

ABSTRACT
The UL47 gene product, VP8, is the most abundant tegument protein of bovine herpesvirus 1 (BoHV-1). Previously, we demonstrated that a UL47-deleted BoHV-1 mutant (BoHV1-ΔUL47) exhibits 100-fold-reduced virulence in vitro and is avirulent in vivo. In this study, we demonstrated that VP8 expression or BoHV-1 infection inhibits interferon beta (IFN-β) signaling by using an IFN-α/β-responsive plasmid in a luciferase assay. As transducer and activator of transcription (STAT) is an essential component in the IFN-signaling pathways, the effect of VP8 on STAT was investigated. An interaction between VP8 and STAT1 was established by coimmunoprecipitation assays in both VP8-transfected and BoHV-1-infected cells. Two domains of VP8, amino acids 259 to 482 and 632 to 686, were found to be responsible for its interaction with STAT1. The expression of VP8 did not induce STAT1 ubiquitination or degradation. Moreover, VP8 did not reduce STAT1 tyrosine phosphorylation to downregulate IFN-β signaling. However, the expression of VP8 or a version of VP8 (amino acids 219 to 741) that contains the STAT1-interacting domains but not the nuclear localization signal prevented nuclear accumulation of STAT1. Inhibition of nuclear accumulation of STAT1 also occurred during BoHV-1 infection, while nuclear translocation of STAT1 was observed in BoHV1-ΔUL47-infected cells. During BoHV-1 infection, VP8 was detected in the cytoplasm at 2 h postinfection without any de novo protein synthesis, at which time STAT1 was already retained in the cytoplasm. These results suggest that viral VP8 downregulates IFN-β signaling early during infection, thus playing a role in overcoming the antiviral response of BoHV-1-infected cells.
IMPORTANCE Since VP8 is the most abundant protein in BoHV-1 virions and thus may be released in large amounts into the host cell immediately upon infection, we proposed that it might have a function in the establishment of conditions suitable for viral replication. Indeed, while nonessential in vitro, it is critical for BoHV-1 replication in vivo. In this study, we determined that VP8 plays a role in downregulation of the antiviral host response by inhibiting IFN-β signaling. VP8 interacted with and prevented nuclear accumulation of STAT1 at 2 h postinfection in the absence of de novo viral protein synthesis. Two domains of VP8, amino acids 259 to 482 and 632 to 686, were found to be responsible for this interaction. These results provide a new functional role for VP8 in BoHV-1 infection and a potential explanation for the lack of viral replication of the UL47 deletion mutant in cattle.
INTRODUCTION
Bovine herpesvirus 1 (BoHV-1) is responsible for several clinical manifestations, including rhinotracheitis, vulvovaginitis, and conjunctivitis, in cattle (1). BoHV-1 is composed of a double-stranded DNA surrounded by a nucleocapsid, a tegument, and an envelope (2). Although the tegument is a major constituent in the BoHV-1 virion, it is the least studied. The tegument consists of at least 20 virus-encoded proteins (reviewed in reference 3). Herpesvirus infection is mediated by the interaction of glycoproteins such as gB, gC, and gD with cellular proteins (4). The majority of the tegument proteins are then released into the cytoplasm, indicating that these proteins are the first to interact with the intracellular environment (5). Herpesvirus tegument proteins are involved in various functions, including capsid transport, DNA replication, transcriptional and translational regulation, and viral assembly and egress (3). These functions suggest that tegument proteins contribute to the establishment of conditions suitable for viral replication.
The UL47 gene product, VP8, is a 97-kDa tegument protein and the most abundant protein in BoHV-1 virions (6). Although BoHV-1 VP8 is not essential for viral infection, a UL47-deleted mutant (BoHV1-ΔUL47) exhibits a small tegument structure and impaired growth in cell culture and is avirulent in cattle (7). In addition, BoHV-1 VP8 plays a role in induction of humoral and cellular immunity (8). VP8 is monoubiquitinated and interacts with DNA damage binding protein-1 (DDB1) (9), which is a component of the Cul4A-DDB1 E3 ubiquitin ligase complex (10). Furthermore, VP8 remodeled the distribution of promyelocytic leukemia (PML) nuclear bodies (NBs) (11).
Viruses can establish an infection in the host cells by overcoming the antiviral defense mechanisms. The antiviral state is established by secretion of type I interferon (IFN), which is needed for the activation of other cellular genes. Interferons are categorized into type I (IFN-α, IFN-β, IFN-ω, IFN-κ, and IFN-ε), type II (IFN-γ), and type III (IFN-λ) depending on their primary structure (reviewed in references 12 and 13). IFN-α and -β are synthesized by cells in response to viral infections, whereas IFN-γ is secreted by activated T lymphocytes and natural killer cells in virus-infected cells. These types of IFN are involved in limiting the growth of target cells and in influencing cell apoptosis, thereby arresting viral spread. The activity of these IFNs is initiated by the binding of IFN-α/β and IFN-γ to their cell surface receptors (14). Some IFN-mediated cascades are regulated by the Janus tyrosine kinase/signal transducer and activator of transcription (JAK/STAT) pathway, whereas others are regulated by STATs to optimize the transcription regulation of target genes (12). IFN-λs are induced by either interferon response factor 3 (IRF3), IRF7, or NF-κB pathways (13). Recently, IFN-λ was identified as having antiviral properties against numerous viruses.
The activation of the JAK/STAT signaling pathway is initiated by binding of IFN-α/β to their receptors, which are composed of IFNAR1 and IFNAR2 subunits. Upon binding of IFN-α/β to its receptors, the IFNAR1 and IFNAR2 complex activates tyrosine kinase 2 (Tyk2) and Janus kinase 1 (JAK1) by transphosphorylation. Activated Tyk2 phosphorylates IFNAR1 on tyrosine 466, making a platform to bind STAT2. This facilitates phosphorylation of STAT2 by activated Tyk2, which in turn recruits STAT1. The newly recruited STAT1 is phosphorylated on residue tyrosine 701 by JAK1. Phosphorylated STAT1 and STAT2 form a heterodimer, which dissociates from the receptor and then translocates to the nucleus to bind with IRF9 and form a heterotrimeric complex, the IFN-stimulated gene factor 3 (ISGF3) complex. The ISGF3 complex binds to the IFN response element for the induction of IFN-stimulated genes (reviewed in reference 14).
Most viruses have developed specific mechanisms to circumvent the IFN response, either by reducing IFN production or by downregulating the IFN signaling cascade (14). For example, rabies virus P protein (15), simian virus 5 (SV5) V protein (16), respiratory syncytial virus (17), human parainfluenza virus type 1 virus V protein (18), and mumps virus V protein (19) inhibit IFN-β signaling by proteasome-mediated degradation of STAT1 or STAT2, by reducing phosphorylation of STAT1 or STAT2, or by inhibiting nuclear translocation of STAT1. During BoHV-1 infection, inhibition of IFN signaling by infected cell protein bICP0 through degradation of IRF3 was observed (20). In the absence of IRF3 expression, bICP0 inhibits the ability of IRF7 to trans-activate the IFN-β promoter (21). Furthermore, bICP27 inhibits the transcriptional activity of two bovine IFN-β gene promoters (IFN-β1 and IFN-β3) during transient transfection (22). However, no other protein of BoHV-1 has been reported to downregulate IFN-β signaling.
Since BoHV-1 VP8 is essential for viral replication in vivo, we examined its effect on the IFN signaling pathway. We determined that VP8 downregulates the IFN response both in VP8-transfected and in BoHV-1-infected cells. VP8 interacted with STAT1, and this interaction required two distinct domains of VP8. Furthermore, VP8 acted as an IFN antagonist by preventing nuclear translocation of STAT1.
MATERIALS AND METHODS
Cell culture, virus infection, and IFN treatment.
Madin-Darby bovine kidney (MDBK), embryonic bovine tracheal (EBTr), human embryonic kidney HEK293T, and Vero cells were grown in Eagle's minimum essential medium (MEM; Sigma-Aldrich Canada Ltd., Oakville, ON, Canada) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco, Life Technologies, Burlington, ON, Canada), 1% antibiotic-antimycotic (Life Technologies), and 10 mM HEPES buffer (Life Technologies). Cells were cultured with 5% CO2 in a 37°C incubator. Wild-type BoHV-1 108, BoHV1-ΔUL47, and BoHV1-UL47R were propagated in MDBK cells (23). MDBK cells were infected with BoHV-1 at a multiplicity of infection (MOI) of 0.5 unless indicated otherwise. Recombinant human IFN-β was purchased from PeproTech Inc. (Rocky Hill, NJ, USA), and bovine IFN-β was purchased from Kingfisher (Saint Paul, MN, USA).
Antibodies.
VP8-specific mouse monoclonal antibody was used as previously described (8). Rabbit antibodies specific for bICP0 and bICP4 were made in-house and used as described previously (7, 24). Rabbit polyclonal anti-STAT1 (catalog no. sc-345), anti-STAT1 p84/91 (catalog no. sc-346), anti-pSTAT1 (catalog no. sc-8394), anti-STAT2 (catalog no. sc-476), anti-ubiquitin (catalog no. sc-8071), and anti-fibrillarin (H-140) antibodies were obtained from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). Mouse monoclonal anti-FLAG (catalog no. F3165-0.2MG), anti-actin (catalog no. A2228), and anti-tubulin (catalog. no T6199) antibodies were purchased from Sigma-Aldrich. SV5 V5-specific rabbit antibody was purchased from Invitrogen, Life Technologies.
Plasmids.
The UL47 gene (GenBank accession no. AY530215.1) was cloned into pFLAG-CMV2 (Sigma-Aldrich) as described previously (25). The VP8 open reading frame (ORF) was subcloned with an N-terminal FLAG tag into an expression vector (named pCMV4.1k) downstream from a human cytomegalovirus (CMV) promoter with intron A. The resulting plasmid was then used as a template in PCR to generate truncated versions of the FLAG-VP8 ORF, using the primers listed in Table 1. PCR fragments were cloned back into the pCMV4.1k expression vector to create the constructs described in the text. The ORFs of all constructs were verified as correct by DNA sequencing. The pFLAG-CMV-2 plasmid was purchased from Sigma-Aldrich. The IFN-α/β responsive reporter plasmid, pISREluc, and pRL-TK have been described previously (16) and were kindly provided by Danielle Blondel, LVMS, CNRS, France. pISREluc contains the firefly luciferase gene fused with four tandem repeats of the IFN-inducible gene 9-27 interferon-stimulated response element (ISRE). pRL-TK, which contains the herpes simplex virus thymidine kinase promoter region upstream of the Renilla luciferase gene, was used to normalize transfection. A simian virus 5 V expression plasmid, pSV5V, and pHis-Ub plasmids were kindly provided by Richard Randall, University of St. Andrews, School of Biology, St. Andrews, Fife, United Kingdom.
TABLE 1.
Primer list for plasmid construction using PCR (5′ to 3′ end)
| Plasmid | Forward primer sequence | Reverse primer sequence |
|---|---|---|
| VP8 | GAATCTAGAGCCACCATGGACTACAAAGACGATGAC | GGCAGTGAGCGCAACGCAATTAATG |
| VP8 121-741 | GCGGTAGATCTGATTCAAGACTACTTGACGGCCCACCTG | GGCAGTGAGCGCAACGCAATTAATG |
| VP8 219-741 | CGGTAGATCTGATTGAGCGGCTGTCGGAAGGG | GGCAGTGAGCGCAACGCAATTAATG |
| VP8 343-741 | CGGTAGATCTGATTGGCGGCATGTACGTGGGCGCCCCTGAG | GGCAGTGAGCGCAACGCAATTAATG |
| VP8 538-741 | GCGGTAGATCTGATTGCGGCGGCCTTCCGCGAAGTG | GGCAGTGAGCGCAACGCAATTAATG |
| VP8 632-741 | CGGTAGATCTGATTGGCAGCCTGAACCTGCTGCTGAAC | GGCAGTGAGCGCAACGCAATTAATG |
| VP8 1-120 | GAATCTAGAGCCACCATGGACTACAAAGACGATGAC | GAGCTCGAGTCAGCCGTGATTGGGGCCGCGGTTAG |
| VP8 1-258 | GAATCTAGAGCCACCATGGACTACAAAGACGATGAC | GAGCTCGAGTCACTCCCCCGCAGCCGCAGCG |
| VP8 1-482 | GAATCTAGAGCCACCATGGACTACAAAGACGATGAC | GAGCTCGAGTCAGGCCGACTGCAGCCCGGCGCCCGCGTAG |
| VP8 1-631 | GAATCTAGAGCCACCATGGACTACAAAGACGATGAC | GAGCTCGAGTCACAGCCGCTGCGCGATCAGCCC |
| VP8 1-259 | CGCGCCACCAGACATAATAGCTGAC | CAGCGGCACCTCTACCTGAGGCTG |
| VP8 1-371 | CGCGCCACCAGACATAATAGCTGAC | CACGGATCCCCTACGCCTCAGTGGGCGGCA |
| VP8 259-371 | CGCGCCACCAGACATAATAGCTGAC | CACGGATCCCCTACGCCTCAGTGGGCGGCAT |
| VP8 372-483 | CGCGCCACCAGACATAATAGCTGAC | CACGGATCCCTAGTAGCGCTCATTTGCCGTGTAGCC |
| VP8 631-686 | GTGACGGTGCGCGAGGGCACGCT | CACGGATCCCCTACCGCCGCTTGCCGGCCAG |
Luciferase assay.
Vero cells were used as IFN-deficient cells to demonstrate the effect of transient VP8 expression on IFN-treated and nontreated cells. Vero cells were seeded at a concentration of 7 × 104 cells per well in 24-well plates. The next day, the cells were transfected with pRL-TK and pISREluc together with pCMV4.1K (empty vector), pFLAG-EYFP, or pFLAG-VP8 by using Lipofectamine and Plus reagent (Invitrogen, Life Technologies). At 24 h posttransfection, cells were treated with 2,500 units/0.5 ml of human IFN-β or left untreated. While MDBK cells are routinely used for propagation of high-titer BoHV-1, they are very resistant to transfection. EBTr cells have a relatively good transfection efficiency and thus were used to determine the effects of VP8 on IFN during BoHV-1 infection. EBTr cells were transfected with pRL-TK and pISREluc plasmids, and 24 h later the cells were infected with BoHV-1 or BoHV-1-ΔUL47. At 24 h postinfection, the cells were treated with 400 ng/0.5 ml of bovine IFN-β or left untreated. Cells were harvested 6 h after IFN treatment in lysis buffer. Firefly and Renilla luciferase activity were assayed in the cell lysates according to the manufacturer's protocol (dual-luciferase reporter assay system; Promega, Madison, WI, USA). The relative expression levels were determined by dividing the firefly luciferase values by the Renilla luciferase values. Actinomycin D (ActD; Sigma-Aldrich) was used to treat cells before the luciferase assay. EBTr cells were transfected with pRL-TK and pISREluc for 20 h. The transfected cells were treated with ActD at a concentration of 10 μg/ml for 1 h before mock infection or infection with BoHV-1 or BoHV1-UL47R at an MOI of 4 or with BoHV-1-ΔUL47 at an MOI of 10. After 1 h, cells were stimulated with bovine IFN-β for 1 h. ActD was maintained in the medium throughout the infection. Cell lysates were prepared and luciferase assays were performed as described above.
Preparation of cell lysates.
HEK293T and EBTr cells at 80 to 90% confluence were transfected with different plasmids by using Lipofectamine and Plus reagent (Invitrogen, Life Technologies). HEK293T cells were used for coimmunoprecipitation experiments. Cells were incubated with MEM for 48 h, washed with ice-cold PBS (pH 7.3), and lysed in lysis buffer (50 mM Tris, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, pH 7.4) supplemented with 10 μl/ml mammalian cell and tissue extract protease inhibitor cocktail (Sigma-Aldrich). Cells were gently rocked on a nutator for 3 to 4 min and then kept on ice for 30 min before centrifugation at 12,000 × g for 15 min at 4°C. The supernatant was collected in an Eppendorf tube and kept at −80°C for future use. EBTr cells were used for transfection and BoHV-1 infection. To prepare lysates, BoHV-1-infected cells were collected at 24 h postinfection.
Immunoprecipitation and Western blotting.
Cell lysates prepared as described above were incubated with ant-VP8, anti-STAT1, or anti-ubiquitin antibody overnight at 4°C followed by incubation with protein G-Sepharose Fast Flow beads (GE HealthCare, Niskayuna, NY, USA) for 3 h at 4°C; alternatively, anti-FLAG M2 affinity gel (Sigma-Aldrich) was directly added to the cell lysates, and the mixtures were incubated at 4°C overnight. This was followed by three washes with buffer (50 mM Tris-HCl, 250 mM NaCl, 2% Triton X-100, pH 7.4). For Western blotting, 15 to 25 μg of the immune complexes or cell lysates was boiled for 5 min after addition of SDS-PAGE sample buffer. Proteins were separated on 10% or 8 to 16% SDS-PAGE gels and then transferred to nitrocellulose membranes. The nitrocellulose membranes were blocked with 5% skim milk in phosphate-buffered saline-Tween-20 (PBST; 3.2 mM Na2HPO4, 0.5 mM KH2PO4, 1.3 mM KCl, 135 mM NaCl, 0.1% Tween 20, pH 7.4) for 2 h followed by incubation overnight at 4°C with anti-STAT1, anti-FLAG, anti-VP8, and/or anti-ubiquitin antibodies. The membranes were washed three times with PBST and incubated with IRDye680-conjugated anti-mouse IgG or IRDye-800CW-conjugated anti-rabbit IgG (LI-COR Bioscience, Lincoln, NE, USA). The proteins were detected with an Odyssey Infrared Imaging system (LI-COR Bioscience) followed by processing of images by Odyssey 3.0.16 application software (LI-COR Bioscience).
Cell fractionation.
Cytoplasmic and nuclear fractions were obtained as described previously (26). Cell fractionation was performed using Nuclei EZ Prep lysis buffer (Sigma). Briefly, cells were collected by trypsinization followed by lysis for 5 min on ice with the Nuclei EZ lysis buffer. The nuclei were collected by centrifugation at 500 × g for 5 min at 4°C. The incubation in lysis buffer and centrifugation were repeated five times to remove any loosely bound cytoskeletal components from the nuclei. The supernatants were pooled as cytoplasmic fraction and concentrated with an Amicon Ultra-15 Ultracel-10K filter unit (Millipore). For nuclear isolation, the nuclei were resuspended in 3 ml of 0.25 M sucrose, 10 mM MgCl2, and 1 mM phenylmethylsulfonyl fluoride (PMSF), layered over a 3-ml cushion of 0.88 M sucrose, 0.5 mM MgCl2, and 1 mM PMSF, and centrifuged at 2,800 × g for 10 min at 4°C. The nuclear pellet was resuspended in 200 μl of the Nuclei EZ storage buffer. The nuclei were counted with a hemocytometer, and equal numbers of nuclei were lysed by incubation with SDS at 100°C for 5 min. The purity of the nuclei was determined by Western blotting with fibrillarin- and tubulin-specific antibodies.
Immunofluorescence and confocal microscopy.
Vero cells were plated at a concentration of 2 × 105 cells per well in two-chamber Permanox slides (Lab-Tek, Naperville, IL, USA) and mock transfected or transfected with pFLAG-VP8 219-741 or pFLAG-VP8. At 24 h posttransfection, cells were fixed with 4% paraformaldehyde (Sigma-Aldrich) for 20 min at room temperature (RT) followed by permeabilization and blocking with 1% goat serum and 0.1% Triton X-100 in PBS. FLAG-tagged proteins and STAT1 were detected by incubating cells with mouse monoclonal anti-FLAG (diluted 1:1,000) and rabbit anti-STAT1 (diluted 1:50) antibodies for 2 h at RT. MDBK cells were infected with BoHV-1 or BoHV1-UL47R at an MOI of 4 or with BoHV1-ΔUL47 at an MOI of 5 for 14 h and then fixed and stained as described above. For the time course experiment, MDBK cells were infected with BoHV-1 at an MOI of 4, and samples were fixed and blocked overnight. The next morning, cells were incubated with monoclonal anti-VP8 or anti-gB antibodies and rabbit anti-STAT1 antibodies for 2 h. Alexa Fluor 488 goat anti-mouse IgG and Alexa Fluor 633 goat anti-rabbit IgG (diluted 1:500; Invitrogen, Life Technologies) were used as secondary antibodies. Finally, mounting medium containing 4′,6-diamino-2-phenylindinole (DAPI) was added, and the slides were air dried for 24 h at RT. The cells were examined and images taken with a Leica SP5 confocal microscope (Leica Microsystems Inc., Concord, ON, Canada), using green laser excitation at 488 nm (Alexa 488), red laser excitation at 633 nm (Alexa 633), and 461 nm for DAPI. Final images were processed using the Image J browser.
RESULTS
VP8 inhibits IFN-β signaling.
While VP8 has been reported to be nonessential in vitro, it is critical for replication in cattle in vivo, which suggests that it might have a profound impact on the innate antiviral response. As type I IFN signaling constitutes one of the most powerful antiviral defense mechanisms, we examined whether VP8 plays a role in IFN downregulation by conducting luciferase reporter gene assays with Vero cells in the presence or absence of VP8 expression. Since Vero cells are IFN deficient, IFN-β responses were induced by addition of extracellular IFN-β. Vero cells were transfected with luciferase reporter plasmids pRL-TK and pISRE together with pCMV4.1K, pFLAG-EYFP, or pFLAG-VP8, and cells were stimulated with IFN-β at 24 h posttransfection or left untreated. IFN-β treatment of Vero cells resulted in induction of luciferase expression compared to untreated cells (Fig. 1A). When the cells were transfected with CMV4.1K or FLAG-EYFP, no inhibition of IFN-β signaling was observed. However, IFN signaling was reduced to 22% in VP8-expressing cells compared to cells transfected with pCMV4.1K or pFLAG-EYFP. This indicates transcriptional activation of the ISREs due to the formation of ISGF3 transcription complexes in the cells. Treatment of FLAG-VP8-transfected Vero cells with IFN-β demonstrated that the expression of VP8 inhibits IFN-β-responsive transcription. The presence of VP8 in the cell lysates was confirmed by Western blotting with anti-VP8 antibody (Fig. 1B).
FIG 1.

Inhibition of IFN-β signaling by BoHV-1 VP8. (A and B) Vero cells were transfected with pISREluc and pRL-TK together with pCMV4.1K, pFLAG-EYFP, or pFLAG-VP8. (A) At 24 h posttransfection, cells were stimulated with 2,500 units of human IFN-β in 0.5 ml or left untreated. After 6 h of incubation, cell lysates were made and reporter gene activity was measured. (B) Expression of VP8 and EYFP was confirmed by Western blotting with monoclonal anti-FLAG antibody. (C) EBTr cells were transfected with pISREluc and pRL-TK. At 24 h posttransfection cells were mock infected or infected with BoHV-1 or BoHV1-ΔUL47, and another 24 h later cells were treated with 400 ng of bovine IFN-β in 0.5 ml or left untreated. After 6 h of incubation, cell lysates were made and reporter gene activity was measured. (D to G) EBTr cells were transfected with pISREluc and pRL-TK, and at 20 h posttransfection cells were either left untreated (D and E) or were pretreated with ActD (F and G) before mock infection or infection with BoHV-1, BoHV1-ΔUL47, or BoHV1-UL47R. (D and F) At 1 h postinfection, cells were stimulated with IFN-β for 1 h, and at 2 h postinfection cell lysates were collected and reporter gene activity was measured. ActD was maintained in the medium throughout the infection. (E and G) Expression of VP8, bICP0, and bICP4 in untreated cells (E) but not in Act-treated cells (G) was confirmed by Western blotting with monoclonal anti-FLAG antibody and bICP0- and bICP4-specific rabbit antibodies, respectively. IFN-β-induced firefly luciferase reporter values were normalized to the expression of Renilla luciferase. The values are presented as percentages of IFN-stimulated controls and are expressed as means ± standard deviations (SD) for six samples. Statistical significance is indicated by asterisks (***, P < 0.001).
The IFN-β response was also investigated in the context of BoHV-1 infection. EBTr cells were transfected with luciferase reporter plasmids, and at 24 h posttransfection cells were mock infected or infected with BoHV-1 or BHV1-ΔUL47. IFN-β treatment of mock-infected cells resulted in significant production of luciferase, in contrast to what was observed for nontreated cells. However, BoHV-1 infection reduced the induction of luciferase expression by IFN-β to ∼20% compared to mock infection (Fig. 1C), indicating inhibition of IFN signaling. Some luciferase activity was also observed in cells not treated with IFN-β, which can be attributed to the fact that EBTr cells are not IFN deficient. In BoHV1-ΔUL47-infected cells, the luciferase activity was higher than that in BoHV-1-infected cells, further confirming the inhibitory effect of VP8 on the IFN response. These results suggest that VP8 functions as IFN-β antagonist during BoHV-1 infection.
Some residual downregulation of the IFN response was observed in BHV1-ΔUL47-infected cells, which could be attributed to the presence of bICP0. To confirm the inhibition of IFN signaling by VP8 in the absence of any immediate early protein expression, ActD was used to inhibit transcription. EBTr cells were transfected with luciferase reporter plasmids, and at 20 h posttransfection cells were or were not pretreated with ActD before mock infection or infection with BoHV-1, BoHV1-ΔUL47, or BoHV1-UL47R. In BoHV-1- and BoHV-1-UL47R-infected cells, the luciferase signal was reduced to ∼20% without ActD treatment compared to mock-infected cells (Fig. 1D), while infection with BoHV1-ΔUL47 reduced IFN signaling to ∼70%. The expression of VP8 and two immediate early proteins, bICP0 and bICP4, is shown in Fig. 1E. Since ActD is a transcription inhibitor, no expression of immediate early protein was observed in ActD-treated cells as expected. However, VP8 was detected at 2 h postinfection in BoHV-1- and BoHV-1-UL47R-infected cells regardless of ActD treatment (Fig. 1G), representing VP8 released from the virions. In the presence of ActD, IFN signaling was again inhibited by BoHV-1 or BoHV1-UL47R infection to ∼20%, while the mock and BoHV1-ΔUL47 infection did not cause downregulation of IFN signaling (Fig. 1F). These experiments demonstrate that the residual inhibition of IFN signaling induced by BoHV1-ΔUL47 was due to immediate early gene expression. Furthermore, inhibition IFN signaling was caused by VP8 released from the incoming virions in the absence of any immediate early protein expression.
Identification of STAT1 as an interacting target of BoHV-1 VP8.
Since STATs play a critical role in IFN signaling, we determined whether VP8 might interact with STAT1 or STAT2. HEK293T cells were transiently transfected with pFLAG-EYFP or pFLAG-VP8. At 48 h posttransfection, cell lysates were prepared and incubated with anti-FLAG resin (Fig. 2A) and anti-STAT1 antibody followed by protein G-Sepharose (Fig. 2B). STAT1 was precipitated from pFLAG-VP8-transfected cells but not from pFLAG-EYFP- or mock-transfected cells (Fig. 2A). Anti-STAT1 antibody precipitated STAT1 from mock-, FLAG-EYFP-, and FLAG-VP8-transfected lysates (Fig. 2B), whereas it precipitated VP8 from the FLAG-VP8- but not from mock- or FLAG-EYFP-transfected cell lysates (Fig. 2B). Expression of VP8 and EYFP in transfected cells is confirmed by Fig. 2C. As further evidence of VP8 interaction with endogenous STAT1, MDBK cells were infected with BoHV-1 or BoHV1-ΔUL47. At 24 h postinfection, the cells were lysed, and the proteins were precipitated with anti-bovine STAT1 antibody followed by protein G-Sepharose and analyzed by Western blotting. As shown in Fig. 2D, VP8 was precipitated with cellular STAT1 in BoHV-1-infected cells, whereas no VP8 was pulled down from mock- or BoHV1-ΔUL47-infected cell lysates. The expression of VP8 and STAT1 in BoHV-1-infected cells is shown in Fig. 2F. To determine interactions between endogenous STAT1 and VP8, mock-, BoHV-1-, or BoHV1-ΔUL47-infected cell lysates were incubated with anti-VP8 antibody followed by protein G-Sepharose. STAT1 was precipitated with VP8-specific antibody in BoHV-1-infected cells but not in mock- or BoHV1-ΔUL47-infected cells (Fig. 2E). To identify interactions between VP8 and STAT2, mock-, FLAG-EYFP-, and FLAG-VP8-transfected lysates (Fig. 2G) were subjected to immunoprecipitation with anti-FLAG resin. STAT2 was precipitated neither by FLAG-EYFP nor by FLAG-VP8. The expression of STAT1, VP8, and EYFP is shown in Fig. 2H. These results demonstrate that VP8 interacts with STAT1 in both transiently transfected and BoHV-1-infected cells. However, VP8 did not interact with STAT2.
FIG 2.

BoHV-1 VP8 interacts with STAT1. (A to C) HEK293T cells were transfected with pFLAG-EYFP or pFLAG-VP8. (A and B) At 48 h posttransfection, cell lysates were generated and incubated with anti-FLAG resin (A) or anti-STAT1 antibody (B) followed by protein G-Sepharose. (C) Input lysates of mock-, pFLAG-EYFP-, and pFLAG-VP8-transfected cells that were used in the immunoprecipitation assays illustrated in panels A and B. (D to F) MDBK cells were mock infected or infected with BoHV-1 or BHV1-ΔUL47. (D and E) At 24 h postinfection, cell lysates were made and incubated with anti-STAT1 antibody (D) and anti-VP8 antibody (E), followed by protein G-Sepharose. (F) Expression of VP8 in BoHV-1-infected cells and STAT1 in mock-, BoHV-1-, or BoHV1-ΔUL47-infected cells. (G and H) HEK293T cells were transfected with pFLAG-EYFP or pFLAG-VP8. (G) At 48 h posttransfection, cell lysates were collected and incubated with anti-FLAG resin. (H) Input lysates of mock-, pFLAG-EYFP-, and pFLAG-VP8-transfected cells that were used in the immunoprecipitation assay illustrated in panel G. VP8, EYFP, STAT1, and STAT2 were detected by Western blotting with monoclonal anti-VP8 and anti-FLAG antibodies and rabbit anti-STAT1 and anti-STAT2 antibodies, respectively.
Identification of interacting domains of VP8 with STAT1.
Since the central and C-terminal parts of VP8 are conserved between herpesviruses while the N-terminal part is not (Fig. 3), it was of interest to determine which domain of VP8 interacts with STAT1. Nine plasmids containing different N-terminally and C-terminally truncated, FLAG-tagged VP8-coding sequences were generated. First, plasmids encoding N-terminally truncated FLAG-tagged VP8 were used to investigate the role of the C-terminal domain of VP8 in the interaction with STAT1. Anti-STAT1 antibody precipitated all N-terminally truncated versions of VP8 (VP8 segment consisting of amino acids 121 to 741 [VP 121-741], VP8 219-741, VP8 343-741, VP8 538-741, and VP 632-741) as well as full-length VP8 (Fig. 4A) from transfected cells. As all of the N-terminally truncated VP8 versions contain amino acids 632 to 741, this suggests that a STAT1-binding domain is located within this region of VP8. To investigate the involvement of the N-terminal domain of VP8 in the interaction with STAT1, cell lysates with C-terminally truncated VP8 (VP8 1 to 120 [VP8 1-120], VP8 1-258, VP8 1-482, and VP8 1-631) and full-length VP8 were analyzed by immunoprecipitation with anti-STAT1 antibody (Fig. 4C). This experiment demonstrated that VP8 was precipitated by anti-STAT1 antibody only when amino acids 259 to 482 were retained, and this occurred even though amino acids 632 to 741 were absent. Input cell lysates are presented in Fig. 4B and D. These results indicate that the conserved central and C-terminal parts of VP8 contain two domains, aa 259 to 483 and aa 632 to 741, that mediate its interaction with STAT1. To further define the interacting regions of VP8, four additional VP8 truncations consisting of amino acids 1 to 371, 372 to 483, 631 to 686, and 687 to 741 were generated. These four truncations also contained amino acids 1 to 258, a domain shown not to interact with STAT1, at the N-terminal region to facilitate expression. VP8 259-371, VP8 372-483, and VP8 631-686, but not VP8 687-741, were immunoprecipitated with anti-STAT1 antibody (Fig. 4E). Input lysates demonstrating expression of these truncated VP8 versions and STAT1 are shown in Fig. 4F. This result demonstrates that domains within the 259 to 482 and 631 to 686 regions of VP8 are required for its interaction with STAT1. A schematic presentation summarizing VP8-interacting domains with STAT1 is provided in Fig. 5.
FIG 3.

Comparison of the amino acid sequences of BoHV-1 UL47 and its homologues. The sequence labels are bovine herpesvirus 1 (BoHV1), human herpesvirus 1 (HHV1), equine herpesvirus 1 (EHV1), suid herpesvirus 1 (SHV1), and gallid herpesvirus 3 (GHV3) UL47 protein. The consensus secondary structure prediction for the UL47 family is shown above the sequences. The prediction was carried out using PROFphd software (47, 48). Invariant residues are highlighted in bold letters with white boxes, and highly conserved residues are in white bold text surrounded by black boxes. The alignment was generated using UniPro (citation), and the figure was generated using the ESPript server (48).
FIG 4.

Mapping of STAT1 interacting domains in BoHV-1 VP8. HEK293T cells were transfected with plasmids containing FLAG, FLAG-VP8, or different N-terminally and C-terminally truncated FLAG-tagged VP8 versions as indicated. At 48 h posttransfection, cell lysates were made and incubated with anti-STAT1 antibodies (ab), followed by incubation with protein G-Sepharose. Immune complexes were separated by SDS-PAGE and detected by Western blotting. Immunoprecipitation of N-terminally (A) and C-terminally (C) truncated FLAG-tagged VP8 with anti-STAT1 antibody. Input lysates of cells transfected with N-terminally (B) and C-terminally (D) truncated FLAG-tagged VP8. (E) Immunoprecipitation of VP8 259-371, 372-483, 631-686, and 687-741 with anti-STAT1 antibody. (F) Input lysates of cells transfected with VP8 259-371, 372-483, 631-686, or 687-741. Truncated and full-length VP8 and STAT1 were detected using antibodies specific for FLAG and STAT1, respectively. It should be noted that anti-STAT1 antibody detects both STAT1α and STAT1β. Molecular wei markers (× 10−3) are indicated in the left margins.
FIG 5.

Schematic representation of the interacting domains of VP8 with STAT1. The solid lines represent the presence of VP8 amino acids, and empty spaces represent the deleted portions of VP8. Interaction of (domains of) VP8 with STAT1 is shown by the plus sign (+), and lack of interaction is indicated by the minus sign (−). The first and last amino acids of each VP8 mutant are indicated. The red lines indicate STAT1-interacting domains of VP8.
Effects of VP8 on STAT1 ubiquitination and degradation.
STAT1 is a well-known transcriptional regulator. Some viral proteins, such as SV5 V protein, interact with STAT1 and mediate ubiquitination and subsequent degradation of STAT1 (16). Since BoHV-1 VP8 caused downregulation of IFN-β signaling and interacted with STAT1, we investigated whether STAT1 is ubiquitinated and degraded in the presence of VP8 or SV5 protein V, which was used as a positive control for detection of STAT1 ubiquitination. HEK293T cells were cotransfected with pFLAG/pHis-Ub, pFLAG-VP8/pHis-Ub, and pSV5V/pHis-Ub plasmids in the presence or absence of the proteasomal inhibitor MG132. Since ubiquitin proteases cleave off ubiquitin from polyubiquitinated STAT1, His-Ub plasmid was used for transfection in each sample. To prevent cleavage of ubiquitin from ubiquitinated STAT1, the ubiquitin protease inhibitor guanidium hydrochloric acid was used at a final concentration of 6 M in the lysis buffer. At 24 h posttransfection, cell lysates were generated and incubated with His-60 nickel beads, and protein complexes were analyzed by Western blotting with anti-STAT1 antibody. Figure 6A shows that ubiquitinated STAT1 was not precipitated from mock- and FLAG-VP8-transfected lysates but was pulled down from SV5 V-transfected lysates. Input lysates are presented in Fig. 6B. In the absence of MG132, STAT1 was not detected (Fig. 6D) and thus not pulled down by His-Ub (Fig. 6C). VP8 was precipitated by anti-ubiquitin antibody (Fig. 6E) and anti-VP8 antibody pulled down ubiquitinated VP8 from FLAG-VP8-transfected lysates but not from FLAG-transfected lysates (Fig. 6F), which confirms that VP8 is ubiquitinated as shown previously and validates the ubiquitin-specific antibody. This demonstrates that, although BoHV-1 VP8 interacts with STAT1, STAT1 is not ubiquitinated by the presence of VP8. Accordingly, STAT1 was not degraded by the proteasome (data not shown). Taken together, these results suggest that BoHV-1 VP8 interacts with STAT1 without mediating ubiquitination or degradation of STAT1.
FIG 6.

Cellular STAT1 is not ubiquitinated by the presence of BoHV-1 VP8. (A to D) HEK293T cells were transfected with pFLAG/pHis-Ub, pFLAG-VP8/pHis-Ub, and pSV5V/pHis-Ub in the presence or absence of 10 μM MG132. At 24 h posttransfection, cell lysates were generated and incubated with His-60 nickel resin in the presence (A) or absence (C) of MG132 followed by Western blotting with anti-STAT1 antibody. Input lysates of pFLAG/pHis-Ub-, pFLAG-VP8/pHis-Ub-, and pSV5V/pHis-Ub-transfected cells are shown in panels B and D. (E and F) HEK293T cells were transfected with pFLAG or pFLAG-VP8. At 24 h posttransfection, cell lysates were generated and incubated with antiubiquitin antibody (E) and anti-VP8 antibody (F), followed by protein G-Sepharose. Monoubiquitinated VP8 was detected by Western blotting with anti-FLAG antibody (E) or with anti-Ub antibody (F).
Effect of VP8 on STAT1 tyrosine phosphorylation.
IFN-α and -β signaling is stimulated by binding of IFN-α and -β to the cell surface receptors. Engagement of the IFN receptor leads to phosphorylation of STAT1, STAT2, and several other cellular kinases (reviewed in reference 14). To investigate whether VP8 impairs this stage in IFN-β signaling, IFN-β-induced STAT1 tyrosine phosphorylation was examined (Fig. 7). Vero cells were transfected with pFLAG-EYFP or pFLAG-VP8. At 24 h posttransfection, cells were stimulated with human IFN-β for 4 h or left untreated. Cell lysates were separated by SDS-PAGE and transferred to nitrocellulose membrane, and STAT1 and STAT1 phosphorylated at tyrosine residue Y701 were detected using anti-STAT1 and anti-pSTAT1 antibodies, respectively. As a loading control, actin was detected by anti-actin antibody. STAT1 started to be phosphorylated within 10 min of IFN-β treatment, in contrast to nontreated cells, in the presence of both FLAG-EYFP and FLAG-VP8. The total STAT1 and phosphorylated STAT1 amounts were similar in the presence and absence of VP8 in the cells, which indicates that the expression of VP8 does not reduce STAT1 phosphorylation.
FIG 7.
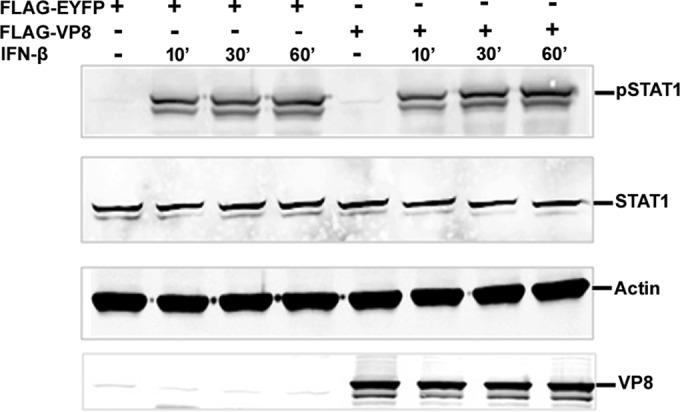
VP8 does not affect STAT1 tyrosine phosphorylation. Vero cells were transfected with pFLAG-EYFP or pFLAG-VP8. At 24 h posttransfection, cells were stimulated with IFN-β (2,500 units/0.5 ml) for 10, 30, and 60 min, and cell lysates were generated. Twenty-five micrograms of cellular protein were loaded and separated on a 10% gel for Western blot analysis. Phosphorylated STAT1, STAT1, and VP8 were detected by anti-pSTAT1, anti-STAT1, and anti-VP8 antibodies, respectively. As a protein loading control, actin was detected by anti-actin antibody.
VP8 inhibits IFN-β-induced nuclear accumulation of STAT1.
In virus-infected cells, nuclear translocation of STAT1 is stimulated by IFN-β. In the nucleus, newly imported STAT1 together with STAT2 and IRF9 forms a multiprotein complex, ISGF3, which functions as transcriptional activator (reviewed in reference 14). To further examine which step of IFN-β signaling is inhibited by VP8, we examined nuclear accumulation of STAT1 by immunofluorescence. Vero cells were transfected with pFLAG, pFLAG-VP8, or pFLAG-VP8 219-741, which expresses VP8 without nuclear localization signal (NLS). VP8 and STAT1 were detected with anti-FLAG and anti-STAT1 antibodies. As shown in Fig. 8A, IFN-β treatment of Vero cells redistributed STAT1 from the cytoplasm to the nucleus, whereas in nontreated cells STAT1 was cytoplasmic. Since the nuclear localization signal of VP8 was removed, VP8 219-741 was completely cytoplasmic and clearly prevented nuclear accumulation of STAT1 following IFN-β treatment (Fig. 8B). Without IFN-β treatment of VP8-expressing cells, STAT1 was also cytoplasmic as expected. The translocation of STAT1 to the nucleus was also studied in the context of expression of full-length VP8, which is found mostly in the nucleus, while some of it is cytoplasmic. In almost all of the cells that expressed VP8, STAT1 remained cytoplasmic (Fig. 8C).
FIG 8.

VP8 prevents IFN-β-induced nuclear accumulation of STAT1. Vero cells were transfected with pFLAG (A), pFLAG-VP8 219-741 (B), or pFLAG-VP8 (C). At 24 h posttransfection, cells were stimulated with 2,500 units of human IFN-β for 30 min or left untreated, followed by fixation with paraformaldehyde and permeabilization. Truncated and full-length VP8 proteins were incubated with monoclonal anti-FLAG antibody and Alexa Fluor 488-conjugated goat anti-mouse IgG. STAT1 was detected with rabbit anti-STAT1 antibody and Alexa Fluor 633-conjugated anti-rabbit IgG. The nucleus was stained with Prolong gold DAPI. The cells were examined with a Leica SP5 confocal microscope.
To examine the translocation of STAT1 in the context of infection, MDBK cells were mock infected or infected with BoHV-1, BoHV1-ΔUL47, or BoHV-1-UL47R for 14 h. Cells were left untreated or were treated with IFN-β and incubated with anti-VP8, anti-gB, and bovine anti-STAT1 antibodies. As shown in Fig. 9A, IFN-β treatment of mock-infected cells resulted in the transport of STAT1 into the nucleus, while without IFN-β treatment STAT1 was cytoplasmic. However, in BoHV-1- and BoHV-1-UL47R-infected cells (Fig. 9B), IFN-β treatment did not result in nuclear accumulation of STAT1. To confirm the role of VP8 in inhibition of STAT1 nuclear translocation, MDBK cells were infected with BoHV1-ΔUL47 (Fig. 9C). Infection with BoHV1-ΔUL47 was confirmed by incubation with gB-specific monoclonal antibody. STAT1 was localized in the nucleus of BoHV1-ΔUL47-infected cells, which confirms the role of VP8 in inhibition of STAT1 nuclear transport. These experiments demonstrated that nuclear accumulation of STAT1was inhibited by the presence of VP8.
FIG 9.

BoHV-1 infection prevents IFN-β-induced nuclear accumulation of STAT1. MDBK cells were mock infected (A) or infected with BoHV-1 at an MOI of 4 (B), BoHV-1ΔUL47 at an MOI of 5 (C), or BoHV-1-UL47R at an MOI of 4 (D). At 14 h postinfection, cells were stimulated with bovine IFN-β for 30 min or left untreated. Cells were then fixed with paraformaldehyde, permeabilized, and incubated with monoclonal anti-VP8 or anti-gB antibodies and Alexa Fluor 488-conjugated goat anti-mouse IgG. STAT1 was detected with anti-STAT1 antibodies and Alexa Fluor 633-conjugated anti-rabbit IgG. DNA was labeled with Prolong gold DAPI. The cells were examined with a Leica SP5 confocal microscope.
The subcellular distribution of VP8 is correlated with STAT1 translocation.
To determine how early after infection VP8 might be able to retain STAT1 in the cytoplasm, the subcellular localization of VP8 at early and late time points in the context of STAT1 retention was determined. MDBK cells were mock infected or infected with BoHV-1. At 2, 4, 6, or 14 h postinfection, cells were stimulated with IFN-β or left untreated followed by VP8 and STAT1 detection (Fig. 10A and B). VP8 was detected as early as 2 h in the cytoplasm, while STAT1 was retained in the cytoplasm. As BoHV-1 infection progressed to 4 h, some VP8 was localized to the nucleus, but VP8 was also still present in the cytoplasm, as was STAT1. At 6 h postinfection, most of the VP8 was detected in the nucleus, while STAT1 was still cytoplasmic. VP8 was again observed in the cytoplasm at 14 h postinfection. Throughout the period postinfection, STAT1 was detected in the cytoplasm, confirming that the retention of STAT1 was initiated with the incoming VP8 as early as 2 h.
FIG 10.


Subcellular localization of BoHV-1 VP8 at different times postinfection. MDBK cells were infected with BoHV-1 at an MOI of 4 for 2, 4, 6, and 14 h. The infected cells were left untreated (A) or stimulated with bovine IFN-β for 30 min (B). Cells were then fixed with paraformaldehyde, permeabilized, and incubated with monoclonal anti-VP8 and Alexa Fluor 488-conjugated goat anti-mouse IgG. STAT1 was detected with anti-STAT1 antibodies and Alexa Fluor 633-conjugated anti-rabbit IgG. DNA was labeled with Prolong gold DAPI. The cells were examined with a Leica SP5 confocal microscope.
To further confirm the subcellular distribution of VP8 at different stages of infection, cytoplasmic and nuclear fractions were prepared from BoHV-1-infected MDBK cells at 2, 4, 6, and 14 h (Fig. 11) and then examined by Western blotting. The fractionation was validated based on the location of tubulin and fibrillarin, which were used as cytoplasmic and nuclear markers, respectively. In agreement with the microscopy data, VP8 was first detected in the cytoplasm at 2 h. At 4 h postinfection, VP8 gradually started to localize to the nucleus, and at 6 h there was more VP8 in the nucleus than in the cytoplasm. However, at 14 h postinfection most of the VP8 was present in the cytoplasm. According to these data, incoming viral VP8 is present in the cytoplasm and thus is capable of retaining STAT1 in the cytoplasm immediately after infection. Although VP8 then migrated from the cytoplasm to the nucleus, some VP8 was present in the cytoplasm at all times, and at late stages of infection it was again more prevalent in the cytoplasm. Since VP8 is capable of STAT1 retention immediately after infection, this explains its ability to inhibit IFN signaling at the onset of BoHV-1 infection.
FIG 11.
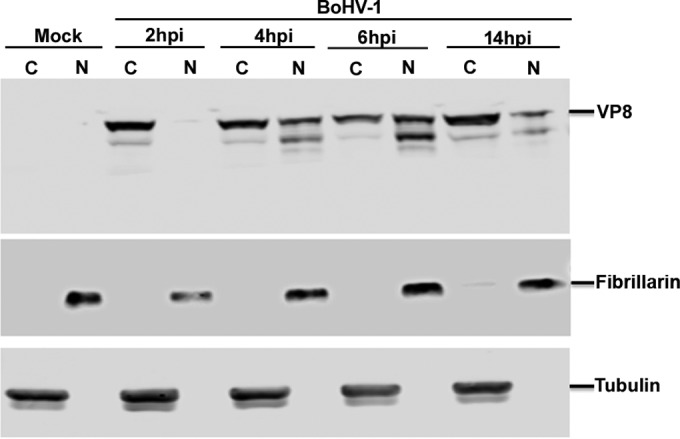
Subcellular fractionation of BoHV-1 VP8 at different times during infection. MDBK cells were mock infected or infected with BoHV-1 at an MOI of 4 for 2, 4, 6, and 14 h. The cells were collected by trypsinization at the indicated time points, followed by cytoplasmic and nuclear fractionation of proteins isolation as described in Materials and Methods. The resulting fractionations were analyzed by SDS-PAGE followed by detection of VP8 with anti-VP8 antibody. The fractionation procedure was validated by incubation with antibodies specific for the cytoplasmic and nuclear proteins tubulin and fibrillarin, respectively.
DISCUSSION
In response to viral infection, cells establish an antiviral state through type I IFN signaling; however, many viruses have evolved unique mechanisms to evade this response. With this study, we demonstrated that VP8 of BoHV-1 plays an important biological function by downregulating IFN-induced responses. Transient expression of BoHV-1 VP8 inhibited IFN-β-induced transcriptional responses in transfected cells. Inhibition of IFN signaling was also observed in BoHV-1-infected cells, while BoHV1-ΔUL47-infected cells showed partial recovery of IFN signaling compared to BoHV-1-infected cells. The presence of bICP0 and possibly bICP27 can also downregulate the IFN responses (20–22), which might explain the incomplete recovery of IFN signaling in BoHV1-ΔUL47-infected cells. To test this, ActD was added during BoHV-1 infection, which prevented immediate early gene expression. In ActD-treated cells, downregulation of IFN signaling was observed in BoHV-1- or BoHV1-UL47R-infected cells, but not in mock- or BoHV1-ΔUL47-infected cells, which further confirmed the role of VP8 in downregulation of IFN signaling.
Despite some homology between the BoHV-1 and human herpesvirus 1 (HHV-1) UL47 gene products, they differ in several respects. The HHV-1 UL47 gene product, VP13/14, interacts with polyadenylate-binding protein (PABP) and disrupts the association of PABP with PABP-interacting protein 2 (Paip2) jointly with its binding partner, ICP27 (27). Since Paip2 remains in the cytoplasm, Dobrikova et al. (27) mentioned that it was unclear whether the Paip2 dissociation is a primary event that happens in the cytoplasm after HHV-1 infection. Shu et al. demonstrated that the HHV-1 UL47 interacts with vhs-RNase and attenuates the degradation of all kinetic classes of viral mRNA and thereby regulates the expression of β and γ genes by controlling the expression of α gene products (28). Thus far, these functions have not been investigated for BoHV-1 VP8. Liu et al. presented data suggesting that HHV-1 VP13/VP14 forms a complex with UL31, UL34, and US3, which are critical for viral nuclear egress, and plays a regulatory role in HHV-1 primary envelopment (29). However, deletion of BoHV-1 UL47 did not result in impaired nuclear egress (7). HHV-1 UL47 (VP13/14) interacts with VP16 or alpha-transinducing factor (αTIF), and UL47-deleted HHV-1 demonstrated impaired ability to induce immediate early promoter-regulated expression of a reporter gene (30, 31), but this effect was not observed for BoHV-1 VP8, as immediate early bICP4 mRNA transcripts were detected at similar levels in wild-type (WT), BoHV1-ΔUL47, and BoHV1-UL47R infected cells (7). In addition, when expressed in Escherichia coli or insect cells, HHV-1 VP13/14, but not BoHV-1 VP8, bound directly to RNA (32). In contrast, VP8 expressed in mammalian cells interacts with bICP0, gB, gC, and gD mRNAs (24).
We identified cellular STAT1 as an interacting partner of BoHV-1 VP8 in both VP8-transfected and BoHV-1 infected cells. The domains of VP8 that interact with STAT1 were found to be located in two different regions, VP8 259-482 and 632-686. BLAST sequence analysis of BoHV-1 VP8 demonstrated high homology of amino acids 280 to 735 with UL47-encoded proteins of human herpesvirus 1 and other herpesviruses and much less sequence similarity in the N-terminal region of amino acids 1 to 259 (Fig. 3). Furthermore, the predicted secondary structure for amino acids 280 to 735 was mostly alpha-helical, a feature that has previously been implicated in protein-protein interactions (24, 33). The crystal structure of STAT1 revealed that it has an alpha-helical coiled-coil domain starting at residue 130 and a DNA binding domain that is in the center of the STAT1 protein. The coiled-coil domain presents extensive possibilities for protein-protein interaction and, indeed, has been documented as interacting with other proteins (reviewed in reference 34). For example, although the rabies virus P protein binding site on STAT1 could not be precisely determined, the P protein-interacting domain resides in the coiled-coil and DNA binding domain of STAT1 (15). This suggests that the BoHV-1 VP8 binding site on STAT1 may be present on the coiled-coil or DNA-binding domain.
Viruses have evolved different mechanisms to antagonize the IFN signaling pathway through STAT1 or STAT2 interaction with viral proteins. For example, several paramyxovirus family members such as human parainfluenza virus 2 and SV5 induce polyubiquitination and degradation of STAT1 to block IFN signaling through the P and V proteins, respectively (35). The C protein of Sendai virus downregulates IFN signaling by inhibiting STAT1 phosphorylation and STAT1 degradation (36). The V protein of measles virus, also belonging to the Paramyxoviridae family, blocks IFN signaling by reducing STAT1 and STAT2 phosphorylation (37). In contrast, mumps virus V protein antagonizes IFN-induced antiviral effects by both degradation of STAT1 and prevention of nuclear translocation of STAT1 (19). Similarly, the V proteins of Nipah virus and Hendra virus, members of the Henipavirus genus, inhibit IFN-α/β and IFN-γ signaling by preventing both STAT1 phosphorylation and nuclear translocation (38, 39). The P protein of rabies virus, belonging to the Rhabdoviridae, neither induces STAT1 degradation nor inhibits STAT1 phosphorylation but prevents STAT1 nuclear accumulation (15). The viral protein pM27 of mouse CMV (MCMV) inhibits IFN signaling by inducing proteasomal degradation of STAT2 (40). These examples show that viral proteins antagonize the IFN-induced host antiviral effects through the JAK-STAT signaling pathway in different ways.
VP8 is the first BoHV-1 protein identified as interacting with STAT1, thus contributing to prevention of IFN signaling. Although the expression of BoHV-1 VP8 inhibited IFN signaling, VP8 induced neither ubiquitination nor degradation or phosphorylation of STAT1. However, translocation of STAT1 to the nucleus following IFN-β treatment was inhibited by full-length VP8 as well as a truncated form of VP8, which contains the domains for interaction with STAT1. After exposure to IFN, ligand-dependent tyrosine phosphorylation and STAT1 dimerization take place, followed by the accumulation of STAT1s to the nucleus (41). The transport of the large protein complexes to the nucleus is facilitated by binding of importin-α with the nuclear localization signal (NLS) of STAT1 (42). Other residues in the coiled-coil domain are also responsible for nuclear import of some STAT proteins (43). A mutation in the STAT1 leucine residue at 407 (L407A) located in the DNA binding domain inhibited nuclear translocation of STAT1 (42). Similarly, McBridge et al. showed that STAT1 protein defective in DNA binding failed to accumulate in to the nucleus (42). The integrity of the DNA binding domain also determines nuclear retention of STAT1 (44). Since point mutations in the arginine- and lysine-rich residues in the DNA binding domain resulted in defective nuclear import of STAT1 (45), a lack of DNA binding is possibly associated with cytoplasmic retention of STAT1. Thus, an interaction between VP8 and the DNA binding domain or coiled-coil domain of STAT1 could impede the nuclear transport machinery as well as the DNA binding function. The NLS of VP8, amino acids 51 to 54 (RRPR), regulates nuclear localization of VP8 (46). A truncated version of VP8 consisting of amino acids 219 to 741, which lacks the NLS but contains two VP8-STAT1 interacting domains, was completely cytoplasmic and also able to retain STAT1 in the cytoplasm.
Accumulation of STAT1 to the nucleus was not observed in BoHV1- and BoHV1-UL47R-infected cells, while BoHV1-ΔUL47 allowed translocation of STAT1 to the nucleus. BoHV1 VP8 is the most abundant tegument protein of the virion. Previously, the full-length VP8 was observed in the cytoplasm as early as 2 h and was visualized in the nucleus at 5 h postinfection (9). Thus, interaction of VP8 with STAT1 in the cytoplasm might interfere with the nuclear import of STAT1 early after initiation of infection. We demonstrated that VP8 is present in the cytoplasm at 2 h postinfection, at which time STAT1 was retained in the cytoplasm. Since VP8 is a late protein, this suggests that viral VP8 released into the cytoplasm immediately after initiation of infection counteracts the establishment of an antiviral state by interacting with STAT1 to inhibit IFN signaling.
In summary, our data provide evidence that BoHV-1 VP8 inhibits IFN signaling early after initiation of infection in the absence of immediate early protein synthesis. Inhibition of IFN signaling appeared to occur through interference with nuclear translocation of STAT1 in both VP8-transfected and BoHV-1 infected cells. This is the first BoHV-1 protein shown to interact with cellular STAT1 to inhibit IFN-β signaling before the onset of virus replication. These results provide a new functional role for VP8 in BoHV-1 infection and a potential explanation for the lack of viral replication of the UL47 deletion mutant in cattle.
ACKNOWLEDGMENTS
We acknowledge Danielle Blondel (Laboratoire de Virologie Moléculaire et Structurale, LVMS, CNRS, France) for kindly providing plasmids pISREluc and pRL-TK and Richard Randall, University of St. Andrews, School of Biology, St. Andrews, Fife, United Kingdom, for kindly proving us with pSV5V and pHis-Ub plasmids. The confocal microscopy was performed in the College of Veterinary Medicine, University of Saskatchewan.
Funding Statement
This work was funded by Natural Sciences and Engineering Research Council grant 90887-2010 RGPIN, awarded to Sylvia van Drunen Littel van den Hurk. S.A. was partially supported by a Canadian Institutes of Health Research Training Grant in Health Research Using Synchrotron Techniques (CIHR-THRUST).
Footnotes
This is VIDO manuscript number 744.
REFERENCES
- 1.Nandi S, Kumar M, Manohar M, Chauhan RS. 2009. Bovine herpes virus infections in cattle. Anim Health Res Rev 10:85–98. doi: 10.1017/S1466252309990028. [DOI] [PubMed] [Google Scholar]
- 2.Nii S, Uno F, Yoshida M, Akatsuka K. 1998. Structure and assembly of human beta herpesviruses. Nihon Rinsho 56:22–28. (In Japanese.) [PubMed] [Google Scholar]
- 3.Kelly BJ, Fraefel C, Cunningham AL, Diefenbach RJ. 2009. Functional roles of the tegument proteins of herpes simplex virus type 1. Virus Res 145:173–186. doi: 10.1016/j.virusres.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 4.Spear PG. 2004. Herpes simplex virus: receptors and ligands for cell entry. Cell Microbiol 6:401–410. doi: 10.1111/j.1462-5822.2004.00389.x. [DOI] [PubMed] [Google Scholar]
- 5.Granzow H, Klupp BG, Mettenleiter TC. 2005. Entry of pseudorabies virus: an immunogold-labeling study. J Virol 79:3200–3205. doi: 10.1128/JVI.79.5.3200-3205.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carpenter DE, Misra V. 1991. The most abundant protein in bovine herpes 1 virions is a homologue of herpes simplex virus type 1 UL47. J Gen Virol 72(Part 12):3077–3084. [DOI] [PubMed] [Google Scholar]
- 7.Lobanov VA, Maher-Sturgess SL, Snider MG, Lawman Z, Babiuk LA, van Drunen Littel-van den Hurk S. 2010. A UL47 gene deletion mutant of bovine herpesvirus type 1 exhibits impaired growth in cell culture and lack of virulence in cattle. J Virol 84:445–458. doi: 10.1128/JVI.01544-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Drunen Littel-van den Hurk S, Garzon S, van den Hurk JV, Babiuk LA, Tijssen P. 1995. The role of the major tegument protein VP8 of bovine herpesvirus-1 in infection and immunity. Virology 206:413–425. doi: 10.1016/S0042-6822(95)80057-3. [DOI] [PubMed] [Google Scholar]
- 9.Vasilenko NL, Snider M, Labiuk SL, Lobanov VA, Babiuk LA, van Drunen Littel-van den Hurk S. 2012. Bovine herpesvirus-1 VP8 interacts with DNA damage binding protein-1 (DDB1) and is monoubiquitinated during infection. Virus Res 167:56–66. doi: 10.1016/j.virusres.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 10.Barry M, Fruh K. 2006. Viral modulators of cullin RING ubiquitin ligases: culling the host defense. Sci STKE 2006(335):pe21. [DOI] [PubMed] [Google Scholar]
- 11.Zhang K, Afroz S, Brownlie R, Snider M, van Drunen Littel-van den Hurk S. 2015. Regulation and function of phosphorylation on VP8, the major tegument protein of bovine herpesvirus 1. J Virol 89:4598–4611. doi: 10.1128/JVI.03180-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Platanias LC. 2005. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol 5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 13.Kotenko SV. 2011. IFN-lambdas. Curr Opin Immunol 23:583–590. doi: 10.1016/j.coi.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goodbourn S, Didcock L, Randall RE. 2000. Interferons: cell signalling, immune modulation, antiviral response and virus countermeasures. J Gen Virol 81:2341–2364. doi: 10.1099/0022-1317-81-10-2341. [DOI] [PubMed] [Google Scholar]
- 15.Vidy A, Chelbi-Alix M, Blondel D. 2005. Rabies virus P protein interacts with STAT1 and inhibits interferon signal transduction pathways. J Virol 79:14411–14420. doi: 10.1128/JVI.79.22.14411-14420.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Didcock L, Young DF, Goodbourn S, Randall RE. 1999. The V protein of simian virus 5 inhibits interferon signalling by targeting STAT1 for proteasome-mediated degradation. J Virol 73:9928–9933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramaswamy M, Shi L, Monick MM, Hunninghake GW, Look DC. 2004. Specific inhibition of type I interferon signal transduction by respiratory syncytial virus. Am J Respir Cell Mol Biol 30:893–900. doi: 10.1165/rcmb.2003-0410OC. [DOI] [PubMed] [Google Scholar]
- 18.Andrejeva J, Young DF, Goodbourn S, Randall RE. 2002. Degradation of STAT1 and STAT2 by the V proteins of simian virus 5 and human parainfluenza virus type 2, respectively: consequences for virus replication in the presence of alpha/beta and gamma interferons. J Virol 76:2159–2167. doi: 10.1128/jvi.76.5.2159-2167.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kubota T, Yokosawa N, Yokota S, Fujii N, Tashiro M, Kato A. 2005. Mumps virus V protein antagonizes interferon without the complete degradation of STAT1. J Virol 79:4451–4459. doi: 10.1128/JVI.79.7.4451-4459.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saira K, Zhou Y, Jones C. 2007. The infected cell protein 0 encoded by bovine herpesvirus 1 (bICP0) induces degradation of interferon response factor 3 and, consequently, inhibits beta interferon promoter activity. J Virol 81:3077–3086. doi: 10.1128/JVI.02064-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saira K, Zhou Y, Jones C. 2009. The infected cell protein 0 encoded by bovine herpesvirus 1 (bICP0) associates with interferon regulatory factor 7 and consequently inhibits beta interferon promoter activity. J Virol 83:3977–3981. doi: 10.1128/JVI.02400-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.da Silva LF, Sinani D, Jones C. 2012. ICP27 protein encoded by bovine herpesvirus type 1 (bICP27) interferes with promoter activity of the bovine genes encoding beta interferon 1 (IFN-beta1) and IFN-beta3. Virus Res 169:162–168. doi: 10.1016/j.virusres.2012.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Misra V, Babiuk LA, Darcel CL. 1983. Analysis of bovine herpes virus-type 1 isolates by restriction endonuclease fingerprinting. Arch Virol 76:341–354. doi: 10.1007/BF01311201. [DOI] [PubMed] [Google Scholar]
- 24.Islam A, Schulz S, Afroz S, Babiuk LA, van Drunen Littel-van den Hurk S. 2015. Interaction of VP8 with mRNAs of bovine herpesvirus-1. Virus Res 197:116–126. doi: 10.1016/j.virusres.2014.12.017. [DOI] [PubMed] [Google Scholar]
- 25.Labiuk SL, Babiuk LA, van Drunen Littel-van den Hurk S. 2009. Major tegument protein VP8 of bovine herpesvirus 1 is phosphorylated by viral US3 and cellular CK2 protein kinases. J Gen Virol 90:2829–2839. doi: 10.1099/vir.0.013532-0. [DOI] [PubMed] [Google Scholar]
- 26.Lobanov VA, Zheng C, Babiuk LA, van Drunen Littel-van den Hurk S. 2010. Intracellular trafficking of VP22 in bovine herpesvirus-1 infected cells. Virology 396:189–202. doi: 10.1016/j.virol.2009.10.022. [DOI] [PubMed] [Google Scholar]
- 27.Dobrikova E, Shveygert M, Walters R, Gromeier M. 2010. Herpes simplex virus proteins ICP27 and UL47 associate with polyadenylate-binding protein and control its subcellular distribution. J Virol 84:270–279. doi: 10.1128/JVI.01740-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shu M, Taddeo B, Zhang W, Roizman B. 2013. Selective degradation of mRNAs by the HSV host shutoff RNase is regulated by the UL47 tegument protein. Proc Natl Acad Sci U S A 110:E1669–E1675. doi: 10.1073/pnas.1305475110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Z, Kato A, Shindo K, Noda T, Sagara H, Kawaoka Y, Arii J, Kawaguchi Y. 2014. Herpes simplex virus 1 UL47 interacts with viral nuclear egress factors UL31, UL34, and Us3 and regulates viral nuclear egress. J Virol 88:4657–4667. doi: 10.1128/JVI.00137-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y, Sirko DA, McKnight JL. 1991. Role of herpes simplex virus type 1 UL46 and UL47 in alpha TIF-mediated transcriptional induction: characterization of three viral deletion mutants. J Virol 65:829–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y, McKnight JL. 1993. Herpes simplex virus type 1 UL46 and UL47 deletion mutants lack VP11 and VP12 or VP13 and VP14, respectively, and exhibit altered viral thymidine kinase expression. J Virol 67:1482–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Donnelly M, Verhagen J, Elliott G. 2007. RNA binding by the herpes simplex virus type 1 nucleocytoplasmic shuttling protein UL47 is mediated by an N-terminal arginine-rich domain that also functions as its nuclear localization signal. J Virol 81:2283–2296. doi: 10.1128/JVI.01677-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stites WE. 1997. Protein-protein interactions: interface structure, binding thermodynamics, and mutational analysis. Chem Rev 97:1233–1250. doi: 10.1021/cr960387h. [DOI] [PubMed] [Google Scholar]
- 34.Levy DE, Darnell JE Jr. 2002. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol 3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 35.Ulane CM, Horvath CM. 2002. Paramyxoviruses SV5 and HPIV2 assemble STAT protein ubiquitin ligase complexes from cellular components. Virology 304:160–166. doi: 10.1006/viro.2002.1773. [DOI] [PubMed] [Google Scholar]
- 36.Komatsu T, Takeuchi K, Yokoo J, Gotoh B. 2002. Sendai virus C protein impairs both phosphorylation and dephosphorylation processes of Stat1. FEBS Lett 511:139–144. doi: 10.1016/S0014-5793(01)03301-4. [DOI] [PubMed] [Google Scholar]
- 37.Takeuchi K, Kadota SI, Takeda M, Miyajima N, Nagata K. 2003. Measles virus V protein blocks interferon (IFN)-alpha/beta but not IFN-gamma signaling by inhibiting STAT1 and STAT2 phosphorylation. FEBS Lett 545:177–182. doi: 10.1016/S0014-5793(03)00528-3. [DOI] [PubMed] [Google Scholar]
- 38.Rodriguez JJ, Parisien JP, Horvath CM. 2002. Nipah virus V protein evades alpha and gamma interferons by preventing STAT1 and STAT2 activation and nuclear accumulation. J Virol 76:11476–11483. doi: 10.1128/JVI.76.22.11476-11483.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodriguez JJ, Wang LF, Horvath CM. 2003. Hendra virus V protein inhibits interferon signaling by preventing STAT1 and STAT2 nuclear accumulation. J Virol 77:11842–11845. doi: 10.1128/JVI.77.21.11842-11845.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Le VT, Trilling M, Wilborn M, Hengel H, Zimmermann A. 2008. Human cytomegalovirus interferes with signal transducer and activator of transcription (STAT) 2 protein stability and tyrosine phosphorylation. J Gen Virol 89:2416–2426. doi: 10.1099/vir.0.2008/001669-0. [DOI] [PubMed] [Google Scholar]
- 41.Schindler C, Shuai K, Prezioso VR, Darnell JE Jr. 1992. Interferon-dependent tyrosine phosphorylation of a latent cytoplasmic transcription factor. Science 257:809–813. doi: 10.1126/science.1496401. [DOI] [PubMed] [Google Scholar]
- 42.McBride KM, Banninger G, McDonald C, Reich NC. 2002. Regulated nuclear import of the STAT1 transcription factor by direct binding of importin-alpha. EMBO J 21:1754–1763. doi: 10.1093/emboj/21.7.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ma J, Zhang T, Novotny-Diermayr V, Tan AL, Cao X. 2003. A novel sequence in the coiled-coil domain of Stat3 essential for its nuclear translocation. J Biol Chem 278:29252–29260. doi: 10.1074/jbc.M304196200. [DOI] [PubMed] [Google Scholar]
- 44.Meyer T, Marg A, Lemke P, Wiesner B, Vinkemeier U. 2003. DNA binding controls inactivation and nuclear accumulation of the transcription factor Stat1. Genes Dev 17:1992–2005. doi: 10.1101/gad.268003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Melen K, Kinnunen L, Julkunen I. 2001. Arginine/lysine-rich structural element is involved in interferon-induced nuclear import of STATs. J Biol Chem 276:16447–16455. doi: 10.1074/jbc.M008821200. [DOI] [PubMed] [Google Scholar]
- 46.Zheng C, Brownlie R, Babiuk LA, van Drunen Littel-van den Hurk S. 2004. Characterization of nuclear localization and export signals of the major tegument protein VP8 of bovine herpesvirus-1. Virology 324:327–339. doi: 10.1016/j.virol.2004.03.042. [DOI] [PubMed] [Google Scholar]
- 47.Rost B, Sander C. 1994. Combining evolutionary information and neural networks to predict protein secondary structure. Proteins 19:55–72. doi: 10.1002/prot.340190108. [DOI] [PubMed] [Google Scholar]
- 48.Robert X, Gouet P. 2014. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42:W320–W324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]