

ABSTRACT
The potyviral RNA genome encodes two polyproteins that are proteolytically processed by three viral protease domains into 11 mature proteins. Extensive molecular studies have identified functions for the majority of the viral proteins. For example, 6K2, one of the two smallest potyviral proteins, is an integral membrane protein and induces the endoplasmic reticulum (ER)-originated replication vesicles that target the chloroplast for robust viral replication. However, the functional role of 6K1, the other smallest protein, remains uncharacterized. In this study, we developed a series of recombinant full-length viral cDNA clones derived from a Canadian Plum pox virus (PPV) isolate. We found that deletion of any of the short motifs of 6K1 (each of which ranged from 5 to 13 amino acids), most of the 6K1 sequence (but with the conserved sequence of the cleavage sites being retained), or all of the 6K1 sequence in the PPV infectious clone abolished viral replication. The trans expression of 6K1 or the cis expression of a dislocated 6K1 failed to rescue the loss-of-replication phenotype, suggesting the temporal and spatial requirement of 6K1 for viral replication. Disruption of the N- or C-terminal cleavage site of 6K1, which prevented the release of 6K1 from the polyprotein, either partially or completely inhibited viral replication, suggesting the functional importance of the mature 6K1. We further found that green fluorescent protein-tagged 6K1 formed punctate inclusions at the viral early infection stage and colocalized with chloroplast-bound viral replicase elements 6K2 and NIb. Taken together, our results suggest that 6K1 is required for viral replication and is an important viral element of the viral replication complex at the early infection stage.
IMPORTANCE Potyviruses account for more than 30% of known plant viruses and consist of many agriculturally important viruses. The genomes of potyviruses encode two polyproteins that are proteolytically processed into 11 mature proteins, with the majority of them having been at least partially functionally characterized. However, the functional role of a small protein named 6K1 remains obscure. In this study, we showed that deletion of 6K1 or a short motif/region of 6K1 in the full-length cDNA clones of plum pox virus abolishes viral replication and that mutation of the N- or C-terminal cleavage sites of 6K1 to prevent its release from the polyprotein greatly attenuates or completely inhibits viral replication, suggesting its important role in potyviral infection. We report that 6K1 forms punctate structures and targets the replication vesicles in PPV-infected plant leaf cells at the early infection stage. Our data reveal that 6K1 is an important viral protein of the potyviral replication complex.
INTRODUCTION
To establish their infection, positive-sense single-stranded RNA viruses remodel and recruit host cellular membranes to assemble the viral replication complex (VRC), which consists of both viral proteins and host factors, for robust viral replication (1–4). Therefore, the molecular identification and functional characterization of viral proteins or host factors essential for viral multiplication directly contribute to the elucidation of the viral replication mechanism and to the development of novel antiviral strategies in the long run.
Potyviruses belong to the family Potyviridae and include many agriculturally and economically important pathogens, such as Plum pox virus (PPV), Soybean mosaic virus (SMV), Turnip mosaic virus (TuMV), Tobacco etch virus (TEV), Potato virus A (PVA), and Potato virus Y (PVY), and cause significant losses in a wide range of crops (5, 6). In the past decade, extensive studies have been carried to understand many aspects, particularly the molecular biology, of potyviruses (5, 6). The potyviral RNA genome codes for a long open reading frame (ORF) and another relatively short ORF that results from RNA polymerase slippage in the P3-coding sequence (7–10). Upon translation, these two polyproteins are proteolytically processed by three viral protease domains into 11 mature viral proteins, namely (from the N terminus), P1, HC-Pro, P3, P3N-PIPO, 6K1, CI, 6K2, VPg, NIa-Pro, NIb, and CP (6, 7, 11–13). Most potyviral proteins are known to play multifunctional roles in potyviral infection. For example, as the first two products of the potyviral polyprotein, P1 and HC-Pro function as proteinases to efficiently catalyze their own cleavage sites for their release from the viral polyprotein (14). HC-Pro is also a well-known RNA-silencing suppressor (15). P1 enhances viral suppression of the RNA silencing mediated by HC-Pro (16, 17) and interacts with host ribosomal proteins to stimulate the translation of viral proteins in infected cells (18). P1 likely modulates viral replication and the host defense response via host-dependent regulation of its viral protease activity (19). Another good example is the multifunctional protein CI, whose functions have been reviewed recently (20).
Of the 11 potyviral proteins, 6K1 and 6K2 are the two smallest proteins, which have similar molecular weights, and both contain hydrophobic stretches (21). 6K2 is an integral membrane protein (22, 23) and induces the endoplasmic reticulum (ER)-derived replication vesicles that target the chloroplast for robust viral replication (24, 25). Viral proteins P3, CI, VPg, NIa-Pro, and NIb are associated with 6K2-induced VRCs, which have been identified to be elements essential for viral genome replication (25–30). Nevertheless, not much work has been devoted to the functional characterization of 6K1, and no defined roles have been assigned to this small peptide.
In this study, a series of full-length infectious PPV clones was developed, and subsequently, a variety of 6K1 mutants was created to investigate the functional role of the potyviral 6K1 protein in viral infection. We show that deletion of the entire 6K1 or any short motif/region of 6K1 abolishes viral replication and that mutation of the NIa-Pro cleavage sites at both the N and C termini of 6K1 to prevent the release of the mature 6K1 from the polyprotein greatly attenuates or completely inhibits viral multiplication, suggesting that mature 6K1 alone plays an important role in viral replication. We demonstrate that the loss-of-replication phenotype of the 6K1 deletion mutant could not be rescued by expressing 6K1 in trans or coexpressing a dislocated 6K1 in cis. We further discovered that the 6K1 protein forms punctate structures (inclusions) in PPV-infected Nicotiana benthamiana leaf cells and subcellularly colocalizes with the VRC-containing viral proteins 6K2 and NIb. Collectively, these results suggest that 6K1 is required for viral replication as an important viral element of the viral replication complex at the early infection stage.
MATERIALS AND METHODS
Virus source and plant materials.
Peach leaf tissues infected by PPV (the PPV-D strain) were collected from an experimental orchard at Jordan Station, Ontario, Canada, and used for cloning of the full-length genome and construction of infectious cDNA clones. This PPV isolate was designated VPH. The PPV experimental host N. benthamiana and its natural host, the peach tree (Prunus persica L. cv. Loring), were maintained in a greenhouse with 16 h of light at 21 to 23°C and 8 h of darkness at 20°C. Peach tree seedlings were prepared essentially as described previously (31).
Cloning, sequencing, and plasmid construction.
The near-full-length genomic sequence of PPV isolate VPH was obtained by reverse transcription-PCR (RT-PCR), cloning, and sequencing essentially as described previously (32, 33). The 5′- or 3′-terminal cDNAs were cloned using a 5′ or 3′ rapid amplification of cDNA ends kit (Invitrogen). The full-length cDNA (Fig. 1A) was cloned into a modified mini-binary vector, pCB301 (34), and placed between a 35S promoter and a Nos terminator to create the plasmid pVPH (Fig. 1B).
FIG 1.

Development of full-length infectious PPV cDNA clones. (A) Schematic representation of the genomic organization of PPV isolate VPH. Circle, the genome-linked viral protein VPg; two short horizontal lines, 5′ and 3′ untranslated regions, respectively; large box, the long ORF (nucleotides 148 to 9570); smaller boxes, the mature proteins resulting from the proteolytic processing of the large polyprotein; short gray bar, PIPO (nucleotides 2907 to 3218) derived from RNA polymerase slippage on the P3 cistron; (A)n, the poly(A) tail. (B) Diagram showing the VPH-derived T-DNA constructs. For plasmid pVPH, the entire genomic cDNA of VPH was introduced within the T-DNA borders of a mini-binary plant expression vector, pCB301 (34). For plasmid pVPH-GFP, the GFP-coding sequence was integrated between P1 and HC-Pro of pVPH. For plasmid pVPH-GFP//mCherry (designated the WT), the red fluorescent protein mCherry gene expression cassette fused with a luminal ER retention signal (mCherry-HDEL) was introduced within the T-DNA borders adjacent to the expression cassette for the GFP-tagged PPV clone pVPG-GFP. RB, right border; 35S, the cauliflower mosaic virus 35S promoter; NOS, the nopaline synthase terminator; LB, left border. (C, D) Infectivity of the pVPH clone for N. benthamiana (C) and P. persica cv. Loring (D). (E) GFP fluorescence in N. benthamiana plants at 18 dpi (left) and P. persica cv. Loring plants at 23 dpb (right) under UV light. (F) (Top) Western blot detection of GFP in infected N. benthamiana plants at 18 dpi and P. persica cv. Loring plants at 23 dpb. Arrows, free GFP (∼27.7 kDa). (Bottom) A Coomassie brilliant blue-stained gel used as a loading control. Lanes 1 and 2 were loaded with two protein samples from plants (indicated on top) infected by the virus derived from pVPH; lanes 3 and 4 were from those infected by the virus derived from pVPH-GFP. (G) Time course analysis of viral intercellular movement at the periphery of the area infiltrated with the WT plasmid in N. benthamiana. The green and red fluorescence came from GFP-tagged PPV and the mCherry expression cassette, respectively. In the merged images, yellow regions resulting from overlapping GFP and mCherry fluorescence represent initially infected foci, and green fluorescence only represents secondarily infected cells. Bars, 150 μm.
To develop a recombinant infectious PPV clone tagged by green fluorescent protein (GFP), a GFP-coding region was amplified from the vector pCambiaTunos/GFP (28) and inserted into the P1/HC-Pro junction of pVPH to produce the plasmid pVPH-GFP (Fig. 1B).
To modify pVPH-GFP for the inclusion of an independent gene expression cassette for coexpression of an mCherry recombinant protein fused with an ER retention signal, the mCherry-HDEL fragment was amplified from the vector pCambiaTuMV/6K2::mCherry//GFP-HDEL (35). The resulting fragment was inserted between the 35S promoter and the Nos terminator of the binary plant expression vector pCaMterX (36). The mCherry-HDEL expression cassette in the resulting vector was amplified and finally integrated into pVPH-GFP via several intermediate cloning steps to produce the plasmid pVPH-GFP//mCherry. Standard DNA manipulation techniques were used to generate various mutant clones using pVPH-GFP//mCherry as the parental wild-type (WT) PPV clone.
The Gateway technology was used to construct plant expression vectors for transient expression of the genes of interest in planta (37). The 6K1 or 6K2 cistron was amplified by PCR from the PPV clone pVPH. The Gateway technology with the entry vector pDONR221 and destination vector pEarleyGate103 or pGWB454 was used to produce the plasmid pEarleyGate103-6K1 for transient expression of the 6K1-GFP fusion protein or pGWB454-6K2 for expression of the 6K2-red fluorescent protein (RFP) fusion protein in planta. To construct the vector pCaMterX-NIb-mCherry for transient expression of the NIb-mCherry fusion protein, the NIb- and mCherry-coding sequences were amplified from the WT parent vector. Then, the fused NIb-mCherry fragment was obtained by overlapping PCR and inserted into pCaMterX to produce the vector pCaMterX-NIb-mCherry. A similar strategy was used to generate the plant expression vectors pCaMterX-6K1 and pCaMterX-P36K1. The sequences of all the vectors mentioned above were confirmed by DNA sequencing.
Agroinfiltration and biolistic bombardment.
The fully expanded leaves of N. benthamiana plants were used for Agrobacterium tumefaciens (strain GV3101)-mediated transient expression as described previously (31, 38). Biolistic bombardment of peach tree seedlings with full-length PPV cDNA clones and their derived mutants was performed essentially as previously described (31).
DAS-ELISA, RT-PCR and real-time qRT-PCR.
After agroinfiltration or biolistic bombardment, PPV in the treated plants was detected by double-antibody sandwich enzyme-linked immunosorbent assay (DAS-ELISA), RT-PCR, or real-time quantitative RT-PCR (qRT-PCR) at the time points indicated below. DAS-ELISA was conducted with an ELISA kit (Agdia) following the manufacturer's protocol. For RT-PCR and real-time qRT-PCR, total RNAs were extracted from agroinfiltrated or newly emerging leaves using the TRIzol reagent (Invitrogen) and treated with DNase I (Invitrogen) to remove contaminated DNA. For RT-PCR detection of PPV, the first-strand cDNAs were generated by reverse transcription reactions with a random hexamer primer (New England BioLabs) and SuperScript III reverse transcriptase (Invitrogen), and PCR was carried out to amplify the partial coat protein (CP)-coding region using the primer set P1 (5′-CAGACTACAGCCTCGCCAGA) and P2 (5′-ACCGAGACCACTACACTCCC). For real-time qPCR for determination of the viral positive-sense RNA accumulation level, first-strand cDNA synthesis and real-time qPCR were performed with iScript reverse transcription supermix and SsoFast EvaGrean supermix (Bio-Rad), respectively. For qRT-PCR to quantify the viral negative-sense RNA accumulation level, reverse transcription was performed with primers qCP101F (5′-GAGAAGGCGAGGAGGAAGTT) and mCherry115R (5′-TGGTGTAGTCCTCGTTGTGG) and SuperScript III reverse transcriptase (Invitrogen). qRT-PCR was performed and the products were analyzed with a CFX96 real-time PCR detection system (Bio-Rad) following the manufacturer's instructions. The relative level of PPV accumulation was determined by real-time qPCR using CP-specific primers qCP101F and qCP101R (5′-TGCAGGCTGTATGACTGGAG) and the mCherry transcript as an internal control.
Western blotting.
Plant leaf tissues were homogenized in a TissueLyser disrupter (Qiagen). Total proteins were extracted in 50 mM Tris-HCl (pH 6.8), 50 mM dithiothreitol, 4% (wt/vol) SDS, 10% (vol/vol) glycerol, 1% (wt/vol) polyvinylpyrrolidone 40, and 5% (vol/vol) phenylmethylsulfonyl fluoride and centrifuged at 4°C and 12,000 rpm for 15 min to remove the cell debris. Total proteins were separated by SDS-PAGE, transferred onto a polyvinylidene difluoride (PVDF) membrane, and detected by immunoblotting using an anti-GFP polyclonal antibody (N-terminal antibody; Sigma-Aldrich) or an anti-6×His monoclonal antibody (Abcam). Goat anti-rabbit or goat anti-mouse immunoglobulin antibody (Sigma-Aldrich) conjugated to horseradish peroxidase was used as the secondary antibody. The blots were treated with enhanced chemiluminescence detection reagents (GE Healthcare) and exposed to films for visualization.
Confocal microscopy.
For confocal microscopy analysis, epidermal cells of N. benthamiana were examined using a Leica TCS SP2 inverted confocal microscope with a 10× dry immersion or 63× water immersion objective. The autofluorescence of GFP, monomeric red fluorescent protein/mCherry, and chlorophyll was analyzed as described previously (25). Images were captured digitally and handled using Leica LAS AF Lite software.
Nucleotide sequence accession numbers.
The genomic sequence of the Canadian PPV isolate VPH (PPV-D strain) from P. persica was deposited in the GenBank database with accession number KP998124.
RESULTS
Development of a series of recombinant full-length infectious PPV cDNA clones.
To explore the functional roles of 6K1, we developed a series of recombinant full-length infectious PPV cDNA clones. We first determined the genomic sequence of the Canadian PPV isolate VPH (PPV-D strain) from P. persica by cloning and sequencing of overlapping VPH cDNAs. The full-length genomic cDNA sequence of VPH is 9,787 nucleotides in length, excluding the 3′-terminal poly(A) sequence (Fig. 1A). The two deduced polyproteins contain predicted cleavage sites and conserved domains for 11 mature proteins (Fig. 1A). The entire cDNA of VPH was integrated into a mini-binary vector, pCB301 (Fig. 1B). To test the infectivity of the resulting full-length cDNA clone, pVPH, the cDNA plasmid was biolistically introduced into eight peach tree seedlings. The newly emerging leaves of all the bombarded plants showed severe vein yellowing, mosaic, and V-shaped symptoms at 13 days postbombardment (dpb) (Fig. 1D). The typical PPV symptoms, such as ring spot and mosaic, were shown at about 8 weeks postbombardment. The presence of PPV in the upper newly emerging leaves of all peach plants bombarded with the pVPH clone was confirmed by DAS-ELISA and RT-PCR. To test whether the pVPH clone was also infectious in a PPV experimental host, N. benthamiana, 14 plants at the 3- to 4-leaf stage were infiltrated with cells from an agrobacterial culture (optical density at 600 nm [OD600], 0.5) harboring pVPH. DAS-ELISA and RT-PCR revealed the presence of PPV in the inoculated and upper leaves of N. benthamiana plants at 9 days postinfiltration (dpi), and the infected plants showed mosaic and green island symptoms during the late infection stage (35 dpi) (Fig. 1C). Similar results were obtained from at least two additional independent experiments. These results suggest that the full-length cDNA clone of PPV isolate VPH is infectious in its natural and experimental hosts and the pathogenicity discrepancy in peach and N. benthamiana possibly results from viral host adaptation, as previously reported for PPV (39–41).
For potyviruses, the P1/HC-Pro and NIb/CP intercistronic positions have been used or have have been suggested for use for expression of heterologous proteins (42–44). To create a GFP-tagged PPV clone, the foreign GFP cistron was integrated between the P1- and HC-Pro-coding regions of pVPH (Fig. 1B). In the resulting clone, pVPH-GFP, the P1 self-cleavage peptide NEIIH(Y/S)D was kept at the P1/GFP junction and the NIa-Pro cleavage site sequence NVVVH(Q/A)D at the NIb/CP position was engineered into the GFP and HC-Pro junction. To examine the infectivity of pVPH-GFP, 15 N. benthamiana plants (3- to 4-leaf stage) were agroinfiltrated with the pVPH-GFP clone, and the GFP fluorescence signals were recorded at different time points. At 10 dpi, 10 out of 15 plants showed GFP signals along the veins of newly developed leaves under UV light (data not shown). The strong fluorescence signals were observed in the stems, petioles, and leaves of all infiltrated plants at 18 dpi (Fig. 1E). In all five peach plants bombarded with the pVPH-GFP clone, strong GFP fluorescence signals were evident along the veins of newly expanded leaves at 23 dpb (Fig. 1E). These observations were confirmed in two additional independent experiments using both the natural and the experimental hosts, in which similar observations were made. Furthermore, the leaf tissues of systemically infected N. benthamiana and peach plants were subjected to Western blot analysis of GFP expression. A major band corresponding to the predicted size for the recombinant GFP (27.7 kDa) was detected, indicating that the free GFP protein was efficiently processed and released from the virus genome-encoded polyprotein (Fig. 1F).
To establish a system to monitor viral early replication and intercellular movement, the RFP mCherry gene expression cassette fused with a luminal ER retention signal (mCherry-HDEL) was introduced within the transfer DNA (T-DNA) borders and adjacent to the expression cassette of the GFP-tagged VPH, and the resulting clone was designated pVPH-GFP//mCherry (Fig. 1B). For the convenience of description, this clone was designated the WT. Upon introduction of the WT into the N. benthamiana leaf cells via agroinfiltration, the cells initially infected emitted green and red fluorescence signals, whereas the cells secondarily infected as a result of viral intercellular movement emitted green fluorescence only. To test the infectivity of this clone, cells from an agrobacterial culture harboring the WT at a concentration with an OD600 of 0.1 were infiltrated into the expanded leaves of N. benthamiana. The periphery of the infiltrated area was observed under a confocal microscope. At 48 and 72 h postinfiltration (hpi), the mCherry and GFP fluorescent signals were clearly observed in the agroinfiltrated cells (Fig. 1G). At 96 hpi, some cells adjacent to those emitting both green and red fluorescence signals started to emit green fluorescence signals only (Fig. 1G), indicating that they were secondarily infected cells. In addition, this clone induced in N. benthamiana symptoms similar to those induced by pVPH-GFP and pVPH.
Deletion of PPV 6K1 abolishes viral infectivity, and disruption of the NIa-Pro cleavage sites at the N and C termini of 6K1 partially or completely inhibits viral RNA multiplication.
To dissect the functional roles of 6K1 in viral infection, two 6K1 deletion mutants were generated using the WT parental clone. For the deletion clone WTΔ6K1 (designated the Δ6K1 mutant), the majority of the 6K1 sequence was deleted from the WT clone;, i.e., a sequence of 45 amino acids that did not contain the first and last 6 amino acids of 6K1, which are conserved cleavage site sequences recognized by the proteinase NIa-Pro, were deleted (Fig. 2A). For the deletion clone WTΔc6K1 (designated the Δc6K1 mutant), the entire 6K1 sequence was removed (Fig. 2A). In addition, a GDD deletion clone (designated the ΔGDD mutant) was created by deletion of the highly conserved GDD motif from the infectious WT clone. GDD is essential for RNA polymerase activity, and deletion of the GDD motif abolishes viral replication (45). The two 6K1 deletion mutants, the Δ6K1 and Δc6K1 mutants, as well as the parental infectious WT clone and the replication-defective mutant ΔGDD, were introduced into N. benthamiana leaves to evaluate viral cell-to-cell movement and viability. For viral intercellular movement analysis, cells from an agrobacterial culture (OD600, 0.1) harboring the corresponding plasmid were infiltrated into fully expanded leaves of N. benthamiana and the periphery of the infiltrated region was observed under a confocal microscope. At 96 hpi, viral intercellular movement was observed only in the leaves infiltrated with the WT and not in the leaves infiltrated with the 6K1 deletion mutants (the Δ6K1 and Δc6K1 mutants) or the GDD deletion mutant (Fig. 2B). No cell-to-cell movement of these mutants was evident for at least 1 week after agroinfiltration. For viral replication assays, cells from each agrobacterial culture (OD600, 0.1) were infiltrated into the fully expanded leaves of 15 uniform N. benthamiana plants at the 6- to 7-leaf stage. At 54 hpi (note that viral intercellular movement, if any, did not occur until 96 hpi, as shown in Fig. 1G), the inoculated tissues of these plants were collected, pooled, and subjected to real-time qPCR analysis of viral RNA accumulation, using the mCherry transcripts as an internal control. The levels of both positive-sense and negative-sense viral RNA were determined. The results showed that the level of accumulation of either positive-sense or negative-sense viral RNA resulting from plasmids carrying the Δ6K1 and Δc6K1 mutants was similar to that resulting from the plasmid carrying the ΔGDD mutant and significantly lower than that resulting from the plasmid carrying the WT (Fig. 2C). These results suggest that 6K1 is indispensable for viral multiplication and cell-to-cell movement. The small amount of positive-sense and negative-sense viral RNA transcripts of the GDD deletion and 6K1 deletion mutants resulted from the activity of the 35S promoter and cellular RNA-dependent RNA polymerase, respectively.
FIG 2.

Deletion of the PPV 6K1-coding region abolishes viral viability, and disruption of the NIa-Pro cleavage site at the P3/6K1 junction significantly reduces viral genome multiplication. (A) Diagram showing the PPV mutants which resulted from the deletion of the entire 6K1 sequence or partial 6K1 sequences or replacement of 6K1 with the Thosea asigna virus 2A self-cleaving peptide (T2A) (47). The amino acid sequences of 6K1, including its flanking NIa-Pro cleavage heptapeptides, are shown. The 6K1 cistron is defined by two vertical arrows. Horizontal dashed lines, the deletion of amino acids; (•45•), a 45-amino-acid sequence. Dots at the beginning or end of the sequences represent the P3 sequence (excluding its last six residues) and the CI sequence excluding the first residue, respectively. Curved arrows represent T2A. (B) Confocal microscopic observation of viral intercellular movement of PPV mutants, the wild type, and nonreplicating controls under a low magnification. All the images were taken at 4 dpi. Green and red fluorescence resulted from the GFP-tagged PPV genome and the mCherry-HDEL expression cassette, respectively. For merged images, yellow regions indicate overlapping GFP and mCherry fluorescence, whereas mCherry fluorescence and green fluorescence represent initially and secondarily infected foci, respectively. Arrows, direction of viral movement. Bars, 150 μm. (C, D) Detection by qRT-PCR of positive-sense viral RNA [(+)RNA] or negative-sense viral RNA [(−)RNA] accumulation in N. benthamiana plants agroinfiltrated with PPV clones. Cells from each agrobacterial culture (OD600, 0.1) harboring the corresponding plasmid were infiltrated into 15 (C) or 9 (D) uniform N. benthamiana plants at the 6- to 7-leaf stage. Agroinfiltrated leaves were collected and pooled at 54 hpi for RNA purification. Real-time qRT-PCR was performed to quantify viral genome accumulation (by analysis of the CP RNA level) using the mCherry transcript level as an internal control. Error bars denote standard errors from at least three biological replicates. Statistically significant differences, determined by an unpaired two-tailed Student's t test, are indicated by brackets and asterisks. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (E) GFP fluorescence in N. benthamiana agroinfiltrated with the WT or Δ1CS mutant at 25 dpi under UV light. (I and II) Systemic infection of the WT and the Δ1CS mutant in whole plants (I) and newly developed leaves (II).
To further test if the proteolytic processing of 6K1 is essential for viral intercellular movement and replication, two more 6K1-derived PPV mutants, WTΔ1CS (the Δ1CS mutant) and WTΔ2CS (the Δ2CS mutant), in which the first amino acid or the last 6 amino acids of 6K1 were deleted, respectively, were constructed (Fig. 2A). Deletion of the first amino acid or the last 6 amino acids of 6K1 was expected to destroy the conserved NIa-Pro cleavage site at the P3/6K1 or 6K1/CI junction, respectively, and to result in the proteolytically nonprocessable P3-6K1 or 6K1-CI protein, respectively. Like the 6K1 deletion and GDD deletion mutants, the Δ2CS mutant lost the ability to replicate or move intercellularly (Fig. 2B and C). Interestingly, the Δ1CS mutant replicated in N. benthamiana leaves but did so at a significantly lower level than the WT (Fig. 2C). The Δ1CS mutant also moved to neighboring cells remarkably more slowly than the WT (Fig. 2D). Systemic infection by the Δ1CS mutant was significantly attenuated (Fig. 2E). The genomic sequence surrounding the P3/6K1 junction of the progeny of the Δ1CS mutant, including the C terminus of P3 (110 nucleotides), the entire 6K1 sequence, and the N terminus of CI (369 nucleotides), was sequenced at 12 dpi. No compensatory mutations were found in the progeny of the Δ1CS mutant. Altogether, these data suggest that the 6K1 mature protein is important for maintaining the high infectivity of PPV, that the nonprocessable P3-6K1 can partially complement the function of 6K1 and P3, and that the nonprocessable 6K1-CI does not functionally overlap 6K1 and CI.
In a previous study, the proteolytic processing of the P3/CI site was shown to be compromised in insect cells in a mutant of PVA from which 6K1 was deleted and the mutant was shown to lose infectivity in plants or protoplasts (46). On the basis of this observation, it has been suggested that the 6K1 peptide might function as a spacer contributing to the proteolytic processing of P3 and CI from the polyprotein (46). To test this idea, two PPV mutants, WTΔ6K1T2A (the Δ6K1T2A mutant) and WTΔc6K1T2A (the Δc6K1T2A mutant), in which the 45 amino acids of 6K1 or the entire 6K1 sequence was replaced by the Thosea asigna virus 2A self-cleaving peptide (T2A) (47), respectively, were constructed (Fig. 2A). Both mutants were noninfectious, similar to the nonreplicating ΔGDD mutant (Fig. 2B and D). Therefore, 6K1 is not likely a spacer.
All motifs/regions of 6K1 are required for viral viability.
To find conserved motifs/regions of 6K1, the 6K1 sequences of 16 additional potyviruses were retrieved from the GenBank database and subjected to multiple-sequence alignment with the PPV 6K1 sequence. It was found that the potyviral 6K1 proteins are relatively conserved (Fig. 3A). 6K1 was predicted to be membrane associated by three computer-based prediction algorithms available online, including TMHMM (http://www.cbs.dtu.dk/services/TMHMM/), the ΔG Prediction Server (http://dgpred.cbr.su.se/), and Split (http://split4.pmfst.hr/split/4/). The predicted hydrophobic helix domain (residues 7 to 26) of PPV 6K1 is located at the N-terminal moiety (Fig. 3A). Five motif/region deletion mutants of 6K1, i.e., mutants with deletions of amino acids 2 to 6, 7 to 16, 17 to 26, 27 to 39, and 40 to 46 (the Δ2-6, Δ7-16, Δ17-26, Δ27-39, and Δ40-46 mutants, respectively) were constructed (Fig. 3A). In planta assays of these mutants for viral replication and intercellular movement revealed that deletion of any of the five domains/motifs abolished viral multiplication (Fig. 3B) and intercellular movement (data not shown). These data further support the indispensable role of 6K1 in viral infection.
FIG 3.
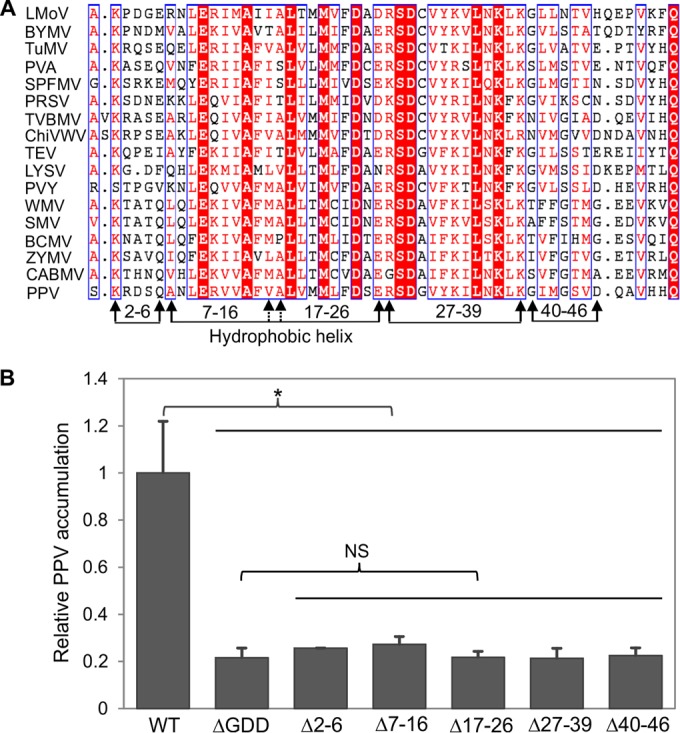
Evaluation of different motifs/regions of PPV 6K1 contributing to viral viability. (A) A multiple-sequence alignment of the 6K1 protein sequences of 17 potyviruses. Except for the 6K1 sequence of the PPV VPH isolate obtained from this study, the sequences of the other isolates were retrieved from the NCBI GenBank database. The sequences of the following viruses (GenBank accession numbers) are shown: Lily mottle virus (LMoV; AB570195), Bean yellow mosaic virus (BYMV; JX173278), Turnip mosaic virus (TuMV; AF169561), Potato virus A (PVA; AJ131402), Sweet potato feathery mottle virus (SPFMV; AB465608), Papaya ringspot virus (PRSV; X97251), Tobacco vein banding mosaic virus (TVBMV; EU734432), Chilli veinal mottle virus (ChiVWV; AJ972878), Tobacco etch virus (TEV; M11458), Leek yellow stripe virus (LYSV; AB194622), Potato virus Y (PVY; AJ439544), Watermelon mosaic virus (WMV; EU660581), Soybean mosaic virus (SMV; AF241739), Bean common mosaic virus (BCMV; AY863025), Zucchini mosaic virus (ZYMV; AF014811), and Cowpea aphid-borne mosaic virus (CABMV; HQ880242. The hydrophobic helix domains of potyviral 6K1 proteins are shown, and the amino acids are numbered according to the PPV 6K1 sequence. The different motifs/regions subjected to deletion analyses are indicated by arrows. Identical residues are shown as white letters in red background, whereas conserved substitutions are colored in red in blue boxes. (B) Detection of viral RNA accumulation in N. benthamiana plants agroinfiltrated with PPV mutants, the WT, and the nonreplicating ΔGDD mutant controls using qRT-PCR. Cells from each agrobacterial culture (OD600, 0.1) harboring the corresponding plasmid were infiltrated into nine uniform N. benthamiana plants at the 6- to 7-leaf stage. Agroinfiltrated leaves were collected and pooled at 54 hpi for RNA purification. Real-time qPCR was performed to quantify viral genome accumulation by analysis of the CP RNA level using the mCherry transcript level as an internal control. Error bars denote standard errors from three biological replicates. Statistically significant differences, determined by an unpaired two-tailed Student's t test, are indicated by brackets. NS, no significant differences; *, P < 0.05.
The loss of infectivity of mutants with a 6K1 deletion cannot be rescued by transient expression of 6K1 in trans or a translocated 6K1 in the PPV genome.
As shown above, the 6K1 deletion in PPV was lethal. To test if this phenotype could be complemented by 6K1 expression in trans, we constructed a binary plant expression vector, pCaMterX-6K1, expressing a recombinant histidine-tagged 6K1 (6K1-7×His). A mix of cells from agrobacterial cultures (OD600, 0.3) harboring the Δ6K1 mutant (or the Δc6K1 mutant) and pCaMterX-6K1 at a ratio of 1:2 was infiltrated into N. benthamiana plants and analyzed essentially as described above. The result showed that the Δ6K1 mutant (or the Δc6K1 mutant) supplemented with pCaMterX-6K1 remained noninfectious, similar to the findings for the nonreplicating ΔGDD mutant control and the Δ6K1 or Δc6K1 mutant when infiltrated alone (Fig. 4B). No intercellular movement of the Δ6K1 mutant (or the Δc6K1 mutant) supplemented with pCaMterX-6K1 was observed either. Transient expression of the histidine-tagged 6K1 was confirmed by Western blotting using anti-6×His monoclonal antibodies (Fig. 4E). Thus, the replication-defective phenotype of the 6K1 deletion mutants could not be rescued by 6K1 provided in trans.
FIG 4.

The loss-of-infectivity phenotype of PPV 6K1 deletion mutants cannot be rescued by the trans expression of 6K1 or P3-6K1 through transient expression or the cis expression of a dislocated 6K1 in the modified PPV genome. (A) Schematic representation of the PPV clones constructed for the functional complementation assays. The small gray symbol marked with Δc6K1 in the PPV cassette represents the deletion of the entire 6K1, and the small gray symbol marked with Δ6K1 represents the deletion of the entire 6K1 sequence, excluding its first amino acid and last 6 amino acids. The small rectangles, defined by two vertical dashed lines and marked with 6K1GFP, represent the fused 6K1-GFP sequence located between the P1 and HC-Pro cistrons in the PPV genome. (B) qRT-PCR detection of relative viral accumulation levels in N. benthamiana plants coagroinfiltrated with PPV 6K1 deletion mutants Δ6K1 (or PPV 6K1 deletion mutant Δc6K1) and pCaMterX-6K1 (6K1), as well as agroinfiltrated with the Δ6K1 mutant (or the Δc6K1 mutant), the WT, and the ΔGDD mutant. Agroinfiltrated leaves were collected and pooled at 54 hpi for RNA purification. Real-time qPCR was performed to quantify the relative level of viral genome accumulation by analysis of the CP RNA level, and the mCherry transcript level was used as the internal control. Error bars denote standard errors from three biological replicates. Statistically significant differences, determined by an unpaired two-tailed Student's t test, are indicated by brackets. NS, no significant differences; ***, P < 0.001. (C) qRT-PCR detection of the viral accumulation level in N. benthamiana plants agroinfiltrated with PPV clone 6K1::Δ6K1 or 6K1::Δc6K1 as well as the WT and the ΔGDD mutant. Analyses were conducted as described in the legend to panel B. (D) qRT-PCR detection of relative viral accumulation levels in N. benthamiana plants coagroinfiltrated with PPV 6K1 deletion mutants Δc6K1 and pCaMterX-P36K1 (P36K1), as well as agroinfiltrated with the Δc6K1 mutant, the WT, and the ΔGDD mutant. Agroinfiltrated leaves were collected and pooled at 48 hpi for RNA purification. Analyses were conducted as described in the legend to panel B. (E) (Top) Western blot analysis of the transient expression of the 6K1 protein in N. benthamiana. Samples of N. benthamiana leaves infiltrated with cells from an agrobacterial culture (OD600, 0.3) harboring the plasmid pCaMterX-6K1 clone or buffer (Mock) were collected at 2 dpi and subjected to Western blot analysis using anti-6×His monoclonal antibodies. Arrow, the His-tagged 6K1 protein with a predicted size of 6.8 kDa. (Bottom) A Coomassie brilliant blue-stained gel used as a loading control. (F) Confocal microscopic observation of viral intercellular movement of PPV clones WT//6K1 and Δc6K1//6K1 under a low magnification. Cells from an agrobacterial culture harboring the relevant plasmid were highly diluted to an OD600 of 0.0005 and infiltrated into completely expanded N. benthamiana leaves to initiate infection from single cells. The images were taken at 4 dpi. Arrows, the direction of viral spread. Bars, 100 μm. (G) Confocal microscopic observation of viral intercellular movement of PPV clones WT//P36K1 and Δc6K1//P36K1, as described in the legend to panel F. Bars, 100 μm.
To corroborate this finding, the 6K1-7×His expression cassette was removed from pCaMterX-6K1 and introduced within the T-DNA borders adjacent to the expression cassette of wild-type PPV or PPV from which 6K1 was deleted to generate clones pVPH-GFP//6K1 (WT//6K1) and pVPH-GFPΔc6K1//6K1 (the Δc6K1//6K1 mutant) (Fig. 4A). Similar to the findings for the Δ6K1 mutant (or the Δc6K1 mutant) supplemented with pCaMterX-6K1 (Fig. 4B), the Δc6K1//6K1 mutant remained noninfectious. Viral movement was clearly observed for WT//6K1 but not for the Δc6K1//6K1 mutant (Fig. 4F). Taken together, these results suggest that 6K1 expression in trans could not rescue either the replication or movement defect of PPV 6K1 deletion mutants.
As potyviral NIa-Pro partially cleaves at the P3/6K1 junction, both the mature 6K1 and the polyprotein P3-6K1 are present during PPV infection (48, 49). As shown above (Fig. 2B and C), P3-6K1 partially replaces the function of 6K1 and P3. To test if the expression of P3-6K1 in trans could complement the defects of the 6K1 deletion mutants, coexpression experiments similar to those described above were conducted. The results showed that the trans expression of P3-6K1 also could not rescue the infectivity (both the replication- and intercellular movement-defective phenotypes) of the PPV 6K1 deletion mutants (Fig. 4D and G).
To examine if a translocated 6K1 in the PPV genome could recover viral infectivity, two more clones, pVPH-6K1GFPΔ6K1//mCherry (the 6K1::Δ6K1 mutant) and pVPH-6K1GFPΔc6K1//mCherry (the 6K1::Δc6K1 mutant), in which the original 6K1-coding region in the PPV genome was translocated between P1 and HC-Pro (both tagged by GFP), were constructed (Fig. 4A). The viral multiplication level was examined by real-time qPCR. The 6K1::Δ6K1 or 6K1::Δc6K1 mutant remained noninfectious (Fig. 4C). Collectively, the data presented above suggest that in order to maintain viral infectivity, 6K1 not only is required but also must be expressed in the ordered context.
The 6K1 protein forms punctate inclusions during viral infection.
To localize the distribution pattern of 6K1 in plant cells, cells from an agrobacterial culture (OD600, 0.1) harboring pEarleyGate103-6K1 (Fig. 5A) were infiltrated into fully expanded leaves of N. benthamiana, and 6K1-GFP expression was observed by confocal microscopy at 36 and 48 hpi. 6K1-GFP was found in both the cytoplasm and the nucleus (Fig. 5B, panel VI), consistent with the recent observation that the 6K1 of the potyvirus TuMV is a soluble protein when expressed ectopically (50).
FIG 5.
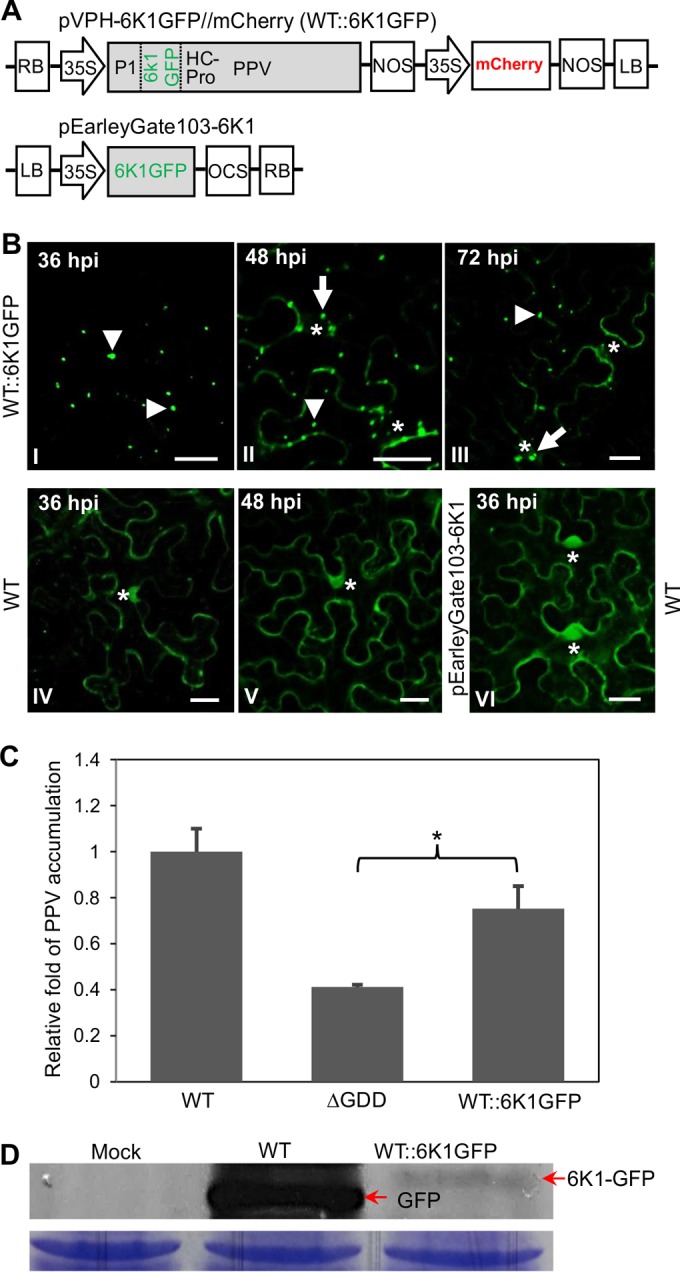
6K1-GFP coproduced from a PPV clone forms punctate inclusions during viral early infection. (A) Schematic diagram showing the T-DNA construct of pVPH-6K1GFP//mCherry (WT::6K1GFP), which contains an additional copy of 6K1 tagged by GFP at its C terminus between the P1 and HC-Pro cistrons. A diagram of pEarleyGate103-6K1 expressing GFP-tagged PPV 6K1 (6K1-GFP) is also shown. OCS, the 3′ sequences of the octopine synthase gene, including polyadenylation and presumptive transcription termination sequences. (B) The 6K1 protein coproduced from PPV clone WT::6K1GFP forms tiny punctate inclusions with a diameter of ≤1 μm at 36 hpi (I) and obvious punctate inclusions with a diameter of ≤2 μm at 48 and 72 hpi (II, III). As a control, the free GFP resulting from the WT or 6K1-GFP from pEarleyGate103-6K1 is distributed in the cytosol and nucleus in N. benthamiana leaf cells (IV to VI). Asterisks, the cellular nucleus; arrowheads and arrows (in association with the nucleus), 6K1-GFP inclusions. Bars, 30 μm. (C) qRT-PCR detection of the viral accumulation level in N. benthamiana plants agroinfiltrated with WT::6K1GFP, the WT, and the ΔGDD mutant. Agroinfiltrated leaves were collected and pooled at 48 hpi for RNA purification. Viral RNA was quantified by qRT-PCR analysis of the CP RNA level using the mCherry transcript level as an internal control. Statistically significant differences from three biological replicates, determined by an unpaired two-tailed Student's t test, are indicated by brackets and an asterisk. *,0.01 < P < 0.05. (D) (Top) Western blot analysis of the expression of 6K1-GFP or free GFP in N. benthamiana plants using anti-GFP antibodies (against the N terminus of GFP). Cells from an agrobacterial culture (OD600, 0.3) harboring the plasmid WT::6K1GFP or the WT were infiltrated into N. benthamiana leaves. Samples collected at 54 hpi were used for Western blot analysis. Arrows, the fused 6K1-GFP band (∼33.5 kDa) or free GFP (∼27.7 kDa) produced from WT::6K1GFP or the WT. (Bottom) A Coomassie brilliant blue-stained gel used as a loading control.
To further investigate the subcellular distribution of 6K1 in the presence of viral infection, the plasmid pVPH-6K1GFP//mCherry (WT::6K1GFP) was generated by replacement of GFP in the WT plasmid (Fig. 1B) with a GFP-tagged 6K1 (Fig. 5A). This clone as well as its parental WT clone was agroinfiltrated into N. benthamiana leaves. The infectivity of this clone was as competent as that of the WT (Fig. 5C), indicating that the introduction of a second copy of 6K1 did not significantly affect viral infectivity. The 6K1-GFP fluorescence signals produced from the modified viral genome (WT::6K1GFP) in the initially infected cells were observed at different time points under a confocal microscope. At 36 hpi, 6K1-GFP formed many punctate inclusions, most of which were ≤1 μm in diameter (Fig. 5B, panel I). At 48 hpi, more and slightly larger punctate inclusions of 6K1-GFP appeared (Fig. 5B, panel II). At 72 hpi, the amount of 6K1-GFP inclusions was largely reduced (Fig. 5B, panel III), and very little GFP fluorescence was evident at 96 hpi (data not shown), suggesting that 6K1-GFP is not stable in infected cells. In contrast, the free GFP derived from the WT was evenly distributed in the cytoplasm and nucleus at 36 hpi (Fig. 5B, panel IV) and at 48 hpi (Fig. 5B, panel V), and the GFP signals were stable at 72 and 96 hpi. The expression levels of 6K1-GFP and GFP at 54 hpi were examined by Western blot analysis using anti-GFP polyclonal antibodies. Both 6K1-GFP (∼33.5 kDa) from the modified WT::6K1GFP virus and GFP (∼27.7 kDa) from the parental WT virus were detected (Fig. 5D). The amount of 6K1-GFP was much less than that of free GFP (Fig. 5D). Taken together, these findings indicate that 6K1 forms punctate structures which do not seem to be stable in virus-infected cells.
6K1 punctate inclusions colocalize with 6K2-induced VRCs.
In potyvirus-infected cells, the ER-derived 6K2 vesicles predominantly traffic from the ER to the periphery of chloroplasts, where the vesicular structures dock and induce chloroplast membrane invaginations (25). The potyviral 6K2-induced vesicles contain viral and cellular components and viral genomic RNA, which are required for viral replication (25, 51, 52). To determine if 6K1 is involved in viral replication, we first checked whether 6K1 punctate structures are associated with chloroplasts in PPV-infected leaf tissues. In N. benthamiana leaves infiltrated with WT::6K1GFP, about 80% of the 6K1-GFP punctate inclusions were indeed associated with chloroplasts at 36, 48, and 72 hpi (Fig. 6A).
FIG 6.

PPV 6K1 punctate inclusions are associated with chloroplasts and colocalize with VRC elements 6K2 and NIb during viral early infection. (A) Time course of 6K1 punctate inclusions (36, 48, and 72 hpi [panels I to III, respectively]) in association with chloroplasts in N. benthamiana leaves infiltrated with WT::6K1GFP. Green and red fluorescence signals represent 6K1-GFP and chlorophyll autofluorescence, respectively. Arrowheads, 6K1-GFP punctate inclusions. Bars, 10 μm. (B) 6K1 punctate inclusions colocalize with 6K2 in association with chloroplasts in N. benthamiana leaves infiltrated with pVPH::6K1GFP (which was constructed by removal of the mCherry expression cassette of WT::6K1GFP) and further infiltrated with pGWB451-6K2. Green fluorescence and red fluorescence represent 6K1-GFP and 6K2-RFP, respectively. Chl, chlorophyll autofluorescence (blue). Asterisk, the nucleus. Bars, 5 μm. (C) 6K1 punctate inclusions colocalize with the NIb protein (viral RNA-dependent RNA polymerase) in association with chloroplasts in N. benthamiana leaves infiltrated with pVPH::6K1GFP and further infiltrated with pCaMterX-NIb-mCherry. Green fluorescence and red fluorescence represent 6K1-GFP and NIb-mCherry, respectively. Chl, chlorophyll autofluorescence (blue). Arrowheads and arrows, 6K1-GFP inclusions and NIb punctate structures, respectively. Bars, 10 μm.
As has been well established, the potyviral VRC consists of several viral proteins, such as 6K2, NIb, P3, VPg, NIa-Pro, and CI, which have been shown to be essential for viral replication (25, 28, 30, 51). To test if 6K1 is colocalized with 6K2, fully expanded leaves of N. benthamiana were infiltrated with cells from an agrobacterial culture (OD600, 0.2) harboring pVPH::6K1GFP (which was created by removal of the mCherry expression cassette from WT::6K1GFP), and 24 h later, these leaves were infiltrated with cells from an agrobacterial culture (OD600, 0.1) harboring pGWB454-6K2 (for transient expression of the fused 6K2-RFP). At 48 hpi with pVPH::6K1GFP, the punctate inclusions of 6K1-GFP were observed to subcellularly colocalize chloroplasts and 6K2-RFP structures (Fig. 6B). To examine if 6K1 punctate inclusions also colocalize with NIb, the RNA-dependent RNA polymerase (which is absolutely required for viral replication), cells from an agrobacterial culture (OD600, 0.2) harboring pVPH::6K1GFP were infiltrated into fully expanded leaves of N. benthamiana, which, after 6 h, were reinfiltrated with cells from an agrobacterial culture (OD600, 0.1) harboring pCaMterX-NIb-mCherry for transient expression of the fused NIb-mCherry protein. At 54 hpi with pVPH-6K1GFP, the punctate inclusions of 6K1-GFP were observed to colocalize with NIb inclusions in association with chloroplasts (Fig. 6C). Taken together, 6K1 punctate inclusions colocalize with chloroplast-bound 6K2 and NIb, suggesting that 6K1 is part of the VRC at the early infection stage.
DISCUSSION
In this study, we developed a series of recombinant full-length infectious PPV cDNA clones for the functional characterization of 6K1, one of a few of the least-studied potyviral proteins. This set of infectious clones, being infectious for both the model plant N. benthamiana and the natural host, the peach tree (Fig. 1), is thus a useful tool for potyvirus research on both herb and woody plants. Using this system, we created a variety of 6K1 mutants and found that deletion of any short motifs of 6K1 (each of which ranged from 5 to 13 amino acids), most of the 6K1 sequence (but with the conserved sequence of the cleavage sites being retained), or the entire 6K1 sequence abolished viral replication (Fig. 2C and 3B). It is worth mentioning that all mutants in this study in which the replication ability was abolished also lost the ability to move intercellularly, consistent with our recent observation that potyviral replication and intercellular movement are coupled processes (45). As the deletion of 6K1 may affect the efficient proteolytic processing at the newly formed P3/CI junction, we inserted a self-cleaving peptide, T2A, at the P3/CI junction of 6K1 deletion mutants. The mutations were also lethal for the resulting clones (Fig. 2D). Based on these data, we conclude that 6K1 is indispensable for viral genome multiplication. In agreement with this conclusion, deletion of 6K1 was also lethal for PVA (46). Moreover, Kekarainen and colleagues generated a genomic 15-bp insertion mutant library on PVA and identified four PVA mutants with an insertion in the N-terminal region of 6K1 that were noninfectious in a tobacco protoplast-based replication assay (21).
The potyviral 6K1 protein was first identified to be a mature protein by the discovery of processing of the proteolytic sites between P3 and 6K1 and between 6K1 and CI in vitro over 2 decades ago (48). The mature 6K1 protein was indeed detected in PPV-infected N. benthamiana plants (49). Extensive in vitro studies suggest that cleavage at the N and C termini of the intermediate precursor, P3-6K1, by HC-Pro (self-cleavage) and NIa-Pro, respectively, is efficient, whereas cleavage at the P3 and 6K1 junction is very slow, leading to the generation of three viral proteins, P3, 6K1, and P3-6K1 (53). In agreement with this assumption, both the P3 and P3-6K1 proteins were detected in Tobacco vein mottling virus (TVMV)-infected plants (54). In this study, we provide evidence that knockout of the cleavage between P3 and 6K1 reduced PPV infectivity and knockout of the cleavage between 6K1 and CI was deleterious (Fig. 2C). Consistent with our findings, artificial inactivation of the NIa-Pro cleavage site at the P3/6K1 junction of PPV (the Rankovic isolate in the Czech Republic) greatly delayed the time to disease and the infected plants became asymptomatic (53). Different from the findings for the mutant of the Rankoyic isolate whose progeny underwent a second mutation in 6K1, which restored the high infection efficiency of the isolate (53), no compensatory mutations were found around the mutated P3/6K1 junction in the viral progeny of the Δ1CS mutant. Taken together, these data suggest that the nonprocessable P3-6K1 can partially complement the function of the mature P3 and 6K1 proteins.
On the basis of the suggestion made above, it would be logical to analyze the sequence of 6K1 and examine the subcellular localization of 6K1. Clearly, all 17 potyviral 6K1 proteins analyzed in this study contained a hydrophobic stretch and are predicted to be integral membrane proteins (Fig. 3A). Among the 11 potyviral proteins, 6K2 and P3 are experimentally proven membrane proteins (22, 23, 29). 6K2 remodels the ER to form the replication vesicles that target the chloroplast for robust viral replication (24, 25). As an ER membrane protein, P3 forms punctate inclusions, traffics along the actin filaments, and colocalizes with the chloroplast-bound 6K2-induced replication vesicles (29). Although potyviral 6K1 sequences were predicted to be membrane associated, the distribution pattern of the GFP-tagged PPV 6K1 protein resembled that of the free form of GFP when expressed alone (Fig. 5B, panels IV to VI). A similar observation was also reported for mCherry-tagged TuMV 6K1 in a recent study (50). However, in the presence of viral infection, 6K1 formed punctate structures (Fig. 5B, panels I to III). These 6K1 structures were also associated with chloroplasts (Fig. 6A) and colocalized with chloroplast-bound 6K2 vesicles and the chloroplast-associated NIb (Fig. 6B and C). As these 6K2-induced vesicles are recognized to be potyviral replication sites (25, 52), it is reasonable to suggest that, like other VRC-containing proteins (such as 6K2, NIb, P3, and CI), 6K1 is also an essential element of the VRC. This suggestion is further supported by a recent proteomic study showing that 6K1, together with other viral replicase elements, such as CI, HC-Pro, P3, NIb, and VPg, is evident in the purified 6K2 protein complex (51). It is possible that 6K1 is recruited to the VRC either through P3-6K1 (due to inefficient processing by NIa-Pro) or via interactions with other viral replicase elements. As shown previously, the PPV 6K1 protein interacts with 6K2 and CI in planta (55).
In this study, we show that the trans expression of 6K1 through transient expression or cis expression of a dislocated 6K1 in the modified PPV genome failed to rescue PPV mutants with a 6K1 deletion (Fig. 4). Moreover, the trans expression of P3-6K1 also could not complement the viral replication defect of PPV mutants with a 6K1 deletion (Fig. 4D). These data suggest that a functional 6K1 must be expressed in an orchestrated context and a precisely timely manner. In agreement with this, we found that 6K1-derived punctate inclusions were greatly attenuated after 48 hpi (Fig. 5B). Indeed, the mature 6K1 protein level is very low in PPV-infected N. benthamiana plants, and it is detectable only after affinity purification (49). In view of its colocalization with 6K2-induced VRCs, requirement for viral replication, and temporal and spatial expression-dependent function, it is possible that the 6K1 protein joins two other viral membrane proteins, 6K2 and P3, to mediate VRC assembly at the early infection stage. This assumption is supported by the fact that the nonprocessable P3-6K1 can partially complement the function of the mature P3 and 6K1 proteins. The mechanistic roles of these viral proteins in the formation of VRC for viral replication remain to be elucidated.
ACKNOWLEDGMENTS
We are indebted to David J. Oliver (Iowa State University) for providing the mini-binary vector pCB301 and Lorne Stobbs (Agriculture and Agri-Food Canada [AAFC]) for providing PPV-positive peach tree samples, Xiaofei Cheng (AAFC) for helpful discussions and assistance with confocal microscopy work, Huaiyu Wang and Jamie McNeil (AAFC) for technical support, and Alex Molnar (AAFC) for assistance with artwork submission.
REFERENCES
- 1.den Boon JA, Diaz A, Ahlquist P. 2010. Cytoplasmic viral replication complexes. Cell Host Microbe 8:77–85. doi: 10.1016/j.chom.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nagy PD, Pogany J. 2012. The dependence of viral RNA replication on co-opted host factors. Nat Rev Microbiol 10:137–149. doi: 10.1038/nrmicro2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.García JA, Pallás V. 2015. Viral factors involved in plant pathogenesis. Curr Opin Virol 11:21–30. doi: 10.1016/j.coviro.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 4.Wang A. 2015. Dissecting the molecular network of virus-plant interactions: the complex roles of host factors. Annu Rev Phytopathol 53:45–66. doi: 10.1146/annurev-phyto-080614-120001. [DOI] [PubMed] [Google Scholar]
- 5.Ivanov KI, Eskelin K, Lõhmus A, Mäkinen K. 2014. Molecular and cellular mechanisms underlying potyvirus infection. J Gen Virol 95:1415–1429. doi: 10.1099/vir.0.064220-0. [DOI] [PubMed] [Google Scholar]
- 6.Revers F, García JA. 2015. Molecular biology of potyviruses. Adv Virus Res 92:101–199. doi: 10.1016/bs.aivir.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 7.Chung BYW, Miller WA, Atkins JF, Firth AE. 2008. An overlapping essential gene in the Potyviridae. Proc Natl Acad Sci U S A 105:5897–5902. doi: 10.1073/pnas.0800468105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rodamilans B, Valli A, Mingot A, San León D, Baulcombe D, López-Moya JJ, García JA. 2015. RNA polymerase slippage as a mechanism for the production of frameshift gene products in plant viruses of the Potyviridae family. J Virol 89:6965–6967. doi: 10.1128/JVI.00337-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olspert A, Chung BYW, Atkins JF, Carr JP, Firth AE. 2015. Transcriptional slippage in the positive-sense RNA virus family Potyviridae. EMBO Rep 16:995–1004. doi: 10.15252/embr.201540509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.White KA. 2015. The polymerase slips and PIPO exists. EMBO Rep 16:885–886. doi: 10.15252/embr.201540871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Riechmann JL, Laín S, García JA. 1992. Highlights and prospects of potyvirus molecular biology. J Gen Virol 73:1–16. doi: 10.1099/0022-1317-73-1-1. [DOI] [PubMed] [Google Scholar]
- 12.Carrington JC, Freed DD, Sanders TC. 1989. Autocatalytic processing of the potyvirus helper component proteinase in Escherichia coli and in vitro. J Virol 63:4459–4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.García JA, Glasa M, Cambra M, Candresse T. 2014. Plum pox virus and sharka: a model potyvirus and a major disease. Mol Plant Pathol 15:226–241. doi: 10.1111/mpp.12083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Verchot J, Koonin EV, Carrington JC. 1991. The 35-kDa protein from the N-terminus of a potyviral polyprotein functions as a third virus-encoded proteinase. Virology 185:527–535. doi: 10.1016/0042-6822(91)90522-D. [DOI] [PubMed] [Google Scholar]
- 15.Kasschau KD, Carrington JC. 1998. A counterdefensive strategy of plant viruses: suppression of posttranscriptional gene silencing. Cell 95:461–470. doi: 10.1016/S0092-8674(00)81614-1. [DOI] [PubMed] [Google Scholar]
- 16.Rajamäki ML, Kelloniemi J, Alminaite A, Kekarainen T, Rabenstein F, Valkonen JP. 2005. A novel insertion site inside the potyvirus P1 cistron allows expression of heterologous proteins and suggests some P1 functions. Virology 342:88–101. doi: 10.1016/j.virol.2005.07.019. [DOI] [PubMed] [Google Scholar]
- 17.Valli A, Martín-Hernández AM, López-Moya JJ, García JA. 2006. RNA silencing suppression by a second copy of the P1 serine protease of Cucumber vein yellowing ipomovirus, a member of the family Potyviridae that lacks the cysteine protease HCPro. J Virol 80:10055–10063. doi: 10.1128/JVI.00985-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martínez F, Daròs JA. 2014. Tobacco etch virus protein P1 traffics to the nucleolus and associates with the host 60S ribosomal subunits during infection. J Virol 88:10725–10737. doi: 10.1128/JVI.00928-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pasin F, Simón-Mateo C, García JA. 2014. The hypervariable amino-terminus of P1 protease modulates potyviral replication and host defense responses. PLoS Pathog 10:e1003985. doi: 10.1371/journal.ppat.1003985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sorel M, Garcia JA, German-Retana S. 2014. The Potyviridae cylindrical inclusion helicase: a key multipartner and multifunctional protein. Mol Plant Microbe Interact 27:215–226. doi: 10.1094/MPMI-11-13-0333-CR. [DOI] [PubMed] [Google Scholar]
- 21.Kekarainen T, Savilahti H, Valkonen JP. 2002. Functional genomics on potato virus A: virus genome-wide map of sites essential for virus propagation. Genome Res 12:584–594. doi: 10.1101/gr.220702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Restrepo-Hartwig MA, Carrington JC. 1994. The tobacco etch potyvirus 6-kilodalton protein is membrane associated and involved in viral replication. J Virol 68:2388–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schaad MC, Jensen PE, Carrington JC. 1997. Formation of plant RNA virus replication complexes on membranes: role of an endoplasmic reticulum-targeted viral protein. EMBO J 16:4049–4059. doi: 10.1093/emboj/16.13.4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wei T, Wang A. 2008. Biogenesis of cytoplasmic membranous vesicles for plant potyvirus replication occurs at endoplasmic reticulum exit sites in a COPI- and COPII-dependent manner. J Virol 82:12252–12264. doi: 10.1128/JVI.01329-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wei T, Huang TS, McNeil J, Laliberté JF, Hong J, Nelson RS, Wang A. 2010. Sequential recruitment of the endoplasmic reticulum and chloroplasts for plant potyvirus replication. J Virol 84:799–809. doi: 10.1128/JVI.01824-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beauchemin C, Boutet N, Laliberté JF. 2007. Visualization of the interaction between the precursors of VPg, the viral protein linked to the genome of turnip mosaic virus, and the translation eukaryotic initiation factor iso 4E in planta. J Virol 81:775–782. doi: 10.1128/JVI.01277-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dufresne PJ, Ubalijoro E, Fortin MG, Laliberté JF. 2008. Arabidopsis thaliana class II poly(A)-binding proteins are required for efficient multiplication of turnip mosaic virus. J Gen Virol 89:2339–2348. doi: 10.1099/vir.0.2008/002139-0. [DOI] [PubMed] [Google Scholar]
- 28.Cotton S, Grangeon R, Thivierge K, Mathieu I, Ide C, Wei T, Wang A, Laliberté JF. 2009. Turnip mosaic virus RNA replication complex vesicles are mobile, align with microfilaments, and are each derived from a single viral genome. J Virol 83:10460–10471. doi: 10.1128/JVI.00819-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cui X, Wei T, Chowda-Reddy RV, Sun G, Wang A. 2010. The Tobacco etch virus P3 protein forms mobile inclusions via the early secretory pathway and traffics along actin microfilaments. Virology 397:56–63. doi: 10.1016/j.virol.2009.11.015. [DOI] [PubMed] [Google Scholar]
- 30.Wan J, Cabanillas DG, Zheng H, Laliberté JF. 2015. Turnip mosaic virus moves systemically through both phloem and xylem as membrane-associated complexes. Plant Physiol 167:1374–1388. doi: 10.1104/pp.15.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cui H, Hong N, Wang G, Wang A. 2013. Genomic segments RNA1 and RNA2 of Prunus necrotic ringspot virus codetermine viral pathogenicity to adapt to alternating natural Prunus hosts. Mol Plant Microbe Interact 26:515–527. doi: 10.1094/MPMI-12-12-0282-R. [DOI] [PubMed] [Google Scholar]
- 32.Cui H, Hong N, Wang G, Wang A. 2012. Detection and genetic diversity of Prunus necrotic ringspot virus in the Niagara Fruit Belt, Canada. Can J Plant Pathol 34:104–113. doi: 10.1080/07060661.2012.661768. [DOI] [Google Scholar]
- 33.Cui H, Hong N, Wang G, Wang A. 2012. Molecular characterization of two Prunus necrotic ringspot virus isolates from Canada. Arch Virol 157:999–1001. doi: 10.1007/s00705-012-1247-5. [DOI] [PubMed] [Google Scholar]
- 34.Xiang C, Han P, Lutziger I, Wang K, Oliver DJ. 1999. A mini binary vector series for plant transformation. Plant Mol Biol 40:711–717. doi: 10.1023/A:1006201910593. [DOI] [PubMed] [Google Scholar]
- 35.Agbeci M, Grangeon R, Nelson RS, Zheng H, Laliberté JF. 2013. Contribution of host intracellular transport machineries to intercellular movement of turnip mosaic virus. PLoS Pathog 9:e1003683. doi: 10.1371/journal.ppat.1003683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang DJ, Brandsma M, Yin Z, Wang A, Jevnikar AM, Ma S. 2008. A novel platform for biologically active recombinant human interleukin-13 production. Plant Biotechnol J 6:504–515. doi: 10.1111/j.1467-7652.2008.00337.x. [DOI] [PubMed] [Google Scholar]
- 37.Earley KW, Haag JR, Pontes O, Opper K, Juehne T, Song K, Pikaard CS. 2006. Gateway-compatible vectors for plant functional genomics and proteomics. Plant J 45:616–629. doi: 10.1111/j.1365-313X.2005.02617.x. [DOI] [PubMed] [Google Scholar]
- 38.Sparkes IA, Runions J, Kearns A, Hawes C. 2006. Rapid, transient expression of fluorescent fusion proteins in tobacco plants and generation of stably transformed plants. Nat Protoc 1:2019–2025. doi: 10.1038/nprot.2006.286. [DOI] [PubMed] [Google Scholar]
- 39.Salvador B, Delgadillo MO, Sáenz P, García JA, Simón-Mateo C. 2008. Identification of Plum pox virus pathogenicity determinants in herbaceous and woody hosts. Mol Plant Microbe Interact 21:20–29. doi: 10.1094/MPMI-21-1-0020. [DOI] [PubMed] [Google Scholar]
- 40.Carbonell A, Maliogka VI, Pérez JDJ, Salvador B, León DS, García JA, Simón-Mateo C. 2013. Diverse amino acid changes at specific positions in the N-terminal region of the coat protein allow Plum pox virus to adapt to new hosts. Mol Plant Microbe Interact 26:1211–1224. doi: 10.1094/MPMI-04-13-0093-R. [DOI] [PubMed] [Google Scholar]
- 41.Calvo M, Malinowski T, García JA. 2014. Single amino acid changes in the 6K1-CI region can promote the alternative adaptation of Prunus- and Nicotiana-propagated Plum pox virus C isolates to either host. Mol Plant Microbe Interact 27:136–149. doi: 10.1094/MPMI-08-13-0242-R. [DOI] [PubMed] [Google Scholar]
- 42.Majer E, Salvador Z, Zwart MP, Willemsen A, Elena SF, Daròs JA. 2014. Relocation of the NIb gene in the tobacco etch potyvirus genome. J Virol 88:4586–4590. doi: 10.1128/JVI.03336-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fernández-Fernández R, Mouriño M, Rivera J, Plana-Durán J, García JA. 2001. Protection of rabbits against rabbit hemorrhagic disease virus by immunization with the VP60 protein expressed in plants with a potyvirus-based vector. Virology 280:283–291. doi: 10.1006/viro.2000.0762. [DOI] [PubMed] [Google Scholar]
- 44.Beauchemin C, Bougie V, Laliberté JF. 2005. Simultaneous production of two foreign proteins from a potyvirus-based vector. Virus Res 112:1–8. doi: 10.1016/j.virusres.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 45.Deng P, Wu Z, Wang A. 2015. The multifunctional protein CI of potyviruses plays interlinked and distinct roles in viral genome replication and intercellular movement. Virol J 12:141. doi: 10.1186/s12985-015-0369-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Merits A, Rajamäki ML, Lindholm P, Runeberg-Roos P, Kekarainen T, Puustinen P, Mäkeläinen K, Valkonen JP, Saarma M. 2002. Proteolytic processing of potyviral proteins and polyprotein processing intermediates in insect and plant cells. J Gen Virol 83:1211–1221. doi: 10.1099/0022-1317-83-5-1211. [DOI] [PubMed] [Google Scholar]
- 47.Donnelly ML, Hughes LE, Luke G, Mendoza H, ten Dam E, Gani D, Ryan MD. 2001. The ‘cleavage’ activities of foot-and-mouth disease virus 2A site-directed mutants and naturally occurring ‘2A-like’ sequences. J Gen Virol 82:1027–1041. doi: 10.1099/0022-1317-82-5-1027. [DOI] [PubMed] [Google Scholar]
- 48.García JA, Martín MT, Cervera MT, Riechmann J. 1992. Proteolytic processing of the plum pox potyvirus polyprotein by the NIa protease at a novel cleavage site. Virology 188:697–703. doi: 10.1016/0042-6822(92)90524-S. [DOI] [PubMed] [Google Scholar]
- 49.Waltermann A, Maiss E. 2006. Detection of 6K1 as a mature protein of 6 kDa in plum pox virus-infected Nicotiana benthamiana. J Gen Virol 87:2381–2386. doi: 10.1099/vir.0.81873-0. [DOI] [PubMed] [Google Scholar]
- 50.Jiang J, Patarroyo C, Cabanillas DG, Zheng H, Laliberté JF. 2015. The vesicle-forming 6K2 protein of turnip mosaic virus interacts with the COPII coatomer Sec24a for viral systemic infection. J Virol 89:6695–6710. doi: 10.1128/JVI.00503-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lõhmus A, Varjosalo M, Mäkinen K. 17 November 2015. Protein composition of 6K2-induced membrane structures formed during Potato virus A infection. Mol Plant Pathol. doi: 10.1111/mpp.12341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li Y, Cui H, Cui X, Wang A. 2015. The altered photosynthetic machinery during compatible virus infection. Curr Opin Virol 17:19–24. doi: 10.1016/j.coviro.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 53.Riechmann JL, Cervera MT, García JA. 1995. Processing of the plum pox virus polyprotein at the P3-6K1 junction is not required for virus viability. J Gen Virol 76:951–956. doi: 10.1099/0022-1317-76-4-951. [DOI] [PubMed] [Google Scholar]
- 54.Rodríguez-Cerezo E, Shaw JG. 1991. Two newly detected nonstructural viral proteins in potyvirus-infected cells. Virology 185:572–579. doi: 10.1016/0042-6822(91)90527-I. [DOI] [PubMed] [Google Scholar]
- 55.Zilian E, Maiss E. 2011. Detection of plum pox potyviral protein-protein interactions in planta using an optimized mRFP-based bimolecular fluorescence complementation system. J Gen Virol 92:2711–2723. doi: 10.1099/vir.0.033811-0. [DOI] [PubMed] [Google Scholar]