anti-TIP60, anti-H2AX, and anti-H2B. Nondamaged cells and damaged cells (2 min after 12 Gy of IR) were used for the analysis. (E) The purified eH2AX complex was subjected to glycerol gradient (10% to 35%) fractionation. Immunoblot analyses using anti-PARP-1, anti-TIP60, anti-H2AX, and anti-H2B were performed. The even-number fractions (Fr) were subjected to Western blotting analysis.
We next examined whether this interaction occurs within cells in the presence or absence of DNA damage by an in situ PLA in which the protein-protein interactions are visualized as red PLA spots. As a result, red PLA spots for the interaction between TIP60 and PARP-1 were observed before and after DNA damage. The induction of DNA damage slightly increased the number of these spots within the cells, and the spots partially colocalized with γ-H2AX (S139-phosphorylated H2AX) foci (Fig. 1B and C). These findings indicated that PARP-1 was associated with TIP60 within cells and their interaction increased after the induction of DNA damage.
We have previously shown that TIP60 associates with soluble H2AX tagged with a Flag-HA epitope (C-terminal tag for eH2AX), which represents H2AX evicted from chromatin, only after DNA damage (9, 15). Here, we also found that the association of PARP-1 with eH2AX purified from the soluble nuclear extract of HeLa cells was DNA damage dependent (Fig. 1D). It should be noted that the association of PARP-1 with TIP60 was not disrupted by DNase I treatment, suggesting that these interactions were not mediated by DNA (see Fig. S1 in the supplemental material).
Furthermore, to confirm whether TIP60, PARP-1, and H2AX are in the same complex, the purified eH2AX complex after DNA damage was separated on a 10% to ∼35% glycerol gradient by ultracentrifugation (Fig. 1E). Western blotting with anti-H2AX and anti-H2B antibodies revealed that the eH2AX complex has at least two peaks with higher and lower molecular weights together with H2B. Further Western blot analyses with anti-PARP-1 and anti-TIP60 showed the cofractionation of PARP-1 and TIP60, with an eH2AX peak at a higher molecular weight. Interestingly, the fraction of PARP-1 that comigrated with TIP60 was associated with the hypershift, which is reminiscent of the poly(ADP-ribosyl)ated form of PARP-1. Together, these results suggested that TIP60 and PARP-1 cooperatively regulate histone H2AX to facilitate its exchange at DNA damage sites.
PARP-1 regulates histone H2AX exchange at microirradiated areas.
Histone binding to DNA becomes highly dynamic during the DNA damage response (9, 30, 31). We previously established a method combining iFRAP with microirradiation to analyze the dynamic binding of GFP-tagged histones to damaged chromatin. Using this method, we have shown that H2AX binding to chromatin becomes more dynamic at DNA damage sites than in nondamaged areas (9). It should be noted that the dynamic behavior of GFP-H2AX likely is not caused by gross movement of chromatin, as canonical histones (GFP-H2A, -H2B, -H3, and -H4) were not dynamic (9). It also should be noted that we previously confirmed that GFP-H2AX is functional, as it is efficiently incorporated into mitotic chromosomes in a manner similar to that of the endogenous H2AX (9, 15).
To examine whether PARP-1 is involved in the eviction of GFP-H2AX from DNA damage sites, we photobleached GFP-H2AX after microirradiation in mock shRNA- or PARP-1 shRNA-expressing cells and pursued the remaining fluorescent signal of GFP-H2AX (immunoblot of shPARP-1-expressing cells is shown in Fig. 2A). The cells first were microirradiated (Fig. 2B, yellow boxes) and then photobleached (Fig. 2B, red box, excluding small interior boxes), and the diffusion of the fluorescent signal from the unbleached area was quantified. As a result, in the mock short hairpin RNA (shRNA)-expressing cells, the remaining GFP-H2AX fluorescent signal in the irradiated areas but not that in the unirradiated areas diffused away from the unbleached areas (Fig. 2B and C). This indicated that the eviction of GFP-H2AX is facilitated at DNA damage sites (9). In contrast, in the PARP-1 shRNA-expressing cells, the diffusion of the remaining GFP-H2AX fluorescent signal from the unbleached area became slower in the irradiated areas compared to that in the mock short interfering RNA (siRNA)-expressing cells (Fig. 2B and C). These findings indicated that PARP-1 is involved in the eviction of H2AX from the microirradiated area.
FIG 2.

PARP-1 is required for rapid exchange of H2AX at DNA damage sites. (A) Confirmation of the knockdown of PARP-1 by two different shRNAs (1 and 2). Immunoblots with anti-PARP-1 and anti-GAPDH are shown. (B) iFRAP analysis to monitor the eviction of H2AX from DNA damage sites. GM0637 cells were microirradiated (yellow box) and then photobleached (red box; excluding small boxes inside) as previously described (9). Either mock shRNA- or shPARP-1-expressing cells were subjected to the analysis. The diffusion of the unbleached signal in the small red boxes was quantified. (C) Quantification of the remaining signals shown in panel B. Fluorescent intensities were plotted. (D) FRAP analysis to monitor the incorporation of H2AX at damage sites. GM0637 cells first were microirradiated (yellow boxes) and then photobleached (red boxes). (E) The fluorescence recovery of the cells in panel D was monitored as previously described (9). (Left) The fluorescence recovery in the bleached area (for the damaged area, line 1; for the nondamaged area, line 2) was quantified. (Right) The quantification of the fluorescence recovery was plotted. Line 1 represents fluorescence recovery at the damaged area. Mock shRNA (left), fluorescence recovery of 33.3% ± 9.1% (n = 11); shTIP60 (right), 20.9% ± 6.7% (n = 11), with a P value of 0.0016 between mock shRNA and shPARP-1. Line 2 represents fluorescence recovery at the nondamaged area. In shTIP60 cells, fluorescence recovery was 8.6% ± 3.4% (n = 11); in mock shRNA, it was 9.8% ± 3.7% (n = 11). (F) eH2AX complexes were purified from mock- or siPARP-1-expressing cells before and after gamma IR (12 Gy). Immunoblot analyses using anti-PARP-1, anti-TIP60, anti-H2AX K5 acetylation (Ac), and anti-H2AX were performed. (G) Confirmation of inhibition of poly(ADP-ribosyl)ation of PARP-1 by olaparib. Immunoprecipitation (IP) with an anti-PAR antibody was performed with nuclear extracts of GM0637 cells treated with 10 μM olaparib for 24 h. An anti-PARP-1 immunoblot is shown. DMSO, dimethyl sulfoxide. (H) The inhibition of H2AX dynamics by olaparib was monitored by a FRAP analysis after microirradiation. The fluorescence recovery was quantified and plotted. Line 1 represents fluorescence recovery at the damaged area, namely, 23.6% ± 6.0% (no olaparib, n = 10) and 17.2% ± 5.9% (with olaparib, n = 10; P value of 0.0038 for comparisons with and without olaparib), and line 2 represents that at the nondamaged area, namely, 7.0% ± 4.2% (no olaparib, n = 10) and 5.9% ± 2.9% (with olaparib, n = 10; P value of 0.34 for comparisons with and without olaparib). Standard deviations are shown. Bars, 10 μm.
We then examined whether PARP-1 regulates the incorporation of GFP-H2AX into the microirradiated area by monitoring fluorescence recovery. The cells first were microirradiated (Fig. 2D, yellow box) and then photobleached (Fig. 2D, red box). The recovery of the fluorescent signal in the bleached area was quantified. The fluorescence recovery of the GFP-H2AX signal after microirradiation was observed (Fig. 2E, red line) (fluorescence recovery, 33.3% ± 9.1% [n = 11]), but was not observed in the unirradiated areas (Fig. 2E, blue line) (fluorescence recovery, 9.8% ± 3.7% [n = 11]), in the mock shRNA-expressing cells (Fig. 2D and E). In contrast, in the PARP-1 shRNA-expressing cells, the fluorescence recovery of the GFP-H2AX signal after microirradiation was strongly inhibited (fluorescence recovery at the damaged area is 20.9% ± 6.7% [n = 11], with a P value of 0.0016 between mock shRNA and shPARP-1, and fluorescent recovery at the nondamaged area is 8.6% ± 3.4% [n = 11]) (Fig. 2D and E).
The dynamic binding of H2AX to chromatin at DNA damage sites is dependent on K5 acetylation of H2AX by TIP60 (9). To determine whether the defect in PARP-1 could compromise the K5 acetylation of H2AX, we purified eH2AX from the nuclear extract from cells in which siPARP-1 was expressed and subjected the extract to Western blot analyses using antibodies against PARP-1, TIP60, and K5-acetylated H2AX. As expected, siPARP-1 reduced the K5 acetylation of eH2AX in the nuclear extract (Fig. 2F, left). In the purified eH2AX complex, we also observed a reduction in the amount of TIP60 protein as well as reduced K5 acetylation of eH2AX regardless of DNA damage (Fig. 2F, right).
The catalytic activity of PARP-1 can be inhibited by the anticancer drug olaparib. Indeed, we confirmed that the exposure of cells to olaparib reduces the poly(ADP-ribosyl)ation of PARP-1. Briefly, immunoprecipitation by an anti-poly(ADP-ribosyl)ation antibody (anti-PAR) captured poly(ADP-ribosyl)ated PARP-1, and the amount was greatly reduced both before and after DNA damage when the cells were treated with olaparib (Fig. 2G). We further examined the exchange of H2AX at damaged chromatin, in the presence of olaparib, using FRAP analysis after microirradiation. In the cells exposed to olaparib, the fluorescence recovery of the GFP-H2AX signal after microirradiation was strongly inhibited in the microirradiated area (fluorescence recovery at damaged areas was 23.6% ± 6.0% [no olaparib; n = 10] and 17.2% ± 5.9% [with olaparib; n = 10], with a P value of 0.0038 between olaparib treatment and no olaparib treatment, and fluorescence recovery at nondamaged areas was 7.0% ± 4.2% [no olaparib; n = 10] and 5.9% ± 2.9% [with olaparib; n = 10], with a P value of 0.34 between olaparib treatment and no olaparib treatment) (Fig. 2H).
Considering these results together, we concluded that PARP-1 regulates histone H2AX exchange at DNA damage sites. In contrast to our findings, Heo et al. reported that PARP-1 inhibits the exchange of histone H2AX by the ADP-ribosylation of FACT in vitro. This discrepancy might arise from the different assay systems used (27).
Histone H2AX acetylation at Lys5 by TIP60 is required for auto-poly(ADP-ribosyl)ation activity of PARP-1 upon DNA damage.
PARP-1 itself is known to become autoribosylated upon DNA damage. As shown above, PARP-1 is required for H2AX exchange (Fig. 2). We previously reported that H2AX exchange at DNA damage sites requires the TIP60-dependent acetylation of H2AX at Lys5 (9). Therefore, we next examined the relationship between TIP60, or the acetylation of H2AX at Lys5, with the auto-poly(ADP-ribosyl)ation activity of PARP-1 after the induction of DNA damage. To clarify this, we first performed immunoprecipitation using an anti-PAR antibody with nuclear extracts prepared from gamma-irradiated cells expressing either mock shRNA or TIP60 shRNA, and the precipitated proteins were analyzed by immunoblotting with anti-PARP-1. The results showed the reduction of auto-poly(ADP-ribosyl)ation of PARP-1 in TIP60 shRNA-expressing cells compared to that in mock shRNA-expressing cells, both with and without DNA damage (Fig. 3A). The defect in the auto-poly(ADP-ribosyl)ation could be rescued by expressing the shRNA-resistant eTIP60 but not the shRNA-resistant eTIP60 HAT mutant, TIPM (Fig. 3B). These results indicated that the acetyltransferase activity of TIP60 likely is required for the auto-poly(ADP-ribosyl)ation activity of PARP-1 after the induction of DNA damage.
FIG 3.

K5 acetylation of H2AX is required for efficient auto-poly(ADP-ribosyl)ation of PARP-1 upon DNA damage response. (A) Anti-PAR antibody immunoprecipitates were subjected to an immunoblot analysis using an anti-PARP-1 antibody. Unirradiated or irradiated (12 Gy) HeLa cells stably expressing mock shRNA or shTIP60 were used for immunoprecipitation. Amounts of PARP-1, TIP60, and control GAPDH in input materials were detected by immunoblotting with the respective antibodies. (B) Anti-PAR antibody immunoprecipitates were subjected to an immunoblot analysis using an anti-PARP-1 antibody. Unirradiated or irradiated (12 Gy) HeLa cells stably expressing shTIP60 and reconstituted with either eTIP60 or eTIPM were used for immunoprecipitation. Input nuclear extracts immunoblotted with anti-PARP-1, an anti-HA antibody to detect eTIP60, and anti-GAPDH antibodies also are shown. (C) Immunopurified H2AX complex by sequential immunoprecipitation using anti-Flag and anti-HA antibodies. Immunoblots using anti-PAR and anti-H2AX antibodies are presented. (D) The H2AX complex immunopurified by an anti-Flag antibody was subjected to immunoblot analyses using anti-PAR, anti-PARP-1, anti-TIP60, anti-K5Ac H2AX, and anti-H2AX antibodies. HeLa cells, in which the endogenous H2AX was knocked down, were reconstituted with pOZ-C eH2AX WT or K5R (9). Cells were irradiated (12 Gy) before harvest. (E) Immunopurified H2AX complexes by anti-Flag antibody shown in panel D subsequently were immunoprecipitated using the anti-PAR antibody and subjected to immunoblot analyses using anti-PARP-1, anti-PAR, and anti-TIP60 antibodies.
Since TIP60 acetylates histone H2AX at Lys5 and is essential for histone H2AX exchange at DNA damage sites, we next examined whether TIP60 enhances the auto-(ADP)ribosylation activity of PARP-1 through the acetylation of histone H2AX at Lys5. As shown in Fig. 1D, PARP-1 bound to the soluble H2AX-H2B dimer after the induction of DNA damage. At first, we determined whether DNA damage could facilitate the auto-poly(ADP-ribosyl)ation of histone-bound PARP-1. H2AX was immunopurified from unirradiated or irradiated (12 Gy) HeLa cells stably expressing eH2AX and was subjected to sequential immunopurifications using anti-Flag and anti-HA antibodies. The immunopurified H2AX complexes were analyzed by immunoblotting using anti-PAR antibodies to detect auto-poly(ADP-ribosyl)ation. As expected, a stronger auto-poly(ADP-ribosyl)ation signal was detected after irradiation (Fig. 3C).
We then compared the degree of the auto-poly(ADP-ribosyl)ation of PARP-1 after irradiation (12 Gy) between cells expressing H2AX WT and H2AX K5R. First, H2AX was immunopurified with an anti-Flag antibody from a nuclear extract of irradiated HeLa cells. Immunoblotting by the anti-PAR antibody showed an apparent decrease in the amount of poly(ADP-ribosyl)ation in the H2AX K5R complex compared to that of the H2AX WT complex (Fig. 3D). The amount of TIP60 associated with H2AX also was significantly reduced in H2AX K5R (Fig. 3D). It should be noted that the K5R mutation of H2AX did not compromise the interaction with PARP-1; instead, an increased amount of PARP-1 was detected (Fig. 3D).
The purified histone H2AX complex subsequently was immunoprecipitated using an anti-poly(ADP-ribosyl)ation antibody. The precipitated proteins were analyzed by immunoblotting using anti-PARP-1; thus, the auto-poly(ADP-ribosyl)ation of PARP-1 was compared between irradiated cells expressing H2AX WT and H2AX K5R (Fig. 3E). The auto-poly(ADP-ribosyl)ation signal of PARP-1 was not observed in the complex purified from the H2AX K5R lysate, in contrast to that from the H2AX WT lysate. We also observed that the anti-PAR immunoprecipitation of the H2AX WT complex includes TIP60, unlike the anti-PAR immunoprecipitation of the H2AX K5R complex (Fig. 3E). Therefore, we concluded that the TIP60-mediated acetylation of histone H2AX at Lys5 is required for the auto-poly(ADP-ribosyl)ation activity of PARP-1 after the induction of DNA damage; however, we do not exclude the possibility that TIP60 regulates PARP-1 independently of the K5 acetylation of H2AX.
The dynamic binding of PARP-1 to damaged chromatin is regulated by its auto-poly(ADP-ribosyl)ation in vivo.
GFP-tagged PARP-1 (PARP-1–GFP) binds dynamically to chromatin upon DNA damage, as shown by a FRAP analysis in combination with microirradiation (21). We established PARP-1–GFP-expressing PARP-1 KO MEF cells (Fig. 4A and B) and confirmed that PARP-1–GFP rapidly accumulated at the microirradiated area in these cells. The fluorescence recovery of PARP-1–GFP was observed after photobleaching at a microirradiated (damaged) area in these cells (71.1% ± 16.3% [n = 12]) (Fig. 4C and D), but it was less than that at a nondamaged area (96.6% ± 13.6% [n = 10]) (Fig. 4D). These results suggested that DNA damage induced the localization of PARP-1 to DNA damage sites, but its binding is dynamic. We next investigated whether this dynamic binding of PARP-1–GFP to damaged chromatin is dependent on its auto(ADP-ribosyl)ation activity. The central glutamic acid at amino acid position 988 (Glu 988) of PARP-1 is reportedly a catalytic amino acid residue that is responsible for PAR chain elongation and also is essential for the dissociation of PARP-1 from chromatin (21). Thus, we mutated Glu 988 to Lys in PARP-1 (PARP-1 E988K) and confirmed the absence of the auto-poly(ADP-ribosyl)ation activity of the PARP-1 E988K mutant compared to that of the PARP-1 wild type using an in vitro poly(ADP-ribosyl)ation assay (Fig. 4E; also see Fig. S2 in the supplemental material). We then established the PARP-1 E988K–GFP-expressing PARP-1 KO MEF cell line (Fig. 4B) and performed a FRAP/microirradiation analysis. Consistent with a previous study (21), the fluorescence recovery of the PARP-1 E988K–GFP mutant after photobleaching of the microirradiated area was inhibited compared to that of the PARP-1–GFP wild type in our FRAP/microirradiation system (E988K in nondamaged area, 96.0% ± 7.9% [n = 13]; in damaged area, 41.0% ± 17.6% [n = 11]; P value of 0.00034 between the WT and the E988K mutant in the damaged area) (Fig. 4C and D). This could be interpreted as the exchange of PARP-1 rather than the defective binding of PARP-1 to damaged chromatin by the PARP-1 E988K mutant. Thus, the results indicated that the auto-poly(ADP-ribosyl)ation activity of PARP-1 is required for the dynamic binding of PARP-1 to damaged chromatin, as previously reported (21).
FIG 4.

FRAP analysis using GFP–PARP-1. (A) Immunoblot analysis to confirm PARP-1 KO cells. (B) Reconstitution of either GFP-tagged PARP-1 WT or PARP-1 with the E988K mutation (E988K) was subjected to immunoblot analyses using anti-GFP, anti-PARP-1, and anti-beta-actin antibodies. (C) FRAP analysis to monitor the dynamics of PARP-1–GFP. The PARP-1–GFP WT or that with the E988K mutation accumulated at the microirradiated area was photobleached. Bar, 10 μm. (D) The fluorescence recovery of the GFP signal of PARP-1–GFP WT or E988K was quantified and plotted. (Left) Nonmicroirradiated (nondamaged) area. WT, 96.6% ± 13.6% (n = 10); E988K, 96.0% ± 7.9% (n = 13). (Right) Microirradiated area (damaged area). WT, 71.1% ± 16.3% (n = 12); E988K, 41.0% ± 17.6% (n = 11). Standard deviations are shown. (E) In vitro analysis of auto-poly(ADP-ribosyl)ation activity of PARP-1. Immunoblot analyses using anti-poly(ADP-ribosyl)ation antibody (top) and anti-PARP-1 antibody (bottom) are shown.
TIP60 is required for the dynamic binding of PARP-1 to damaged chromatin.
As discussed above, we found that the acetylation of H2AX at Lys5 by TIP60 is required for the auto-poly(ADP-ribosyl)ation activity of PARP-1 after the induction of DNA damage (Fig. 3). Since the poly(ADP-ribosyl)ation activity of PARP-1 is necessary for the dynamic binding of PARP-1 to chromatin, and as it was proposed that auto-poly(ADP-ribosyl)ation could promote the rapid dissociation of PARP-1 from DNA damage sites (Fig. 4) (21), we examined the effect of the TIP60-mediated acetylation of H2AX at Lys5 on the dynamic binding of PARP-1 at the sites of DNA damage, using our FRAP/microirradiation assay, in cells stably expressing PARP-1–GFP (Fig. 5A).
FIG 5.

Acetyltransferase activity of TIP60 is required for PARP-1 dynamics on DNA damage sites. (A) Immunoblot analysis of TIP60 knockdown by shTIP60 in MEF cells in which PARP-1–GFP was stably expressed. Further reconstitution by eTIP60 or by eTIPM was monitored by immunoblot analysis. Anti-GFP and anti-PARP-1 were used to monitor PARP-1–GFP, anti-HA was used for eTIP60 or eTIPM, and anti-TIP60 was used for endogenous TIP60. Anti-beta-actin also was used. (B) FRAP analysis to monitor the dynamics of PARP-1–GFP. One side of the PARP-1–GFP accumulated at the microirradiated area was photobleached. TIP60 knockdown cells that were reconstituted with eTIP60 or eTIPM were used. Bar, 10 μm. (C) Fluorescent recovery of the GFP signal of PARP-1–GFP in either the unirradiated (left; eTIP60 in nondamaged area, 97.9% ± 5.5% [n = 15]; eTIPM, 99.3% ± 8.2% [n = 15]) or microirradiated area (right; eTIP60 in damaged area, 72.2% ± 8.7% [n = 15]; eTIPM, 45.2% ± 18.6% [n = 14]; P value of 4.4 × 10−5 between eTIP60 and eTIPM at the damaged area) was quantified and plotted.
We compared the fluorescence recovery of PARP-1–GFP in cells expressing the TIP60 shRNA to that in cells expressing the mock shRNA. Although the accumulation of PARP-1–GFP at the microirradiated area in TIP60 shRNA-expressing cells was comparable to that in the mock shRNA-expressing cells, the fluorescence recovery of PARP-1–GFP at the damaged area after photobleaching in cells expressing the TIP60 shRNA (54.5% ± 10.5% [n = 12]) was inhibited compared to that in cells expressing the mock shRNA (79.0% ± 10.6% [n = 12]; P value of 1.1 × 10−5 between mock shRNA and shTIP60) (see Fig. S3A and B in the supplemental material). Thus, TIP60 is required for the dynamic binding of PARP-1 to chromatin.
We next examined whether the acetyltransferase activity of TIP60 is required for the PARP-1 dynamics. Either WT TIP60 or TIPM was reconstituted into TIP60 shRNA-expressing MEF cells in which PARP-1–GFP was stably expressed (Fig. 5A). The fluorescence recovery of PARP-1–GFP after microirradiation then was monitored. The fluorescent signal of PARP-1–GFP in the microirradiated area rapidly recovered in the eTIP60 WT-reconstituted cells (72.2% ± 8.7% [n = 15]), while the recovery apparently was reduced in the eTIPM-reconstituted cells (45.3% ± 18.6% [n = 14], P value of 4.4 × 10−5 between eTIP60 and eTIPM reconstituted cells) (Fig. 5B and C). Thus, we concluded that the acetyltransferase activity of TIP60 is required for the PARP-1 dynamics at DNA damage sites.
H2AX acetylation at Lys5 is required for the dynamic binding of PARP-1 to damaged chromatin.
TIP60 acetylates K5 of H2AX after the induction of DNA damage (9, 15–17). Therefore, we first determined whether H2AX is required for the dynamic binding of PARP-1–GFP to chromatin at sites of DNA damage using our FRAP/microirradiation assay. PARP-1–GFP was stably expressed in WT MEF cells or MEF cells in which H2AX was knocked out (H2AX KO) (Fig. 6A). As shown in Fig. S4 in the supplemental material, the fluorescent signal of PARP-1–GFP rapidly recovered in the WT MEF cells (fluorescence recovery, 71.1% ± 14.6% [n = 11]), while its recovery apparently was reduced in the H2AX KO cells (47.3% ± 17.4% [n = 12]; P value of 0.002 between WT and H2AX KO). Thus, H2AX is required for the dynamics of PARP-1.
FIG 6.

K5 acetylation, but not S139 phosphorylation, of H2AX is required for dynamic binding of PARP-1 to DNA damage sites. (A) Immunoblot analysis of H2AX KO MEF cells and that reconstituted with either the WT, K5R, or S139A version of H2AX. PARP-1–GFP also was stably expressed. Anti-GFP, anti-PARP-1, anti-beta-actin, and anti-H2AX were used. (B) FRAP analysis to monitor the dynamics of PARP-1–GFP. PARP-1–GFP accumulated at the microirradiated area was photobleached. H2AX KO MEF cells that were reconstituted with either H2AX WT, H2AX K5R, or H2AX S139A were used. Bar, 10 μm. (C) Fluorescence recovery of the GFP signal of PARP-1–GFP in either the nondamaged (left; H2AX WT, 98.7% ± 9.1% [n = 15]; H2AX K5R, 95.2% ± 6.2% [n = 13]; H2AX S139A, 98.4% ± 7.4% [n = 16]) or damaged area (right; H2AX WT, 71.4% ± 13.0% [n = 14]; H2AX K5R, 49.4% ± 18.3% [n = 14]; H2AX S139A, 72.1% ± 12.6% [n = 16]; P value of 0.88 between H2AX WT- and H2AX S139A-expressing cells and P value of 0.00042 between H2AX K5R- and H2AX S139A-expressing cells) was quantified and plotted.
We then determined whether the K5 acetylation of H2AX is necessary for the dynamic binding of PARP-1–GFP to chromatin in DNA damage sites. We compared the fluorescence recovery of PARP-1–GFP after photobleaching in cells expressing H2AX WT to that in cells expressing H2AX K5R using H2AX KO cells reconstituted with either H2AX WT or H2AX mutants (Fig. 6A). In this experiment, we also included H2AX S139A for the analysis, and it should be noted that for H2AX, the acetylation of K5, but not the phosphorylation of S139, facilitates its exchange at DNA damage sites.
As expected, the fluorescence recovery of PARP-1–GFP in H2AX K5R-expressing cells was inhibited (49.4% ± 18.3% [n = 14]) compared to that in H2AX WT-expressing cells (71.4% ± 13.1% [n = 14]; P value of 0.0011 between H2AX WT- and H2AX K5R-expressing cells) (Fig. 6B and C). However, the recovery of PARP-1–GFP in H2AX S139A-expressing cells was comparable to that in H2AX WT-expressing cells (S139A; 72.1% ± 12.6% [n = 16]; P value of 0.88 between H2AX WT- and H2AX S139A-expressing cells, P value of 0.00042 between H2AX K5R- and H2AX S139A-expressing cells) (Fig. 6B and C). Together, these results implied that the K5 acetylation, but not the S139 phosphorylation, of H2AX by TIP60 activates the ADP-ribosylation activity of PARP-1 in its complex and in turn facilitates the dynamics of PARP-1 at DNA damage sites.
DISCUSSION
Cooperation of PARP-1 and H2AX dynamics during the DNA damage response.
Upon the DNA damage response, PARP-1 is immediately recruited to damaged chromatin. PARP-1 is believed to play important roles in DNA damage recognition, chromatin modulation, and DNA damage signaling. Although PARP-1 is recognized as a crucial target molecule for anticancer therapy, its underlying mechanism in vivo has remained undefined (19, 25, 32). In this study, we found a mutual relationship between PARP-1 activation and K5 acetylation by TIP60 that is dependent on the dynamic exchange of H2AX at DNA damage sites, which might be a crucial link coupling the DNA damage response and chromatin modulation (Fig. 7).
FIG 7.
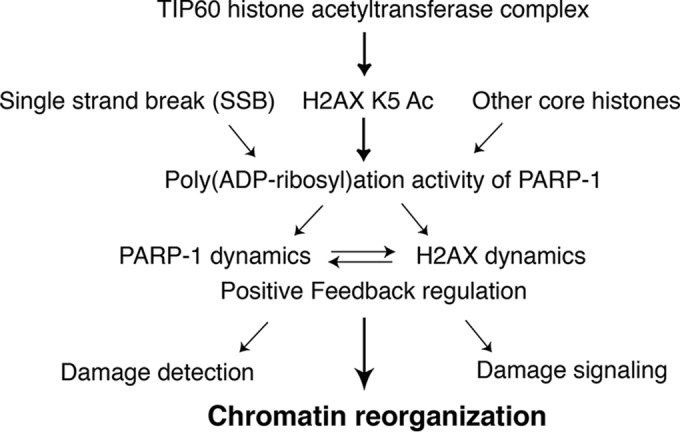
Schematics of cooperation between TIP60-dependent K5 acetylation of H2AX and PARP-1.
We previously reported that H2AX K5 is acetylated by TIP60 (9). We also demonstrated that the acetylation of H2AX (K5), but not its phosphorylation (S139), facilitates the incorporation/eviction of H2AX to/from chromatin at DNA damage sites (9). In this study, we identified PARP-1 as a component of the TIP60 histone acetyltransferase complex and found that they interact regardless of DNA damage. Interestingly, PARP-1 was previously reported to be included in the histone variant H2AX complex (27), and we also detected PARP-1 associated with our purified H2AX in amounts that increase upon the DNA damage response. The increase could be explained by the fact that H2AX associates with the TIP60–PARP-1 complex upon the induction of the DNA damage response. When the eH2AX complex, which was purified from the nuclear extract of gamma-irradiated cells, was subjected to glycerol gradient analysis, copurified PARP-1 comigrated with TIP60. Interestingly, PARP-1 that comigrated with TIP60 ran as a hypershifted form in SDS-PAGE, which is reminiscent of poly(ADP-ribosyl)ation (Fig. 1E). Moreover, when the eH2AX complex was further immunoprecipitated with an anti-PAR antibody, TIP60 was coprecipitated (Fig. 3E). Together, these results suggested that a functional relationship exists between H2AX, TIP60, and PARP-1.
Interestingly, PARP-1 could affect the dynamic behavior of H2AX, as the knockdown of PARP-1 or the treatment with a catalytic inhibitor of PARP-1, olaparib, reduced the exchange rate of H2AX at DNA damage sites. At the same time, the K5 acetylation of H2AX could affect the poly(ADP-ribosyl)ation activity of PARP-1. We found compromised auto-poly(ADP-ribosyl)ation of PARP-1 in cells expressing the mutant eTIP60 that lacks acetyltransferase activity (eTIPM) and in cells that expressed eH2AX K5R (Fig. 3B and D). These results were further confirmed by the FRAP analysis combined with microirradiation. The eH2AX K5R- and eTIPM-expressing cells both presented the compromised fluorescent recovery of PARP-1 at DNA damage sites, indicating that a dynamic property of PARP-1 at DNA damage sites is lost in eH2AX K5R- and eTIPM-expressing cells. Since the dynamic mobility of PARP-1 at DNA damage sites was proposed to be caused by the rapid auto-poly(ADP-ribosyl)ation of PARP-1 (21), we concluded that the loss of the PARP-1 dynamics at DNA damage sites in H2AX K5 acetylation-defective cells is due to the reduction of the poly(ADP-ribosyl)ation activity of PARP-1. These results strongly indicate the close relationship between PARP-1 activity and K5 acetylation-dependent H2AX dynamics. Importantly, the S139 phosphorylation of H2AX in mammalian cells is not required for the dynamics of either H2AX or PARP-1 (9 and this study). Although this contradicts the results obtained from the study in a fly system, where the phosphorylation of the H2A variant (H2Av) controlled PARP-1 upon transcriptional activation, the discrepancy may be due to differences in the biological functions of the H2A variants in the different organisms (33).
Impact of PARP-1 and histone exchange at DNA damage sites.
We previously reported that the K5 acetylation-dependent dynamic exchange of H2AX at DNA damage sites efficiently recruits the DNA double-strand break sensor protein NBS1, possibly to promote HR or DNA checkpoint signaling (15). In fact, the efficient accumulation of NBS1 at DNA damage sites also requires the S139 phosphorylation of H2AX (34). However, these acetylation- or phosphorylation-dependent mechanisms that contribute to NBS1 accumulation onto DNA damage sites seem to be independent processes (15). While the K5 acetylation of H2AX facilitates the rapid assembly of NBS1 onto DNA damage sites, its S139 phosphorylation functions to retain NBS1 onto DNA damage sites, either directly or mediated by the MDC1 protein (35, 36). Interestingly, a defect in PARP-1 also compromises NBS1 accumulation (37). Presently, it is unclear which process of NBS1 accumulation is defective in PARP-1-deficient cells. However, in PARP-1-defective cells, the phosphorylation of H2AX reportedly is not abolished and actually is rather increased (38, 39). Furthermore, in this study we also found that shPARP-1-expressing cells, or cells treated with olaparib, are defective in H2AX exchange at DNA damage sites. Consistent with this, siPARP-1 expression reduced the K5 acetylation of H2AX, and this correlated with the reduced binding of TIP60 to H2AX in those cells. Together, these findings imply that the defect in NBS1 accumulation in PARP-1-deficient cells is at least partly due to the compromised K5 acetylation and dynamics of H2AX upon the DNA damage response.
It is widely known that PARP-1 is involved in SSBR to recruit XRCC1 to DNA breaks. However, growing evidence has indicated that PARP-1 also could be involved in various repair processes, including base excision repair, nucleotide excision repair, and HR, in which single-stranded DNA appears as a repair intermediate (19). Alternatively, PARP-1 may participate in chromatin reorganization at DNA damage sites to facilitate their sensing or repair (22, 32). Chromatin loosening at DNA damage sites could be facilitated by the cooperation between PARP-1, H2AX, and TIP60. Indeed, TIP60 is known to promote HR (40), and whether PARP-1 and its cooperation with H2AX dynamics are involved in the same pathway should be clarified in the future.
H2AX K5 acetylation-dependent activation of PARP-1 in DNA damage response.
The role of PARP-1 in chromatin modulation is important for transcriptional activation (20, 41). To facilitate the recruitment of transcription factors, the chromatin must be remodeled. Chromatin decondensation or histone exchange at transcription sites occurs in response to extracellular stress such as heat shock, and the activation of PARP-1 promotes this process (33, 42, 43). PARP-1 activation at transcription sites is likely to require nucleosomes, as they can accommodate PARP-1 (44). In turn, the enzymatic activity of PARP-1 is probably activated by histones, and then it could modulate the chromatin structure (45, 46).
In the case of the DNA damage response, the proposed model is that single-stranded DNA is a potent activator of the poly(ADP-ribosyl)ation activity (19). However, it is possible that nucleosomes or histones facilitate PARP-1 action upon the DNA damage response, as well as upon transcriptional activation. As mentioned above, we have found that the K5 acetylation of H2AX is responsible for PARP-1 activation during the DNA damage response. It is possible that canonical H2A activates PARP-1; however, H2A is much less dynamic at DNA damage sites (9), and in this regard, it is not likely that H2A has a role in activating PARP-1. PARP-1 could become activated by its dynamic assembly onto chromatin, and H2AX K5-dependent dynamic histone exchange itself could promote the poly(ADP-ribosyl)ation activity of PARP-1. Alternatively, the DNA damage-dependent formation of the H2AX-TIP60 complex could be a potent activator of PARP-1. In any case, it is plausible that the cooperative activation of PARP-1 and K5-dependent H2AX exchange could facilitate the DNA repair activity in the context of chromatin in vivo.
PARP-1-dependent histone exchange as a potential target for tumor therapy.
PARP-1 inhibition effectively kills tumor cells that lack the tumor suppressor BRCA1 or BRCA2 and cells with compromised HR activity (47, 48). It was previously thought that compromising PARP-1 causes an increase of DNA SSBs, which could be converted into irreparable DNA DSBs (double-stranded breaks) during DNA replication and become toxic to the HR-defective tumor cells. This model was based on the idea that PARP-1 was involved in SSBR. However, subsequent studies revealed that the cellular level of SSBs does not increase after PARP-1 inhibition (49).
Furthermore, growing evidence has suggested the involvement of PARP-1 upon the reactivation of stalled replication forks (19, 38). For example, PARP-1 could help control the reversed replication forks that appear upon treatments with low doses of topoisomerase poison (50). PARP-1 could protect the HR substrate, which is generated by a collapsed replication fork, from “toxic” nonhomologous end-joining (NHEJ) repair (51, 52). The specific PARP-1 function that affects the lethality in tumor cells still needs to be investigated. However, we propose that chromatin modulating function in the DNA repair process also should be considered. Indeed, in the case of the regressed fork, nucleosomes are immediately assembled on the regressed strand (53), implying that the histone exchange plays an active role in promoting the processing of the regressed end to reactivate the replication fork. The possible functions of histone exchange in tumor cells and its role in DNA metabolism require further clarification. In fact, in this study, we showed that the anticancer drug olaparib inhibits H2AX dynamics (Fig. 2H). Detailed knowledge of how PARP-1 promotes H2AX dynamics and how the K5 acetylation of H2AX by TIP60 affects PARP-1 activity may be the key to the exploration of novel antitumor therapeutic targets.
Supplementary Material
ACKNOWLEDGMENTS
We thank S. Matsuda (Kyoto University) and A. Kakino (Shinshu University) for discussions and comments on the manuscript and C. Asano for assistance. We also thank M. Masutani (Nagasaki University) for providing the PARP-1 KO MEF cells.
This work was supported by grants-in-aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01085-15.
REFERENCES
- 1.van Attikum H, Gasser SM. 2009. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol 19:207–217. doi: 10.1016/j.tcb.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 2.Berger SL. 2002. Histone modifications in transcriptional regulation. Curr Opin Genet Dev 12:142–148. doi: 10.1016/S0959-437X(02)00279-4. [DOI] [PubMed] [Google Scholar]
- 3.van Attikum H, Gasser SM. 2005. The histone code at DNA breaks: a guide to repair? Nat Rev Mol Cell Biol 6:757–765. doi: 10.1038/nrm1737. [DOI] [PubMed] [Google Scholar]
- 4.Mizuguchi G, Shen X, Landry J, Wu WH, Sen S, Wu C. 2004. ATP-driven exchange of histone H2AZ variant catalyzed by SWR1 chromatin remodeling complex. Science 303:343–348. doi: 10.1126/science.1090701. [DOI] [PubMed] [Google Scholar]
- 5.Wu WH, Alami S, Luk E, Wu CH, Sen S, Mizuguchi G, Wei D, Wu C. 2005. Swc2 is a widely conserved H2AZ-binding module essential for ATP-dependent histone exchange. Nat Struct Mol Biol 12:1064–1071. doi: 10.1038/nsmb1023. [DOI] [PubMed] [Google Scholar]
- 6.Kusch T, Florens L, Macdonald WH, Swanson SK, Glaser RL, Yates JR III, Abmayr SM, Washburn MP, Workman JL. 2004. Acetylation by Tip60 is required for selective histone variant exchange at DNA lesions. Science 306:2084–2087. doi: 10.1126/science.1103455. [DOI] [PubMed] [Google Scholar]
- 7.Morrison AJ, Shen X. 2005. DNA repair in the context of chromatin. Cell Cycle 4:568–571. [PubMed] [Google Scholar]
- 8.Park YJ, Luger K. 2008. Histone chaperones in nucleosome eviction and histone exchange. Curr Opin Struct Biol 18:282–289. doi: 10.1016/j.sbi.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ikura T, Tashiro S, Kakino A, Shima H, Jacob N, Amunugama R, Yoder K, Izumi S, Kuraoka I, Tanaka K, Kimura H, Ikura M, Nishikubo S, Ito T, Muto A, Miyagawa K, Takeda S, Fishel R, Igarashi K, Kamiya K. 2007. DNA damage-dependent acetylation and ubiquitination of H2AX enhances chromatin dynamics. Mol Cell Biol 27:7028–7040. doi: 10.1128/MCB.00579-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soria G, Polo SE, Almouzni G. 2012. Prime, repair, restore: the active role of chromatin in the DNA damage response. Mol Cell 46:722–734. doi: 10.1016/j.molcel.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 11.Scully R, Xie A. 2013. Double strand break repair functions of histone H2AX. Mutat Res 750:5–14. doi: 10.1016/j.mrfmmm.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stucki M, Jackson SP. 2006. γH2AX and MDC1: anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair (Amst) 5:534–543. doi: 10.1016/j.dnarep.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 13.Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. 2005. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 123:1213–1226. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 14.Lou Z, Minter-Dykhouse K, Franco S, Gostissa M, Rivera MA, Celeste A, Manis JP, van Deursen J, Nussenzweig A, Paull TT, Alt FW, Chen J. 2006. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol Cell 21:187–200. doi: 10.1016/j.molcel.2005.11.025. [DOI] [PubMed] [Google Scholar]
- 15.Ikura M, Furuya K, Matsuda S, Matsuda R, Shima H, Adachi J, Matsuda T, Shiraki T, Ikura T. 2015. Acetylation of histone H2AX at Lys5 by the TIP60 histone acetyltransferase complex is essential for the dynamic binding of NBS1 to damaged chromatin. Mol Cell Biol 35:4147–4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamagata K, Kitabayashi I. 2009. Sirt1 physically interacts with Tip60 and negatively regulates Tip60-mediated acetylation of H2AX. Biochem Biophys Res Commun 390:1355–1360. doi: 10.1016/j.bbrc.2009.10.156. [DOI] [PubMed] [Google Scholar]
- 17.Hu R, Wang E, Peng G, Dai H, Lin SY. 2013. Zinc finger protein 668 interacts with Tip60 to promote H2AX acetylation after DNA damage. Cell Cycle 12:2033–2041. doi: 10.4161/cc.25064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang L, Xie L, Ramachandran S, Lee Y, Yan Z, Zhou L, Krajewski K, Liu F, Zhu C, Chen DJ, Strahl BD, Jin J, Dokholyan NV, Chen X. 2015. Non-canonical bromodomain within DNA-PKcs promotes DNA damage response and radioresistance through recognizing an IR-induced acetyl-lysine on H2AX. Chem Biol 22:849–861. doi: 10.1016/j.chembiol.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caldecott KW. 2014. Protein ADP-ribosylation and the cellular response to DNA strand breaks. DNA Repair (Amst) 19:108–113. doi: 10.1016/j.dnarep.2014.03.021. [DOI] [PubMed] [Google Scholar]
- 20.D'Amours D, Desnoyers S, D'Silva I, Poirier GG. 1999. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J 342(Part 2):249–268. [PMC free article] [PubMed] [Google Scholar]
- 21.Mortusewicz O, Ame JC, Schreiber V, Leonhardt H. 2007. Feedback-regulated poly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA damage in living cells. Nucleic Acids Res 35:7665–7675. doi: 10.1093/nar/gkm933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caldecott KW. 2007. Mammalian single-strand break repair: mechanisms and links with chromatin. DNA Repair (Amst) 6:443–453. doi: 10.1016/j.dnarep.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 23.Zahradka P, Ebisuzaki K. 1982. A shuttle mechanism for DNA-protein interactions. The regulation of poly(ADP-ribose) polymerase. Eur J Biochem 127:579–585. [PubMed] [Google Scholar]
- 24.Ferro AM, Olivera BM. 1982. Poly(ADP-ribosylation) in vitro. Reaction parameters and enzyme mechanism. J Biol Chem 257:7808–7813. [PubMed] [Google Scholar]
- 25.Langelier MF, Pascal JM. 2013. PARP-1 mechanism for coupling DNA damage detection to poly(ADP-ribose) synthesis. Curr Opin Struct Biol 23:134–143. doi: 10.1016/j.sbi.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hassler M, Ladurner AG. 2012. Towards a structural understanding of PARP1 activation and related signalling ADP-ribosyl-transferases. Curr Opin Struct Biol 22:721–729. doi: 10.1016/j.sbi.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 27.Heo K, Kim H, Choi SH, Choi J, Kim K, Gu J, Lieber MR, Yang AS, An W. 2008. FACT-mediated exchange of histone variant H2AX regulated by phosphorylation of H2AX and ADP-ribosylation of Spt16. Mol Cell 30:86–97. doi: 10.1016/j.molcel.2008.02.029. [DOI] [PubMed] [Google Scholar]
- 28.Ikura T, Ogryzko VV, Grigoriev M, Groisman R, Wang J, Horikoshi M, Scully R, Qin J, Nakatani Y. 2000. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell 102:463–473. doi: 10.1016/S0092-8674(00)00051-9. [DOI] [PubMed] [Google Scholar]
- 29.Nakatani Y, Ogryzko V. 2003. Immunoaffinity purification of mammalian protein complexes. Methods Enzymol 370:430–444. doi: 10.1016/S0076-6879(03)70037-8. [DOI] [PubMed] [Google Scholar]
- 30.Tsukuda T, Fleming AB, Nickoloff JA, Osley MA. 2005. Chromatin remodelling at a DNA double-strand break site in Saccharomyces cerevisiae. Nature 438:379–383. doi: 10.1038/nature04148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nishibuchi I, Suzuki H, Kinomura A, Sun J, Liu NA, Horikoshi Y, Shima H, Kusakabe M, Harata M, Fukagawa T, Ikura T, Ishida T, Nagata Y, Tashiro S. 2014. Reorganization of damaged chromatin by the exchange of histone variant H2A.Z-2. Int J Radiat Oncol Biol Phys 89:736–744. doi: 10.1016/j.ijrobp.2014.03.031. [DOI] [PubMed] [Google Scholar]
- 32.Kim MY, Zhang T, Kraus WL. 2005. Poly(ADP-ribosyl)ation by PARP-1: “PAR-laying” NAD+ into a nuclear signal. Genes Dev 19:1951–1967. doi: 10.1101/gad.1331805. [DOI] [PubMed] [Google Scholar]
- 33.Kotova E, Lodhi N, Jarnik M, Pinnola AD, Ji Y, Tulin AV. 2011. Drosophila histone H2A variant (H2Av) controls poly(ADP-ribose) polymerase 1 (PARP1) activation in chromatin. Proc Natl Acad Sci U S A 108:6205–6210. doi: 10.1073/pnas.1019644108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Celeste A, Fernandez-Capetillo O, Kruhlak MJ, Pilch DR, Staudt DW, Lee A, Bonner RF, Bonner WM, Nussenzweig A. 2003. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat Cell Biol 5:675–679. doi: 10.1038/ncb1004. [DOI] [PubMed] [Google Scholar]
- 35.Lukas C, Melander F, Stucki M, Falck J, Bekker-Jensen S, Goldberg M, Lerenthal Y, Jackson SP, Bartek J, Lukas J. 2004. Mdc1 couples DNA double-strand break recognition by Nbs1 with its H2AX-dependent chromatin retention. EMBO J 23:2674–2683. doi: 10.1038/sj.emboj.7600269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kobayashi J, Tauchi H, Sakamoto S, Nakamura A, Morishima K, Matsuura S, Kobayashi T, Tamai K, Tanimoto K, Komatsu K. 2002. NBS1 localizes to gamma-H2AX foci through interaction with the FHA/BRCT domain. Curr Biol 12:1846–1851. doi: 10.1016/S0960-9822(02)01259-9. [DOI] [PubMed] [Google Scholar]
- 37.Haince JF, McDonald D, Rodrigue A, Dery U, Masson JY, Hendzel MJ, Poirier GG. 2008. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J Biol Chem 283:1197–1208. doi: 10.1074/jbc.M706734200. [DOI] [PubMed] [Google Scholar]
- 38.Helleday T. 2011. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol 5:387–393. doi: 10.1016/j.molonc.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fisher AE, Hochegger H, Takeda S, Caldecott KW. 2007. Poly(ADP-ribose) polymerase 1 accelerates single-strand break repair in concert with poly(ADP-ribose) glycohydrolase. Mol Cell Biol 27:5597–5605. doi: 10.1128/MCB.02248-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang J, Cho NW, Cui G, Manion EM, Shanbhag NM, Botuyan MV, Mer G, Greenberg RA. 2013. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat Struct Mol Biol 20:317–325. doi: 10.1038/nsmb.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kraus WL, Hottiger MO. 2013. PARP-1 and gene regulation: progress and puzzles. Mol Aspects Med 34:1109–1123. doi: 10.1016/j.mam.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 42.Petesch SJ, Lis JT. 2012. Activator-induced spread of poly(ADP-ribose) polymerase promotes nucleosome loss at Hsp70. Mol Cell 45:64–74. doi: 10.1016/j.molcel.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O'Donnell A, Yang SH, Sharrocks AD. 2013. PARP1 orchestrates variant histone exchange in signal-mediated transcriptional activation. EMBO Rep 14:1084–1091. doi: 10.1038/embor.2013.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clark NJ, Kramer M, Muthurajan UM, Luger K. 2012. Alternative modes of binding of poly(ADP-ribose) polymerase 1 to free DNA and nucleosomes. J Biol Chem 287:32430–32439. doi: 10.1074/jbc.M112.397067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pinnola A, Naumova N, Shah M, Tulin AV. 2007. Nucleosomal core histones mediate dynamic regulation of poly(ADP-ribose) polymerase 1 protein binding to chromatin and induction of its enzymatic activity. J Biol Chem 282:32511–32519. doi: 10.1074/jbc.M705989200. [DOI] [PubMed] [Google Scholar]
- 46.Kim MY, Mauro S, Gevry N, Lis JT, Kraus WL. 2004. NAD+-dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP-1. Cell 119:803–814. doi: 10.1016/j.cell.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 47.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. 2005. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 48.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A. 2005. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 49.Gottipati P, Vischioni B, Schultz N, Solomons J, Bryant HE, Djureinovic T, Issaeva N, Sleeth K, Sharma RA, Helleday T. 2010. Poly(ADP-ribose) polymerase is hyperactivated in homologous recombination-defective cells. Cancer Res 70:5389–5398. doi: 10.1158/0008-5472.CAN-09-4716. [DOI] [PubMed] [Google Scholar]
- 50.Ray Chaudhuri A, Hashimoto Y, Herrador R, Neelsen KJ, Fachinetti D, Bermejo R, Cocito A, Costanzo V, Lopes M. 2012. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat Struct Mol Biol 19:417–423. doi: 10.1038/nsmb.2258. [DOI] [PubMed] [Google Scholar]
- 51.Hochegger H, Dejsuphong D, Fukushima T, Morrison C, Sonoda E, Schreiber V, Zhao GY, Saberi A, Masutani M, Adachi N, Koyama H, de Murcia G, Takeda S. 2006. Parp-1 protects homologous recombination from interference by Ku and Ligase IV in vertebrate cells. EMBO J 25:1305–1314. doi: 10.1038/sj.emboj.7601015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Patel AG, Sarkaria JN, Kaufmann SH. 2011. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc Natl Acad Sci U S A 108:3406–3411. doi: 10.1073/pnas.1013715108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sogo JM, Lopes M, Foiani M. 2002. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 297:599–602. doi: 10.1126/science.1074023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.