

Abstract
Cell fate specification requires precise coordination of transcription factors and their regulators to achieve fidelity and flexibility in lineage allocation. The transcriptional repressor growth factor independence 1 (GFI1) is comprised of conserved Snail/Slug/Gfi1 (SNAG) and zinc finger motifs separated by a linker region poorly conserved with GFI1B, its closest homolog. Moreover, GFI1 and GFI1B coordinate distinct developmental fates in hematopoiesis, suggesting that their functional differences may derive from structures within their linkers. We show a binding interface between the GFI1 linker and the SP-RING domain of PIAS3, an E3-SUMO (small ubiquitin-related modifier) ligase. The PIAS3 binding region in GFI1 contains a conserved type I SUMOylation consensus element, centered on lysine-239 (K239). In silico prediction algorithms identify K239 as the only high-probability site for SUMO modification. We show that GFI1 is modified by SUMO at K239. SUMOylation-resistant derivatives of GFI1 fail to complement Gfi1 depletion phenotypes in zebrafish primitive erythropoiesis and granulocytic differentiation in cultured human cells. LSD1/CoREST recruitment and MYC repression by GFI1 are profoundly impaired for SUMOylation-resistant GFI1 derivatives, while enforced expression of MYC blocks granulocytic differentiation. These findings suggest that SUMOylation within the GFI1 linker favors LSD1/CoREST recruitment and MYC repression to govern hematopoietic differentiation.
INTRODUCTION
The molecular machinery governing multipotential cell fate is generally characterized by flexibility and combinatorial diversity to achieve distinct outcomes from a limited collection of tools. This principle is observed commonly in hematopoiesis, where factors may contribute to maintenance of stem cells and uncommitted progenitors yet also may be critical determinants of terminal differentiation. The growth factor independence (GFI) family of transcriptional regulators, comprised of GFI1 and GFI1B, typifies this principle.
GFI1 was identified in a Moloney murine leukemia virus (MoMuLV) insertional mutagenesis screen for factors conferring interleukin-2 (IL-2)-independent growth upon cultured lymphocytes (1). GFI1B was discovered from its homology to GFI1 (2, 3). Both proteins harbor nearly identical Snail/Slug/Gfi1 (SNAG) domains and a highly conserved concatemer of six C2H2-type zinc fingers (ZnFs) at their N and C termini, respectively. Linker regions with limited conservation separate the SNAG and ZnF motifs (4). SNAG domains confer recruitment of LSD1 and its binding partner CoREST, which figure prominently in transcriptional repression by GFI proteins (5). Sequence-specific DNA binding to a common response element [TAAATCAC(A/T)GCA; response element in boldface] is coordinated by ZnFs 3, 4, and 5 in both proteins (2, 6, 7), while ZnFs 1, 2, and 6 and the linkers provide interacting surfaces for transcriptional partners and epigenetic regulators. Despite extraordinary conservation within their SNAG and ZnF regions, GFI1 and GFI1B support distinct fates in hematopoietic differentiation (4, 8, 9).
GFI1 regulates hematopoietic stem cell (HSC) self-renewal and maintains HSC quiescence (10–12). Early stages in B-cell and T-cell lymphopoiesis (B- and T-lymphopoiesis, respectively) and T-cell subset allocation in the adaptive immune response also require GFI1 (13, 14). Within the myelo-erythroid compartment, GFI1 is required for both qualitative and quantitative aspects of granulocyte development. Two dominant mutations in GFI1, N382S and K403R, cause severe congenital neutropenia (SCN) type 2 (15, 16), while compound heterozygosity for these mutations has been described in cyclic neutropenia (17). Additionally, reduced GFI1 expression cooperates with mutant C/EBPε in specific granule deficiency (SGD) (18). Outside the hematopoietic compartment, GFI1 plays crucial roles in sensorineural, neuroendocrine, and intestinal secretory lineage development (19–21). Gfi1−/−mice are viable and notably recapitulate these human phenotypes (14, 19, 21–23). In contrast, Gfi1b expression appears largely limited to hematopoietic tissues, and its effects are complementary or mutually exclusive to those of Gfi1 (24). Gfi1b is required for erythroid and megakaryocyte fate specification, and nullizygosity for Gfi1b is lethal by embryonic day 15 (E15) due to failure of definitive erythropoiesis (25). Enforced expression of GFI1B induces T-cell lymphopenia (T-lymphopenia) and impairs granulocytic differentiation in vitro (2). These findings suggest that GFI1 and GFI1B can have oppositional roles in lineage allocation (26–28).
Features that functionally distinguish GFI1 from GFI1B are poorly understood, and their distinct roles in hematopoiesis are not completely explained by their patterns of expression. Reporter mouse strains reveal complex expression patterns for both factors (24, 26). During hematopoiesis, both coexpression and lineage-restricted, mutually exclusive expression are observed for Gfi1 and Gfi1b, suggesting that there are points in development when cells must distinguish between them. For example, despite coexpression in HSCs, Gfi1b does not complement the stem cell defect in Gfi1−/− mice (10). Likewise, hematopoietic defects in Gfi1−/− mice are only partially reversed by GFI1B expression from the endogenous Gfi1 promoter, and the defect in sensorineural development is unaffected (23). These findings imply a surprising complexity in systems governing GFI family member function and suggest that elements of primary structure unique to each protein may provide a platform for differential regulation and function.
The human genome encodes three small ubiquitin-related modifier (SUMO) proteins (29, 30) that can be attached to target substrates to modulate their functions. SUMOylation regulates diverse cellular processes, including protein turnover, nucleocytoplasmic transport, subnuclear organization, chromatin structure, and transcriptional control. SUMOs are covalently linked to lysine ε-amino groups in target substrates through coordinated actions of three enzymes (29, 31). An E1 SUMO-activating enzyme links the SUMO C terminus to its active-site cysteine and then transfers SUMO to UBC9, the single E2 SUMO-conjugating enzyme. E3 SUMO ligases then facilitate SUMO transfer to target proteins. Prominent among E3 SUMO ligases are PIAS (protein inhibitor of activated STAT) family proteins (32). PIAS proteins contain a SIZ/PIAS (SP)-RING domain, analogous to the RING domain of E3 ubiquitin ligases, as well as predicted SUMO-interacting motifs (SIM) (29, 33, 34). Through simultaneous binding of UBC9, its bound SUMO, and the target protein, E3 ligases confer substrate and SUMO paralog specificity. Approximately 75% of SUMO-conjugated lysines are found within type I consensus elements, ψKX(D/E), where ψ is a large hydrophobic residue and X is any amino acid (35–37).
A growing roster of transcriptional regulators is being recognized as targets for SUMO conjugation, either via confirmed SUMO attachments or inferred from binding relationships with the SUMOylation machinery (29, 30). We show that GFI1 is modified by SUMO. Mutation of lysine-239 (K239) within the GFI1 linker profoundly limits SUMO modification. GFI1 K239 exists within a type I SUMOylation consensus element embedded in its PIAS3 binding site. Reciprocal mapping studies reveal a binding interface between the PIAS3 SP-RING domain and the C-terminal half of the GFI1 linker. K239-dependent SUMO modification impacts GFI1's role in transcriptional repression and supports both zebrafish primitive erythropoiesis and granulocyte differentiation in HL-60 cells in response to all-trans retinoic acid (ATRA). We show that MYC expression declines during granulocytic differentiation yet remains elevated with GFI1 depletion. Wild-type GFI1 expression restores both MYC suppression and granulocytic differentiation to HL-60 cells depleted of GFI1, yet SUMOylation-resistant GFI1 derivatives fail to do so. Likewise, SUMOylation-resistant GFI1 is impaired in LSD1 and CoREST binding, which figures prominently in GFI1-mediated transcriptional repression. These results define a structural motif in GFI1 that alters its recruitment of LSD1/CoREST to govern its transcriptional repressor function in hematopoietic development.
MATERIALS AND METHODS
Reagents and antibodies.
Mouse monoclonal anti-Flag (M2), antitubulin (clone B-5-1-2), anti-PIAS3 (clone PIA3), rabbit anti-Flag (F7425), normal mouse serum (NMS), and normal goat serum (NGS) were obtained from Sigma. Mouse monoclonal anti-myc (9E10) was acquired from Vanderbilt University Medical Center and rabbit polyclonal anti-myc (A-14) was obtained from Santa Cruz Biotechnology, Inc. Mouse monoclonal anti-green fluorescent protein (anti-GFP) (7.1/13.1) was obtained from Roche Applied Science, Inc. Rabbit polyclonal anti-SUMO (ab30258) and anti-GFI1 (ab55108) antibodies were bought from Abcam. Horseradish peroxidase (HRP)-conjugated anti-mouse, anti-rabbit, anti-rat, and anti-goat IgG were obtained from Jackson ImmunoResearch. Alexa Fluor 488-conjugated anti-rabbit IgG was purchased from ThermoFisher Scientific, and anti-CoREST monoclonal antibody (D612U) was purchased from Cell Signaling Technologies. Lipofectamine, Mirus, and To-Pro-3 iodide were obtained from Invitrogen. Protein G-Sepharose was purchased from Sigma-Aldrich. Fugene-6 and a Dual-Luciferase assay kit were purchased from Promega. HCI-2509 synthesis, purification, and characterization have been previously described (38). Puromycin (puro), hygromycin (hygro), o-dianisidine, and Polybrene were purchased from Sigma. Restriction endonucleases, polymerases, and ligases were purchased from New England Biosciences. All other materials were of reagent grade.
Plasmids and subcloning.
Expression plasmid pCS3-6×-myc:PIAS3α (with six copies of the myc tag) and its derivative fragments consisting of PIAS3 amino acids 1 to 273, 274 to 584, 274 to 392, and 393 to 584 have been previously described (39). Plasmids expressing Flag-tagged PIAS1, PIASxα, PIASxβ, PIAS3α, PIASy, HDAC1, and β-catenin have been described previously (39, 40). The GFI1:3×Flag (with three copies of the Flag tag) expression vector was a generous gift from Tarik Moroy and has been previously described (28, 41). The pcDNA3.1+-6×-myc expression vector was created by subcloning the 6×myc coding sequence, bounded by BamH1 and EcoR1 restriction sites, into BamH1/EcoR1-restricted pcDNA3.1+. Rat Gfi1 cDNA was subcloned into EcoR1/XbaI-restricted pcDNA3.1+-6×-myc. cDNAs for N-terminal and C-terminal deletion (ΔN and ΔC, respectively) derivatives of rat Gfi1 were generated by PCR. Amplimers were restricted and subcloned into pcDNA3.1+-6×-myc. The K239R and E241Q substitutions in rat GFI1 were created by two-stage PCR and splicing by overlap extension (42). Expression constructs for 3×Flag-tagged SUMO1, SUMO2, and SUMO3 were generated by PCR and subcloned into pCMV5 and eMIG′+. Retroviruses expressing GFI1:3×Flag, GFI1-K239R:3×-Flag, and GFI1-E241Q:3×Flag were produced using Gateway technology with the pDONR221 and pMIP.RFA plasmids. Retroviruses expressing Flag-tagged SUMO and GFI1 constructs were collected from the supernatants of transfected HEK293T cells. The pEIZ-MYC and pEIT vector control were generous gifts of Don Ayer. All constructs were confirmed by automated dideoxy sequencing at the University of Utah sequencing core facility. Primer sequences and further details of subcloning strategies used to generate constructs are available upon request.
Cell culture, transient transfections, and retroviral transduction.
COS7L, HEK293T (American Type Culture Collection), and 293-T-REx-5×Gal-luciferase cells (generously provided by Raphael Margueron) were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). NIH 3T3 fibroblasts were propagated in DMEM supplemented with 10% bovine calf serum (BCS). HL-60 cells were maintained in Iscove's modified Dulbecco's medium (IMDM) supplemented with 20% FBS. All cell culture media were supplemented with 2 mM l-glutamine, 50 units/ml penicillin, and 50 μg/ml streptomycin. Transient transfections in COS7L cells were performed with Lipofectamine per the manufacturer's instructions. For retrovirus production in HEK293T cells, monolayers were transfected using Fugene-6 or Mirus, and then supernatants containing viral particles were collected on consecutive days and clarified by centrifugation. HL-60 cells were transduced with retroviral particles by successive spinoculation in the presence of 8 μg/ml Polybrene. Selection in 4 μg/ml puromycin was conducted to generate stable cells. For differential RNA interference (RNAi) experiments, HL-60 cells were transduced with pMKO1-puro expressing a GFI1-targeted small hairpin RNA (shRNA) or a scrambled control and selected in puromycin to establish stable isolates. Expression of Flag-tagged GFI1 or its K239R or E241Q derivatives was restored in GFI1-depleted HL-60 cells by retroviral transduction using a pMSCV-hygro (where MSCV is murine stem cell virus) vector containing the appropriate cDNAs, followed by selection in hygromycin.
Immune precipitation and immunoblotting.
Following enforced expression of the proteins indicated in the figure legends or those expressed endogenously, immune precipitation and/or immunoblotting was performed essentially as described previously using antibodies indicated in figure legends (43).
Transcriptional reporter assays.
293-T-REx-5×Gal-luciferase cells were transfected with pRL and expression plasmids for Gal4 fusion constructs as shown in figures and referred to in the figure legends. LSD1 inhibitor HCI-2509 was employed at 100 nM in the indicated experiments. Luciferase activities were determined essentially as described previously (44). Statistical significance was determined via an unpaired Student t test in GraphPad Prism, version 6.0.
TCA precipitation/SUMOylation assays.
COS7L cells expressing Flag-tagged GFI1, GFI1-K239R, GFI1-E241Q, β-catenin, or HDAC1 were harvested in ice-cold phosphate-buffered saline (PBS) and collected by centrifugation. Cell pellets were washed with PBS and then snap-frozen in liquid nitrogen (LN2). Cell pellets were thawed into 20% trichloroacetic acid (TCA) and subjected to sonication. Precipitating material was collected by centrifugation and then solubilized in 1× SDS-PAGE buffer supplemented with 0.5 mg/ml bovine serum albumin. This solution was neutralized to pH 7.5 with Tris base and then boiled. Flag-tagged proteins were analyzed by SDS-PAGE and immunoblotting or were collected by immune precipitation with rabbit anti-Flag antibody and protein G-Sepharose beads. Immune complexes were fractionated by SDS-PAGE and then subjected to immunoblotting.
Immunofluorescence immunohistochemistry.
NIH 3T3 fibroblasts stably expressing Flag-tagged GFI1 or GFI1-K239R were grown on coverslips, fixed in buffered 3.7% formalin, washed with PBS, made permeable with 1% Triton X-100, again washed with PBS, and blocked with 10% NGS in PBS. GFI1 or GFI1-K239R was detected with rabbit anti-Flag followed by Alexa Fluor 488-conjugated goat anti-rabbit secondary antibody and epifluorescence microscopy. Nuclei were revealed with To-Pro-3 iodide.
Morpholino injection.
Endogenous gfi1aa depletion in zebrafish was achieved using a splice-blocking, antisense morpholino oligonucleotide (5′-CCAATCTAGCCTGAAAATGGCACAA-3′) (Gene Tools, LLC) targeting the intron 1/exon 2 boundary of the primary transcript. Morpholinos were injected at the one-cell stage using 1 nl of a 20 μM solution. Wild-type and mutant rescue constructs were subcloned into pCS2+, and RNA was synthesized through in vitro mMessage mMachine transcription reactions (Ambion, Inc.). Rescue RNA constructs were coinjected with the morpholino at 100 ng/μl. Expression of GFI1 and its derivatives in zebrafish embryos was determined by Western blotting directed at the Flag epitope tag at the C terminus of each protein. Experiments with zebrafish were conducted under University of Utah IACUC approved protocol 14-07007.
Reverse transcriptase PCR.
Total RNAs were isolated from zebrafish embryos, treated as shown in Fig. 4, at 48 h postfertilization (hpf) using Trizol and reverse transcribed. Primers hybridized within intron 1 (sense) and exon 2 (antisense) to amplify a 169-bp sequence spanning the intron 1-exon 2 boundary in unspliced gfi1aa mRNA. A second sense primer in exon 1 paired with the exon 2 antisense primer was used to amplify a 263-bp fragment in spliced gfi1aa mRNA. Primer sequences are available by request.
FIG 4.

GFI1 SUMOylation on K239 is required for zebrafish primitive erythropoiesis. (A) One-cell-stage zebrafish embryos were injected with gfi1aa splice-blocking morpholino, alone or in combination with RNA expressing wild-type GFI1, GFI1-K239R, or GFI1-E241Q, each with a C-terminal Flag epitope tag. At 48 h postfertilization (hpf), primitive erythropoiesis was revealed by o-dianisidine staining. Four representative embryos are shown out of 200 embryos scored for each knockdown/rescue combination. Arrows in uninjected controls show the zone of primitive erythropoiesis. Primitive erythropoiesis was quantified using a graded scoring system. Scores of 1 to 4 were assigned for each embryo to indicate none, modest, moderate, or complete hemoglobinization, respectively. Statistical significance was assessed using a Wilcoxon-Mann-Whitney test (***, P < 0.0005). (B) Morpholino-induced splicing blockade of gfi1aa in zebrafish. Total RNA was isolated from uninjected (U) zebrafish embryos or after injection of a scrambled control (Sc) or gfi1aa splice-blocking morpholino (SB) targeting the boundary between intron 1 and exon 2, as shown (thick line). Unspliced gfi1aa cDNA was amplified with primers a and b spanning the intron 1-exon 2 boundary to yield a 169-bp amplimer. Spliced gfi1aa mRNA was amplified using primers c and b to yield a 263-bp amplimer. (C) Expression of Flag-tagged GFI1, GFI1-K239R (K>R), and GFI1-E241Q (E>Q) derivatives in extracts prepared from the equivalent of eight zebrafish embryos at 24 h postinjection.
o-Dianisidine staining.
Hemoglobinized cells of the developing zebrafish were visualized using o-dianisidine staining. Briefly, embryos were collected and dechorionated by pronase digestion at 3 mg/ml. Embryos were anesthetized with 0.2% tricaine for 40 min. Embryos at 48 hpf were stained in the dark on a rolling rotisserie for 2 h in a freshly prepared solution composed of 0.6 mg/ml o-dianisidine in 40% ethanol (EtOH), 0.65% H2O2, and 0.01 M sodium acetate. Embryos were thoroughly washed with PBS-Tween 20 (PBS-T) and photographed on an Olympus SZX16 dissecting microscope equipped with an Olympus DP71 camera. Scores from 1 to 4, representing none to complete hemoglobinization, respectively, were assigned for 200 embryos by an investigator blinded to the experimental conditions, and statistical significance was determined by Wilcoxon-Mann-Whitney testing in GraphPad Prism, version 6.0.
HL-60 cell differentiation.
Naive HL-60 cells, scrambled controls, cells having undergone GFI1 depletion with or without rescue with GFI1 or its K239R or E241Q derivative, or those with enforced MYC expression were treated with 0.1 μM ATRA or vehicle for 4 days. Aliquots were removed during treatment to analyze CD11B cell surface expression by flow cytometry and at the end of the experiment to confirm expression from rescue constructs. On day 4, cells were transferred to glass slides by cytospin, subjected to Wright staining, and scored in a blinded fashion for morphological maturation. Promyelocyte, myelocyte, and metamyelocyte morphologies were scored as immature, while cells whose nuclei displayed band form or segmented morphologies were scored as mature. In parallel, total RNA was collected from stable isolates using an RNeasy minikit (Qiagen). Changes in CEBPA, MYC, and AZU gene expression levels were assessed by quantitative reverse transcription-PCR (qRT-PCR) using the ΔΔCT (where CT is threshold cycle) method with β-glucuronidase gene (GUS) expression as an internal control and normalized to untreated vector controls. Statistical significance was assessed by an unpaired Student's t test using GraphPad Prism, version 6.0. Primer sequences employed for RT-PCR analysis of GFI1 target genes are available upon request.
RESULTS
PIAS3 SP-RING domain binds the GFI1 linker.
Through its interaction with PIAS3, GFI1 reverses the inhibitory effect of PIAS3 toward STAT3-dependent transcription (45). To further our understanding of the GFI1-PIAS3 relationship, we first confirmed the GFI1-PIAS3 interaction (Fig. 1A) (45) and then defined the boundaries of their binding interface using transiently expressed, myc-tagged GFI1 or deletion derivatives with Flag-tagged PIAS3 by immune precipitation and Western blotting (Fig. 1B and C). An interaction between full-length GFI1 and PIAS3 was clearly visible. The SNAG and zinc finger domains of GFI1 were dispensable for PIAS3 binding. Instead, a motif between residues 136 and 254 of GFI1 was required to bind PIAS3. Additional contributions were made by the N-terminal half of the linker as deleting residues 21 to 135 diminished PIAS3 binding. However, this region was neither necessary nor sufficient for GFI1 to interact with PIAS3.
FIG 1.

The C-terminal half of the GFI1 linker binds the PIAS3 SP-RING domain. (A) PIAS3 binds GFI1. Myc-tagged GFI1 and Flag-tagged PIAS3 were expressed as shown. PIAS3 was immunopurified from whole-cell lysates (WCL), and coprecipitation of GFI1 was determined by immunoblot (Western blotting [WB]) analysis of immune complexes (IC). Green fluorescent protein (GFP) and tubulin were employed as controls for transfection efficiency and gel loading, respectively. (B) Structures of GFI1 and PIAS3 derivatives employed in the experiments shown in panels C and D. SNAG and ZnF regions are shown for GFI1. SAP, SP-RING, AD, and S/T-rich domains are shown for PIAS3. Amino acid residues corresponding to fragment boundaries are indicated in parentheses. Hatched boxes represent regions forming the GFI1-PIAS3 binding interface. Open circles indicate the 6×myc tag. (C and D) Mapping the GFI1-PIAS3 binding interface. PIAS3 and GFI1 forms, with accompanying epitope tags, were expressed as shown. Coprecipitating GFI1 (C) and PIAS3 (D) forms were identified by Western blotting. WT, wild type; FL, Flag epitope tag; IP, immunoprecipitation.
To gain additional insights into the relationship between GFI1 and PIAS3, we employed a collection of fragments (Fig. 1B) spanning the PIAS3 primary structure to map the GFI1 binding site (39). Myc-tagged PIAS3 fragments (Fig. 1D) were transiently expressed in COS7L cells with Flag-tagged GFI1, and coprecipitating PIAS3 fragments were detected in anti-Flag immune complexes by anti-myc Western blotting. A C-terminal fragment of PIAS3 containing the SP-RING domain, acidic domain (AD), and the serine/threonine (S/T)-rich tail readily bound GFI1. Deleting the SP-RING domain (amino acids 274 to 322) and 71 additional residues before the AD completely abolished GFI1 binding. We also observed interactions between GFI1 and PIASxα, PIASxβ, PIASy, and PIAS1 (data not shown). Primary structure alignment for PIAS proteins revealed rapidly declining homology past the SP-RING domain, suggesting that the conserved SP-RING domain of PIAS family proteins contributes significantly to GFI1 binding.
GFI1 SUMOylation involves a SUMO consensus motif within its linker region.
Interactions between GFI1 and PIAS family E3 SUMO ligases suggest that GFI1 could be modified by SUMO. To address this hypothesis, we first screened for high-molecular-weight (HMW) forms of GFI1 captured under instantly denaturing conditions, suggesting conjugation by ubiquitin family proteins. COS7L cells transiently expressing Flag-tagged GFI1 were snap-frozen in LN2, thawed into 20% trichloroacetic acid (TCA), and made soluble by boiling in buffer containing 1% SDS. Solubilized proteins were analyzed by immunoblotting with anti-Flag antibody to detect HMW GFI1 derivatives (Fig. 2A). In parallel, Flag-tagged HDAC1 and β-catenin were expressed as controls for SUMOylation and ubiquitination, respectively. Both controls showed evidence of anti-Flag immune reactivity in the HMW range, suggesting that both SUMO- and ubiquitin-modified targets could be visualized by this technique. For GFI1, a prominent HMW population was observed, as well as several GFI1 fragments consistent with proteolysis. To determine if HMW forms of GFI1 contained SUMO and/or ubiquitin, we purified Flag-tagged GFI1 from proteins solubilized after isolation in TCA as described above. GFI1-containing immune complexes subjected to Western blotting with anti-SUMO and antiubiquitin antibodies clearly showed SUMO and ubiquitin conjugation (Fig. 2B).
FIG 2.

GFI1 is SUMOylated and ubiquitinated. (A) High-molecular-weight (HMW) derivatives of GFI1 identified under instantly denaturing conditions. COS7L cells were transfected with empty vector or expression constructs for Flag-tagged GFI1, HDAC1, and β-catenin. Cells were collected in-ice cold PBS, snap-frozen in liquid nitrogen, and suspended in 20% TCA. Precipitated proteins were solubilized by boiling in solubilizing buffer. Aliquots were analyzed by immunoblotting with anti-Flag antibody M2. (B) HMW derivatives of GFI1 contain SUMO and ubiquitin modifications. Flag-tagged GFI1 was expressed in COS7L cells, collected, and processed as described for panel A. Anti-Flag immune complexes (IC) and whole-cell lysate (WCL) were analyzed by SDS-PAGE and Western blotting (WB) as shown. (C) Each SUMO paralog can conjugate GFI1. Flag-tagged SUMO1, SUMO2, or SUMO3 was expressed in COS7L cells with myc-tagged GFI1 as shown. Cells were harvested as described for panel A. SUMOylated proteins were collected by anti-Flag immune precipitation. Immune complexes and whole-cell lysates were analyzed by Western blotting (WB). (D) Endogenously expressed GFI1 is SUMOylated. HL-60 cells in the quantities shown, stably expressing Flag-tagged SUMO2, SUMO3, or vector control, were collected and processed as described for panel A and then immunopurified via the Flag epitope tag (FL). Immune complexes (IC) were probed for GFI1 by Western blotting (WB). Expression of GFI1 and SUMO proteins was confirmed in whole-cell lysates (WCL) by Western blotting. Ub, ubiquitin.
The apparent molecular masses of HMW GFI1 derivatives approach 250 kDa, suggesting that GFI1 may be conjugated by multiple SUMO and/or ubiquitin moieties. Among SUMO family proteins, only SUMO2 and SUMO3 can form poly-SUMO chains, owing to the presence of a SUMOylation consensus element in both proteins (30). SUMO1 lacks a SUMOylation consensus motif, enabling mono-SUMOylation and restricting its presence in poly-SUMO chains to the terminal SUMO modification (30). To address the possibility that GFI1 can be modified by SUMO1, -2, or -3, we expressed Flag-tagged versions of each SUMO with myc-tagged GFI1, isolated SUMOylated targets by anti-Flag immune precipitation from TCA-precipitated, resolubilized proteins, and subjected immune complexes to anti-myc Western blotting. GFI1 conjugation by each SUMO paralog could be clearly demonstrated (Fig. 2C). To confirm SUMOylation at endogenous levels of GFI1 expression, we created HL-60 cells stably expressing Flag-tagged SUMO2 or SUMO3 and immunopurified SUMO2- or SUMO3-conjugated proteins and probed for GFI1 by Western blotting (Fig. 2D). As expected, GFI1 signal was detected in the HMW range for both SUMO2- and SUMO3-conjugated proteins. Similar results were observed for HL-60 cells stably expressing SUMO1 (data not shown). These findings indicate that endogenously expressed GFI1 can be modified by each SUMO paralog and collectively suggest an intimate relationship between GFI1 and the SUMOylation machinery.
GFI1 SUMO modification requires K239 within an intact SUMOylation consensus element.
Approximately 75% of experimentally confirmed SUMO modifications occur within ψKX(D/E) SUMOylation consensus elements. The GFI1 linker contains a motif (GVKVES; motif is underlined) within the PIAS3 binding site, centered on K239, that matches a type I SUMOylation consensus sequence (Fig. 3A). This motif is absolutely conserved in mammalian GFI1 orthologs (46). Yet given imperfect prediction by the SUMOylation consensus alone, we utilized three in silico prediction algorithms of SUMOylation, SUMOplot (Abgent) (47), SUMOsp2.0 (37), and SUMOhydro (36), to identify high-probability sites for SUMO conjugation in GFI1 (data not shown). Among GFI1 lysine residues, only K239 met the criteria of being conserved among mammalian GFI1 orthologs, being absent from GFI1B, and being identified as a high-probability site in each algorithm. To assess K239 in SUMO modification of GFI1, we created an arginine substitution at this site (K239R). Because type I SUMOylation consensus elements have an invariant aspartate or glutamate at the +2 position, we also created a glutamate (E)-to-glutamine (Q) substitution at E241 (E241Q), leaving the acceptor lysine (K239) intact. Using instantly denaturing LN2/TCA/SDS conditions followed by anti-Flag immune precipitation and anti-SUMO immunoblotting, SUMO modification of GFI1 was readily observed. However, neither the K239R nor E241Q derivative of GFI1 showed appreciable SUMO conjugation (Fig. 3B). Notably, GFI1 and its K239R and E241Q derivatives displayed comparable PIAS3 binding and nuclear localizations (Fig. 3C and D), implying that SUMO conjugation failure was not due to altered subcellular localization or interaction with PIAS3. These data indicate that GFI1 SUMOylation depends upon K239 within an intact, type I SUMOylation consensus element.
FIG 3.

GFI1 SUMOylation requires a type I SUMOylation consensus element in the GFI1 linker. (A) Alignment and conservation of a type I SUMOylation consensus sequence in the GFI1 linker in selected mammalian species. The putative SUMO acceptor lysine (K239 in rat) is indicated by an asterisk. (B) SUMO modification of GFI1 is abolished by K239R or E241Q substitution. Flag-tagged GFI1 or its K239R or E241Q derivative was expressed and then isolated by anti-Flag immune precipitation (IP) from total cellular protein harvested under instantly denaturing conditions as described in the legend to Fig. 2. Immune complexes (IC) and whole-cell lysates (WCL) were subjected to immunoblotting with anti-SUMO or anti-Flag antibodies as shown. (C) GFI1 and its K239R or E241Q derivative display PIAS3 binding. A 6×myc-tagged GFI1 or its K239R or E241Q derivative was expressed with Flag-tagged PIAS3. Presence of GFI1, GFI1-K239R, or GFI1-E241Q in anti-Flag immune complexes was determined by Western blotting. (D) GFI1 and its K239R derivative localize to the nucleus. NIH 3T3 cells were transduced with retrovirus expressing Flag-tagged GFI1 or its K239R derivative, and then subcellular localization was determined in stable polyclonal populations by epifluorescence detection. Nuclei were counterstained with To-Pro-3-iodide.
GFI1 SUMOylation supports cell fate determination in hematopoiesis.
GFI1 serves multiple roles in developmental hematopoiesis, involving both myeloid and lymphoid lineages. The GFI1 homolog in zebrafish, gfi1aa, is required for primitive erythropoiesis (48). We leveraged this requirement to determine the impact of GFI1 SUMOylation in vivo. Hemoglobinized cells characteristic of primitive erythropoiesis were readily revealed by o-dianisidine staining of zebrafish embryos at 48 hpf (Fig. 4A). To render observations quantitative across a continuum, we used a scoring system to represent none to complete hemoglobinization, with scores assigned to each of 200 embryos by an investigator blinded to the experimental conditions. Using a morpholino oligonucleotide designed to block splicing of the gfi1aa mRNA (Fig. 4B), we confirmed that gfi1aa depletion could abort primitive erythropoiesis and that wild-type rat Gfi1 could complement this deficiency. However, despite expression comparable to that of wild-type GFI1 (Fig. 4C) neither Gfi1-K239R nor Gfi1-E241Q could do so (29, 30). These data indicate that GFI1 SUMOylation supports primitive erythropoiesis in zebrafish.
GFI1 is also required for mammalian neutrophil differentiation in vivo, and mutations in GFI1 cause SCN type 2. To extend our findings to mammalian hematopoiesis, we leveraged granulocytic differentiation of HL-60 human promyelocytic leukemia cells in response to all-trans retinoic acid (ATRA). HL-60 cells were transduced with retroviruses expressing either a GFI1-targeted shRNA or a scrambled control, and GFI1 depletion in stable isolates was confirmed by Western blotting (Fig. 5A). Stable cells were exposed to either ATRA or vehicle and then examined for granulocytic differentiation by CD11B expression (Fig. 5B) and cell morphology (Fig. 5C and D). CD11B expression rose spontaneously in transduced HL-60 cells with time in culture, increased significantly in response to ATRA in control cells, and was reduced in the context of GFI1 depletion. However, when assessed morphologically, granulocytic differentiation was nearly absent with GFI1 depletion. To determine the contribution of GFI1 SUMOylation to this phenotype, we rescued expression with GFI1 or GFI1-K239R in GFI1-depleted HL-60 cells, treated stable isolates with ATRA for 4 days, and then assessed granulocytic differentiation morphologically (Fig. 5E). Restoring GFI1 expression complemented the defect in ATRA-mediated granulocyte differentiation, but GFI1-K239R failed to do so despite comparable levels of expression. Collectively, these findings point toward an essential role for GFI1 SUMOylation in hematopoietic differentiation.
FIG 5.

GFI1 SUMOylation supports HL-60 cell granulocytic differentiation in response to all-trans retinoic acid (ATRA). (A) GFI1 depletion from HL-60 cells. Whole-cell extracts (WCLs) were prepared from either naive HL-60 cells or those infected with retrovirus expressing small hairpin RNA targeting GFI1 (sh-Gfi1) or a content-matched scrambled control shRNA (Scr. Ctl.). GFI1 levels were determined by Western blotting. Actin levels were used to confirm equal loading. (B) CD11B expression following stimulation with ATRA. HL-60 cells were treated with 0.1 μM ATRA or vehicle following shRNA-mediated depletion of GFI1 versus a content-matched scrambled control shRNA. CD11B+ cells were determined by flow cytometry. (C) Granulocytic differentiation of HL-60 cells requires GFI1. HL-60 cells were treated with ATRA or vehicle for 4 days following shRNA-mediated depletion of GFI1 or a scrambled control. One thousand cells from randomly selected fields for each condition were visually scored as immature (promyelocyte, myelocyte, or metamyelocyte morphology) or mature (band form or multisegmented nuclei). (D) Morphology of immature versus mature HL-60 cells following ATRA treatment. Arrows indicate mature cells with segmented nuclei reminiscent of granulocyte differentiation. (E) GFI1-K239R expression fails to complement the granulocyte maturation defect brought on by GFI1 depletion. HL-60 cells were subjected to scrambled control or GFI1-depleting shRNA, then rescued with expression constructs for Flag-tagged wild type (WT:FL) or GFI1-K239R (K>R:FL) as shown. After 4 days of ATRA exposure, cells were scored visually for granulocyte maturation as described for panel C. Expression of Flag-tagged GFI1 variants was confirmed by Western blotting.
GFI1 SUMOylation regulates MYC expression during granulocytic differentiation.
GFI1 is transactivated by CAAT/enhancer binding protein α (C/EBPα), a critical determinant of granulocytic differentiation, and both GFI1 and C/EBPα are implicated in repression of MYC (49–51). Moreover, MYC expression declines precipitously during ATRA-mediated differentiation of HL-60 cells, and enforced expression of MYC blocks terminal differentiation in multiple settings (52–54). To gain mechanistic insights for GFI1 SUMOylation in granulocytic differentiation, we assessed expression of CEBPA, MYC, and azurocidin gene (AZU) in HL-60 cells with and without GFI1, in the absence or presence of ATRA (Fig. 6A). Expression changes for these factors in HL-60 cells treated with ATRA and occupancy of their promoters by GFI1 have been previously described (52). HL-60 cells were transduced with retroviruses expressing either a GFI1-targeted shRNA or a scrambled control, and then GFI1 expression was restored relative to the level with empty vector rescue. CEBPA expression did not change appreciably with GFI1 depletion or enforced GFI1 expression and fell modestly under each condition following ATRA exposure. MYC expression fell precipitously with ATRA exposure in control cells but rose significantly with GFI1 depletion, and in response to ATRA MYC expression did not fall below the level seen for undifferentiated controls. Moreover, MYC repression was restored by enforced expression of GFI1 and further potentiated by ATRA exposure. AZU expression fell in HL-60 cells exposed to ATRA, failed to do so in GFI1-depleted cells, but was again repressed when GFI1 expression was restored. To address contributions from GFI1 SUMOylation to transcriptional control, we again depleted HL-60 cells of GFI1, restored expression of GFI1 or its SUMOylation-deficient forms, K239R and E241Q, and then assessed expression of CEBPA, MYC, and AZU in the absence or presence of ATRA (Fig. 6B). None of these GFI1 forms showed a significant impact upon CEBPA expression. However, while GFI1 restored repression of MYC and AZU in response to ATRA, the K239R and E241Q derivatives of GFI1 were impaired. These data indicate that GFI1-mediated repression of MYC and AZU correlates with its effects on granulocytic differentiation and that SUMOylation favors this function of GFI1.
FIG 6.

SUMOylation modulates GFI1-dependent MYC expression to direct ATRA-mediated granulocyte maturation in HL-60 cells. (A) GFI1 modulates MYC expression in ATRA-mediated granulocytic differentiation. GFI1-targeted shRNA (sh-Gfi1) or a content-matched scrambled control (Scr) was used to deplete GFI1 in HL-60 cells, followed by rescue with a vector control (Vect) or GFI1 as shown. Levels of CEBPA, MYC, and AZU (azurocidin) mRNAs were determined by qRT-PCR relative to that of the GUS internal control. Fold change in expression is shown relative to that of the untreated, scrambled or vector control cells. (B) GFI1 was depleted from HL-60 cells using GFI1-targeted shRNA, followed by restored expression of GFI1, GFI1-K239R, or GFI1-E241Q and then treatment with vehicle or ATRA as shown. Expression of CEBPA, MYC, and AZU was measured as described for panel A. Statistical significance was determined by Wilcoxon-Mann-Whitney testing (*, P < 0.05; **, P < 0.005; ***, P < 0.0005).
In multiple settings, enforced expression of MYC impairs terminal differentiation (53, 54). To determine the impact of elevated MYC expression resulting from GFI1 depletion, we expressed MYC constitutively in HL-60 cells and then assessed granulocytic differentiation in response to ATRA relative to that of naive and vector control cells (Fig. 7). While granulocytic maturation was readily apparent in naive and vector control cells following ATRA exposure, it was noticeably impaired in the context of enforced MYC expression. These data suggest that increases in MYC expression can block HL-60 cell differentiation in response to ATRA and support the possibility that MYC repression by SUMOylation-competent GFI1 enables this differentiated phenotype.
FIG 7.
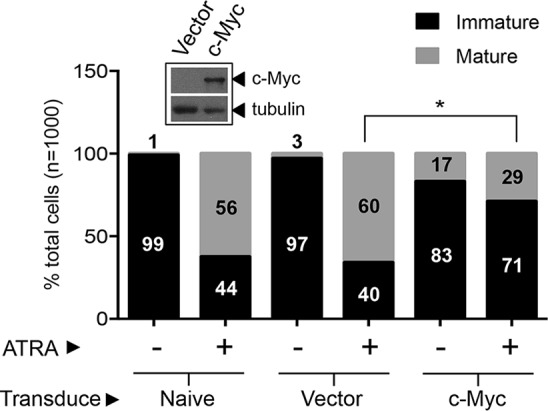
Enforced MYC expression blocks granulocytic differentiation of HL-60 cells in response to ATRA. Naive HL-60 cells and those transduced with a MYC-expressing retrovirus or with a control vector were treated with 0.1 μM ATRA for 4 days and scored for immature versus mature granulocytic morphology. The Western blot shows c-Myc expression in transduced cells relative to that in vector control cells (inset) using anti-myc rabbit polyclonal antibody, A-14. Tubulin serves as a loading control. Statistical significance was determined by Wilcoxon-Mann-Whitney testing (*, P < 0.05).
SUMOylation favors GFI1-LSD1/CoREST binding and transcriptional repression.
LSD1 binds the GFI1 SNAG domain, and factors that disrupt GFI1-LSD1 binding impair GFI1-mediated transcriptional repression (4, 5, 55). Similarly, the demethylase activity of LSD1 toward mono- and dimethylated histone H3 lysine 4 (H3K4me1/2) in nucleosomes is augmented by its interaction with CoREST and is further favored by SUMO2 and SUMO3 (SUMO2/3) binding to a SIM in CoREST (56–58). These findings suggest that the SUMOylation of transcription factors that partner with LSD1-CoREST may favor LSD1 activity at target promoters. To explore contributions from SUMOylation to transcriptional repression by GFI1, we used the reporter cell line 293-T-REx-5×Gal-TK-luciferase (59–61) and fusion proteins comprised of GFI1 sequences fused to the DNA binding region of Gal4 (Fig. 8A). Eliminating GFI1 ZnFs 4 and 5 prevents binding at GFI1-regulated endogenous promoters, instead directing the GFI1 repressor function to the 5×Gal-upstream activation sequence (UAS) of the integrated reporter construct. Thus, the GFI1 repressor function on endogenous promoters can be assessed in a chromatinized context independent of partnerships with other DNA binding proteins. In this model system, GFI1-Δ1C:Gal4 displayed dose-dependent transcriptional repression. SNAG-WT:Gal4 (where WT is wild type) and GFI1-Δ1C/K239R:Gal4 also showed dose-dependent reductions in reporter output that were comparable but moderately and consistently less than that of GFI1-Δ1C:Gal4. This difference became less apparent at higher plasmid doses. These findings mirror those showing significant but submaximal transcriptional repression attributable to the GFI1 SNAG domain in prior studies (55). Repression was antagonized by the LSD1 inhibitor HCI-2509 (Fig. 8B) and by the P2A substitution that abolishes SNAG-LSD1 binding (Fig. 8C) (38). These data indicate that LSD1 recruitment dominates transcriptional repression by GFI1, that SUMOylation supports this repressor function, and that when SUMOylation is impaired, repression by GFI1 resembles that achieved by the SNAG domain alone.
FIG 8.

SUMOylation supports LSD1/CoREST binding and transcriptional repression by GFI1. (A) SUMOylation contributes to GFI1-mediated repression. GFI1-Δ1C, the SNAG domain, or GFI1-Δ1C with the K239R substitution was expressed as a fusion with Gal4 in HEK293-T-REx-5×Gal-luciferase cells grown in six-well plates. Firefly luciferase activity was determined relative to that of Renilla luciferase using a dual-luciferase assay kit. (B and C) LSD1 activity contributes to transcriptional repression by GFI1. GFI1-Δ1C, the SNAG domain, or GFI1-Δ1C/K239R was expressed as a Gal4 fusion protein in the presence or absence of LSD1 inhibitor, HCI-2509 (LSD1i). Luciferase activity was scored as described for panel A. Similarly, transcriptional repression by GFI1-Δ1C or the SNAG domain, each harboring the P2A mutation that abolishes LSD1 binding, was compared to repression produced by its wild-type configurations at this position. (D) GFI1-K239R displays impaired LSD1/CoREST binding. Flag-tagged GFI1 or GFI1-K239R was expressed in COS7L cells and immunopurified from nuclear extracts (NE) by anti-Flag immune precipitation. Endogenously expressed LSD1 and CoREST coprecipitating with GFI1 or GFI1-K239R were determined by Western blotting (WB) of anti-Flag immune complexes (IC). (E) A model hypothesizing how GFI1 SUMOylation could influence transcriptional repression by the GFI1-LSD1/CoREST complex. RLU, relative light units.
The GFI1 SNAG domain interacts directly with LSD1 and indirectly recruits its corepressor, CoREST (5). In light of impaired transcriptional repression by GFI1-K239R, we hypothesized impaired LSD1/CoREST binding by this mutant. In coprecipitation assays, we found that endogenously expressed LSD1 and CoREST interacted readily with wild-type GFI1 (Fig. 8D). However, despite its intact SNAG domain, GFI1-K239R was noticeably impaired in LSD1 and CoREST binding. These findings indicate that GFI1 SUMOylation favors LSD1/CoREST binding, providing a functional link between SUMO conjugation and transcriptional repression.
DISCUSSION
GFI1 is a transcriptional repressor that controls growth, differentiation, and survival in normal and malignant hematopoiesis. Yet we have only a limited understanding of factors governing GFI1 function. By focusing on the GFI1 linker region, we have identified a type I SUMOylation motif unique to GFI1 and embedded within its PIAS3 binding site. K239R and E241Q substitutions within this site abolish GFI1 SUMOylation, and unlike wild-type GFI1, these derivatives fail to complement GFI1 depletion phenotypes. Likewise, these SUMOylation-deficient derivatives are impaired in transcriptional repression at GFI1 target genes. These data suggest that SUMOylation at K239 favors GFI1-mediated transcriptional control and cell fate determination.
It is likely that SUMOylation occurs directly on K239 as the E241Q substitution is similarly impaired yet retains the K239 ε-amino group for alternative posttranslational modifications. However, these functional defects could also reflect modification(s) elsewhere in GFI1 that depends upon K239 and/or E241, including conjugation by SUMO or other ubiquitin family members. SUMOylation on multiple lysines has been indicated for other transcriptional regulators, including c-Jun, p53, and Nkx2.5 (62, 63). Notably, studies of a 25-amino-acid peptide from SENS, the Drosophila melanogaster ortholog of GFI1, show that K509 can be SUMOylated in vitro. Yet a K509R derivative of SENS is still SUMOylated when expressed in S2 cells (64), indicating alternative sites for SUMO conjugation. K509 is analogous to K403 in GFI1, which is mutated in SCN type 2 (15, 16), suggesting that a defect in K403 SUMOylation might impair GFI1 function in neutrophil development. However, K403 in GFI1 does not occupy a consensus SUMOylation site, nor has K403 SUMOylation ever been demonstrated. Defining the full complement of GFI1 modifications by SUMO/ubiquitin family members and the contexts in which these modifications occur will be important to understanding their contributions to GFI1-mediated transcriptional control and cell fate specification.
Our findings indicate that GFI1 SUMOylation correlates with transcriptional repression, as has been seen for other DNA binding proteins (65–67). LSD1 is the dominant effector of GFI1-mediated transcriptional repression. Its demethylase activity toward H3K4me1/2 peptide substrates is enhanced by CoREST, while CoREST-LSD1 binding favors demethylation of H3K4me1/2 in nucleosomes. Moreover, an interaction between SUMO2/3 and the SIM in CoREST potentiates its activation of LSD1 demethylase activity. SUMO1 does not do so. Since LSD1 function at target promoters should be proportional to its recruitment and its activation state, factors that favor either should support transcriptional repression while those that compromise either should impair it. We observed a prominent decline in both LSD1 and CoREST binding to GFI1-K239R relative to that of GFI1, and this correlated with impaired transcriptional repression and complementation failure in functional assays. Based upon these findings, we speculate that SUMO-CoREST binding provides an additional tether for LSD1 bound to the GFI1 SNAG domain and augments LSD1 demethylase activity. Therefore, SUMOylation-deficient derivatives of GFI1 may display relatively less LSD1 binding and activity, causing functional impairment.
The finding that GFI1 can be modified by SUMO1, -2, or -3 and ubiquitin suggests considerable complexity in GFI1 regulation by ubiquitin family members. For example, whether GFI1 is modified by SUMO1 or SUMO2/3 could influence CoREST recruitment and stimulation of intrinsic LSD1 activity at GFI1-regulated promoters. Distinct patterns of SUMO/ubiquitin conjugation could impact composition, activity, and integrity of GFI1 complexes, thereby expanding its functional repertoire. Moreover, SUMO/ubiquitin modifications potentially occur on the same residues under different conditions. These modifications would be mutually exclusive and may also compete with lysine modifications by other small molecules derived from metabolic pathways to modulate gene expression based upon cellular needs (68–70). As such, our results may reflect one of many potential complexes assembled to regulate GFI1-responsive genes, each favored by a discrete constellation of posttranslational modifications.
Changes in gene expression are both energetically costly and impactful. Thus, requiring dual inputs before executing changes seems not only reasonable but advantageous. With respect to the GFI1-LSD1/CoREST axis, LSD1 binding to the SNAG domain would serve as the first input, with GFI1 SUMOylation and CoREST recruitment serving as the second (Fig. 8E). This model predicts that transcriptional repression by GFI1 would be most favored by concurrent SNAG-LSD1 and SUMO2/3-CoREST binding. The former would deliver LSD1 to GFI1-regulated genes while the latter would stabilize LSD1 binding and augment its demethylase activity. Should CoREST engage LSD1 in solution, its stimulating effect toward LSD1 would be limited by the need for a secondary input from SUMO2/3 binding. LSD1 actions could then be limited by reversal of either event or by GFI1 turnover. This fail-safe strategy could help ensure fidelity when a change in gene expression is initiated and yet provide mechanisms to modulate and ultimately terminate it. Thus, SUMO/ubiquitin modifications to GFI1 may offer additional inputs governing transcriptional control and supporting distinct, context-dependent cell fate decisions in hematopoiesis. We are only at the beginning of unraveling the potential regulatory complexity for this deceptively simple transcriptional repressor.
ACKNOWLEDGMENTS
We are indebted to colleagues Srividya Bhaskara, Trudy Oliver, Thomas O'Hare, Don Ayer, Stephen Lessnick, Kimble Frazer, and Nicholas Trede for critical review and helpful suggestions regarding the manuscript. We gratefully acknowledge support from the oligonucleotide synthesis, DNA sequencing, and Centralized Zebrafish Animal Resource (CZAR) core facilities of the University of Utah and the Huntsman Cancer Institute.
The Huntsman Cancer Foundation and the National Institutes of Health under award numbers P30CA042014, K08DK080190 (M.E.E.), and T32DK007115-39 (L.M.) provided support for the research presented in this publication. Additional support was provided via a Career Development Award from the St. Baldrick's Foundation (M.E.E.) and from the Office of the Vice President for Research of the University of Utah School of Medicine.
Funding Statement
The funders had no role in the design of experiments, collection of data, or interpretation of study results. Likewise, they had no role in the decision to submit the work for publication
REFERENCES
- 1.Gilks CB, Bear SE, Grimes HL, Tsichlis PN. 1993. Progression of interleukin-2 (IL-2)-dependent rat T cell lymphoma lines to IL-2-independent growth following activation of a gene (Gfi-1) encoding a novel zinc finger protein. Mol Cell Biol 13:1759–1768. doi: 10.1128/MCB.13.3.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tong B, Grimes HL, Yang TY, Bear SE, Qin Z, Du K, El-Deiry WS, Tsichlis PN. 1998. The Gfi-1B proto-oncoprotein represses p21WAF1 and inhibits myeloid cell differentiation. Mol Cell Biol 18:2462–2473. doi: 10.1128/MCB.18.5.2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rodel B, Wagner T, Zornig M, Niessing J, Moroy T. 1998. The human homologue (GFI1B) of the chicken GFI gene maps to chromosome 9q34.13-A locus frequently altered in hematopoietic diseases. Genomics 54:580–582. doi: 10.1006/geno.1998.5601. [DOI] [PubMed] [Google Scholar]
- 4.van der Meer LT, Jansen JH, van der Reijden BA. 2010. Gfi1 and Gfi1b: key regulators of hematopoiesis. Leukemia 24:1834–1843. doi: 10.1038/leu.2010.195. [DOI] [PubMed] [Google Scholar]
- 5.Saleque S, Kim J, Rooke HM, Orkin SH. 2007. Epigenetic regulation of hematopoietic differentiation by Gfi-1 and Gfi-1b is mediated by the cofactors CoREST and LSD1. Mol Cell 27:562–572. doi: 10.1016/j.molcel.2007.06.039. [DOI] [PubMed] [Google Scholar]
- 6.Zweidler-Mckay PA, Grimes HL, Flubacher MM, Tsichlis PN. 1996. Gfi-1 encodes a nuclear zinc finger protein that binds DNA and functions as a transcriptional repressor. Mol Cell Biol 16:4024–4034. doi: 10.1128/MCB.16.8.4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee S, Doddapaneni K, Hogue A, McGhee L, Meyers S, Wu Z. 2010. Solution structure of Gfi-1 zinc domain bound to consensus DNA. J Mol Biol 397:1055–1066. doi: 10.1016/j.jmb.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 8.Phelan JD, Shroyer NF, Cook T, Gebelein B, Grimes HL. 2010. Gfi1-cells and circuits: unraveling transcriptional networks of development and disease. Curr Opin Hematol 17:300–307. doi: 10.1097/MOH.0b013e32833a06f8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kazanjian A, Gross EA, Grimes HL. 2006. The growth factor independence-1 transcription factor: new functions and new insights. Crit Rev Oncol Hematol 59:85–97. doi: 10.1016/j.critrevonc.2006.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeng H, Yucel R, Kosan C, Klein-Hitpass L, Moroy T. 2004. Transcription factor Gfi1 regulates self-renewal and engraftment of hematopoietic stem cells. EMBO J 23:4116–4125. doi: 10.1038/sj.emboj.7600419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hock H, Hamblen MJ, Rooke HM, Schindler JW, Saleque S, Fujiwara Y, Orkin SH. 2004. Gfi-1 restricts proliferation and preserves functional integrity of haematopoietic stem cells. Nature 431:1002–1007. doi: 10.1038/nature02994. [DOI] [PubMed] [Google Scholar]
- 12.Liu Y, Elf SE, Miyata Y, Sashida G, Liu Y, Huang G, Di Giandomenico S, Lee JM, Deblasio A, Menendez S, Antipin J, Reva B, Koff A, Nimer SD. 2009. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell 4:37–48. doi: 10.1016/j.stem.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spooner CJ, Cheng JX, Pujadas E, Laslo P, Singh H. 2009. A recurrent network involving the transcription factors PU.1 and Gfi1 orchestrates innate and adaptive immune cell fates. Immunity 31:576–586. doi: 10.1016/j.immuni.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yucel R, Karsunky H, Klein-Hitpass L, Moroy T. 2003. The transcriptional repressor Gfi1 affects development of early, uncommitted c-Kit+ T cell progenitors and CD4/CD8 lineage decision in the thymus. J Exp Med 197:831–844. doi: 10.1084/jem.20021417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Person RE, Li FQ, Duan Z, Benson KF, Wechsler J, Papadaki HA, Eliopoulos G, Kaufman C, Bertolone SJ, Nakamoto B, Papayannopoulou T, Grimes HL, Horwitz M. 2003. Mutations in proto-oncogene GFI1 cause human neutropenia and target ELA2. Nat Genet 34:308–312. doi: 10.1038/ng1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xia J, Bolyard AA, Rodger E, Stein S, Aprikyan AA, Dale DC, Link DC. 2009. Prevalence of mutations in ELANE, GFI1, HAX1, SBDS, WAS and G6PC3 in patients with severe congenital neutropenia. Br J Haematol 147:535–542. doi: 10.1111/j.1365-2141.2009.07888.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Armistead PM, Wieder E, Akande O, Alatrash G, Quintanilla K, Liang S, Molldrem J. 2010. Cyclic neutropenia associated with T cell immunity to granulocyte proteases and a double de novo mutation in GFI1, a transcriptional regulator of ELANE. Br J Haematol 150:716–719. doi: 10.1111/j.1365-2141.2010.08274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khanna-Gupta A, Sun H, Zibello T, Lee HM, Dahl R, Boxer LA, Berliner N. 2007. Growth factor independence-1 (Gfi-1) plays a role in mediating specific granule deficiency (SGD) in a patient lacking a gene-inactivating mutation in the C/EBPε gene. Blood 109:4181–4190. doi: 10.1182/blood-2005-05-022004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wallis D, Hamblen M, Zhou Y, Venken KJ, Schumacher A, Grimes HL, Zoghbi HY, Orkin SH, Bellen HJ. 2003. The zinc finger transcription factor Gfi1, implicated in lymphomagenesis, is required for inner ear hair cell differentiation and survival. Development 130:221–232. doi: 10.1242/dev.00190. [DOI] [PubMed] [Google Scholar]
- 20.Kazanjian A, Wallis D, Au N, Nigam R, Venken KJ, Cagle PT, Dickey BF, Bellen HJ, Gilks CB, Grimes HL. 2004. Growth factor independence-1 is expressed in primary human neuroendocrine lung carcinomas and mediates the differentiation of murine pulmonary neuroendocrine cells. Cancer Res 64:6874–6882. doi: 10.1158/0008-5472.CAN-04-0633. [DOI] [PubMed] [Google Scholar]
- 21.Shroyer NF, Wallis D, Venken KJ, Bellen HJ, Zoghbi HY. 2005. Gfi1 functions downstream of Math1 to control intestinal secretory cell subtype allocation and differentiation. Genes Dev 19:2412–2417. doi: 10.1101/gad.1353905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karsunky H, Zeng H, Schmidt T, Zevnik B, Kluge R, Schmid KW, Duhrsen U, Moroy T. 2002. Inflammatory reactions and severe neutropenia in mice lacking the transcriptional repressor Gfi1. Nat Genet 30:295–300. doi: 10.1038/ng831. [DOI] [PubMed] [Google Scholar]
- 23.Fiolka K, Hertzano R, Vassen L, Zeng H, Hermesh O, Avraham KB, Duhrsen U, Moroy T. 2006. Gfi1 and Gfi1b act equivalently in haematopoiesis, but have distinct, non-overlapping functions in inner ear development. EMBO Rep 7:326–333. doi: 10.1038/sj.embor.7400618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vassen L, Okayama T, Moroy T. 2007. Gfi1b:green fluorescent protein knock-in mice reveal a dynamic expression pattern of Gfi1b during hematopoiesis that is largely complementary to Gfi1. Blood 109:2356–2364. doi: 10.1182/blood-2006-06-030031. [DOI] [PubMed] [Google Scholar]
- 25.Saleque S, Cameron S, Orkin SH. 2002. The zinc-finger proto-oncogene Gfi-1b is essential for development of the erythroid and megakaryocytic lineages. Genes Dev 16:301–306. doi: 10.1101/gad.959102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yucel R, Kosan C, Heyd F, Moroy T. 2004. Gfi1:green fluorescent protein knock-in mutant reveals differential expression and autoregulation of the growth factor independence 1 (Gfi1) gene during lymphocyte development. J Biol Chem 279:40906–40917. doi: 10.1074/jbc.M400808200. [DOI] [PubMed] [Google Scholar]
- 27.Doan LL, Porter SD, Duan Z, Flubacher MM, Montoya D, Tsichlis PN, Horwitz M, Gilks CB, Grimes HL. 2004. Targeted transcriptional repression of Gfi1 by GFI1 and GFI1B in lymphoid cells. Nucleic Acids Res 32:2508–2519. doi: 10.1093/nar/gkh570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vassen L, Fiolka K, Mahlmann S, Moroy T. 2005. Direct transcriptional repression of the genes encoding the zinc-finger proteins Gfi1b and Gfi1 by Gfi1b. Nucleic Acids Res 33:987–998. doi: 10.1093/nar/gki243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilkinson KA, Henley JM. 2010. Mechanisms, regulation and consequences of protein SUMOylation. Biochem J 428:133–145. doi: 10.1042/BJ20100158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gareau JR, Lima CD. 2010. The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat Rev Mol Cell Biol 11:861–871. doi: 10.1038/nrm3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hochstrasser M. 2009. Origin and function of ubiquitin-like proteins. Nature 458:422–429. doi: 10.1038/nature07958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shuai K, Liu B. 2005. Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat Rev Immunol 5:593–605. doi: 10.1038/nri1667. [DOI] [PubMed] [Google Scholar]
- 33.Hecker CM, Rabiller M, Haglund K, Bayer P, Dikic I. 2006. Specification of SUMO1- and SUMO2-interacting motifs. J Biol Chem 281:16117–16127. doi: 10.1074/jbc.M512757200. [DOI] [PubMed] [Google Scholar]
- 34.Stehmeier P, Muller S. 2009. Phospho-regulated SUMO interaction modules connect the SUMO system to CK2 signaling. Mol Cell 33:400–409. doi: 10.1016/j.molcel.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 35.Xu J, He Y, Qiang B, Yuan J, Peng X, Pan XM. 2008. A novel method for high accuracy sumoylation site prediction from protein sequences. BMC Bioinformatics 9:8. doi: 10.1186/1471-2105-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen YZ, Chen Z, Gong YA, Ying G. 2012. SUMOhydro: a novel method for the prediction of sumoylation sites based on hydrophobic properties. PLoS One 7:e39195. doi: 10.1371/journal.pone.0039195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ren J, Gao X, Jin C, Zhu M, Wang X, Shaw A, Wen L, Yao X, Xue Y. 2009. Systematic study of protein sumoylation: development of a site-specific predictor of SUMOsp 2.0. Proteomics 9:3409–3412. doi: 10.1002/pmic.200800646. [DOI] [PubMed] [Google Scholar]
- 38.Sorna V, Theisen ER, Stephens B, Warner SL, Bearss DJ, Vankayalapati H, Sharma S. 2013. High-throughput virtual screening identifies novel N′-(1-phenylethylidene)-benzohydrazides as potent, specific, and reversible LSD1 inhibitors. J Med Chem 56:9496–9508. doi: 10.1021/jm400870h. [DOI] [PubMed] [Google Scholar]
- 39.Long J, Wang G, Matsuura I, He D, Liu F. 2004. Activation of Smad transcriptional activity by protein inhibitor of activated STAT3 (PIAS3). Proc Natl Acad Sci U S A 101:99–104. doi: 10.1073/pnas.0307598100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moore AC, Amann JM, Williams CS, Tahinci E, Farmer TE, Martinez JA, Yang G, Luce KS, Lee E, Hiebert SW. 2008. Myeloid translocation gene family members associate with T-cell factors (TCFs) and influence TCF-dependent transcription. Mol Cell Biol 28:977–987. doi: 10.1128/MCB.01242-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vassen L, Fiolka K, Moroy T. 2006. Gfi1b alters histone methylation at target gene promoters and sites of gamma-satellite containing heterochromatin. EMBO J 25:2409–2419. doi: 10.1038/sj.emboj.7601124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Horton RM, Cai ZL, Ho SN, Pease LR. 1990. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Biotechniques 8:528–535. [PubMed] [Google Scholar]
- 43.Engel ME, Nguyen HN, Mariotti J, Hunt A, Hiebert SW. 2010. Myeloid translocation gene 16 (MTG16) interacts with Notch transcription complex components to integrate Notch signaling in hematopoietic cell fate specification. Mol Cell Biol 30:1852–1863. doi: 10.1128/MCB.01342-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barrett CW, Smith JJ, Lu LC, Markham N, Stengel KR, Short SP, Zhang B, Hunt AA, Fingleton BM, Carnahan RH, Engel ME, Chen X, Beauchamp RD, Wilson KT, Hiebert SW, Reynolds AB, Williams CS. 2012. Kaiso directs the transcriptional corepressor MTG16 to the Kaiso binding site in target promoters. PLoS One 7:e51205. doi: 10.1371/journal.pone.0051205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rodel B, Tavassoli K, Karsunky H, Schmidt T, Bachmann M, Schaper F, Heinrich P, Shuai K, Elsasser HP, Moroy T. 2000. The zinc finger protein Gfi-1 can enhance STAT3 signaling by interacting with the STAT3 inhibitor PIAS3. EMBO J 19:5845–5855. doi: 10.1093/emboj/19.21.5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, Lopez R. 2010. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res 38:W695–699. doi: 10.1093/nar/gkq313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pichler A. 2008. Analysis of sumoylation. Methods Mol Biol 446:131–138. doi: 10.1007/978-1-60327-084-7_9. [DOI] [PubMed] [Google Scholar]
- 48.Cooney JD, Hildick-Smith GJ, Shafizadeh E, McBride PF, Carroll KJ, Anderson H, Shaw GC, Tamplin OJ, Branco DS, Dalton AJ, Shah DI, Wong C, Gallagher PG, Zon LI, North TE, Paw BH. 2013. Teleost growth factor independence (gfi) genes differentially regulate successive waves of hematopoiesis. Dev Biol 373:431–441. doi: 10.1016/j.ydbio.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lidonnici MR, Audia A, Soliera AR, Prisco M, Ferrari-Amorotti G, Waldron T, Donato N, Zhang Y, Martinez RV, Holyoake TL, Calabretta B. 2010. Expression of the transcriptional repressor Gfi-1 is regulated by C/EBPα and is involved in its proliferation and colony formation-inhibitory effects in p210BCR/ABL-expressing cells. Cancer Res 70:7949–7959. doi: 10.1158/0008-5472.CAN-10-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Johansen LM, Iwama A, Lodie TA, Sasaki K, Felsher DW, Golub TR, Tenen DG. 2001. c-Myc is a critical target for C/EBPα in granulopoiesis. Mol Cell Biol 21:3789–3806. doi: 10.1128/MCB.21.11.3789-3806.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Duan Z, Zarebski A, Montoya-Durango D, Grimes HL, Horwitz M. 2005. Gfi1 coordinates epigenetic repression of p21Cip/WAF1 by recruitment of histone lysine methyltransferase G9a and histone deacetylase 1. Mol Cell Biol 25:10338–10351. doi: 10.1128/MCB.25.23.10338-10351.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Duan Z, Horwitz M. 2003. Targets of the transcriptional repressor oncoprotein Gfi-1. Proc Natl Acad Sci U S A 100:5932–5937. doi: 10.1073/pnas.1031694100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leon J, Ferrandiz N, Acosta JC, Delgado MD. 2009. Inhibition of cell differentiation: a critical mechanism for MYC-mediated carcinogenesis? Cell Cycle 8:1148–1157. doi: 10.4161/cc.8.8.8126. [DOI] [PubMed] [Google Scholar]
- 54.Delgado MD, Leon J. 2010. Myc roles in hematopoiesis and leukemia. Genes Cancer 1:605–616. doi: 10.1177/1947601910377495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grimes HL, Chan TO, Zweidler-McKay PA, Tong B, Tsichlis PN. 1996. The Gfi-1 proto-oncoprotein contains a novel transcriptional repressor domain, SNAG, and inhibits G1 arrest induced by interleukin-2 withdrawal. Mol Cell Biol 16:6263–6272. doi: 10.1128/MCB.16.11.6263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee MG, Wynder C, Cooch N, Shiekhattar R. 2005. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 437:432–435. [DOI] [PubMed] [Google Scholar]
- 57.Shi YJ, Matson C, Lan F, Iwase S, Baba T, Shi Y. 2005. Regulation of LSD1 histone demethylase activity by its associated factors. Mol Cell 19:857–864. doi: 10.1016/j.molcel.2005.08.027. [DOI] [PubMed] [Google Scholar]
- 58.Ouyang J, Shi Y, Valin A, Xuan Y, Gill G. 2009. Direct binding of CoREST1 to SUMO-2/3 contributes to gene-specific repression by the LSD1/CoREST1/HDAC complex. Mol Cell 34:145–154. doi: 10.1016/j.molcel.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sarma K, Margueron R, Ivanov A, Pirrotta V, Reinberg D. 2008. Ezh2 requires PHF1 to efficiently catalyze H3 lysine 27 trimethylation in vivo. Mol Cell Biol 28:2718–2731. doi: 10.1128/MCB.02017-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li G, Margueron R, Ku M, Chambon P, Bernstein BE, Reinberg D. 2010. Jarid2 and PRC2, partners in regulating gene expression. Genes Dev 24:368–380. doi: 10.1101/gad.1886410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mozzetta C, Pontis J, Fritsch L, Robin P, Portoso M, Proux C, Margueron R, Ait-Si-Ali S. 2014. The histone H3 lysine 9 methyltransferases G9a and GLP regulate polycomb repressive complex 2-mediated gene silencing. Mol Cell 53:277–289. doi: 10.1016/j.molcel.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 62.Costa MW, Lee S, Furtado MB, Xin L, Sparrow DB, Martinez CG, Dunwoodie SL, Kurtenbach E, Mohun T, Rosenthal N, Harvey RP. 2011. Complex SUMO-1 regulation of cardiac transcription factor Nkx2-5. PLoS One 6:e24812. doi: 10.1371/journal.pone.0024812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schmidt D, Muller S. 2002. Members of the PIAS family act as SUMO ligases for c-Jun and p53 and repress p53 activity. Proc Natl Acad Sci U S A 99:2872–2877. doi: 10.1073/pnas.052559499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Powell LM, Chen A, Huang YC, Wang PY, Kemp SE, Jarman AP. 2012. The SUMO pathway promotes basic helix-loop-helix proneural factor activity via a direct effect on the Zn finger protein senseless. Mol Cell Biol 32:2849–2860. doi: 10.1128/MCB.06595-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gill G. 2004. SUMO and ubiquitin in the nucleus: different functions, similar mechanisms? Genes Dev 18:2046–2059. doi: 10.1101/gad.1214604. [DOI] [PubMed] [Google Scholar]
- 66.Hay RT. 2005. SUMO: a history of modification. Mol Cell 18:1–12. doi: 10.1016/j.molcel.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 67.Geiss-Friedlander R, Melchior F. 2007. Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol 8:947–956. doi: 10.1038/nrm2293. [DOI] [PubMed] [Google Scholar]
- 68.Walsh CT, Garneau-Tsodikova S, Gatto GJ Jr. 2005. Protein posttranslational modifications: the chemistry of proteome diversifications. Angew Chem Int Ed Engl 44:7342–7372. doi: 10.1002/anie.200501023. [DOI] [PubMed] [Google Scholar]
- 69.Zhang Z, Tan M, Xie Z, Dai L, Chen Y, Zhao Y. 2011. Identification of lysine succinylation as a new post-translational modification. Nat Chem Biol 7:58–63. doi: 10.1038/nchembio.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen Y, Sprung R, Tang Y, Ball H, Sangras B, Kim SC, Falck JR, Peng J, Gu W, Zhao Y. 2007. Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol Cell Proteomics 6:812–819. doi: 10.1074/mcp.M700021-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]