

ABSTRACT
Nonprimate hepacivirus (NPHV), the closest homolog of hepatitis C virus (HCV) described to date, has recently been discovered in horses. Even though the two viruses share a similar genomic organization, conservation of the encoded hepaciviral proteins remains undetermined. The HCV p7 protein is localized within endoplasmic reticulum (ER) membranes and is important for the production of infectious particles. In this study, we analyzed the structural and functional features of NPHV p7 in addition to its role during virus assembly. Three-dimensional homology models for NPHV p7 using various nuclear magnetic resonance spectroscopy (NMR) structures were generated, highlighting the conserved residues important for ion channel function. By applying a liposome permeability assay, we observed that NPHV p7 exhibited liposome permeability features similar to those of HCV p7, indicative of similar ion channel activity. Next, we characterized the viral protein using a p7-based trans-complementation approach. A similar subcellular localization pattern at the ER membrane was observed, although production of infectious particles was likely hindered by genetic incompatibilities with HCV proteins. To further characterize these cross-species constraints, chimeric viruses were constructed by substituting different regions of HCV p7 with NPHV p7. The N terminus and transmembrane domains were nonexchangeable and therefore constitute a cross-species barrier in hepaciviral assembly. In contrast, the basic loop and the C terminus of NPHV p7 were readily exchangeable, allowing production of infectious trans-complemented viral particles. In conclusion, comparison of NPHV and HCV p7 revealed structural and functional homology of these proteins, including liposome permeability, and broadly acting determinants that modulate hepaciviral virion assembly and contribute to the host-species barrier were identified.
IMPORTANCE The recent discovery of new relatives of hepatitis C virus (HCV) enables for the first time the study of cross-species determinants shaping hepaciviral pathogenesis. Nonprimate hepacivirus (NPHV) was described to infect horses and represents so far the closest homolog of HCV. Both viruses encode the same viral proteins; however, NPHV protein functions remain poorly understood. In this study, we aimed to dissect NPHV p7 on a structural and functional level. By using various NMR structures of HCV p7 as templates, three-dimensional homology models for NPHV p7 were generated, highlighting conserved residues that are important for ion channel function. A p7-based trans-complementation approach and the construction of NPHV/HCV p7 chimeric viruses showed that the N terminus and transmembrane domains were nonexchangeable. In contrast, the basic loop and the C terminus of NPHV p7 were readily exchangeable, allowing production of infectious viral particles. These results identify species-specific constraints as well as exchangeable determinants in hepaciviral assembly.
INTRODUCTION
For more than 2 decades, hepatitis C virus (HCV) and GB virus B (GBV-B) were the sole members of the genus Hepacivirus within the Flaviviridae family. Recently, multiple new hepaciviruses have been identified in dogs (1), horses (2), bats (3, 4), rodents (3, 5), nonhuman primates (6), rats (7), and cattle (8, 9). Among them, nonprimate hepacivirus (NPHV), initially described to infect dogs and subsequently horses, is the closest homolog of HCV and thus represents a unique model to study differences in hepacivirus pathogenesis of HCV and HCV-related viruses (10, 11).
HCV is globally distributed, and approximately 146 million people of the world's population are persistently infected (12). Individuals infected with HCV are at high risk of developing liver cirrhosis and hepatocellular carcinoma (13). The development of direct-acting antivirals (DAAs) has significantly improved antiviral treatment options (14). However, a prophylactic vaccine is still lacking. The genome of HCV consists of a single-stranded RNA with positive polarity and encodes 10 viral proteins in an open reading frame (ORF) (15). The small membrane protein p7 is encoded between the structural proteins core, E1, and E2 and the nonstructural proteins. p7 is classified into the group of viroporins since it fulfills major characteristics of this family, for instance, its small size (63 amino acids) and its ability to form oligomeric, hydrophobic ion channels in the endoplasmic reticulum (ER) membrane (16). p7 is composed of two transmembrane passages connected by a short polar loop. The N-terminal helix and C terminus face the lumen of the ER (17); however, another topology whereby the C terminus is exposed toward the cytosol has also been reported (18). p7 monomers assemble to form hexameric or heptameric structures (19–22). By applying single-particle electron microscopy, a three-dimensional model of a p7 hexamer was resolved (20). Additionally, the monomeric and oligomeric structure of p7 of different genotypes was elucidated by nuclear magnetic resonance spectroscopy (NMR) studies in different lipid-mimicking environments (trifluoroethanol [TFE], 1,2-dihexanoyl-sn-glycero-3-phosphocholine [DHPC], dodecylphosphocholine [DPC], or methanol) (23–26), which likely explains the structural discrepancies observed between these models. In vitro analysis revealed that p7 is essential for HCV assembly and release, whereas it is dispensable for viral replication (27, 28). For further details on structural and functional properties of HCV p7, see also recent reviews (16, 29, 30).
After the identification of NPHV, several studies have been conducted to investigate the differences and similarities between NPHV and HCV. A high seroprevalence of anti-NPHV antibodies (30 to 40%) among horses was reported, with 2 to 7% of the horses also carrying viral RNA (10). Similar to HCV, NPHV is also a hepatotropic virus, as was evidenced by accumulation of viral plus and minus strand RNA in liver sections (31). The genomic organization of HCV and NPHV is highly conserved, with one ORF encoding the viral proteins (10, 11). As seen for HCV, the ORF of NPHV is flanked by two untranslated regions (UTRs) at the 5′ and 3′ ends, with the 5′UTR displaying a larger stem-loop I (2). Regarding the function of NPHV viral proteins, individual expression of the NPHV core protein showed that core localizes on lipid droplets as reported for HCV core (32). In addition, the NS3/4A protein of NPHV has been shown to have a function similar to that of the HCV equivalent by cleaving human mitochondrial antiviral signaling protein (MAVS) and Toll-interleukin 1 (IL-1) receptor domain-containing adaptor inducing interferon beta (TRIF) (33). However, a detailed understanding of viral protein function, especially in the context of cross-species determinants shaping hepaciviral pathogenesis, is lacking.
In this study, we discovered that although NPHV p7 shared comparable structural features with its human homolog and exerted an ion channel activity, the protein could not fully replace HCV p7 during virus assembly. Replacement of the basic loop and the C terminus within NPHV p7, however, led to production of infectious HCV particles, thus defining virus species-specific and interchangeable subdomains within p7.
MATERIALS AND METHODS
Sequence and phylogenetic analysis.
Nucleotide sequences of NPHV p7 isolates (GenBank accession numbers KP325401, JQ434002, JQ434003, JQ434004, JQ434005, JQ434006, JQ434007, JQ434008, JX948116; the p7 sequences generated in this study are available as indicated below) were translated and aligned using MEGA6 (34), and a consensus sequence was generated. For phylogenetic analysis, one representative p7 sequence of each HCV genotype was utilized (GenBank accession numbers NC_004102, YP_001469630, NC_009824, NC_009825, NC_009826, NC_009827, EF108306). The HCV p7 consensus sequence was deduced from the ClustalW multiple alignment (35) of p7 sequences from representative HCV strains of confirmed genotypes (as described in reference 23). A maximum likelihood phylogenetic tree was generated using MEGA6 (34).
Structural analysis.
Three-dimensional homology models of NPHV p7 monomer were constructed by the Swiss-Model automated protein structure homology modeling server (http://www.expasy.org/spdbv/ [36]) by using the NMR structures of HCV p7 as templates (23–26). Two models of the NPHV p7 three-dimensional hexamer were generated. The positions of models 1 and 2 relative to the membrane bilayer were deduced from molecular dynamics (MD) simulations of HCV p7 in the 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayer as reported by Chandler et al. (19) and Kalita et al. (37), respectively. Figures were generated from structure coordinates by using VMD (http://www.ks.uiuc.edu/Research/vmd/ [38]) and rendered with POV-Ray (http://www.povray.org/).
Peptide synthesis of HCV and NPHV p7.
The p7 peptides of the JFH-1 or H14 strain were synthesized with a CEM microwave peptide synthesizer. To that end, all required amino acids were dissolved in N,-N,-dimethylformamide (DMF; Rathburn Chemicals Ltd.). As the activator, hydroxybenzotriazole (HoBt) hydrate was used, and as the activator base, N,N′-diisopropylcarbodiimide (DIC; Sigma) was used. Deprotection was conducted in 20% piperidine (Sigma) in DMF (vol/vol). Dichloromethane (Sigma) was used for washing. A 16.4-liter volume of DMF, 150 ml of activator, 200 ml of activator base, 2.8 liters of deprotection product, 0.17 g of resin, and 0.2 M each respective amino acid were placed in a CEM microwave peptide synthesizer, and a program was created to start synthesis from the C terminus. The first amino acid added was arginine (Arg), since alanine (Ala) is attached to the resin. Reactions for all the amino acids were performed as double coupling except for proline. Cycles for Arg, Cys, and His are performed at lower temperature and for a longer time period than cycles for other amino acids to avoid racemization. To avoid any side chain reaction, only Fmoc-Lys (Boc)-OH for lysine (Lys) was used with double coupling. The instrument will automatically stop and collect the resin with synthesized peptide, which is required to cleave the peptide from resin. Peptides were purified by high-performance liquid chromatography (HPLC) on a C4 semipreparative column with a linear acetonitrile gradient. The purity was verified by SDS-PAGE. The sequence of the p7 peptide was confirmed by matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometric analysis.
Liposome permeability assay.
Liposome preparation and permeability assays were conducted as described earlier (39). Briefly, lipids (Avanti Polar Lipids) in chloroform were added in a final mixture containing 0.5 mg l-α-phosphatidic acid, 0.5 mg l-α-phosphatidyl choline, and 0.5% (wt/wt) l-α-phosphatidyl ethanolamine with lissamine rhodamine B-labeled head groups. Chloroform was evaporated from the lipids using a stream of nitrogen before the lipids were placed in a vacuum for 4 h at room temperature (RT). Lipids were rehydrated to 2 mg/ml in a self-quenching concentration of carboxyfluorescein (CF) buffer [50 mM 5(6)-carboxyfluorescein (Sigma), 10 mM HEPES (pH 7.4), 107 mM NaCl] and vigorously shaken overnight at RT. Unilamellar liposomes were produced using an extruder (Avanti Polar Lipids) and a 0.4-μm filter (Whatman). Liposomes were purified via centrifugation at 49,000 rpm for 15 min at 25°C, including 4 washing steps, before the pellet was resuspended in 0.5 ml liposome assay buffer (10 mM HEPES, pH 7.4, 107 mM NaCl). For p7 activity assays, the peptides were reconstituted in 100% dimethyl sulfoxide (DMSO), and the concentration was determined by nanodrop. Liposomes supplemented with 1% (vol/vol) DMSO were used as a baseline for fluorescence. Assays were carried out in black-walled, flat-bottomed black-base 96-well plates at 37°C. Each well contained 50 μM liposomes (calculated from the rhodamine fluorescence) and 1 μl of peptide in DMSO in a total volume of 100 μl with liposome assay buffer. Triton TX-100 (0.5%, vol/vol), which lyses liposomes, was used for gain adjustment, setting a level of 90% fluorescence. The 96-well plate was kept on ice for 2 to 5 min after gain adjustment and while the peptide with or without drug was added. CF release measured by increased fluorescence was taken as an indicator of peptide-induced membrane permeability (activity). A set of 30 readings (excitation wavelength [λ], 485 nm; emission λ, 520 nm) was made over the course of 24 min using a FLUOstar Galaxy plate-reader (BMG Labtech). Reactions for each condition were carried out in duplicate wells with three independent experimental repeats. Endpoint measurements were used for the analysis with the average of the duplicate wells taken. For N-nonyl-deoxynojirimycin (NN-DNJ) inhibition assays, liposomes contained 2% (vol/vol) DMSO with or without 40 μM NN-DNJ, these being the respective background levels for drug-free and drug-treated wells. Concentrations of 9 μM NPHV p7 peptide and 44 μM JFH-1 p7 peptide were used. Peptides with or without inhibitor were incubated for 20 min at RT prior to addition to the gain-adjusted plate on ice. Statistical analysis was conducted by a Welch's corrected unpaired t test. P values of <0.05 were considered statistically significant and are marked with an asterisk in the figures.
Cell culture.
Huh-7.5 cells were cultured in Dulbecco's modified Eagle's medium (Life Technologies) supplemented with 10% fetal bovine serum (FCS), 2 mM l-glutamine, nonessential amino acids (Invitrogen), 100 μg/ml streptomycin (Invitrogen), and 100 IU/ml penicillin (Invitrogen) (DMEM complete) at 37°C and 5% CO2. The packaging cell line Huh-7.5[C][E1E2][NS2]J6 expressing the Jc1 derived proteins C, E1E2 and NS2 was generated by lentiviral gene transfer as described earlier (40). Vectors used for the gene transfer carried a blasticidin-S deaminase resistance gene, and therefore, 5 μg/ml of blasticidin (Invivogen) was added for selection.
Plasmids.
The plasmids pFK-PI-Spp7/J6-EI-NS3-5B/JFH-1, pFK-PI-Sp-HA-HA-L-p7/J6EI-NS3-5B/JFH-1, and pFK-PI-G-Luc-EI-NS3-5B/JFH-1 have been described earlier (41, 42) and are based on the bicistronic helper replicon pFK-PI-EI-NS3-5B/JFH-1. This helper replicon contains a poliovirus-derived internal ribosomal entry site (IRES) (PI) downstream of the JFH-1-derived 5′-nontranslated region (5′NTR) (nucleotides 1 to 341 of JFH-1) and is separated by a spacer region of 72 nucleotides. The second cistron is under the control of an encephalomyocarditis virus IRES (EI) that expresses JFH-1-derived NS3 to NS5B proteins. The p7 sequence of the NPHV isolate H14 and different HCV/NPHV p7 chimeras were chemically synthesized (Integrated DNA Technologies [IDT]). The cloned fragments included a signal peptide (sp) derived from the last 51 bp of the E2 protein (HCV isolate J6) downstream of the p7 sequence or additionally a HAHA tag linked to the p7 sequence with a linker and upstream of the sp. The respective genes were cloned into the first cistron of pFK-PI-EI-NS3-5B/JFH-1 by restriction digest and ligation. In addition to bicistronic helper replicons used for trans-complementation assays, experiments with the HCV full-length virus were also performed. Therefore, the plasmids pFK-Jc1 (43), pFK-Jc1-Δp7half (27), and pFK-Jc1-HA-HA-L-p7/J6 (42) were utilized. The p7 sequence of the NPHV isolate H14 and the p7 sequences of HCV/NPHV chimeras (p7J6-loop-H14, p7J6-C-ter-H14, and p7J6-loop-C-ter-H14) were cloned into pFK-Jc1 and pFK-Jc1-HA-HA-L-p7/J6 by PCR-based insertion. All constructs were confirmed by sequencing prior to use. Further details regarding the cloning strategies and exact nucleotide sequences are available upon request.
In vitro transcription and electroporation.
In vitro transcripts were created according to a protocol described previously (40). DNA was purified by the Qiaquick PCR purification kit (Qiagen), and RNA was purified by the NucleoSpin RNA Extraction kit (Macherey-Nagel) according to the manufacturer's instructions. Concentration was determined by nanodrop. In vitro-transcribed RNA was stored at −80°C until electroporation.
Electroporations were conducted as described earlier (40). Briefly, Huh-7.5 or Huh-7.5[C][E1E2][NS2]J6 cells were trypsinized and taken up in DMEM complete, and the cell number was determined. A final concentration of 1.5 × 107 cells/ml in 400 μl of Cytomix (120 mM KCl, 0.15 mM CaCl2, 10 mM K2HPO4 or KH2PO4 [pH 7.6], 25 mM HEPES, 2 mM EGTA, 5 mM MgCl2, with pH adjusted to 7.6 with KOH) supplemented with 2 mM ATP and 5 mM glutathione per electroporation and 5 to 10 μg of in vitro transcripts were used per electroporation. Transfected cells were directly taken up in 12 to 16 ml DMEM complete and seeded into 6-well plates or 10-cm dishes depending on the application.
Immunofluorescence.
After transfection, cells were seeded into a 24-well plate onto coverslips. Cells were fixed 48 h posttransfection by addition of 3% paraformaldehyde. Staining of intracellular HAHA-tagged p7 and a costaining of E2 or NS3 were performed as described elsewhere (42). In brief, fixed cells were permeabilized with 0.5% Triton X-100 for 10 min at RT. Blocking was conducted for 1 h at RT in blocking buffer (5% goat serum [Sigma] in phosphate-buffered saline [PBS]). The primary antibodies were incubated overnight at room temperature in blocking buffer. The primary mouse anti-hemagglutinin (anti-HA) antibody (Covance) was diluted 1:1,000, the primary rabbit anti-NS3 4949 (44) antibody was diluted 1:400, and the primary human anti-E2 antibody CBH-23 (45) was diluted 1:250 in blocking buffer. The anti-NS3 and anti-E2 antibodies were kind gifts from R. Bartenschlager (University of Heidelberg) and S. Foung (Stanford University), respectively. Species-specific secondary antibodies (Alexa Fluor 488-conjugated anti-mouse IgG, Alexa Fluor 546-conjugated anti-rabbit IgG, and Alexa Fluor 546-conjugated anti-human IgG) were diluted 1:1,000 in blocking buffer and incubated for 1 h at RT in the dark. Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen). Last, coverslips were mounted on glass slides using Fluoromount-G (Southern Biotech). Pictures were taken using a lens with a magnification of ×100 by an inverted confocal laser-scanning microscope (Olympus Fluoview 1000). A sequential acquisition mode with an average of 3 frames for each picture (Kalman n = 3) was applied for the 3 channels used.
Western blot analysis.
Western blot analysis of cell lysates was performed as previously described (42). Briefly, cells were lysed 48 h posttransfection by addition of 1% Triton-X100 supplemented with protease inhibitor (Roche). Nuclei were separated by centrifugation, and reducing sample buffer was added to the supernatant. Samples were incubated at 37°C for 15 min prior to separation by SDS-PAGE. After transfer of the separated proteins on a membrane, the membrane was incubated for 1 h in blocking solution (5% milk in 0.05% Tween–PBS). The primary antibody was incubated overnight at 4°C in blocking solution. The following dilutions were used for the antibodies: mouse anti-HA (Sigma), 1:1,000; mouse anti-NS5A 9E10 (46), 1:1.000; mouse anti-NS2 6H6 (47), 1:1,000; mouse anti-E2 AP33, 1:1,000; mouse anti-β-actin (Sigma), 1:1,000. The anti-NS5A 9E10 and anti-NS2 6H6 antibodies were a generous gift from C. M. Rice (Rockefeller University). The anti-E2 AP33 antibody was provided by Genentech and Arvind Partel (University of Glasgow) (48). The secondary horseradish peroxidase (HRP)-conjugated coupled antibody (anti-mouse; Sigma) was incubated for 1 h at room temperature. It was diluted 1:20,000 except after incubation with the anti-HA antibody (1:2,000). After washing in 0.05% Tween–PBS, chemiluminescence was obtained with the ECL Plus Western blotting detection system (GE Healthcare) and measured using a ChemoCam Imager.
Virus titration.
To determine viral titers in collected supernatants, a limiting dilution assay was conducted on Huh-7.5 cells. The 50% tissue culture infectious dose (TCID50) was determined 72 h postinfection as reported earlier (40).
RESULTS
The NPHV p7 amino acid sequence is highly conserved and shows structural features comparable to those of HCV p7.
To examine the degree of p7 amino acid sequence conservation among different NPHV p7 isolates, 15 distinct NPHV p7 sequences were aligned and a consensus sequence was generated (Fig. 1A). The identification of the respective NPHV p7 sequences was based on cleavage site predictions as reported earlier (11). Overall, the NPHV p7 sequences were highly conserved at the amino acid level, with only 11 positions (over the 63 residues) showing some variations (Fig. 1A). Alignment of globally sampled NPHV and HCV p7 nucleotide sequences and subsequent phylogenetic analysis revealed a high level of divergence between HCV and NPHV, with each virus forming a discrete, well-supported clade (Fig. 1B). Of note, sampled NPHV p7 isolates showed a remarkably lower nucleotide variation than HCV p7 isolates derived from genotypes (GT) 1 to 7, which is indicated by the different branch lengths (Fig. 1B). Database searching for proteins related to NPHV p7 using either BLAST (49) or Fasta (50) revealed that p7 from HCV genotype 4f displays the highest similarity to the NPHV consensus sequence, with 33% identical amino acids and 28% and 13% strongly and weakly similar amino acids. Moreover, similar percentages were observed when comparing the NPHV p7 consensus sequence with p7 consensus sequences from representative HCV strains of confirmed genotypes (51, 52) (19% identical, 24% strongly similar, 16% weakly similar, and 30% different residues [Fig. 2A]). With respect to the nonconserved residues, one can distinguish those exhibiting obvious physicochemical differences (colored dark blue in Fig. 2A) from those for which the hydrophobic or hydrophilic character is conserved (colored light blue). Moreover, most of the latter NPHV p7 residues could be observed as minor variants in the p7 amino acid repertoire of HCV reference genotypes (reported in Fig. 2A in reference 27; residue positions 11, 22, 37, 44, 45, and 51). In total, only 20% of residue positions distributed along the sequence appeared to be clearly specific for NPHV and HCV p7, including positions 1, 5, 7, 9, 13, 14, 16, 29, 33, 39, 43, 46, and 47. Altogether, these data indicate that the overall structure of NPHV p7 should be comparable to that of HCV p7. This was also supported by secondary structure analyses and predictions of transmembrane segments, which exhibited similar patterns for NPHV and HCV p7 (data not shown).
FIG 1.

Sequence and phylogenetic analysis of NPHV p7. (A) Amino acid conservation in NPHV p7. (Top) Sequence logo (61) of the 63-amino-acid NPHV p7 ion channel generated from an alignment of 15 globally sampled strains (GenBank accession numbers: KP325401, JQ434002, JQ434003, JQ434004, JQ434005, JQ434006, JQ434007, JQ434008, JX948116; see Materials and Methods for the p7 sequences generated in this study). Nucleotide sequences were translated and aligned using MEGA6 (34). Individual amino acids are color coded according to physiochemical properties, and individual residue frequencies in the population are proportional to the x axis. The global NPHV p7 consensus sequence is positioned directly below, with asterisks (*) indicating complete conservation at the amino acid level in all sampled strains. (B) Phylogenetic comparison of NPHV and HCV p7. NPHV and HCV p7 nucleotide sequences (189 bp) were aligned, and a maximum likelihood phylogenetic tree was generated using MEGA6 (34). NPHV p7 sequences represent all available reported sequences downloaded from GenBank, in addition to sequences generated in this study (the nine GenBank accession numbers listed above for panel A, and sequences for H10 and H14 to H18 determined in this study). The HCV p7 sequences each represent a single isolate from genotypes (GT) 1 to 7 (GenBank accession numbers NC_004102, YP_001469630, NC_009824, NC_009825, NC_009826, NC_009827, EF108306). Branch lengths are proportional to the scale bar and equivalent to genetic distances measured in numbers of nucleotide substitutions per site.
FIG 2.

Homology structure models of NPHV p7. (A) Comparison of the NPHV p7 consensus sequence (see Fig. 1) to the HCV p7 consensus sequence. The degree of amino acid physicochemical conservation at each position is inferred with the similarity index according to ClustalW conventions (asterisk, invariant, red; colon, highly similar, dark pink; dot, similar, pink). Nonconserved but slightly different residues are colored light blue, while very different residues are colored dark blue. Underlined residues correspond to positions exhibiting conserved aromatic residues. Positions 33 and 35 in the central cytosolic loop are boxed. Note that the C-terminal half of p7 exhibits structural features that are characteristic of signal peptides (62), consisting of an N-terminal region (n domain) encompassing 1 to 3 positively charged residues (Arg), a hydrophobic core region (h domain) forming an alpha-helix, and a more polar, flexible region (c domain) containing a signal peptidase cleavage site; residues at positions −1 and −3 relative to the cleavage site are small neutral residues (Ala) and form the recognition site for signal peptidases (63), whereas alpha-helix-destabilizing residues (Pro, Gly) are present at position −6 and/or in the middle of the h domain. (B) Ribbon representations of the three-dimensional homology models of NPHV p7 monomer. Four previously published NMR-based structures were used as the templates for modeling (23–26). Left, p7 monomer structure determined by NMR in 50% TFE and molecular dynamic (MD) simulations (23) (PDB 2K8J); middle left, NMR-based structure of p7 monomer determined in 125 mM DHPC (25) (PDB 2MTS); middle right, Flag-p7 monomer structure determined in 100% methanol (MeOH) (24) (PDB 3ZD0; the Flag tag and C-terminal extension are not shown); right, one subunit of hexamer p7 NMR structure model determined in 200 mM DPC (26) (PDB 2M6X). N and C termini are indicated by “N” and “C,” respectively. Alpha-helical segments are indicated, and residues are color coded as described for panel A. Residues 33 and 35 side chain atoms are represented as van der Waals spheres and illustrate the location of the central cytosolic loop of p7. (C) Three-dimensional homology models 1 and 2 of NPHV p7 hexamer using the HCV p7 NMR/MD model in POPC of Chandler et al. (19) (model 1) and the HCV p7 NMR model in DPC of Ouyang et al. (26) (model 2) as the templates. Two opposing subunits are shown on the left. Hexameric forms of NPHV p7 models are in surface representations from different viewpoints: middle left, side view of the hexamer surface; middle right, sectional view showing the pore interior with its axis symbolized by the dashed line; right, ER lumen view showing the pore. Residues are color coded as described for panels A and B. The thick green lines shown in the left-hand panels represent the polar membrane bilayer interfaces and hydrophobic core (between the middle two lines).
Several NMR structures have been reported for HCV p7 (19, 23–26), allowing us to construct three-dimensional homology structure models for NPHV p7 by using the Swiss-Model automated protein structure homology modeling server (http://www.expasy.org/spdbv/ [36]) and the consensus NPHV p7 sequence as input. The four resulting homology structure models for the monomeric form of NPHV p7 are depicted in Fig. 2B. The three first models exhibited a “hairpin-like” topology consisting of two transmembrane segments that are connected by a hydrophilic, positively charged cytosolic loop containing residues 33 and 35. According to the corresponding hexameric forms of these models (19, 24, 25) and to the typical oligomeric structure of viroporins (53), NPHV p7 subunits would reside side by side as illustrated by model 1 in Fig. 2C. In contrast, the NPHV p7 homology model based on the NMR structure of hexameric p7 reported by Ouyang et al. (26) would exhibit an unusual architecture wherein part of each p7 subunit crosses over to interact with other p7 subunits that are not its neighbors (Fig. 2C, model 2). Nevertheless, these models allowed the positioning of conserved, very similar, similar, different, and very different residues (colored from red to blue, respectively) along the secondary structure elements for each model (Fig. 2B) as well as at the surface of hexamer models and within their ion channel pores (Fig. 2C).
NPHV p7 exerts an ion channel activity in a liposome permeability assay.
As the NPHV p7 structural analyses revealed features similar to those of HCV p7, we next used the liposome permeability assay previously reported for HCV p7 (39) to evaluate its ion channel features. To produce the respective p7 peptides of NPHV (isolate H14) and HCV (isolate JFH-1), a chemical synthesis was performed (see Materials and Methods). The p7 peptides were reconstituted in DMSO and validated using mass spectrometry and SDS-PAGE, before increasing doses of the peptides were incubated with liposomes previously loaded with carboxyfluorescein (CF) at self-quenching concentrations. In this assay, the increase in membrane permeability was measured by the dye release and dequenching. An increase of fluorescence was observed when the NPHV p7 peptide was added to the liposomes, which reached a plateau with a peptide concentration of 10 to 20 μM (Fig. 3A). The HCV JFH-1-derived peptide demonstrated an ion channel activity in a dose-dependent manner with about 2- to 3-fold-higher fluorescent units (FU) than NPHV p7 (Fig. 3B). As the iminosugar derivative N-nonyl-deoxynojirimycin (NN-DNJ) was reported to block the HCV p7 ion channel activity (54), we analyzed the inhibitory effect of NN-DNJ against NPHV p7. As depicted in Fig. 3C, NPHV p7-dependent permeabilization of liposomes could be blocked with 40 μM NN-DNJ in a fashion similar to that observed with HCV p7 (Fig. 3C). Taken together, these data indicate that similar to HCV p7, NPHV p7 is likely able to exert an ion channel function that can be blocked by iminosugar derivatives.
FIG 3.
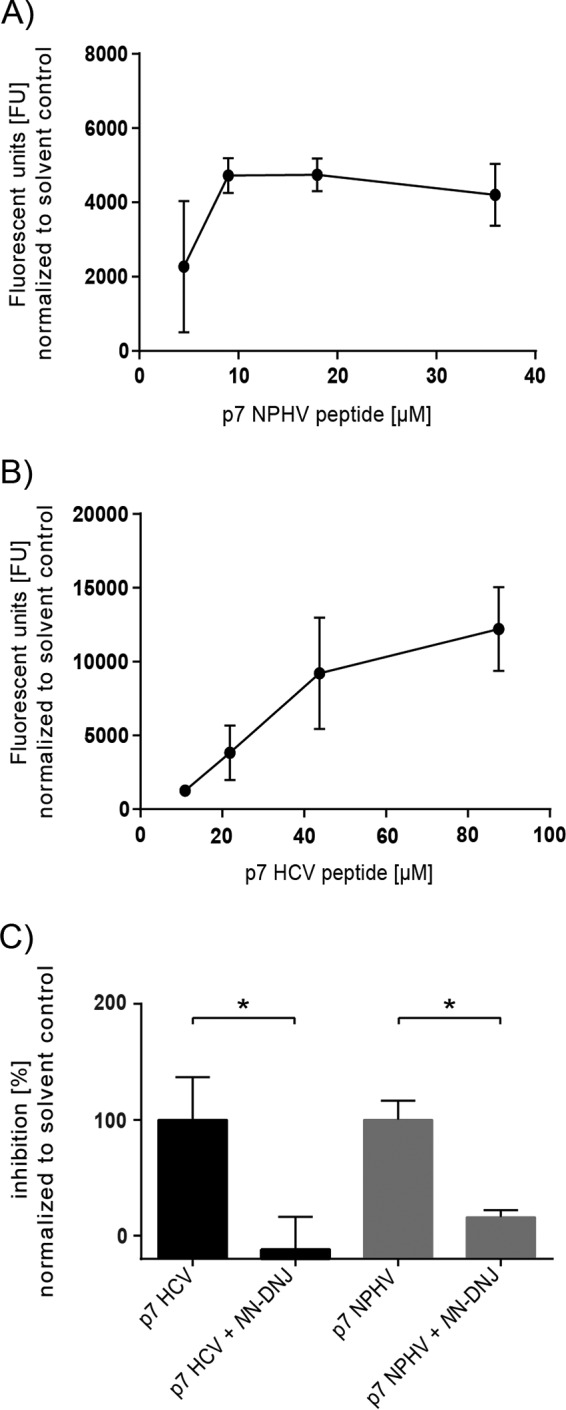
Evaluation of the NPHV p7 ion channel activity in a liposome permeability assay. The HCV and NPHV p7 peptides were chemically synthesized and used for subsequent liposome permeability assays. P7 peptides originating from the NPHV isolate H14 (A) and HCV isolate JFH-1 (B) were reconstituted in DMSO. Different concentrations of peptide were added to 50 μM carboxyfluorescein-loaded liposomes, and the fluorescence units (FU) were determined as an indicator of peptide-induced membrane permeability (activity). Liposomes supplemented with 1% (vol/vol) DMSO were used as solvent control. Three independent experiments in duplicate wells were conducted, and mean values ± standard deviations (SD) are depicted. (C) Inhibition assays with NN-DNJ were conducted. Therefore, liposomes contained 2% (vol/vol) DMSO with or without 40 μM NN-DNJ, these being the respective background levels for drug-free and drug-treated wells. Concentrations of 9 μM NPHV p7 peptide and 44 μM JFH-1 p7 peptide were used. Peptides with or without inhibitor were incubated for 20 min at RT prior to addition to the gain-adjusted plate on ice. Three independent experiments in duplicate wells were conducted. Depicted are the mean values + SD normalized to the respective solvent control. The percentage of inhibition was calculated by normalizing to the DMSO control without inhibitor. Statistical analysis was conducted by Welch's corrected unpaired t test. P values of <0.05 (*) were considered statistically significant.
Cross-species substitutions of the basic loop and the C terminus in p7 lead to production of trans-complemented particles.
To study the capability of NPHV p7 to rescue the production of infectious particles, we made use of a p7-based HCV trans-complementation system (41). This system permits the evaluation of p7 function in virus-producing cells independently of secondary effects on polyprotein processing. The p7 sequence originating from the NPHV isolate H14 was cloned into the first cistron of a bicistronic JFH-1 helper replicon (Fig. 4A). The p7 sequence was located downstream of a signal peptide (sp) sequence encompassed within the last 51 amino acids of the HCV E2 protein derived from the J6 isolate. Additionally and to facilitate p7 detection, another construct containing a sp, a HAHA tag, and a short linker sequence (GGGGSG) connected to NPHV p7 H14 was created. The analogous constructs containing HCV p7 of the isolate J6 or the Gaussia luciferase gene (G-Luc), both previously described (41, 42), were utilized as positive and negative controls, respectively. The second cistron encoded the nonstructural proteins NS3-NS5B from the HCV isolate JFH-1. In vitro transcripts of these constructs were individually transfected into a packaging cell line encoding the remaining viral proteins, core (C), E1E2, and NS2 from the HCV isolates J6 and JFH-1 (Fig. 4A). To confirm the expression of HAHA-tagged p7, we performed Western blot analysis showing that both HAHA-tagged p7 J6 and HAHA-tagged p7 H14 were expressed (Fig. 4B). However, in the lysate of HAHA-tagged p7 H14, additional proteins with a higher molecular weight were detected (Fig. 4B), indicating SDS-resistant oligomeric forms of the HAHA-tagged p7. Next, we analyzed the subcellular localization of HAHA-tagged p7 in fixed cells by indirect fluorescence microscopy using antibodies recognizing the HA tag, HCV NS3, or E2 proteins in order to permit the assessment of colocalization between these polypeptides (Fig. 4C). The two HAHA-tagged p7 proteins, J6 and H14, showed a similar localization in the cytoplasm at ER membranes by colocalizing with E2 and NS3. The rescue of HCV particle production by NPHV p7 was assessed after transfection of the packaging cell line with the different p7 variants and infectivity released into the cell culture supernatant of transfected cells. The J6-derived p7 construct could be rescued with peak titers of 5 × 104 TCID50/ml, while the double HA-tagged genome produced smaller amounts of viral progeny as previously reported (42). In contrast, NPHV p7 (with or without epitope tag) could not replace the HCV p7 function in this trans-complementation setting, suggesting genetic incompatibilities between NPHV p7 and HCV proteins.
FIG 4.

Analysis of NPHV p7 in a trans-complementation setup. (A) Experimental setup. The p7 sequences of the HCV isolate J6 and of the NPHV isolate H14 (see Materials and Methods) were cloned either untagged or linked to a HAHA tag into the first cistron of a bicistronic helper replicon as previously reported (42). The signal peptide-coding sequence (sp) corresponding to the last 51 bp from the E2 J6 sequence was cloned downstream of p7. The insertion of a Gaussia luciferase (G-Luc) served as a negative control. The second cistron encodes the nonstructural proteins NS3-NS5B originating from the HCV isolate JFH-1. These bicistronic helper replicons were individually transfected into a packaging cell line expressing the remaining viral proteins core (C), E1E2, and NS2 from the HCV isolate J6. (B) The expression of HAHA-tagged p7 in cell lysates was confirmed 48 h posttransfection by Western blotting. Additionally, the viral protein NS5A and a cellular protein β-actin were stained. (C) Costaining of NS3 and HAHA-tagged p7 (upper panel) and of E2 and HAHA-tagged p7 (lower panel) was performed 48 h posttransfection on fixed cells. Depicted are single stainings with DAPI, anti-HA, anti-NS3, and anti-E2 in gray as well as colored merge pictures. Here, cell nuclei are shown in blue, anti-HA staining is shown in green, and anti-NS3 and anti-E2 are shown in red. Pictures were taken using a lens with a magnification of ×100. (D) Viral titers in supernatants 24 h, 48 h, and 72 h posttransfection were determined by TCID50. The mean of three independent experiments is depicted as log10 TCID50/ml + standard deviation (SD).
To explore if NPHV p7 and HCV p7 contain virus-specific but potentially also interchangeable (thus functionally conserved) subdomains, we replaced parts of HCV p7 with sequences of NPHV p7. To this end, we constructed 11 distinct chimeras by dividing p7 into the N-terminal transmembrane helix 1 (TM1), basic loop, transmembrane helix 2 (TM2), and C-terminal subdomain (Fig. 5A). All chimeric constructs were N-terminally tagged with a double HA tag connected by a short linker and preceded by a signal peptide sequence. These sequences were cloned into the first cistron of a bicistronic JFH-1 helper replicon, and in vitro transcripts were transfected into Huh-7.5[C][E1E2][NS2]J6 packaging cells analogous to those shown in Fig. 4A. Expression of HAHA-tagged p7 variants was assessed by immunofluorescence analysis, which showed the expression of chimeras 1, 2, 3, 5, and 11 and low or undetectable expression for the remaining constructs (Fig. 5B), indicating an early degradation or a general incompatibility between HCV p7 and NPHV p7 parts. Next, we investigated the capability of these p7 chimeras to trans-complement the production of infectious particles. The p7 chimeras 3 (replacement of the loop, subsequently termed p7J6-loop-H14), 5 (replacement of the C terminus, subsequently termed p7J6-C-ter-H14), and 11 (replacement of the loop and C terminus, subsequently termed p7J6-loop-C-ter-H14) were able to produce infectious particles (Fig. 5C). Viral titers about 1 or 2 orders of magnitude lower than those of p7J6 were observed, with p7J6-C-ter-H14 showing the highest titers. Replacement of the loop decreased the viral titers about 50-fold more drastically and delayed the virus kinetics. To further examine the functionality of these chimeras, the bicistronic replicons encoding p7J6, p7H14, p7J6-loop-H14, p7J6-C-ter-H14, and p7J6-loop-C-ter-H14 N-terminally linked to a signal peptide were cotransfected into Huh-7.5 cells with Jc1Δp7half, an HCV full-length mutant described to completely abrogate viral particle production (27) (Fig. 5D). As shown in Fig. 5E, p7J6, p7J6-loop-H14, p7J6-C-ter-H14, and p7J6-loop-C-ter-H14 were able to rescue infectious particle production (Fig. 5E). In conclusion, cross-species determinants in the basic loop and the C terminus of hepaciviral p7 could be identified by using a p7-based trans-complementation system.
FIG 5.

Construction of HCV/NPHV p7 chimeras in a trans-complementation system. (A) Eleven distinct HCV/NPHV p7 chimeras were constructed by dividing p7 into 5 parts (N terminus, transmembrane helix 1 [TM1], loop, transmembrane helix 2 [TM2], and C terminus). The HCV isolate J6 and the NPHV isolate H14 were utilized as templates, and their respective amino acid sequences are depicted. Amino acid sequences originating from H14 are shown as light gray bars, whereas amino acids originating from J6 are shown as dark gray bars. P7 chimeras were cloned into the bicistronic helper replicon, including a signal peptide, a HAHA tag, and a short linker (see Fig. 4A). Constructed bicistronic helper replicons were transfected into Huh-7.5[C][E1E2][NS2]J6 by electroporation. (B) Cells were fixed 48 h posttransfection, and immunofluorescence analysis by staining for anti-HA and DAPI was performed. Shown are the respective merge pictures with the cell nuclei in blue and the anti-HA staining in green. Pictures were taken using a lens with a magnification of ×100. (D) Jc1 Δp7half was cotransfected into Huh-7.5 cells with in vitro-transcribed bicistronic JFH-1 helper replicons encoding a signal peptide (sp) downstream of p7J6, p7H14, p7J6-loop-H14, p7J6-C-ter-H14, and p7J6-loop-C-terH14 in the first cistron as well as a G-Luc. (E) Viral titers in supernatants 48 h posttransfection were determined by TCID50, and the means from three independent experiments are depicted as log10 TCID50 + SD.
Cross-species determinants of NPHV p7 are important in late steps of the viral life cycle.
After the identification of cross-species determinants in hepaciviral virion production by creating HCV/NPHV p7 chimeras, we next validated their functionality in the context of full-length HCV cell culture-derived particles (HCVcc). Hence, we cloned p7J6-loop-H14, p7J6-C-ter-H14, p7J6-loop-C-ter-H14, and p7H14 into Jc1 and Jc1 HAHA-L-p7 (Fig. 6A). As a positive control, we included the HCV constructs Jc1 and Jc1 HAHA-L-p7J6. These genomes were transfected into Huh-7.5 cells, and expression of epitope-tagged p7 was visualized 48 h later by Western blotting. For the positive control, Jc1 HAHA-L-p7J6 HAHA-tagged p7 could be detected as well as the precursor proteins p7-NS2 and E2-p7-NS2 (Fig. 6B). In the case of HAHA-L-p7H14, cleavage defects were noted with signals at a molecular mass of approximately 24 kDa and 50 kDa, suggesting processing defects due to the insertion of H14 p7 into Jc1 (Fig. 4B). These proteins were also detected for p7J6-loop-H14 and p7J6-loop-C-ter-H14, but here also free p7 was visible. The p7J6-C-ter-H14 chimera showed an HA detection pattern like the parental construct (Fig. 6B), indicating processing defects possibly at the E2/p7 junction or different oligomerization forms. In addition, Western blot analysis to visualize NS2 and E2 in the same cell lysates was conducted and showed that only the precursor p7NS2 and no free NS2 can be detected for Jc1 HAHA-L-p7H14 (Fig. 6C).
FIG 6.

Characterization of HCV/NPHV p7 chimeras in recombinant HCV cell culture viruses. (A) The p7 sequences from the HCV isolate J6 and the NPHV isolate H14 as well as the designated chimeras were cloned into Jc1 or Jc1 HAHA-L-p7 as depicted. (B) In vitro transcripts of the Jc1 HAHA-L-p7 constructs as well as Jc1 wild type were transfected into Huh-7.5 cells, and the expression of HAHA-tagged p7 in cell lysates was visualized by Western blotting 48 h posttransfection. Additionally, the viral protein NS5A and the cellular protein β-actin were stained. (C) Expression of NS2 and E2 in lysates of cells transfected with in vitro transcripts of the Jc1 HAHA-L-p7 constructs was visualized 48 h posttransfection by Western blotting. (D) Viral titers of the Jc1 p7 constructs were determined in supernatants 24 h, 48 h, and 72 h after transfection into Huh-7.5 cells. Shown are log10 TCID50/ml as mean values from three independent experiments + SD. (E) Intra- and extracellular core amounts were measured 48 h posttransfection of the Jc1 p7 constructs into Huh-7.5 cells. The background level of extracellular core amounts was set to the upper value of Jc1 p7H14, since this construct does not produce any infectious particles. Two independent experiments were performed, and the mean values + SD as log10 core femtomoles/liter are shown. (F) The specific infectivity was calculated based on the viral titers shown in panel C and extracellular core release shown in panel D.
Next, the release of infectious viral particles for the Jc1 constructs (Fig. 6D) and Jc1 HAHA-L-p7 (data not shown) constructs was determined by TCID50 at different time points posttransfection. All HCV/NPHV epitope-tagged p7 chimeras led to the production of infectious particles while displaying delayed time kinetics and low titers compared to Jc1 HAHA-L-p7J6 (data not shown). Moreover, as reported previously (42), these titers were around 1 order of magnitude lower than those of the untagged constructs shown in Fig. 6D. Jc1 p7J6-loop-C-ter-H14 showed the lowest production of infectious particles, whereas Jc1 p7H14 was not able to produce infectious particles, which is in concordance with the results in the trans-complementation system. As HCV p7 was reported to be important for the assembly and release step of the viral life cycle (27, 28, 41), we investigated whether this function was also conserved in the context of the p7 chimeric genomes. To this end, we determined extra- and intracellular core amounts 48 h posttransfection (Fig. 6E) and calculated the specific infectivity for each construct (Fig. 6F). The intracellular core amounts were comparable for all constructs, whereas the recombinant chimeric constructs displayed a reduction in extracellular levels of core with Jc1 p7H14 at background levels, in line with the results from the infection assay (Fig. 6E). Therefore, the specific infectivity of Jc1 p7J6-loop-H14 and Jc1 p7J6-C-ter-H14 was comparable to that of Jc1, demonstrating an importance of NPHV p7 in viral assembly and release of infectious particles rather than in virus entry. In the case of Jc1 p7J6-loop-C-ter-H14, the infectivity levels and extracellular core amounts were minimal; therefore, no specific infectivity could be calculated. Taken together, HCV/NPHV p7 chimeras defining cross-species determinants of virion assembly were functional in the context of HCV cell culture particles and were crucial for the late steps of the viral life cycle.
Virion production of HCV/NPHV p7 chimeras can be inhibited by prototypic ion channel blockers.
Inhibitors, including rimantadine and iminosugar derivatives, blocking the ion channel function or oligomerization of HCV p7 have been described (55), but their detailed mechanism of action is not well defined. To facilitate the understanding of p7 inhibitor functions and evaluate the ion channel activity of the HCV/NPHV p7 chimeras in the context of the complete viral life cycle, we next tested the antiviral activity of the prototypic ion channel inhibitors rimantadine (Fig. 7A) and two iminosugars, N-nonyl-deoxygalactonojirimycin (NN-DGJ) (Fig. 7B) and N-nonyl-deoxynojirimycin (NN-DNJ) (Fig. 7C), against Jc1 p7J6-loop-H14 and Jc1 p7J6-C-ter-H14. Inhibitors were added to cells 4 h posttransfection, and viral titers were determined 48 h posttransfection. Jc1 p7J6-loop-H14 was inhibited by rimantadine, NN-DGJ, and NN-DGJ to a level similar to that of the Jc1 wild type (Fig. 7A, B, and C). In contrast, Jc1 p7J6-C-ter-H14 showed a slightly higher resistance profile to rimantadine and NN-DGJ than did the parental Jc1 construct (Fig. 7A and B), indicating a lower binding affinity and less inhibitory activity of these inhibitors when structural changes occur at the C terminus of p7. These results indicate that in the context of HCV/NPHV p7 chimeric viruses, functional ion channels that can be inhibited by specific p7 inhibitors are formed.
FIG 7.
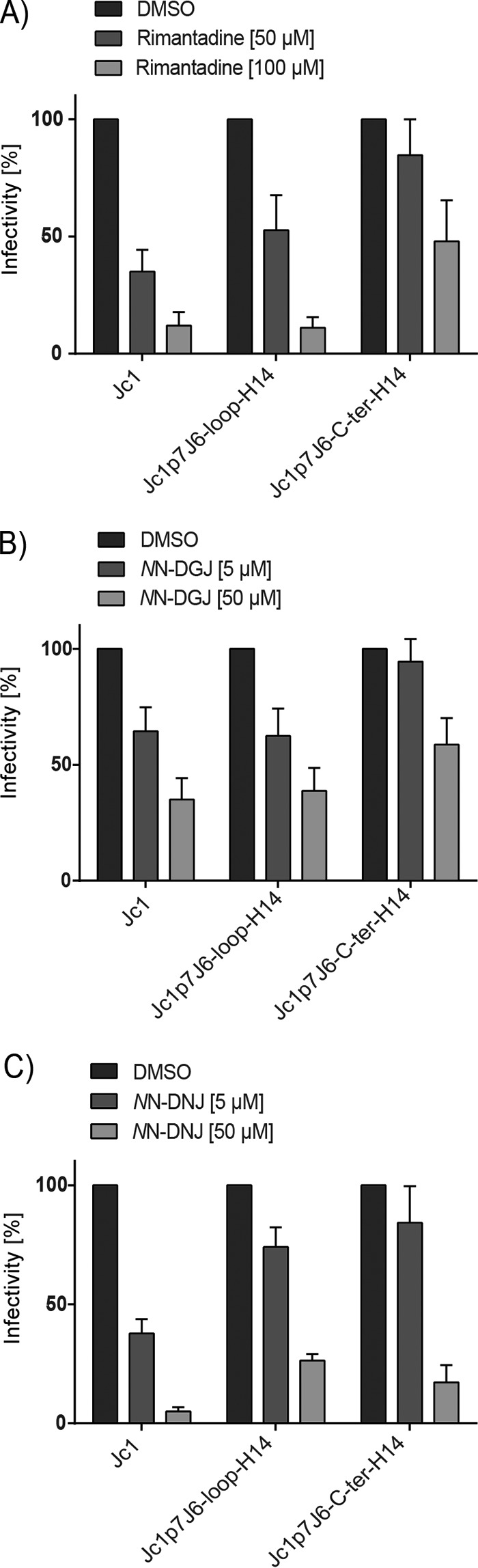
Effects of p7 inhibitors on infectivity of HCV/NPHV p7 chimeras. In vitro transcripts of Jc1, Jc1 p7J6-loop-H14, and Jc1 p7J6-C-ter-H14 were electroporated into Huh-7.5 cells. Four hours posttransfection, the respective drug in two different concentrations or DMSO was added. The percentage of infectivity was calculated by determining the viral titer by TCID50 and normalizing to the DMSO control. Three independent experiments were performed. Test results of the inhibitory effect of rimantadine (50 μM, 100 μM) (A), NN-DGJ (5 μM, 50 μM) (B), and NN-DNJ (5 μM, 50 μM) (C).
DISCUSSION
In this study, we performed a comparison of NPHV and HCV p7 on a structural and functional level. The sequence alignment of reported and novel p7 isolates revealed a high level of conservation among all isolates, especially compared to the variation apparent among HCV isolates. This is in accordance with the overall high conservation of the full NPHV genome between different isolates, among which a diversity of approximately 15% was reported in contrast to a 30% diversity between HCV isolates (10). Amino acid sequence similarities indicate that the overall structure of NPHV p7 is comparable to that of HCV p7, allowing us to construct NPHV p7 homology models using reported three-dimensional NMR-based HCV p7 structures (19, 23–26) as the templates for monomeric and hexameric models. A greater degree of amino acid identity was found in the C-terminal p7 segment of amino acids at positions 48 to 63 (“segment 48–63”), including conservation of the upstream cleavage site of the signal peptidase at the p7-NS2 junction. Despite the amino acid variability in segment 37–47 compared to HCV p7, the overall C-terminal half of p7 NPHV exhibits the characteristic structural features of a signal peptide (Fig. 2A) and thus likely acts as a signal for the reinitialization of translocation of the C-terminal part of p7. Interestingly, most of the different residues in this segment are located at the surface of the hexamer homology models and thus should not disturb ion channeling function of p7 but could potentially play a role in p7 interactions with other viral or cellular partners. Additionally, a high degree of amino acid similarity was found in segment 17–32, which is thought to be the main structural element involved in p7 pore formation and function. An interesting difference is the presence of only one basic residue at position 35 in the putative cytosolic loop of NPHV p7 instead of two fully conserved basic residues at positions 33 and 35 in p7 of all HCV genotypes (17). This suggests that the basic residue at position 33 in HCV might be not essential for p7 functioning. Moreover, the N-terminal segment 1–16 exhibited lower similarity with several different residues located both at the surface of the hexamer models or facing the pore lumen. These features suggest that this relatively poorly homologous segment should not play a critical role in ion channeling but could be important for specific interactions with other viral and/or host-specific cellular factors. Moreover, as observed in HCV p7, this N-terminal segment also might play an essential role in the complex mechanism of E2-p7 processing by signal peptidase (56, 57). Together, all these structural features indicate a possible ion channel function of NPHV p7 similar to HCV p7, which is supported by the results of the liposome permeability assay. For chemically synthesized p7 peptides of NPHV and HCV, an increase of CF release with a concurrent increase of the peptide concentration was observed. However, due to the property of the NPHV p7 peptide to form aggregates at high concentrations, the effect was observed at low peptide concentrations and at an early peak compared to the HCV p7 peptide. The observed CF release could be blocked by addition of the iminosugar NN-DNJ, indicating an ion channel function of NPHV p7. The dibasic motif K33-R35 of HCV p7 was reported earlier to be important to maintain the ion channel activity in liposomes (58). Importantly and in line with the structural analysis of NPHV p7, only the basic residue at position 35 is conserved in NPHV p7 and not the basic residue at position 33, leading to the assumption that specifically residue 35 is essential to preserve p7 function.
We next investigated NPHV p7 determinants specific to NPHV or conserved between HCV and NPHV. Moreover, we analyzed its subcellular localization and its role in the viral life cycle. By using a p7-based trans-complementation approach, which is independent of viral polyprotein processing, we were able to show that NPHV p7 was successfully expressed despite the detection of oligomeric forms of NPHV p7. These oligomeric intermediates have not been observed for HCV p7 so far. Nevertheless, the successful expression of NPHV p7 allowed us to investigate the subcellular localization in infected cells. The highest degree of colocalization of HCV p7 with viral proteins has been described for E2, whereas also colocalization with NS2, NS3, NS5A, and in parts core was reported (42). This is in accordance with our localization analysis showing that epitope-tagged NPHV p7 colocalizes with NS3 and E2 in a pattern similar to that of epitope-tagged HCV p7 in the cytoplasm of the cell. Despite the expression of NPHV p7 and subcellular localization similar to HCV p7, no infectious particles were produced in the trans-complementation system, probably due to genetic incompatibilities of viral proteins. We were able to demonstrate previously that even the replacement of genotype 2a J6-p7 by another HCV isolate Con1 (genotype 1b) fully abrogated HCV particle production and even that replacement by p7 of another genotype 2a isolate (JFH-1) did significantly reduce the release of infectious particles (41). To overcome these cross-species genetic incompatibilities, HCV/NPHV p7 chimeras were constructed demonstrating that the basic cytosolic loop and C terminus of NPHV p7 as well as the combination of the two are interchangeable between NPHV and HCV, thus restoring HCV infectious particle production. For HCV, intergenotypic p7 chimeras were also analyzed for being infectious in vivo when the N-terminal and C-terminal tails of p7 from a genotype 1a virus were maintained but other parts were replaced by genotype 2a sequences (59). Moreover, we observed that the basic loop could be replaced without abolishing particle formation, underlying the importance of the basic residue at position 35 to maintain NPHV p7 functions. Lastly, we tested the generated chimeras as well as the full NPHV p7 sequence in an HCV full-length virus (HCVcc), which revealed that virion production was comparable to that of the trans-complementation system and that the complete NPHV p7 sequence could not compensate for HCV p7 function due to cross-species incompatibilities. In addition, Western blot analysis showed an absence of mature NPHV p7 with the correct molecular weight. However, the molecular weights of the detected proteins coincide with the molecular weights of the proteins detected in the trans-complementation system, where no precursor proteins are produced, indicating an oligomerization of NPHV p7 rather than a cleavage defect. Nevertheless, the novel chimeras were fully functional and were shown to be important for viral assembly and release. Thus, virus-specific determinants of hepaciviral virion assembly are located in the N terminus and transmembrane regions of p7.
Inhibitor experiments with rimantadine and the iminosugar derivatives NN-DGJ and NN-DNJ were conducted because these prototypic ion channel inhibitors were reported to target HCV p7 and reduce particle production (60). These experiments showed that the HCV/NPHV p7 chimera carrying the C terminus of NPHV p7 was more resistant especially to rimantadine and NN-DGJ at high concentrations. The interaction sites of rimantadine with HCV p7 were analyzed previously (24), showing key interacting residues at positions 46, 48, and 52, which are present in the chimeric construct. However, the change of the C terminus of p7 could influence the overall p7 folding, which could compress the rimantadine binding cavity, leading to a less favorable fit for the molecule and resulting in a more-drug-resistant phenotype. The iminosugar derivative NN-DNJ demonstrated an inhibition of all viruses at the highest concentration, which may result not only from the p7 ion channel inhibition but also from the blockage of ER α-glucosidases required for folding and maturation of the HCV glycoproteins (60).
In conclusion, the identification of viruses closely related to HCV allowed for the first time a cross-species comparison of two naturally occurring hepaciviral species. We were able to show that the overall structure of NPHV p7 is highly conserved compared to HCV p7 and that NPHV p7 most likely exhibits an ion channel activity. Molecular analysis revealed a similar subcellular localization of NPHV p7 with an ER-like pattern. Moreover, although NPHV p7 could not fully replace HCV p7 function, the basic loop and C terminus could be replaced, leading to the production of infectious particles. The results implicate a role of NPHV p7 in viral pathogenesis similar to that seen for its human homolog and identify broad hepaciviral protein determinants for virus assembly.
ACKNOWLEDGMENTS
We are grateful to Takaji Wakita for the gift of the JFH1 isolate, to Jens Bukh for the J6CF strain, to Charles Rice for Huh7.5 cells and the 9E10 and 6H6 monoclonal antibodies, to Ralf Bartenschlager for the NS3-specific antibody, to Steven Foung for the E2-specific antibodies, and to Genentech and Arvind Patel for providing the AP33 antibody. We thank Daniel Todt for the statistical analysis and all members of the Institute of Experimental Virology, TWINCORE, for helpful suggestions and discussions.
TWINCORE is a joint venture between the Hannover Medical School (MHH) and the Helmholtz Centre for Infection Research (HZI), Hannover, Germany.
Funding Statement
Stephanie Walter was supported by a stipend from the Helmholtz Centre for Infection Research and by the Hannover Biomedical Research School (HBRS) and the Centre for Infection Biology (ZIB). Eike Steinmann was supported by the DFG (STE 1954/1-1) and by an Intramural Young Investigator Award from the Helmholtz Centre for Infection Research. Abhinav Kumar and Nicole Zitzmann are supported by the Oxford Glycobiology Institute. François Penin was supported by the Mapping project (ANR-11-BINF-003) and the French agency ANRS. Thomas Pietschmann was supported by a grant from the Helmholtz Association (SO-024) and by a grant from the European Research Council (ERC-2011-StG_281473-VIRAFRONT).
REFERENCES
- 1.Kapoor A, Simmonds P, Gerold G, Qaisar N, Jain K, Henriquez JA, Firth C, Hirschberg DL, Rice CM, Shields S, Lipkin WI. 2011. Characterization of a canine homolog of hepatitis C virus. Proc Natl Acad Sci U S A 108:11608–11613. doi: 10.1073/pnas.1101794108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burbelo PD, Dubovi EJ, Simmonds P, Medina JL, Henriquez JA, Mishra N, Wagner J, Tokarz R, Cullen JM, Iadarola MJ, Rice CM, Lipkin WI, Kapoor A. 2012. Serology-enabled discovery of genetically diverse hepaciviruses in a new host. J Virol 86:6171–6178. doi: 10.1128/JVI.00250-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Drexler JF, Corman VM, Muller MA, Lukashev AN, Gmyl A, Coutard B, Adam A, Ritz D, Leijten LM, van Riel D, Kallies R, Klose SM, Gloza-Rausch F, Binger T, Annan A, Adu-Sarkodie Y, Oppong S, Bourgarel M, Rupp D, Hoffmann B, Schlegel M, Kummerer BM, Kruger DH, Schmidt-Chanasit J, Setien AA, Cottontail VM, Hemachudha T, Wacharapluesadee S, Osterrieder K, Bartenschlager R, Matthee S, Beer M, Kuiken T, Reusken C, Leroy EM, Ulrich RG, Drosten C. 2013. Evidence for novel hepaciviruses in rodents. PLoS Pathog 9:e1003438. doi: 10.1371/journal.ppat.1003438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quan PL, Firth C, Conte JM, Williams SH, Zambrana-Torrelio CM, Anthony SJ, Ellison JA, Gilbert AT, Kuzmin IV, Niezgoda M, Osinubi MO, Recuenco S, Markotter W, Breiman RF, Kalemba L, Malekani J, Lindblade KA, Rostal MK, Ojeda-Flores R, Suzan G, Davis LB, Blau DM, Ogunkoya AB, Alvarez Castillo DA, Moran D, Ngam S, Akaibe D, Agwanda B, Briese T, Epstein JH, Daszak P, Rupprecht CE, Holmes EC, Lipkin WI. 2013. Bats are a major natural reservoir for hepaciviruses and pegiviruses. Proc Natl Acad Sci U S A 110:8194–8199. doi: 10.1073/pnas.1303037110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kapoor A, Simmonds P, Scheel TK, Hjelle B, Cullen JM, Burbelo PD, Chauhan LV, Duraisamy R, Sanchez Leon M, Jain K, Vandegrift KJ, Calisher CH, Rice CM, Lipkin WI. 2013. Identification of rodent homologs of hepatitis C virus and pegiviruses. mBio 4:e00216-13. doi: 10.1128/mBio.00216-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lauck M, Sibley SD, Lara J, Purdy MA, Khudyakov Y, Hyeroba D, Tumukunde A, Weny G, Switzer WM, Chapman CA, Hughes AL, Friedrich TC, O'Connor DH, Goldberg TL. 2013. A novel hepacivirus with an unusually long and intrinsically disordered NS5A protein in a wild Old World primate. J Virol 87:8971–8981. doi: 10.1128/JVI.00888-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Firth C, Bhat M, Firth MA, Williams SH, Frye MJ, Simmonds P, Conte JM, Ng J, Garcia J, Bhuva NP, Lee B, Che X, Quan PL, Lipkin WI. 2014. Detection of zoonotic pathogens and characterization of novel viruses carried by commensal Rattus norvegicus in New York City. mBio 5:e01933-14. doi: 10.1128/mBio.01933-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corman VM, Grundhoff A, Baechlein C, Fischer N, Gmyl A, Wollny R, Dei D, Ritz D, Binger T, Adankwah E, Marfo KS, Annison L, Annan A, Adu-Sarkodie Y, Oppong S, Becher P, Drosten C, Drexler JF. 2015. Highly divergent hepaciviruses from African cattle. J Virol 89:5876–5882. doi: 10.1128/JVI.00393-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baechlein C, Fischer N, Grundhoff A, Alawi M, Indenbirken D, Postel A, Baron AL, Offinger J, Becker K, Beineke A, Rehage J, Becher P. 2015. Identification of a novel hepacivirus in domestic cattle from Germany. J Virol 89:7007–7015. doi: 10.1128/JVI.00534-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scheel TK, Simmonds P, Kapoor A. 2015. Surveying the global virome: identification and characterization of HCV-related animal hepaciviruses. Antiviral Res 115:83–93. doi: 10.1016/j.antiviral.2014.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pfaender S, Brown RJ, Pietschmann T, Steinmann E. 2014. Natural reservoirs for homologs of hepatitis C virus. Emerg Microbes Infect 3:e21. doi: 10.1038/emi.2014.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Global Burden of Disease Study Collaborators. 2015. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 386:743–800. doi: 10.1016/S0140-6736(15)60692-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoofnagle JH. 1997. Hepatitis C: the clinical spectrum of disease. Hepatology 26:15S–20S. doi: 10.1002/hep.510260703. [DOI] [PubMed] [Google Scholar]
- 14.Pawlotsky JM. 2014. New hepatitis C therapies: the toolbox, strategies, and challenges. Gastroenterology 146:1176–1192. doi: 10.1053/j.gastro.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 15.Bartenschlager R, Frese M, Pietschmann T. 2004. Novel insights into hepatitis C virus replication and persistence. Adv Virus Res 63:71–180. doi: 10.1016/S0065-3527(04)63002-8. [DOI] [PubMed] [Google Scholar]
- 16.Scott C, Griffin S. 2015. Viroporins: structure, function and potential as antiviral targets. J Gen Virol 96:2000–2027. doi: 10.1099/vir.0.000201. [DOI] [PubMed] [Google Scholar]
- 17.Carrere-Kremer S, Montpellier-Pala C, Cocquerel L, Wychowski C, Penin F, Dubuisson J. 2002. Subcellular localization and topology of the p7 polypeptide of hepatitis C virus. J Virol 76:3720–3730. doi: 10.1128/JVI.76.8.3720-3730.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Isherwood BJ, Patel AH. 2005. Analysis of the processing and transmembrane topology of the E2p7 protein of hepatitis C virus. J Gen Virol 86:667–676. doi: 10.1099/vir.0.80737-0. [DOI] [PubMed] [Google Scholar]
- 19.Chandler DE, Penin F, Schulten K, Chipot C. 2012. The p7 protein of hepatitis C virus forms structurally plastic, minimalist ion channels. PLoS Comput Biol 8:e1002702. doi: 10.1371/journal.pcbi.1002702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luik P, Chew C, Aittoniemi J, Chang J, Wentworth P Jr, Dwek RA, Biggin PC, Venien-Bryan C, Zitzmann N. 2009. The 3-dimensional structure of a hepatitis C virus p7 ion channel by electron microscopy. Proc Natl Acad Sci U S A 106:12712–12716. doi: 10.1073/pnas.0905966106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Griffin SD, Beales LP, Clarke DS, Worsfold O, Evans SD, Jaeger J, Harris MP, Rowlands DJ. 2003. The p7 protein of hepatitis C virus forms an ion channel that is blocked by the antiviral drug, Amantadine. FEBS Lett 535:34–38. doi: 10.1016/S0014-5793(02)03851-6. [DOI] [PubMed] [Google Scholar]
- 22.Clarke D, Griffin S, Beales L, Gelais CS, Burgess S, Harris M, Rowlands D. 2006. Evidence for the formation of a heptameric ion channel complex by the hepatitis C virus p7 protein in vitro. J Biol Chem 281:37057–37068. doi: 10.1074/jbc.M602434200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Montserret R, Saint N, Vanbelle C, Salvay AG, Simorre JP, Ebel C, Sapay N, Renisio JG, Bockmann A, Steinmann E, Pietschmann T, Dubuisson J, Chipot C, Penin F. 2010. NMR structure and ion channel activity of the p7 protein from hepatitis C virus. J Biol Chem 285:31446–31461. doi: 10.1074/jbc.M110.122895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Foster TL, Thompson GS, Kalverda AP, Kankanala J, Bentham M, Wetherill LF, Thompson J, Barker AM, Clarke D, Noerenberg M, Pearson AR, Rowlands DJ, Homans SW, Harris M, Foster R, Griffin S. 2014. Structure-guided design affirms inhibitors of hepatitis C virus p7 as a viable class of antivirals targeting virion release. Hepatology 59:408–422. doi: 10.1002/hep.26685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cook GA, Dawson LA, Tian Y, Opella SJ. 2013. Three-dimensional structure and interaction studies of hepatitis C virus p7 in 1,2-dihexanoyl-sn-glycero-3-phosphocholine by solution nuclear magnetic resonance. Biochemistry 52:5295–5303. doi: 10.1021/bi4006623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.OuYang B, Xie S, Berardi MJ, Zhao X, Dev J, Yu W, Sun B, Chou JJ. 2013. Unusual architecture of the p7 channel from hepatitis C virus. Nature 498:521–525. doi: 10.1038/nature12283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steinmann E, Penin F, Kallis S, Patel AH, Bartenschlager R, Pietschmann T. 2007. Hepatitis C virus p7 protein is crucial for assembly and release of infectious virions. PLoS Pathog 3:e103. doi: 10.1371/journal.ppat.0030103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones CT, Murray CL, Eastman DK, Tassello J, Rice CM. 2007. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J Virol 81:8374–8383. doi: 10.1128/JVI.00690-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Madan V, Bartenschlager R. 2015. Structural and functional properties of the hepatitis C virus p7 viroporin. Viruses 7:4461–4481. doi: 10.3390/v7082826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Steinmann E, Pietschmann T. 2010. Hepatitis C virus p7—a viroporin crucial for virus assembly and an emerging target for antiviral therapy. Viruses 2:2078–2095. doi: 10.3390/v2092078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pfaender S, Cavalleri JM, Walter S, Doerrbecker J, Campana B, Brown RJ, Burbelo PD, Postel A, Hahn K, Anggakusuma Riebesehl N, Baumgartner W, Becher P, Heim MH, Pietschmann T, Feige K, Steinmann E. 2015. Clinical course of infection and viral tissue tropism of hepatitis C virus-like nonprimate hepaciviruses in horses. Hepatology 61:447–459. doi: 10.1002/hep.27440. [DOI] [PubMed] [Google Scholar]
- 32.Tanaka T, Kasai H, Yamashita A, Okuyama-Dobashi K, Yasumoto J, Maekawa S, Enomoto N, Okamoto T, Matsuura Y, Morimatsu M, Manabe N, Ochiai K, Yamashita K, Moriishi K. 2014. Hallmarks of hepatitis C virus in equine hepacivirus. J Virol 88:13352–13366. doi: 10.1128/JVI.02280-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parera M, Martrus G, Franco S, Clotet B, Martinez MA. 2012. Canine hepacivirus NS3 serine protease can cleave the human adaptor proteins MAVS and TRIF. PLoS One 7:e42481. doi: 10.1371/journal.pone.0042481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Cassarino TG, Bertoni M, Bordoli L, Schwede T. 2014. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42:W252–W258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kalita MM, Griffin S, Chou JJ, Fischer WB. 2015. Genotype-specific differences in structural features of hepatitis C virus (HCV) p7 membrane protein. Biochim Biophys Acta 1848:1383–1392. doi: 10.1016/j.bbamem.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Humphrey W, Dalke A, Schulten K. 1996. VMD: visual molecular dynamics. J Mol Graphics 14:33–38, 27–28. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 39.StGelais C, Tuthill TJ, Clarke DS, Rowlands DJ, Harris M, Griffin S. 2007. Inhibition of hepatitis C virus p7 membrane channels in a liposome-based assay system. Antiviral Res 76:48–58. doi: 10.1016/j.antiviral.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steinmann E, Brohm C, Kallis S, Bartenschlager R, Pietschmann T. 2008. Efficient trans-encapsidation of hepatitis C virus RNAs into infectious virus-like particles. J Virol 82:7034–7046. doi: 10.1128/JVI.00118-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brohm C, Steinmann E, Friesland M, Lorenz IC, Patel A, Penin F, Bartenschlager R, Pietschmann T. 2009. Characterization of determinants important for hepatitis C virus p7 function in morphogenesis by using trans-complementation. J Virol 83:11682–11693. doi: 10.1128/JVI.00691-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vieyres G, Brohm C, Friesland M, Gentzsch J, Wolk B, Roingeard P, Steinmann E, Pietschmann T. 2013. Subcellular localization and function of an epitope-tagged p7 viroporin in hepatitis C virus-producing cells. J Virol 87:1664–1678. doi: 10.1128/JVI.02782-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pietschmann T, Kaul A, Koutsoudakis G, Shavinskaya A, Kallis S, Steinmann E, Abid K, Negro F, Dreux M, Cosset FL, Bartenschlager R. 2006. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc Natl Acad Sci U S A 103:7408–7413. doi: 10.1073/pnas.0504877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Appel N, Zayas M, Miller S, Krijnse-Locker J, Schaller T, Friebe P, Kallis S, Engel U, Bartenschlager R. 2008. Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog 4:e1000035. doi: 10.1371/journal.ppat.1000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Keck ZY, Xia J, Wang Y, Wang W, Krey T, Prentoe J, Carlsen T, Li AY, Patel AH, Lemon SM, Bukh J, Rey FA, Foung SK. 2012. Human monoclonal antibodies to a novel cluster of conformational epitopes on HCV E2 with resistance to neutralization escape in a genotype 2a isolate. PLoS Pathog 8:e1002653. doi: 10.1371/journal.ppat.1002653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 47.Dentzer TG, Lorenz IC, Evans MJ, Rice CM. 2009. Determinants of the hepatitis C virus nonstructural protein 2 protease domain required for production of infectious virus. J Virol 83:12702–12713. doi: 10.1128/JVI.01184-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Owsianka A, Tarr AW, Juttla VS, Lavillette D, Bartosch B, Cosset FL, Ball JK, Patel AH. 2005. Monoclonal antibody AP33 defines a broadly neutralizing epitope on the hepatitis C virus E2 envelope glycoprotein. J Virol 79:11095–11104. doi: 10.1128/JVI.79.17.11095-11104.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pearson WR. 1991. Searching protein sequence libraries: comparison of the sensitivity and selectivity of the Smith-Waterman and FASTA algorithms. Genomics 11:635–650. doi: 10.1016/0888-7543(91)90071-L. [DOI] [PubMed] [Google Scholar]
- 51.Smith DB, Bukh J, Kuiken C, Muerhoff AS, Rice CM, Stapleton JT, Simmonds P. 2014. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology 59:318–327. doi: 10.1002/hep.26744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Simmonds P, Bukh J, Combet C, Deleage G, Enomoto N, Feinstone S, Halfon P, Inchauspe G, Kuiken C, Maertens G, Mizokami M, Murphy DG, Okamoto H, Pawlotsky JM, Penin F, Sablon E, Shin IT, Stuyver LJ, Thiel HJ, Viazov S, Weiner AJ, Widell A. 2005. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology 42:962–973. doi: 10.1002/hep.20819. [DOI] [PubMed] [Google Scholar]
- 53.Nieva JL, Madan V, Carrasco L. 2012. Viroporins: structure and biological functions. Nat Rev Microbiol 10:563–574. doi: 10.1038/nrmicro2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pavlovic D, Neville DC, Argaud O, Blumberg B, Dwek RA, Fischer WB, Zitzmann N. 2003. The hepatitis C virus p7 protein forms an ion channel that is inhibited by long-alkyl-chain iminosugar derivatives. Proc Natl Acad Sci U S A 100:6104–6108. doi: 10.1073/pnas.1031527100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Griffin S. 2014. “Too little, too late?” Will inhibitors of the hepatitis C virus p7 ion channel ever be used in the clinic? Future Med Chem 6:1893–1907. doi: 10.4155/fmc.14.121. [DOI] [PubMed] [Google Scholar]
- 56.Carrere-Kremer S, Montpellier C, Lorenzo L, Brulin B, Cocquerel L, Belouzard S, Penin F, Dubuisson J. 2004. Regulation of hepatitis C virus polyprotein processing by signal peptidase involves structural determinants at the p7 sequence junctions. J Biol Chem 279:41384–41392. doi: 10.1074/jbc.M406315200. [DOI] [PubMed] [Google Scholar]
- 57.Scull MA, Schneider WM, Flatley BR, Hayden R, Fung C, Jones CT, van de Belt M, Penin F, Rice CM. 2015. The N-terminal helical region of the hepatitis C virus p7 ion channel protein is critical for infectious virus production. PLoS Pathog 11:e1005297. doi: 10.1371/journal.ppat.1005297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.StGelais C, Foster TL, Verow M, Atkins E, Fishwick CW, Rowlands D, Harris M, Griffin S. 2009. Determinants of hepatitis C virus p7 ion channel function and drug sensitivity identified in vitro. J Virol 83:7970–7981. doi: 10.1128/JVI.00521-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sakai A, Claire MS, Faulk K, Govindarajan S, Emerson SU, Purcell RH, Bukh J. 2003. The p7 polypeptide of hepatitis C virus is critical for infectivity and contains functionally important genotype-specific sequences. Proc Natl Acad Sci U S A 100:11646–11651. doi: 10.1073/pnas.1834545100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Steinmann E, Whitfield T, Kallis S, Dwek RA, Zitzmann N, Pietschmann T, Bartenschlager R. 2007. Antiviral effects of amantadine and iminosugar derivatives against hepatitis C virus. Hepatology 46:330–338. [DOI] [PubMed] [Google Scholar]
- 61.Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res 14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.von Heijne G. 1990. The signal peptide. J Membr Biol 115:195–201. doi: 10.1007/BF01868635. [DOI] [PubMed] [Google Scholar]
- 63.von Heijne G. 1998. Life and death of a signal peptide. Nature 396:111–113. doi: 10.1038/24036. [DOI] [PubMed] [Google Scholar]