

ABSTRACT
The highly conserved H3 poxvirus protein is a major target of the human antibody response against poxviruses and is likely a key contributor to protection against infection. Here, we present the crystal structure of H3 from vaccinia virus at a 1.9-Å resolution. H3 looks like a glycosyltransferase, a family of enzymes that transfer carbohydrate molecules to a variety of acceptor substrates. Like glycosyltransferases, H3 binds UDP-glucose, as shown by saturation transfer difference (STD) nuclear magnetic resonance (NMR) spectroscopy, and this binding requires Mg2+. Mutation of the glycosyltransferase-like metal ion binding motif in H3 greatly diminished its binding to UDP-glucose. We found by flow cytometry that H3 binds to the surface of human cells but does not bind well to cells that are deficient in surface glycosaminoglycans. STD NMR experiments using a heparin sulfate decasaccharide confirmed that H3 binds heparin sulfate. We propose that a surface of H3 with an excess positive charge may be the binding site for heparin. Heparin binding and glycosyltransferase activity may be involved in the function of H3 in the poxvirus life cycle.
IMPORTANCE Poxviruses are under intense research because of bioterrorism concerns, zoonotic infections, and the side effects of existing smallpox vaccines. The smallpox vaccine using vaccinia virus has been highly successful, but it is still unclear why the vaccine is so effective. Studying the antigens that the immune system recognizes may allow a better understanding of how the vaccine elicits immunity and how improved vaccines can be developed. Poxvirus protein H3 is a major target of the immune system. The H3 crystal structure shows that it has a glycosyltransferase protein fold. We demonstrate that H3 binds the sugar nucleotide UDP-glucose, as do glycosyltransferases. Our experiments also reveal that H3 binds cell surface molecules that are involved in the attachment of poxviruses to cells. These structural and functional studies of H3 will help in designing better vaccines and therapeutics.
INTRODUCTION
Smallpox is a highly contagious disease caused by variola virus, a member of the Orthopoxvirus genus in the Chordopoxvirinae subfamily of the Poxviridae family. Smallpox has a mortality rate of >30%, and few specific treatments other than vaccination exist (1). Vaccinia virus, also in the Orthopoxvirus genus, was used for mass vaccination against smallpox by the World Health Organization (WHO) and is considered the model vaccine, as its efficacy led to the eradication of smallpox (2). Routine vaccination against smallpox worldwide ended in 1980. As it is highly contagious and often fatal, smallpox is considered a potential bioterrorism agent.
After smallpox vaccination, immunity is effective for many years, but how long protective immunity lasts is not clear (3). Persons who were vaccinated before 1980 may have inadequate immunity against variola virus and hence may be susceptible to infection. People who were never vaccinated are highly vulnerable to smallpox (4). Since the smallpox vaccine contains live vaccinia virus, it can cause secondary effects. In immunocompromised individuals, these effects can be life threatening. A better smallpox vaccine that has fewer side effects is needed (5).
Some of the members of the Orthopoxvirus genus are zoonotic and have recently caused poxvirus infections in humans, with literature reports describing outbreaks of monkeypox (6), cowpox (7), buffalopox (8), volepox (9), and vaccinia (10). Orthopoxviruses are immunologically cross-reactive and can be cross-protective (11). Interest in poxvirus research is high due to the threats of bioterrorism, the side effects of the existing smallpox vaccine, and frequent zoonotic infections. Many research groups have focused their efforts to identify new vaccine strategies and to develop antipoxvirus therapeutics (12).
Vaccinia virus is a large, complex virus that replicates entirely in the cytoplasm and carries a linear double-stranded DNA, which carries >200 genes (13). Vaccinia virus has two infectious forms, the intracellular mature virion (MV) and the extracellular enveloped virion (EV). Each form displays a distinct set of proteins on its surface, which can be targets of antibody responses during vaccination (14, 15). The MV is the most abundant and stable virion form, and many of its surface proteins (L1, A27, A17, H3, and D8) have various roles in virus infectivity through the process of entry and/or fusion, and they are important targets for neutralizing antibodies (15–17). The EV differs from the MV in having an additional membrane, which is fragile and can be lost, thus exposing the MV surface. H3 has been shown to be a direct target of neutralizing antibodies in humans immunized by the smallpox vaccine (18). Previous studies showed the production of strong immune responses to H3 in mice and rabbits (17, 19).
Vaccinia virus H3 is an immunodominant antigen, containing 324 amino acid residues encoded by the H3L gene, that is expressed late in infection and found as a membrane protein on the surface of MV particles. H3 inserts into MV membranes posttranslationally and is tethered by a hydrophobic membrane-spanning region near its C terminus (20). H3 homologs are present in both the Chordopoxvirinae and Entomopoxvirinae subfamilies of the Poxviridae family (21). The role of the H3 protein in the virus is not clear. Previous studies have shown that vaccinia virus can attach to cells by binding to cell surface glycosaminoglycans (GAGs) using the H3, A27, and D8 proteins. H3 and A27 were reported to bind to heparan sulfate and the surface of cells (22, 23). D8 was shown to bind to chondroitin sulfate (24). Mutant vaccinia viruses that do not express H3 are less infectious, form small plaques, have low MV and EV titers, and have a defect in virus morphogenesis (22, 25).
Sera from humans vaccinated with the vaccinia virus vaccine recognized H3 as a dominant antigen in the human antibody response (26). H3 is likely a key contributor to protection against poxvirus infection and disease. In animal models, treatment with antibodies against both H3 and the EV antigen B5 has better efficacy against vaccinia virus infection than treatment with individual monoclonal antibodies (MAbs) or vaccinia virus immune globulin antiserum (27).
Here, we report the crystal structure of the H3 protein together with the binding of H3 to UDP-glucose and heparin sulfate. We discuss the implications of the structure and consider the role of H3 in poxviruses. The structure suggests other potential functions of the H3 protein and can help in designing new antipoxvirus therapeutics and better vaccines.
MATERIALS AND METHODS
Cloning and mutagenesis.
We generated the H3 expression construct of residues 1 to 282 (GenBank accession no. AAO89380.1) by PCR using the 5′ primer TTGTTGAATTCTAAGGAGGANATTCATATGGCGGCGGCGAAAACTCCTG, the 3′ primer GATCCTCAGCGGCCGCTCAAAATGAAATCAGTGGAGTAGTAAACGC, and vaccinia virus DNA from the Western Reserve (WR) strain. The H3L gene has been given the locus name VACWR101 or VACV101. The PCR products were ligated into the vector pNAN by using the NdeI and NotI restriction sites (28). Mutants were constructed by using standard site-directed mutagenesis techniques. The E125A mutant was produced with the 5′ primer GTTATTGTAGCGAACGATAACGTTATTG and the 3′ primer CAATAACGTTATCGTTCGCTACAATAAC, and the D127A mutant was produced with the 5′ primer GTTATTGTAGAAAACGCGAACGTTATTG and the 3′ primer CAATAACGTTCGCGTTTTCTACAATAAC. A version of H3 with a C-terminal 6×His tag was produced for use in the flow cytometry experiments. Before protein expression, all constructs were confirmed by DNA sequencing.
Expression, refolding, purification, and characterization.
The H3 plasmid was transformed into either Escherichia coli BL21-CodonPlus(DE3)-RIL cells for native protein expression or BL21-CodonPlus(DE3)-RIL-X cells (Stratagene, La Jolla, CA) for selenomethionine (SeMet) incorporation. Cells were grown at 37°C in 6 liters of LB medium or selenomethionine medium (Molecular Dimensions, Apopka, FL) containing 100 μg/ml ampicillin. l-SeMet (50 mg/liter) (Sigma, St. Louis, MO) was added to SeMet medium. The culture was induced with 1 mM isopropyl-β-d-thiogalactopyranoside at an optical density at 600 nm (OD600) of 0.6 to 0.8 for 4 to 6 h to produce insoluble H3 protein as inclusion bodies. The cells were harvested by centrifugation at 4,000 × g and resuspended in 100 ml of lysis buffer (100 mM Tris-Cl [pH 8.0], 150 mM NaCl, 1% [vol/vol] Triton X-100, and 1% [wt/vol] sodium deoxycholate). Cells were lysed in the presence of lysozyme (1 mg/ml) by repeated freezing and thawing.
H3 inclusion bodies were purified by multiple rounds of centrifugation, homogenization, washing, and suspension using lysis buffer without detergent. The final pellet of inclusion bodies was dissolved in a solubilization buffer consisting of 50 mM Tris-Cl (pH 8.0), 6 M guanidine-HCl, 1 M urea, and 2 mM dithiothreitol (DTT) and then centrifuged at 14,000 × g for 10 min to remove any undissolved material. Purified H3 inclusion bodies were refolded by rapid dilution at 4°C in 1 liter of buffer containing 50 mM Tris-Cl (pH 8.0), 50 mM NaCl, 500 mM arginine-HCl, 2 mM EDTA, 40 mM sucrose, 2 mM DTT, and 5 mM cystamine-HCl (CAS 51-85-4). After refolding for 12 to 15 h, cystamine-HCl (50 mM) was added and allowed to react with free cysteines for 30 min. Refolded H3 protein was then dialyzed two times for 24 h each against 12 liters of 20 mM Tris-HCl (pH 8.0). After dialysis, H3 protein was filtered through a 0.22-μm filter membrane with a 10-kDa-molecular-mass cutoff and purified by using a HiTrap Sepharose Q ion exchange column. High-quality H3 protein was obtained from a Superdex-75 column, dialyzed against 10 mM Tris-HCl (pH 7.5) at 4°C, and concentrated in a centrifugal filter device (Centricon YM10; Millipore, Billerica, MA) to 8 mg/ml. The extent of SeMet incorporation was determined by mass spectrometry. H3 mutants E125A and D127A were produced without SeMet under the same conditions and were concentrated to 8 to 10 mg/ml. Proteins were >95% pure as assessed by SDS-PAGE.
Crystallization and structure determination.
After exhaustive screening of numerous combinations of crystallization screens and additives, a single crystal with cell dimensions of a = 42.36, b = 54.79, and c = 111.99 that belonged to space group P212121 (Table 1) was obtained by vapor diffusion in a hanging drop by mixing 1 μl of protein and 1 μl of precipitant and equilibration against a screen with a precipitant (500 μl) containing 100 mM citric acid (pH 4.0), 10 to 20% polyethylene glycol 6000, 5% 1,3-propanediol (Pdo), and 0.5% of a mixture of low-molecular-weight alcohols. This crystal was used to collect a native data set to 1.9 Å. Subsequent soaking of this irradiated crystal with several heavy atoms did not produce a usable heavy atom derivative.
TABLE 1.
Data collection and refinement statistics
| Parameter | Value(s) for native H3a |
|---|---|
| Data collection statistics | |
| Space group | P212121 |
| Cell dimensions | |
| a, b, c (Å) | 42.36, 54.79, 111.99 |
| Resolution (Å) | 49.22–1.90 |
| I/σI | 18.54 (1.95) |
| Completeness (%) | 99.8 (99.6) |
| Redundancy | 4.79 |
| Refinement | |
| Resolution (Å) | 49.22–1.90 |
| No. of reflections | 21,213 |
| Rwork/Rfree | 19.73/24.00 |
| No. of atoms | |
| Protein | 1,652 |
| Ligand | 103 |
| Water | 91 |
| Mg2+ | 1 |
| B-factor | |
| Protein | 32.59 |
| Ligand/ion | 41.16 |
| Water | 38.40 |
| Mg2+ | 34.50 |
| Root mean square deviation | |
| Bond length (Å) | 0.008 |
| Bond angle (°) | 1.089 |
Values in parentheses are for the highest-resolution shell.
Attempts using the same crystallization conditions to obtain additional native crystals that belonged to the same space group were unsuccessful. Crystals grew, but they were smaller, belonged to space group C2221, and diffracted to a considerably lower resolution. Attempts to obtain a heavy atom derivative in this new space group were unsuccessful.
A SeMet H3 protein was produced, and multiple plate-like crystals were grown under similar conditions. The SeMet crystals diffracted to 3.5 Å at best and belonged to space group C2221. The SeMet crystals were used to collect data at the peak, edge, and remote Se energies. All X-ray data sets were integrated and scaled by using HKL2000 (29) or XDS (30). A multiple-wavelength anomalous diffraction (MAD) experiment was performed to obtain experimental protein phases using SOLVE/RESOLVE (31). Density modification and automated model building were performed by using the RESOLVE software program (32).
Of the seven methionines in the H3 sequence, only four produced relatively high Patterson peaks from the C2221 X-ray data and were used in SOLVE to yield a figure of merit of 0.52. RESOLVE generated several contiguous stretches of protein chain, and manual building extended the model into the solvent-flattened MAD electron density map by using the program O (33). After several cycles of manual model building into new electron density maps, calculated by phase combination using SIGMAA and by the application of SOLVE/RESOLVE, no further improvement of the model could be achieved. The partial model was refined by using CNS (34) and used as a search model for obtaining a molecular replacement solution in the 1.9-Å native P212121 data set. There is one H3 molecule in the asymmetric unit. Multiple cycles of refinement with stepwise increases in the resolution using CNS and the program O for manual model building produced the final model (32, 35). All structural figures were produced with PyMOL (PyMOL molecular graphics system, version 1.7.4; Schrödinger, LLC).
Preparation of NMR samples.
Protein nuclear magnetic resonance (NMR) samples in D2O were prepared by lyophilization of the H3 protein in a buffer containing 20 mM sodium phosphate at pH 6.9 and 50 mM NaCl. The samples were lyophilized three times in 50 μl D2O (99.96% deuterium; Cambridge Isotope Laboratories, Inc., Andover, MA) and subsequently dissolved in 600 μl D2O. Control NMR samples of ligands in the absence of protein were prepared in the same manner. NMR samples containing mixtures of protein and ligand were prepared by the addition of small volumes of a concentrated ligand solution in D2O to the protein NMR sample. Protein-to-ligand molar ratios of either 1:500 (3 μM–1.5 mM) or 1:100 (6 μM–600 μM) were used for binding studies of nucleotide sugars or a heparin sulfate fragment, respectively. The heparin sulfate fragment (catalog no. GT-8051; Neoparin, Inc., Alameda, CA) used in this study was a decasaccharide, -[4-GlcA-4GlcNAc-1-]5, containing five repeats of GlcA (glucuronic acid) and GlcNAc (N-acetylglucosamine). UDP and CMP were obtained from Sigma-Aldrich. All of the nucleotide diphospho-sugars used for the study, UDP-Glc (glucose), UDP-GlcNAc, UDP-Gal (galactose), and UDP-GalNAc (N-acetylgalactosamine), were obtained from Calbiochem. NMR binding studies with nucleotide sugars were performed in the presence and in the absence of MgCl2.
STD NMR spectroscopy.
One-dimensional (1D) saturation transfer difference (STD) NMR experiments are based on the intermolecular transfer of saturation (magnetization) from a protein to a bound ligand, giving rise to intensity attenuation of ligand protons at the protein-ligand binding interface, in cases where the exchange between the free and bound states of the ligand is fast on the NMR relaxation time scale. Practically, the transfer process can be achieved by saturating selected protein resonance with a cascade of Gaussian soft pulses. The magnetization propagates rapidly to all other protein protons as well as to protons of the bound ligand via dipolar interaction (1H-1H cross-relaxation pathway, also called spin diffusion), with the effectiveness having a distance dependence of 1/r6. Subsequently, the bound ligand molecule dissociates back into solution, where the saturated state persists due to its low spin-lattice relaxation rate constants. For a duration equal to the length of the radiofrequency saturation pulse, additional fresh ligand exchanges on and off the protein, thus increasing the population of saturated free ligand molecules in solution. The STD effects are detected as the difference of the saturated (attenuated) ligand resonance intensities from those of the corresponding reference ligand intensities in a spectrum that is acquired with off-resonance irradiation. The off-resonance spectrum results in no protein saturation and is acquired under identical conditions to avoid experimental inconsistencies and radiofrequency-induced sample temperature change. The resulting STD spectrum yields only ligand resonances that have experienced saturation as a result of their binding to the protein.
STD NMR experiments were performed at 15°C, 25°C, and 35°C on a Bruker Avance DRX 500-MHz NMR spectrometer equipped with a cryoprobe. A temperature of 15°C proved to be optimal. Spectra were recorded with 1,024 scans and selective saturation of the protein with a series of 40 Gaussian-shaped pulses of 50 ms with a 1-ms delay between pulses for a total saturation time of 2.04 s, as described previously (36). To ensure adequate saturation of the protein and to avoid potential subtraction artifacts, we obtained STD NMR data for control samples of free ligand, free protein, and their complex, optimizing the on-resonance frequency at −0.15 ppm; the radiofrequency power level for selective irradiation (γB1/2 π = 50 Hz); and the protein-to-ligand molar ratios as described above. Background protein resonances were partially suppressed by the application of a T1ρ filter with a 30-ms spin-lock pulse with a field strength, γB1/2 π, of 4,960 Hz. The free induction decays with on- and off-resonance saturations at −0.15 ppm and 50 ppm, respectively, were collected in an interleaved manner to compensate for the effects of temperature and instrument instability. Data were processed and analyzed by using Bruker TopSpin software (Bruker Biospin Corp., Billerica, MA). The STD NMR spectrum was obtained by subtraction of the reference spectrum collected by using off-resonance irradiation from the saturated, or on-resonance, spectrum.
The STD NMR data for the UDP-Glc binding study were acquired without suppressing the large resonance from water, because such suppression can sometimes saturate the ligand resonances that are being measured. In our experiments, we obtained similar data with water suppression, which was achieved by the application of the 3-9-19 water suppression by gradient tailored excitation (WATERGATE) pulse train. All of the data were used to optimize the STD experimental parameters. No effects on the STD signals were observed as a result of varying the length of the protein saturation time (2 s versus 4 s) or the selective frequency for protein saturation (0.15 ppm, 0.65 ppm, and 6.75 ppm). Although the application of the WATERGATE pulse train resulted in excellent suppression of the water signals, the intensities of some ligand resonances near the water line were partially saturated. Partial saturation of the corresponding resonances was also observed in the 1D NMR data.
Binding of H3 to heparin sulfate on the surface of CHO-K1 cells.
Protein binding to GAGs expressed on the surface of Chinese hamster ovary K1 (CHO-K1) cells (American Type Culture Collection [ATCC], Manassas, VA) was carried out by using flow cytometry as described previously (37). Control cells were CHO-745 cells (ATCC), which are deficient in the biosynthesis of heparin sulfate and other GAGs (38). H3 protein (5 μM) was incubated for 30 min with ∼1 million cells (100 μl) at room temperature. Upon completion of incubation, cells were washed three times in fluorescence-activated cell sorter (FACS) buffer and treated with anti-His MAb (MAB050; R&D Systems, Minneapolis, MN) for 30 min at 4°C. The cells were washed twice in phosphate-buffered saline (PBS) and incubated further with Alexa 488-labeled goat anti-mouse IgG (100 μl) (catalog no. A11029; Invitrogen, Carlsbad, CA) diluted 1:250 in FACS buffer. After 30 min of incubation, the cells were washed, resuspended in 200 μl FACS buffer, and analyzed by using a FACSort cytometer (Becton Dickinson). All incubations and washing steps were performed at 4°C. Events (75,000) were acquired by using CELL Quest software (version 3.3; Becton Dickinson), and FlowJo software (version 6.4.1; Tree Star, San Carlos, CA) was used to analyze the data from the live-cell gating.
Protein structure accession number.
The H3 coordinates and X-ray data have been deposited in the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB) under accession no. 5EJ0.
RESULTS
Overall structure and topology of H3.
The crystal structure of the H3 protein encoded by the vaccinia virus H3L gene revealed a mixed β/α/β-fold comprised of a nine-stranded β-sheet flanked by α-helices (Fig. 1A). The nine-stranded β-sheet is made up of strands in the order β6, β7, β9, β5, β8, β4, β1, β3, and β2, and all of the strands except β7 and β8 are oriented parallel to each other (Fig. 1B). The H3 structure includes residues 4 to 34, residues 60 to 240, and an Mg2+ ion. The H3 protein contains two cysteines, Cys86 and Cys90, that are not disulfide bonded. As is frequently observed, the N-terminal methionine residue was removed by bacterial enzymes during expression. Residues 35 to 59 and 240 to 281 were not located in the electron density, presumably because they were disordered in the crystal. Mass spectrometry determined that the mass of purified H3 was 30,339 Da, smaller than the expected mass of 32,476 Da (281 residues). However, a 30,330-Da H3 protein that resulted from proteolytic cleavage after residue 262 could explain the lack of electron density from residues 263 to 281. The highly purified H3 protein migrated as a single band of ∼30 kDa on SDS-PAGE gels and appeared to be a monomer during size exclusion chromatography.
FIG 1.

H3 structure shown in ribbon and topology diagrams. (A) Two views of H3 in which the helices are red and the strands are blue. The right-hand view is rotated 90° from the left-hand view around the vertical axis. The boundaries of the disordered loop (“34” and “60”) are indicated. Residues 4 to 34 and 60 to 240 are shown (see the text for details). (B) Topology diagram showing the nine-stranded beta sheet (pink) and the helices (red). Numbers at the ends of a secondary-structure element are the locations of the helix or strand termini in the H3 sequence. The N and C termini are identified.
H3 has a glycosyltransferase protein fold.
Searches of the structure database with the DALI (39) and the PDBeFold (40) servers returned lists of structurally similar proteins that are known or proposed to be glycosyltransferases (GTs) of the GT-A group. Like H3, the typical topology of the GT-A family displays a central twisted β-sheet of 6 to 10 strands that is surrounded with helices on both sides of the β-sheet. The top Z value of 11.6 in DALI was rabbit N-acetylglucosaminyltransferase I (PDB accession no. 1FO9) (41), with a root mean square deviation (RMSD) of 3.3 Å for 170 structurally aligned α-carbons. The second highest Z value was 9.7 for the putative viral glycosyltransferase A64R from Paramecium bursaria chlorella virus 1 (PBCV-1) (PDB accession no. 2P6W) (42), with an RMSD of 3.3 Å over 155 α-carbons (Fig. 2A). Another viral protein, the putative glycosyltransferase A197 protein from Sulfolobus turreted icosahedral virus (PDB accession no. 2C0N) (43), was similar to H3, with a Z value of 9.3 and an overall RMSD of 3.0 Å over 141 α-carbons.
FIG 2.

Comparison of H3 and the PBCV-1 protein A64R. (A) Structural superimposition of H3 (red) and the PBCV-1 protein A64R (gray). (B) Closeup view of the metal coordination site in the superimposition. The Mg2+ ion of H3 (red) is close to the Mn2+ ion of A64R (gray). H3 residues E125 and D127 are the “E/DxD” motif.
Conserved carboxylates coordinate a cation.
Glycosyltransferase sequences exhibit conserved residues that are involved with their enzymatic activity. Many glycosyltransferase sequences contain a sequence motif of an Asp or Glu residue followed by an Asp residue 2 residues away (D/ExD motif) (Fig. 3A). The D/ExD motif contributes to the coordination of the divalent cation of the nucleotide sugar. In the PBCV-1 A64R structure, the D/ExD motif interacts with the bound Mn2+ ion and UDP-glucose (42). In H3, the D/ExD motif coordinates an Mg2+ ion that is located within 1.0 Å of the Mn2+ ion in A64R (Fig. 2B). The D/ExD motif is conserved in most members of the GT-A family and in all H3 homologs, even in those like canarypox virus H3, with only 30% sequence identity. In H3, oxygen atoms from the D/ExD motif residues Glu125 and Asp127 coordinate an Mg2+ ion. The Mg2+ ion is also coordinated by Ser186, by a water molecule, and by a molecule of 1,3-propanediol from the buffer (Fig. 3B).
FIG 3.

The E/DxD motifs of H3 and glycosyltransferases involve a cation. (A) Sequence alignment of the motif comparing H3 with glycosyltransferases under PDB accession no. 1FO9 (45) (rabbit GnT I), 2P72 (42) (PBCV-1 A64R protein), 2Y4L (64) (MGS [mannosylglycerate synthase]), 2C0N (43) (Sulfolobus turreted icosahedral virus [STIV] A197), 2VS3 (65) (bovine α3GT [α-1,3-galactosyltransferase]), 1G9R (66) (LgtC [Neisseria meningitidis α-1,4-galactosyl-transferase]), 1OMX (67) (EXTL2 [α-1,4-N-acetyl-hexosaminyltransferase]), and 1FGX (68) (bovine β4Gal-T1 [β-1,4-galactosyl-transferase 1]). (B) Coordination (lavender) of the bound Mg2+ ion (green) through Glu125, Asp127, Ser186, and two buffer molecules, 1,3-propanediol (Pdo) and methanol (Moh). Carbon (black) and oxygen (red) atoms are shown.
Binding studies using H3 for STD NMR spectroscopy.
As glycosyltransferases generally use nucleotide diphospho-sugars as monosaccharide donors, we tested several nucleotides and nucleotide sugars as potential H3 protein binding ligands: UDP, UDP-Glc, UDP-GlcNAc, UDP-Gal, UDP-GalNAc, and CMP. Of the above-mentioned ligands, only UDP-Glc led to significant STD enhancements, which were observed only in the presence of Mg2+ (Fig. 4). Preliminary STD NMR experiments were performed with and without Mg2+, using several protein-to-ligand molar ratios (1:50, 1:100, 1:300, 1:500, and 1:1,000) and three temperatures (15°C, 25°C, and 35°C). A protein-to-ligand molar ratio of 1:500, i.e., 3 μM H3 and 1.5 mM UDP-Glc, in the presence of 1.5 mM Mg2+ at 15°C was optimal for observing STD signals from UDP-Glc (Fig. 4D).
FIG 4.

Vaccinia virus H3 protein binds UDP-glucose and requires Mg2+. (A) 1D 1H NMR spectrum of UDP-glucose (UDP-Glc) in the presence of the H3 protein, at a protein-to-ligand molar ratio of 1:500 (3.0 μM–1.5 mM). UDP-Glc protons, with glucose (G), ribose (R), and uracil (U) subscripts, are identified on the spectrum and on the UDP-Glc chemical structure (inset). (B) No STD effects are observed in the absence of Mg2+. (C) A few STD signals were observed at 0.75 mM Mg2+. (D) Maximal STD signals from UDP-Glc protons were observed at 1.5 mM Mg2+, which are the result of UDP-Glc molecules gaining magnetic saturation while bound to H3. After dissociating from H3, the UDP-Glc molecules retain saturation and contribute to the STD spectrum while unbound (free). (E) At 3 mM Mg2+, there is no further enhancement of the STD signals. The STD data were acquired with a spectral width of 6,000 Hz, 32,768 data points, 2,048 scans, and a receiver gain of 128 and without water suppression. The saturation of the protein was achieved by selective irradiation at −0.15 ppm (see Materials and Methods). “HDO,” residual partially deuterated water resonance.
The observed STD resonances show that all UDP-Glc protons experienced some degree of saturation, which indicates that the entire ligand is bound to the H3 protein (compare Fig. 4A and D). Note that the glucose spectral region is ∼3.2 to 4.6 ppm, with the ribose and uracil proton resonances further up-field. The assigned chemical shifts shown in Fig. 4A for UDP-Glc protons as displayed in the 1D NMR spectrum were confirmed by two-dimensional (2D) total correlation spectroscopy (TOCSY) NMR experiments (data not shown) and are consistent with previously reported assignments for UDP-Glc (44). Comparison of the STD data without Mg2+ (Fig. 4B) with the STD data that were acquired with various concentrations of Mg2+ (Fig. 4C to E) reveals the strong Mg2+ dependence of UDP-Glc binding to H3. At an Mg2+ concentration of 1.5 mM, equal to the UDP-Glc concentration, binding was maximal. Less binding was observed at 0.75 mM Mg2+ and at 3 mM Mg2+. This implies that the binding ligand is the complex of Mg2+ and UDP-Glc, which provides further evidence that the vaccinia virus H3 protein might function as a glycosyltransferase.
STD NMR experiments using UDP, UDP-GlcNAc, or CMP under the same conditions revealed small STD effects of a much lower intensity than with UDP-Glc. Only a few protons were involved in saturation transfer, whether measured with or without Mg2+, implying that such minimal binding is not likely to be of biological significance. Using UDP-Gal or UDP-GalNAc, two other potential ligands, small STD effects were observed whether or not Mg2+ was present, indicative of nonspecific binding (data not shown).
Mutation of the E/DxD motif of H3.
Figure 5A displays a 1D NMR spectrum of UDP-Glc for comparison to the STD spectra that follow in Fig. 5. In Fig. 5B, a control STD spectrum that was acquired under identical experimental conditions using 1.5 mM UDP-Glc in the absence of H3 shows no STD effects for any of the UDP-Glc protons. This control spectrum demonstrates the absence of subtraction artifacts and the lack of any accidental direct radiofrequency power saturation of ligand protons in the absence of H3. We performed STD NMR studies of the binding of the UDP-Glc ligand with wild-type H3 (H3_UDP-Glc) (Fig. 5C) and with H3 with a mutation to alanine at either Glu125 (125H3_UDP-Glc) (Fig. 5D) or Asp127 (127H3_UDP-Glc) (Fig. 5E).
FIG 5.
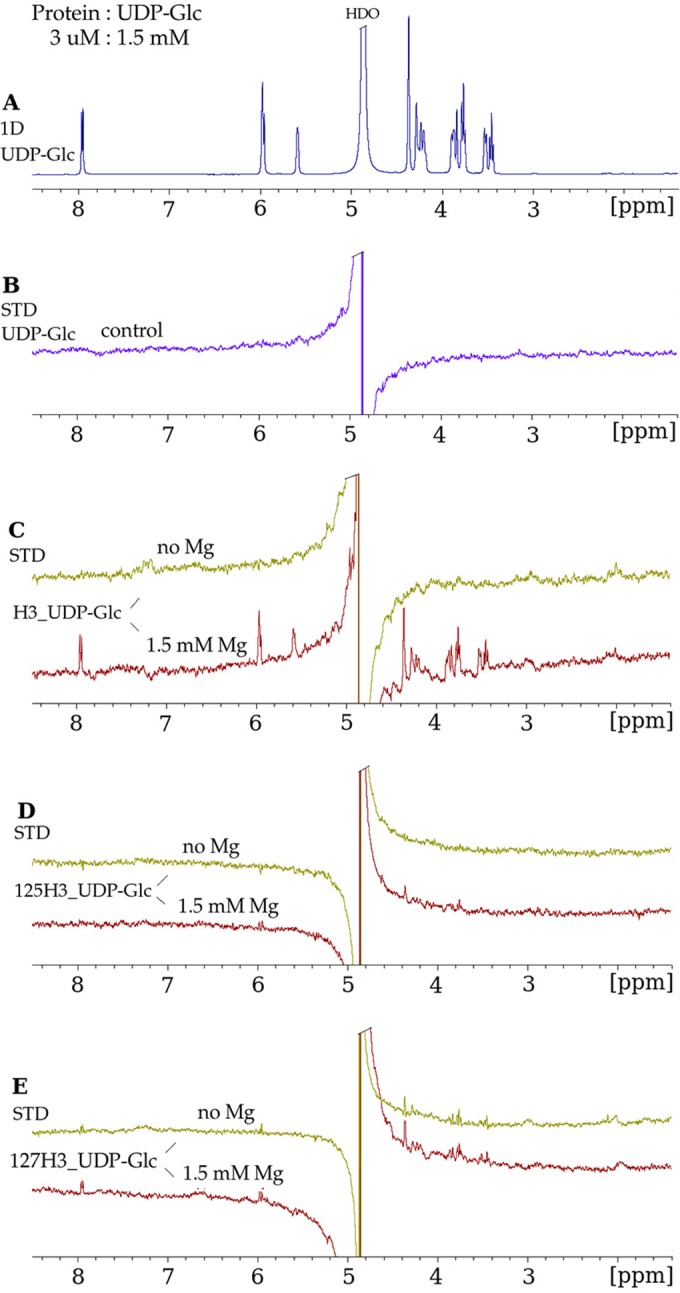
Mutation of the conserved glycosyltransferase D/ExD motif in H3 disrupts UDP-Glc binding. (A) 1D 1H NMR spectrum of UDP-Glc in the presence of H3, at a protein-to-ligand molar ratio of 1:500, under the same conditions as those described in the legend of Fig. 4A. (B) STD NMR spectrum of UDP-Glc in the absence of the H3 protein, in which no STD enhancements were observed for the protons of UDP-Glc. (C) STD NMR spectra in the absence of Mg2+ and in the presence of Mg2+ demonstrating binding of UDP-Glc to wild-type H3. (D) E125A mutant (“125H3_UDP-Glc”). The strong decrease in the STD enhancements for UDP-Glc in the presence of the E125A mutant indicates that the mutation of the H3 protein at residue 125 inhibits the binding of UDP-Glc. (E) D127A mutant (“127H3_UDP-Glc”). The lack of Mg2+ dependence of the STD enhancements in the presence of the D127A mutant suggests nonspecific binding.
The binding of UDP-Glc to the 125H3 alanine mutant shows significant decreases in the STD effects (Fig. 5D) in the sugar spectral region (3.2 to 4.6 ppm) and no STD effects for the H5&6U, H1′R, and H1′G ligand protons (Fig. 5D), compared to the STD of wild-type H3 binding UDP-Glc (Fig. 5C).
The binding of UDP-Glc to the 127H3 alanine mutant exhibits similar STD effects in the absence and in the presence of Mg2+ (Fig. 5E). The STD enhancements are of smaller intensities (Fig. 5E) than the corresponding STD effects observed for H3_UDP-Glc (Fig. 5C), and no STD effect is observed for the H1G ligand protons. The lack of an Mg2+ dependence of the STD effects for the 127H3–UDP-Glc complex implies a nonspecific nature for the binding interaction between 127H3 and UDP-Glc. In conclusion, the mutation at residue 125 inhibits all binding of UDP-Glc to H3, and the mutation at residue 127 results in nonspecific binding of UDP-Glc to H3.
Binding of H3 to the surface of cells as observed by flow cytometry.
It has been reported that the H3 protein binds to heparan sulfate (22). The binding of H3 to the surface of cells was assessed by using flow cytometry. CHO-K1 cells are wild-type cells that express GAGs, including heparan sulfate, on their surface, as do most mammalian cells. The shifted peak of cell fluorescence shown in Fig. 6A demonstrates that H3 bound to the surface of CHO-K1 cells. As a control, we repeated the experiment using instead CHO-745 cells that do not express GAGs on their cell surface. CHO-745 cells have a mutation that leads to a nonfunctional xylose transferase, the enzyme that catalyzes the first step of GAG addition to a GAG core protein. Under identical conditions, H3 bound weakly to the surface of CHO-745 cells (Fig. 6B), suggesting that H3 binding to cell surfaces depended on GAGs.
FIG 6.
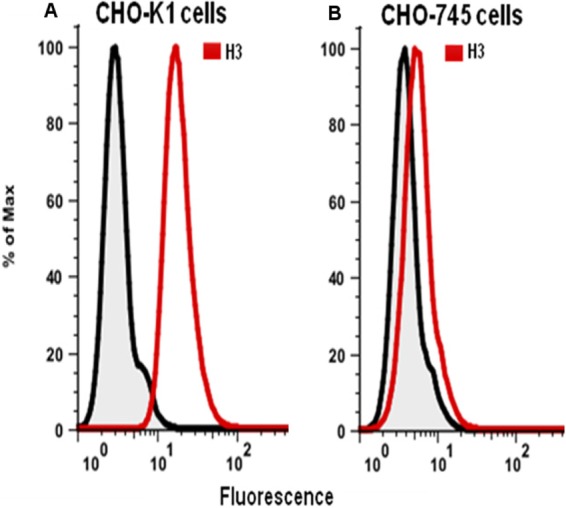
The H3 protein binds to GAGs on the surface of cells. (A) H3 with a His tag sequence at its C terminus was incubated with CHO-K1 cells. Bound H3 (red peak), detected with an anti-His tag antibody and a secondary antibody, yielded higher fluorescence by flow cytometry. (B) Using CHO-745 cells that do not express GAGs, H3 does not bind (red peak). In both panels, the gray peaks are a control in which H3 and anti-His tag antibody were omitted.
H3 binds a heparin sulfate decasaccharide.
The flow cytometry results indicating that H3 binds to GAGs on cell surfaces led us to study the binding of H3 to heparin by saturation transfer NMR techniques. Using a commercially available heparin decasaccharide, we collected NMR data on H3, the decasaccharide, and the mixture of the two. The decasaccharide (∼3,000 Da) has a much higher molecular mass than those of the nucleotide sugars (∼600 Da), and its larger size broadens and weakens its NMR resonances compared to those of the smaller ligands. We found that STD NMR signals from H3 itself were no longer negligible compared to the STD signals from the heparin decasaccharide ligand. We used a double-difference strategy to remove the STD signals due to H3, allowing us to observe the STD signals from the decasaccharide.
In Fig. 7A, a saturation transfer double-difference (STDD) NMR spectrum was used to observe the binding of the heparin decasaccharide to H3 (compare the peaks in Fig. 7A with the 1D spectrum of the decasaccharide alone in Fig. 7B). The STD peaks in Fig. 7A are clearly from only the ligand protons since they are at the same chemical shifts as those in Fig. 7B. The decasaccharide peaks in the STDD spectrum in Fig. 7A are solid evidence that the heparin decasaccharide binds to H3.
FIG 7.
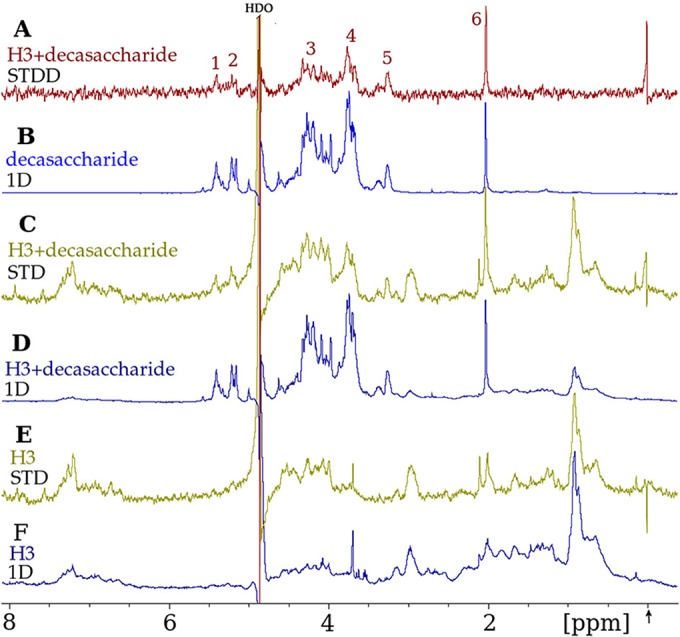
H3 binds a heparin decasaccharide ligand, as observed by STDD NMR spectroscopy. (A) STDD NMR spectrum displaying decasaccharide proton peaks (asterisks) that arise from the transfer of saturation from protons of the H3 protein to protons of the bound decasaccharide. This “double-difference” spectrum is the result of subtracting spectrum “E” (see below) from spectrum “C” and was performed to eliminate background H3 protein resonances. (B) Heparin decasaccharide (600 μM) spectrum in a 1D 1H NMR experiment. Note that these resonances are the same as those in panel A but are at a higher signal-to-noise ratio here. (C) STD NMR spectrum of the H3 protein mixed with the decasaccharide. This STD spectrum was obtained by subtracting the on-resonance (−0.15 ppm) spectrum (not shown) from the off-resonance (50 ppm) spectrum (not shown). (D) 1D 1H NMR spectrum of the mixture of H3 (6 μM) and the decasaccharide (600 μM). (E) STD spectrum of H3 alone as a control, obtained as described above for panel C. (F) 1D 1H NMR spectrum of H3 alone. The arrow (bottom left) denotes that the on-resonance selective irradiation to achieve the saturation of H3 protons was performed at −0.15 ppm, which is distant from all decasaccharide resonances. All spectra were collected with a spectral width of 6,000 Hz and a receiver gain of 128. The 1D 1H NMR spectra in panels B, D, and F were collected by using 4,096 data points and 256 scans with water suppression. The 1D STD NMR on- and off-resonance spectra were collected under identical conditions but using 32,768 data points and 2,048 scans without water suppression.
The STDD spectrum is the result of subtracting the spectrum in Fig. 7E from the spectrum in Fig. 7C. Figure 7C shows an STD spectrum of H3 in the presence of the decasaccharide at a protein-to-ligand molar ratio of 1:100. H3 in the absence of ligand, shown in Fig. 7E, reveals enhancements for the majority of the H3 protein resonances seen in the 1D H3 spectrum in Fig. 7F, demonstrating that there was efficient spin diffusion throughout the H3 protein. Figure 7E also indicates that the application of a T1ρ filter (see Materials and Methods) to the STD NMR data did not result in the complete elimination of the protein resonances. H3 shows STD effects (Fig. 7E) in the same spectral region as that of the decasaccharide resonances (Fig. 7B) and thus would interfere with the interpretation of the decasaccharide STD enhancements. Our use of a double-difference technique overcame this experimental obstacle and resulted in the STDD spectrum shown in Fig. 7A.
The decasaccharide binding data shown in Fig. 7 were collected in the presence of 50 mM NaCl in the NMR samples. In order to probe the effect of the NaCl concentration on binding, we collected additional STD NMR data with 100 mM, 200 mM, and 300 mM NaCl present in the NMR sample (data not shown). While the ligand STD effects at 100 mM NaCl exhibit almost the same intensities as the corresponding ones at 50 mM NaCl shown here, a considerable decrease in the STD effects was observed at 200 mM NaCl, and no STD effects were observed at 300 mM NaCl. These results suggest that the binding of heparin sulfate to the H3 protein is electrostatic in character, which is consistent with the binding of GAG ligands to other proteins (37).
Electrostatic depiction of the surface of H3.
There are 20 lysine and arginine residues of H3 for which electron density for the entire side chain was observed. Six other lysines and arginines were disordered. In Fig. 8, surface representations of H3 are colored in a color ramp indicating the calculated electrostatic potentials from −75 mV (red) to +75 mV (blue). In Fig. 8A, islands of positive charge (blue) may serve to allow heparin binding, as the binding of heparin to proteins is largely electrostatic. Figure 8B shows the location of the bound Mg ion and a Pdo molecule. The broad positive (blue) area that occupies ∼50% of this face of H3 may be a site for an acceptor molecule to bind, proximal to the catalytic site of this putative glycosyltransferase.
FIG 8.

Electrostatic surfaces of H3 detail locations of positive charges and of bound Mg2+. (A) View of the surface of H3, colored by surface positive charge (blue) and negative charge (red), with the darkest red signifying −75 mV and the darkest blue signifying +75 mV. (B) View after a 180° rotation from the view shown in panel A around the vertical axis. All lysines and arginines are identified and were used in the calculation, except for six for which side-chain electron density was absent. Electrostatic calculations were generated with APBS (Adaptive Poisson-Boltzmann Solver) (69).
DISCUSSION
H3 is located in the MV membrane with the domain studied here outside the virus and exposed to the cytoplasm. H3 has no recognizable signal sequence that would traffic it to cellular compartments. In an infection, H3 is produced in the cytoplasm of the infected cell. We produced H3 without its transmembrane region and found that the C-terminal end of recombinant H3 is proteolytically labile. This proteolysis occurred in our preparations and possibly in the bacterial cells during expression. This is consistent with cleavage sites that were estimated to be near Arg248 of H3 when vaccinia virus particles were treated with trypsin (20).
H3 as a glycosyltransferase.
The closest database structural match to H3 is rabbit N-acetylglucosaminyltransferase I (GnT I) (41). Like H3, GnT I has a ExD motif, rather than DxD. GnT I uses UDP-GlcNAc to transfer GlcNAc to oligomannose in a key step in the formation of complex oligosaccharides. The topologies of H3 and GnT I are similar: alternating strands and helices with most of the connections between secondary structure elements being the same. In the structure of GnT I with bound UDP-GlcNAc, residues 318 to 330 form a loop that covers the bound ligand (45). This loop is disordered in the GnT I structure without the ligand. H3 residue 240 superimposes onto GnT I residue 318. H3 residue 240 is the last residue that is ordered in our H3 structure, which is consistent with an unliganded structure. With UDP-Glc being bound, we predict that a loop starting at residue 241 would become ordered and visible in the electron density. Our binding studies show that UDP-Glc binds to H3 and that the binding requires Mg2+. UDP-Glc would be the sugar donor molecule for the H3 glycosyltransferase to transfer glucose to another molecule. It seems unlikely that H3 glycosylates an MV protein, as no viral protein components of the MV are known to be glycosylated (46).
In addition to the H3 putative glycosyltransferase described in this work, another poxvirus sialyltransferase is present in myxoma and fibroma viruses of the Leporipoxvirus genus (47, 48). The sialyltransferase has been found to contribute to immunosuppression during viral infection (49). Sequence analysis shows that there is also an H3 homolog in the viral genomes that have the sialyltransferase.
Some viruses express their own glycosyltransferases for various functions. To evade host antiviral mechanisms, some phages express α- and β-glucosyltransferases, which glucosylate their DNA and make it resistant to host restriction endonucleases (50). Paramecium bursaria chlorella virus 1 (PBCV-1), a member of the virus family Phycodnaviridae, encodes at least five glycosyltransferases (51). One of them, A64R, is involved in the glycosylation of the virus major capsid protein Vp54 (42). Like H3, A64R binds UDP-Glc. Glucose is one of the carbohydrate components of Vp54, unlike the glycan components of mature N- or O-linked eukaryotic glycoproteins that usually lack glucose (52). None of these glycosyltransferases have an identifiable signal peptide that would target them to the endoplasmic reticulum (ER). The Vp54 N-linked glycans do not occur at NX(S/T) sites that are the canonical recognition sites of the eukaryotic cellular enzymes that carry out N-linked protein glycosylation (42). Thus, it was predicted that PBCV-1 encodes most of the enzymes that glycosylate Vp54 (53), and there is evidence that the glycosylation of Vp54 occurs in the cytoplasm (53, 54).
H3 binds heparin sulfate.
Our experimental results demonstrate that the H3 protein binds a heparin sulfate decasaccharide and binds to heparan sulfate expressed on the surface of mammalian cells. Glycosaminoglycans are linear, complex anionic polysaccharides that are universally available in the cell surface glycocalyx and in the extracellular matrix (55). Heparan sulfate is the most complex glycosaminoglycan and is highly expressed on cell surfaces. Some viruses have been shown to use heparan sulfate as a receptor to invade host cells (56). Heparan sulfate may function as a target for poxvirus adhesion or as a coreceptor to capture ligands and enable further interactions with other target proteins.
The binding of H3, A27, and D8 to GAGs on the MV surface has been implicated in mediating the first attachment of the poxvirus particle with the host cell (22–24, 57, 58). However, the binding of the MV to GAGs is not absolutely required for MV invasion of host cells (59), and deletions of the H3, A27, or D8 gene yield mutant viruses that are still able to infect cells (60).
Disruption of the EV membrane must be the first step in virus entry to expose the entry fusion machinery on the MV membrane. It is established that polyanions disrupt the outer membrane of the EV (61). When the EV comes into contact with polyanions, predominantly the negatively charged heparan sulfate of the extracellular matrix, the fragile EV outer membrane may break and expose the MV membrane containing the two heparan sulfate binding proteins H3 and A27. Since the pH of the extracellular matrix is ∼7.0, H3, having an isoelectric point (pI) of 6.1, would have a net negative charge at this pH. The binding of H3, and thus MV, to the heparan sulfate of the extracellular matrix would be unfavorable since both H3 and the heparan sulfate would be negatively charged. On the other hand, when the MV encounters a host cell glycocalyx, with a local pH at the surface that is lower than the pI of H3 (62), H3 would have a net positive charge. This would allow a favorable interaction with the host cell surface via H3 and also A27. Attachment to the cell in this way would be the first step leading to the formation of the fusogenic complex (63).
H3 is involved in attachment to the host cell, contributes to viral morphogenesis, and plays a role in infection (22). Since H3 is a major immune system target that is recognized by neutralizing antibodies, H3 is an important viral protein to be included in new vaccines (18). The H3 crystal structure and ligand binding results presented here shed light on the roles that H3 may play in the vaccinia virus life cycle.
ACKNOWLEDGMENTS
We thank Phuc Nguyen for preparing mutant plasmids, Michael Mohan for carrying out initial expression and refolding trials with the mutants, Satish Madala for helping with flow cytometry, and Mark Garfield for performing mass spectrometry.
X-ray data were collected at the Southeast Regional Collaborative Access Team (22-ID and 22-BM) and Structural Biology Center (19ID) beam lines at the Advanced Photon Source, which is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract W-31-109-Eng-38. This research was supported by the Intramural Research Program of the NIH, NIAID.
REFERENCES
- 1.Henderson DA, Inglesby TV, Bartlett JG, Ascher MS, Eitzen E, Jahrling PB, Hauer J, Layton M, McDade J, Osterholm MT, O'Toole T, Parker G, Perl T, Russell PK, Tonat K. 1999. Smallpox as a biological weapon: medical and public health management. Working Group on Civilian Biodefense. JAMA 281:2127–2137. [DOI] [PubMed] [Google Scholar]
- 2.Fenner F, Henderson DA, Arita I, Jezek Z, Ladnyi ID. 1988. Smallpox and its eradication, p 539–592. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 3.Kennedy RB, Ovsyannikova IG, Jacobson RM, Poland GA. 2009. The immunology of smallpox vaccines. Curr Opin Immunol 21:314–320. doi: 10.1016/j.coi.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gallwitz S, Schutzbank T, Heberling RL, Kalter SS, Galpin JE. 2003. Smallpox: residual antibody after vaccination. J Clin Microbiol 41:4068–4070. doi: 10.1128/JCM.41.9.4068-4070.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shchelkunov SN. 2011. Emergence and reemergence of smallpox: the need for development of a new generation smallpox vaccine. Vaccine 29(Suppl 4):D49–D53. doi: 10.1016/j.vaccine.2011.05.037. [DOI] [PubMed] [Google Scholar]
- 6.Rimoin AW, Mulembakani PM, Johnston SC, Lloyd Smith JO, Kisalu NK, Kinkela TL, Blumberg S, Thomassen HA, Pike BL, Fair JN, Wolfe ND, Shongo RL, Graham BS, Formenty P, Okitolonda E, Hensley LE, Meyer H, Wright LL, Muyembe JJ. 2010. Major increase in human monkeypox incidence 30 years after smallpox vaccination campaigns cease in the Democratic Republic of Congo. Proc Natl Acad Sci U S A 107:16262–16267. doi: 10.1073/pnas.1005769107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vogel S, Sardy M, Glos K, Korting HC, Ruzicka T, Wollenberg A. 2012. The Munich outbreak of cutaneous cowpox infection: transmission by infected pet rats. Acta Derm Venereol 92:126–131. doi: 10.2340/00015555-1227. [DOI] [PubMed] [Google Scholar]
- 8.Gurav YK, Raut CG, Yadav PD, Tandale BV, Sivaram A, Pore MD, Basu A, Mourya DT, Mishra AC. 2011. Buffalopox outbreak in humans and animals in Western Maharashtra, India. Prev Vet Med 100:242–247. doi: 10.1016/j.prevetmed.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 9.Gallardo-Romero NF, Drew CP, Weiss SL, Metcalfe MG, Nakazawa YJ, Smith SK, Emerson GL, Hutson CL, Salzer JS, Bartlett JH, Olson VA, Clemmons CJ, Davidson WB, Zaki SR, Karem KL, Damon IK, Carroll DS. 2012. The pox in the North American backyard: volepox virus pathogenesis in California mice (Peromyscus californicus). PLoS One 7:e43881. doi: 10.1371/journal.pone.0043881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mota BE, Trindade GS, Diniz TC, da Silva-Nunes M, Braga EM, Urbano-Ferreira M, Rodrigues GO, Bonjardim CA, Ferreira PC, Kroon EG. 2010. Seroprevalence of orthopoxvirus in an Amazonian rural village, Acre, Brazil. Arch Virol 155:1139–1144. doi: 10.1007/s00705-010-0675-3. [DOI] [PubMed] [Google Scholar]
- 11.Haller SL, Peng C, McFadden G, Rothenburg S. 2014. Poxviruses and the evolution of host range and virulence. Infect Genet Evol 21:15–40. doi: 10.1016/j.meegid.2013.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Handley L, Buller RM, Frey SE, Bellone C, Parker S. 2009. The new ACAM2000 vaccine and other therapies to control orthopoxvirus outbreaks and bioterror attacks. Expert Rev Vaccines 8:841–850. doi: 10.1586/erv.09.55. [DOI] [PubMed] [Google Scholar]
- 13.Johnson GP, Goebel SJ, Paoletti E. 1993. An update on the vaccinia virus genome. Virology 196:381–401. doi: 10.1006/viro.1993.1494. [DOI] [PubMed] [Google Scholar]
- 14.Smith GL, Vanderplasschen A, Law M. 2002. The formation and function of extracellular enveloped vaccinia virus. J Gen Virol 83:2915–2931. doi: 10.1099/0022-1317-83-12-2915. [DOI] [PubMed] [Google Scholar]
- 15.Condit RC, Moussatche N, Traktman P. 2006. In a nutshell: structure and assembly of the vaccinia virion. Adv Virus Res 66:31–124. doi: 10.1016/S0065-3527(06)66002-8. [DOI] [PubMed] [Google Scholar]
- 16.Benhnia MR, McCausland MM, Su HP, Singh K, Hoffmann J, Davies DH, Felgner PL, Head S, Sette A, Garboczi DN, Crotty S. 2008. Redundancy and plasticity of neutralizing antibody responses are cornerstone attributes of the human immune response to the smallpox vaccine. J Virol 82:3751–3768. doi: 10.1128/JVI.02244-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meng X, Zhong Y, Embry A, Yan B, Lu S, Zhong G, Xiang Y. 2011. Generation and characterization of a large panel of murine monoclonal antibodies against vaccinia virus. Virology 409:271–279. doi: 10.1016/j.virol.2010.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davies DH, McCausland MM, Valdez C, Huynh D, Hernandez JE, Mu Y, Hirst S, Villarreal L, Felgner PL, Crotty S. 2005. Vaccinia virus H3L envelope protein is a major target of neutralizing antibodies in humans and elicits protection against lethal challenge in mice. J Virol 79:11724–11733. doi: 10.1128/JVI.79.18.11724-11733.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crickard L, Babas T, Seth S, Silvera P, Koriazova L, Crotty S. 2012. Protection of rabbits and immunodeficient mice against lethal poxvirus infections by human monoclonal antibodies. PLoS One 7:e48706. doi: 10.1371/journal.pone.0048706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.da Fonseca FG, Wolffe EJ, Weisberg A, Moss B. 2000. Characterization of the vaccinia virus H3L envelope protein: topology and posttranslational membrane insertion via the C-terminal hydrophobic tail. J Virol 74:7508–7517. doi: 10.1128/JVI.74.16.7508-7517.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Theze J, Takatsuka J, Li Z, Gallais J, Doucet D, Arif B, Nakai M, Herniou EA. 2013. New insights into the evolution of Entomopoxvirinae from the complete genome sequences of four entomopoxviruses infecting Adoxophyes honmai, Choristoneura biennis, Choristoneura rosaceana, and Mythimna separata. J Virol 87:7992–8003. doi: 10.1128/JVI.00453-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin CL, Chung CS, Heine HG, Chang W. 2000. Vaccinia virus envelope H3L protein binds to cell surface heparan sulfate and is important for intracellular mature virion morphogenesis and virus infection in vitro and in vivo. J Virol 74:3353–3365. doi: 10.1128/JVI.74.7.3353-3365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chung CS, Hsiao JC, Chang YS, Chang W. 1998. A27L protein mediates vaccinia virus interaction with cell surface heparan sulfate. J Virol 72:1577–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsiao JC, Chung CS, Chang W. 1999. Vaccinia virus envelope D8L protein binds to cell surface chondroitin sulfate and mediates the adsorption of intracellular mature virions to cells. J Virol 73:8750–8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.da Fonseca FG, Wolffe EJ, Weisberg A, Moss B. 2000. Effects of deletion or stringent repression of the H3L envelope gene on vaccinia virus replication. J Virol 74:7518–7528. doi: 10.1128/JVI.74.16.7518-7528.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davies DH, Molina DM, Wrammert J, Miller J, Hirst S, Mu Y, Pablo J, Unal B, Nakajima-Sasaki R, Liang X, Crotty S, Karem KL, Damon IK, Ahmed R, Villarreal L, Felgner PL. 2007. Proteome-wide analysis of the serological response to vaccinia and smallpox. Proteomics 7:1678–1686. doi: 10.1002/pmic.200600926. [DOI] [PubMed] [Google Scholar]
- 27.McCausland MM, Benhnia MR, Crickard L, Laudenslager J, Granger SW, Tahara T, Kubo R, Koriazova L, Kato S, Crotty S. 2010. Combination therapy of vaccinia virus infection with human anti-H3 and anti-B5 monoclonal antibodies in a small animal model. Antivir Ther 15:661–675. doi: 10.3851/IMP1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Su HP, Lin DY, Garboczi DN. 2006. The structure of G4, the poxvirus disulfide oxidoreductase essential for virus maturation and infectivity. J Virol 80:7706–7713. doi: 10.1128/JVI.00521-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode, p 307–326. In Carter CW, Sweet RM (ed), Methods in enzymology, vol 276 Academic Press, New York, NY. [DOI] [PubMed] [Google Scholar]
- 30.Kabsch W. 2010. XDS. Acta Crystallogr D Biol Crystallogr 66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Terwilliger TC, Berendzen J. 1999. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr 55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Terwilliger TC. 2000. Maximum-likelihood density modification. Acta Crystallogr D Biol Crystallogr 56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47:110–119. doi: 10.1107/S0108767390010224. [DOI] [PubMed] [Google Scholar]
- 34.Brunger AT. 2007. Version 1.2 of the crystallography and NMR system. Nat Protoc 2:2728–2733. doi: 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- 35.Terwilliger TC. 2003. Automated main-chain model building by template matching and iterative fragment extension. Acta Crystallogr D Biol Crystallogr 59:38–44. doi: 10.1107/S0907444902018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mayer M, Meyer B. 2001. Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J Am Chem Soc 123:6108–6117. doi: 10.1021/ja0100120. [DOI] [PubMed] [Google Scholar]
- 37.Singh K, Gittis AG, Nguyen P, Gowda DC, Miller LH, Garboczi DN. 2008. Structure of the DBL3x domain of pregnancy-associated malaria protein VAR2CSA complexed with chondroitin sulfate A. Nat Struct Mol Biol 15:932–938. doi: 10.1038/nsmb.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Esko JD. 1991. Genetic analysis of proteoglycan structure, function and metabolism. Curr Opin Cell Biol 3:805–816. doi: 10.1016/0955-0674(91)90054-3. [DOI] [PubMed] [Google Scholar]
- 39.Holm L, Rosenstrom P. 2010. Dali server: conservation mapping in 3D. Nucleic Acids Res 38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krissinel E, Henrick K. 2004. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr 60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 41.Gordon RD, Sivarajah P, Satkunarajah M, Ma D, Tarling CA, Vizitiu D, Withers SG, Rini JM. 2006. X-ray crystal structures of rabbit N-acetylglucosaminyltransferase I (GnT I) in complex with donor substrate analogues. J Mol Biol 360:67–79. doi: 10.1016/j.jmb.2006.04.058. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y, Xiang Y, Van Etten JL, Rossmann MG. 2007. Structure and function of a chlorella virus-encoded glycosyltransferase. Structure 15:1031–1039. doi: 10.1016/j.str.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Larson ET, Reiter D, Young M, Lawrence CM. 2006. Structure of A197 from Sulfolobus turreted icosahedral virus: a crenarchaeal viral glycosyltransferase exhibiting the GT-A fold. J Virol 80:7636–7644. doi: 10.1128/JVI.00567-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haselhorst T, Lamerz AC, Itzstein M. 2009. Saturation transfer difference NMR spectroscopy as a technique to investigate protein-carbohydrate interactions in solution. Methods Mol Biol 534:375–386. doi: 10.1007/978-1-59745-022-5_26. [DOI] [PubMed] [Google Scholar]
- 45.Unligil UM, Zhou S, Yuwaraj S, Sarkar M, Schachter H, Rini JM. 2000. X-ray crystal structure of rabbit N-acetylglucosaminyltransferase I: catalytic mechanism and a new protein superfamily. EMBO J 19:5269–5280. doi: 10.1093/emboj/19.20.5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moss B. 2015. Poxvirus membrane biogenesis. Virology 479–480:619–626. doi: 10.1016/j.virol.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jackson RJ, Hall DF, Kerr PJ. 1999. Myxoma virus encodes an alpha2,3-sialyltransferase that enhances virulence. J Virol 73:2376–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Willer DO, McFadden G, Evans DH. 1999. The complete genome sequence of shope (rabbit) fibroma virus. Virology 264:319–343. doi: 10.1006/viro.1999.0002. [DOI] [PubMed] [Google Scholar]
- 49.Boutard B, Vankerckhove S, Markine-Goriaynoff N, Sarlet M, Desmecht D, McFadden G, Vanderplasschen A, Gillet L. 2015. The alpha2,3-sialyltransferase encoded by myxoma virus is a virulence factor that contributes to immunosuppression. PLoS One 10:e0118806. doi: 10.1371/journal.pone.0118806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Markine-Goriaynoff N, Gillet L, Van Etten JL, Korres H, Verma N, Vanderplasschen A. 2004. Glycosyltransferases encoded by viruses. J Gen Virol 85:2741–2754. doi: 10.1099/vir.0.80320-0. [DOI] [PubMed] [Google Scholar]
- 51.Xiang Y, Baxa U, Zhang Y, Steven AC, Lewis GL, Van Etten JL, Rossmann MG. 2010. Crystal structure of a virus-encoded putative glycosyltransferase. J Virol 84:12265–12273. doi: 10.1128/JVI.01303-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang IN, Li Y, Que Q, Bhattacharya M, Lane LC, Chaney WG, Van Etten JL. 1993. Evidence for virus-encoded glycosylation specificity. Proc Natl Acad Sci U S A 90:3840–3844. doi: 10.1073/pnas.90.9.3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Van Etten JL. 2003. Unusual life style of giant chlorella viruses. Annu Rev Genet 37:153–195. doi: 10.1146/annurev.genet.37.110801.143915. [DOI] [PubMed] [Google Scholar]
- 54.Nandhagopal N, Simpson AA, Gurnon JR, Yan X, Baker TS, Graves MV, Van Etten JL, Rossmann MG. 2002. The structure and evolution of the major capsid protein of a large, lipid-containing DNA virus. Proc Natl Acad Sci U S A 99:14758–14763. doi: 10.1073/pnas.232580699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu D, Esko JD. 2014. Demystifying heparan sulfate-protein interactions. Annu Rev Biochem 83:129–157. doi: 10.1146/annurev-biochem-060713-035314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Spillmann D. 2001. Heparan sulfate: anchor for viral intruders? Biochimie 83:811–817. doi: 10.1016/S0300-9084(01)01290-1. [DOI] [PubMed] [Google Scholar]
- 57.Hsiao JC, Chung CS, Chang W. 1998. Cell surface proteoglycans are necessary for A27L protein-mediated cell fusion: identification of the N-terminal region of A27L protein as the glycosaminoglycan-binding domain. J Virol 72:8374–8379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maa JS, Rodriguez JF, Esteban M. 1990. Structural and functional characterization of a cell surface binding protein of vaccinia virus. J Biol Chem 265:1569–1577. [PubMed] [Google Scholar]
- 59.Carter GC, Law M, Hollinshead M, Smith GL. 2005. Entry of the vaccinia virus intracellular mature virion and its interactions with glycosaminoglycans. J Gen Virol 86:1279–1290. doi: 10.1099/vir.0.80831-0. [DOI] [PubMed] [Google Scholar]
- 60.Moss B. 2006. Poxvirus entry and membrane fusion. Virology 344:48–54. doi: 10.1016/j.virol.2005.09.037. [DOI] [PubMed] [Google Scholar]
- 61.Law M, Carter GC, Roberts KL, Hollinshead M, Smith GL. 2006. Ligand-induced and nonfusogenic dissolution of a viral membrane. Proc Natl Acad Sci U S A 103:5989–5994. doi: 10.1073/pnas.0601025103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Uversk VN. 2015. Protein misfolding in lipid-mimetic environments, p 33–66. In Gursky O. (ed), Lipids in protein misfolding. Springer International Publishing, New York, NY. [DOI] [PubMed] [Google Scholar]
- 63.Senkevich TG, Ojeda S, Townsley A, Nelson GE, Moss B. 2005. Poxvirus multiprotein entry-fusion complex. Proc Natl Acad Sci U S A 102:18572–18577. doi: 10.1073/pnas.0509239102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nielsen MM, Suits MD, Yang M, Barry CS, Martinez-Fleites C, Tailford LE, Flint JE, Dumon C, Davis BG, Gilbert HJ, Davies GJ. 2011. Substrate and metal ion promiscuity in mannosylglycerate synthase. J Biol Chem 286:15155–15164. doi: 10.1074/jbc.M110.199844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tumbale P, Jamaluddin H, Thiyagarajan N, Brew K, Acharya KR. 2008. Structural basis of UDP-galactose binding by alpha-1,3-galactosyltransferase (alpha3GT): role of negative charge on aspartic acid 316 in structure and activity. Biochemistry 47:8711–8718. doi: 10.1021/bi800852a. [DOI] [PubMed] [Google Scholar]
- 66.Persson K, Ly HD, Dieckelmann M, Wakarchuk WW, Withers SG, Strynadka NC. 2001. Crystal structure of the retaining galactosyltransferase LgtC from Neisseria meningitidis in complex with donor and acceptor sugar analogs. Nat Struct Biol 8:166–175. doi: 10.1038/84168. [DOI] [PubMed] [Google Scholar]
- 67.Pedersen LC, Dong J, Taniguchi F, Kitagawa H, Krahn JM, Pedersen LG, Sugahara K, Negishi M. 2003. Crystal structure of an alpha 1,4-N-acetylhexosaminyltransferase (EXTL2), a member of the exostosin gene family involved in heparan sulfate biosynthesis. J Biol Chem 278:14420–14428. doi: 10.1074/jbc.M210532200. [DOI] [PubMed] [Google Scholar]
- 68.Gastinel LN, Cambillau C, Bourne Y. 1999. Crystal structures of the bovine beta4galactosyltransferase catalytic domain and its complex with uridine diphosphogalactose. EMBO J 18:3546–3557. doi: 10.1093/emboj/18.13.3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. 2001. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A 98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]