

ABSTRACT
The human papillomavirus (HPV) life cycle is tightly linked to differentiation of the infected epithelial cell, suggesting a sophisticated interplay between host cell metabolism and virus replication. Previously, we demonstrated in differentiated keratinocytes in vitro and in vivo that HPV type 16 (HPV16) infection caused increased levels of the cellular SR splicing factors (SRSFs) SRSF1 (ASF/SF2), SRSF2 (SC35), and SRSF3 (SRp20). Moreover, the viral E2 transcription and replication factor that is expressed at high levels in differentiating keratinocytes could bind and control activity of the SRSF1 gene promoter. Here, we show that the E2 proteins of HPV16 and HPV31 control the expression of SRSFs 1, 2, and 3 in a differentiation-dependent manner. E2 has the greatest transactivation effect on expression of SRSF3. Small interfering RNA depletion experiments in two different models of the HPV16 life cycle (W12E and NIKS16) and one model of the HPV31 life cycle (CIN612-9E) revealed that only SRSF3 contributed significantly to regulation of late events in the virus life cycle. Increased levels of SRSF3 are required for L1 mRNA and capsid protein expression. Capsid protein expression was regulated specifically by SRSF3 and appeared independent of other SRSFs. Taken together, these data suggest a significant role of the HPV E2 protein in regulating late events in the HPV life cycle through transcriptional regulation of SRSF3 expression.
IMPORTANCE Human papillomavirus replication is accomplished in concert with differentiation of the infected epithelium. Virus capsid protein expression is confined to the upper epithelial layers so as to avoid immune detection. In this study, we demonstrate that the viral E2 transcription factor activates the promoter of the cellular SRSF3 RNA processing factor. SRSF3 is required for expression of the E4^L1 mRNA and so controls expression of the HPV L1 capsid protein. Thus, we reveal a new dimension of virus-host interaction crucial for production of infectious virus. SRSF proteins are known drug targets. Therefore, this study provides an excellent basis for developing strategies to regulate capsid protein production in the infected epithelium and the production of new virions.
INTRODUCTION
Human papillomaviruses (HPVs) infect epithelia causing benign lesions or warts. For the so-called “high-risk” (HR) HPVs such as HPV type 16 (HPV16), the most prevalent HPV, persistent infection causes cervical and other anogenital lesions and head and neck lesions that may progress to cancer (1). Although prophylactic vaccines against the most prevalent HR-HPVs are available, therapies are still required to treat infected individuals who are not vaccinated. A more comprehensive understanding of the HR-HPV replication cycle could help in the development of novel therapeutic approaches.
The HPV16 life cycle is tightly linked to differentiation of the epithelium the virus infects (2). Initial infection is within basal epithelial cells where the episomal viral genome is maintained at around 50 to 100 nuclear copies (3). Differentiation of infected epithelial cells leads to activation of early and then late gene expression (4). The viral replication and transcription factor, E2, is expressed at greatest levels in the middle to upper layers of the epithelium (5, 6) where, together with the viral DNA helicase, E1, it facilitates vegetative viral DNA replication, leading to production of thousands of viral genome copies (7). The viral late proteins, including the capsid proteins L1 and L2, are synthesized in the uppermost granular layer of the epithelium to encapsidate the newly replicated genomes (4). This spatial restriction of production of the highly immunogenic capsid proteins is important since it avoids triggering an immune response as a result of low immune surveillance in the upper epithelial layers.
Expression of virus capsid proteins is known to be controlled not only at the level of transcription initiation but also at various posttranscriptional levels, including polyadenylation, alternative splicing, nuclear export, mRNA stability, and translation (2, 8–12). Notably, at least 13 mRNAs are produced late in the virus life cycle that contain open reading frames encoding the capsid proteins (10). Seven mRNAs contain the L1 but not the L2 open reading frame, and six mRNAs contain both L1 and L2 open reading frames. Alternative splicing regulates the proportions of the various late mRNAs encoding the capsid proteins (12). In particular, viral capsid protein L1 is thought to be encoded by an E4^L1 mRNA produced by splicing from a splice donor site at the end of the E4 open reading frame to the splice acceptor site at the start of the L1 coding region, while the L2 capsid protein is encoded by a readthrough L2L1 RNA (10).
Constitutive splicing is the process whereby introns are removed from pre-mRNAs and exons are spliced together to form a protein-coding mRNA. Alternative splicing is a mechanism used by mammalian and viral genomes to maximize coding potential (13, 14). A gene is transcribed to give a single primary transcript, but from this precursor RNA (pre-mRNA) different mature mRNA isoforms are produced by differential inclusion or exclusion of exons and introns. Each alternatively spliced mRNA isoform can encode a different protein. Alternative splicing is directly regulated by the following two classes of proteins, SR proteins (serine-arginine-rich splicing factors [SRSFs]) and heterogenous ribonucleoproteins (hnRNPs) (15). SR proteins can bind to exonic sequence enhancers (ESEs) to stimulate recognition of adjacent splice sites by the splicing machinery, while hnRNPs recognize exonic splicing silencers (ESSs) to repress splice site utilization (16). Therefore, the combinations of cis-acting ESEs and ESSs in open reading frames, together with the relative concentrations of trans-acting SR and hnRNP proteins that can access these sites, determine the ultimate mRNA isoforms produced from a single gene.
Aside from splicing, SR proteins can potentially regulate other processes that control protein production from an mRNA, including transcription elongation, polyadenylation, nonsense-mediated decay, nuclear export mRNA stability, and translation (17). Nine classical SR proteins have been described: SRSF1 to SRSF9. Previous data have demonstrated the importance of a number of these proteins in production of HPV mRNAs (18). SRSF1 (SF2/ASF), SRSF3 (SRp20), and SRSF9 (SRp30c) have been shown to bind viral mRNAs in the E4 open reading frame (19–22). SRSF2 (SC35) and SRSF3 have been shown to control production of the viral E6 and E7 oncoprotein mRNAs (19, 23). Previous studies that have examined SR protein regulation of viral RNA splicing in tumor cells transiently expressing subgenomic reporter constructs have demonstrated that SRSF1 controls expression of early and late viral mRNAs (20, 22), while SRSF3 can regulate BPV1 and HPV16 early and late gene expression (19). SRSF9 can also control HPV16 late gene expression (21).
Previously, we demonstrated in vitro and in vivo that HPV16 infection upregulated the expression of a specific subset of SR proteins in differentiating epithelial cells, SRSF1, SRSF2, and SRSF3, (24). Subsequently, we showed that the viral transcription factor E2 activated the SRSF1 promoter via its transactivation domain (25). We demonstrate here that HPV16 E2 also specifically transactivates the promoters of SRSF2 and SRSF3. We have extended the work to show that HPV31 E2 can also transcriptionally control SR protein expression in an epithelial differentiation-stage specific manner. Small interfering RNA (siRNA) depletion experiments were used to determine which HPV-regulated SR protein(s) was important for HPV16 late mRNA and capsid protein production. The data revealed that SRSF3 depletion resulted in significantly reduced levels of L1 protein, suggesting that E2-regulated SRSF3 is required for capsid protein expression. SRSF3 was required for expression of the viral E4^L1 spliced mRNA but appeared to repress expression of the L2L1 unspliced readthrough RNA. Taken together, these data indicate that the E2 protein links the viral replication cycle to epithelial differentiation via SRSF3, a key cellular regulator of HR-HPV gene expression.
MATERIALS AND METHODS
Cell lines.
W12E cells are nontumor cervical epithelial cells (clone 20863 [26]) that, if maintained at low passage (<17), contain 50 to 100 nuclear episomal copies of the HPV16 genome. NIKS16 cells are normal immortalized keratinocytes (NIKS) stably transfected with the HPV16 genome. Clone 2L maintains episomal HPV16 genomes (27). CIN612-9E cells are cervical keratinocytes containing episomal HPV31 genomes (28). All three lines form tissues in raft culture that mimic a low-grade cervical lesion. W12E, NIKS, NIKS16, and CIN612-9E cells were cultured in E-medium and differentiated as described (2 × 105 cells per 10-cm plate) on mitomycin C-treated J2 3T3 mouse fibroblasts (10, 26). Cells were differentiated by growing to high density in the presence of 1.88 mM Ca2+ as previously described (26). The 3T3 cells were grown in Dulbecco modified Eagle medium (DMEM) with 10% donor calf serum. Prior to harvesting for RNA or protein preparation, 3T3 cells were removed by trypsinization and cells layers washed twice with phosphate-buffered saline (PBS). U2OS osteosarcoma cells and U2OS clones stably expressing HPV16 E2 (U2OSA4 and U2OSB1) were cultured in DMEM with 10% fetal calf serum. All cells were maintained under humidified 5% CO2 and 95% air at 37°C.
Cell transfection and siRNA depletion.
Cells were seeded at 2 × 105 per 10-cm dish and grown for 4 days on mitomycin C-treated J2 3T3 mouse fibroblasts as described above. At day 4, fibroblasts were removed by brief trypsinization. Keratinocytes were washed twice with PBS and transfected with plasmids using Lipofectamine 3000 according to the manufacturer's protocol. At 8 h after transfection, freshly prepared mitomycin C-treated J2 3T3 mouse fibroblasts were added back to the keratinocyte cultures in fresh E-medium. After 72 h, the cells were harvested into 400 μl of protein loading buffer (125 mM Tris [pH 6.8], 4% sodium dodecyl sulfate [SDS], 20% glycerol, 10% mercaptoethanol, 0.006% bromophenol blue, and fresh protein inhibitor cocktail [Roche, United Kingdom]). Lysates were passed through a 21-gauge needle 15 times and then sonicated in a Sonibath for 3 × 30 s. Cells were seeded at 2 × 105 per well in a six-well plate 24 h prior to transfection in antibiotic-free medium. siRNA (10 nM) and Lipofectamine RNAiMAX (Invitrogen) were diluted in Opti-MEM serum-free medium (Invitrogen). Cells were transfected for 48 h according to the manufacturer's protocol. siGENOME SMART pools (consisting of four siRNAs designed to minimize off-target effects) specific for each of the SRSFs tested were purchased from Dharmacon. Transfection efficiencies as calculated by cotransfection with siGLO (Dharmacon) or a green fluorescent protein (GFP) expression plasmid were between 70 and 80%.
Cloning the SRSF3 expression vector.
pEGFPSRp20 was a gift from R. Sandri-Goldin. The insert fragment from pEGFPSRp20 was cleaved out using BamHI/EcoRI restriction digestion. The fragment was then inserted using the same sites into pcDNA3.1 to give plasmid pcDNA3.1SRSF3.
Cloning the SRSF promoters.
SRSF promoter regions were amplified from HeLa cell DNA. For SRSF1pr, the forward primer was 5′-GATCCTCGAGGTTACGGTTCTCACATCCATTTTGC-3′, and the reverse primer was 5′-GTGCAAGCTTCTCCCGCGGCCCCTCCAAAATG-3′ (amplifying nucleotides [nt] −886 to +143 relative to the transcription initiation site at +1). For SRSF2pr, the forward primer was 5′-GGGTGGTACCGTCAGCTCTCCTCGGGGCGAAG-3′, and the reverse primer was 5′-GTACAAGCTTTCTCAGGCAGTTGCCTTCCGCG-3′ (amplifying nt −985 to +96 relative to the transcription initiation site at +1). For SRSF3pr, the forward primer was 5′-GATCGGTACCGCGGCTCTGTCTTCGTAAGGG-3′, and the reverse primer was 5′-GTGCAAGCTTCTCTCACTCACCCGGCGTCC-3′ (amplifying nt −720 to +81 relative to the transcription initiation site at +1). For SRSF7pr, the forward primer was 5′-CATCCTCGAGACCAACTAGGCCTGCTTTCC-3′, and the reverse primer was 5′-GTGCAAGCTTAAACAGCCAAGAAACGACGC-3′ (amplifying nt −1036 to +69) relative to the transcription initiation site at + 1). Nucleotides in boldface italics indicate the XhoI or HindIII restriction enzyme sites that were used for cloning. A 4-bp overhang was added to the end of each primer sequence to facilitate restriction enzyme cleavage. PCR was performed with high-fidelity Platinum Taq polymerase (Invitrogen). Single amplicons were obtained with the correct theoretical length. PCR products were ligated into pGL3 basic vector (Promega) and transformed into supercompetent Escherichia coli DH5α (Invitrogen). Insert sequences of clones obtained were confirmed by DNA sequencing.
Transcription assays.
U2OS and U2OS clones A4 and B1 cells that stably express HPV16-E2 (29) were seeded in 10-mm-well plates at 105 cells per well without antibiotics 24 h prior to transfection. Transfections were carried out using Lipofectamine 2000 (Invitrogen). An EGFP expression vector (pMAXEGFP) was used as a transfection efficiency control using. Protein was extracted 48 to 72 h posttransfection as follows. Cells were washed twice in ice-cold PBS after the removal of the culture medium. NP-40 lysis buffer (0.5% NP-40, 150 mM NaCl, 50 mM Tris HCl [pH 8]) with protease (Roche Diagnostics) and phosphatase (Roche Diagnostics) inhibitors was added to the cells, and the cells scraped into the buffer on ice. The lysed cells were transferred to a 1.5-ml Eppendorf tube and incubated on ice for 30 min with periodic vortexing. The extracts were centrifuged at 10,000 × g for 10 min, and the supernatant was subsequently stored at −80°C. A luciferase assay system (Promega) and a GloMax–Multi detection system luminometer were used to detect luciferase activity according to the manufacturer's instructions. Samples were normalized by protein concentration. At least three independent experiments were performed.
Cloning the HPV31 genome E2:I73L mutant.
The I73L mutation in the HPV31 genome was created using the QuikChange XL site-directed mutagenesis kit (Agilent) using the plasmid pLit-HPV31 as the template and the primers 5′-GCCAAAGCCTTACAAGCTcTTGAACTACAAATGATGTTGG-3′ and 5′-CCAACATCATTTGTAGTTCAAgAGCTTGTAAGGCTTTGGC-3′ according to the manufacturer's directions. Primary human foreskin keratinocytes were transfected with recircularized HPV31 genomes and expanded after drug selection as previously described (30).
Western blotting.
Cells were scraped into protein-loading buffer (125 mM Tris [pH 6.8], 4% SDS, 20% glycerol, 10% mercaptoethanol, and 0.006% bromophenol blue with fresh protein inhibitor cocktail [Roche, United Kingdom]). Lysates were passed through a 21-gauge needle 15 times and then sonicated in a Sonibath for 3 × 30 s. Subsequently, 10 μg of protein (or 5, 10, or 20 μg where indicated) was resolved by polyacrylamide gel electrophoresis (PAGE) using Novex precast 4 to 12% gradient gels (Invitrogen) and then transferred to nitrocellulose membrane using an i-Blotter (Invitrogen). The membrane was preincubated for 1 h at room temperature with 5% dried milk powder in PBS–0.1% Tween (PBS-T), before overnight incubation at 4°C with diluted primary antibody in PBS-T–1% dried milk powder. The following antibodies were used: SRSF1 (clone 96; Invitrogen, catalog no. 32-4500) at 1:1,000, SRSF2 (BD Pharmingen, catalog no. 556363) at 1:250, SRSF3 (clone 7B4; Invitrogen, catalog no. 33-4200) at 1:250, SRSF7 (clone 98, a gift from James Stevenin, IGBMC, Strasbourg, France) at 1:100, GAPDH (glyceraldehyde-3-phosphate dehydrogenase; clone 6CS; Biodesign International, catalog no. H86504M) at 1:5,000, Involucrin (Sigma, catalog no. 19018) at 1:1,000, HPV16 E2 (clone TVG261; Abcam, catalog no. ab17185) at 1:250, and HPV L1 (clone K1H8; Dako, catalog no. M3528). Mab104 (used neat) detects a phospho-epitope on SRSFs 1 to 6 (an ATCC hybridoma supernatant). Because SRSFs1 and 2 have similar apparent molecular masses on SDS-PAGE, as well as probing with Mab104, we also probed blots with specific monoclonal antibodies against these SR proteins as above. Horseradish peroxidase-conjugated secondary antibodies (Pierce-ECL) were diluted 1:2,000 in PBS-T and incubated for 1 h. Blots were developed using Pierce enhanced chemiluminescence (ECL) kit and exposed to Kodak X-Omat film. Blots were quantified by scanning at 300 dpi and image analysis using ImageJ.
Immunohistochemistry.
Archival paraffin-embedded cervical biopsy samples were obtained with ethical permission (Glasgow Royal Infirmary, permit RN04PC003). Diagnosis was made by a gynecological pathologist. HPV presence was confirmed by PCR. Immunohistochemistry was carried out by the University of Glasgow Veterinary Diagnostic Services. SRSF3 antibody (clone 7B4; Invitrogen, catalog no. 33-4200) was used at a dilution of 1:100.
RNA extraction.
Cells were scraped into TRIzol reagent (Invitrogen) and total RNA was extracted according to the manufacturer's protocol. Polyadenylated RNA was isolated using an oligo(dT)-based mRNA extraction kit (Oligotex; Qiagen) according to the manufacturer's instructions. DNA was removed from all RNAs using the Promega RQ1 DNase kit according to the manufacturer's instructions. DNase-treated RNA was reverse transcribed using the Superscript III kit (Invitrogen) according to the protocol provided.
PCR.
cDNA was amplified using 200 nM primers, 200 μM dNTPs, 1.5 mM MgCl2 and 2 units Taq polymerase (Invitrogen). The primers for E4^L1 amplification were E4 forward (nt 3518 to 3540; 5′-GTTGTTGCACAGAGACTCAGTGG-3′) matched with L1 reverse (nt 6918 to 6997; 5′-GACAAGCAATTGCCTGGGTTAC-3′). The primers for L2L1 amplification were L2 forward (nt 5465 to 5475; 5-GTATCAGGTCCTGATATACCC-3′) matched with L1 reverse B (nt 5690 to 5669; 5′-TACTGGGACAGGAGGCAAGTAG-3′), GAPDH forward (5′-TCCACCACCCTGTTGCTGTA-3′), and GAPDH reverse (5′-ACCACAGTCCATGCCATCAC-3′). PCR products were separated on a 6% acrylamide gel and poststained with ethidium bromide.
qPCR.
cDNA was amplified using an Applied Biosystems 7500 qPCR machine. The Stratagene Brilliant qPCR Mastermix was used for quantitative PCRS (qPCRs). cDNA (100 ng) was amplified in each reaction in triplicate, and three different experiments were performed. Reaction mixtures (25 μl) contained 1× Mastermix (Stratagene), 900 nM concentrations of primers, 100 nM concentrations of probe, and 300 nM concentrations of reference dye (Stratagene) as follows: E4 forward primer (nt 3594 to 3615; 5′-CTGTAATAGTAACACTACACCC-3′), E4^L1 probe (nt 3620 to 3631 and nt 5637 to 5656; 5′-TACATTTAAAAGTGTCTCTTTGGCTGCCTAG-3′), L2 forward primer (nt 5547 to 5569; 5′-CAATTATTGCTGATGCAGGTGAC-3′), L2L1 probe (nt 5600 to 5622; 5′-CGAAAACGACGTAAACGTTTACC-3′), and L1 reverse (nt 5690 to 5669; 5′-TACTGGGACAGGAGGCAAGTAG-3′). The amplification protocol was 95°C for 15 s, followed by 50°C for 60 s for 40 cycles. Expression was quantified by determining the ΔΔCT relative to the GAPDH values.
RESULTS
HPV E2 regulates the promoters of the genes encoding SRSF1, -2, and -3.
Using cell line models of epithelial differentiation and HPV infection and backed up by data from virus-infected patient tissue, we and others demonstrated that SRSF1 (SF2/ASF), SRSF2 (SC35), and SRSF3 (SRp20) are specifically upregulated in HPV-infected, differentiated epithelial cells (24, 25, 31, 32). For SRSF1, we showed previously that HPV16 E2 controlled the promoter of the SRSF1 gene by interacting within a region from nt −565 to nt −363 with respect to the transcription initiation site (this was originally cited as nt −689 to nt −482 in our previous paper [25] according to the numbering of a previously sequenced version of the SRSF1 promoter). Transcription activation was through the E2 transactivation domain (25). E2 was only able to transactivate the SRSF1 promoter in the A4 clone of the osteosarcoma cell line U2OS that stably expresses low E2 levels (Fig. 1A) (25, 29). We proposed previously that E2 may begin to transrepress the SRSF1 promoter at higher concentrations, as it has been shown to do for the BPV4 long control region promoter (25). To examine whether E2 also controlled expression of the genes encoding the other two SR proteins, SRSF2 and SRSF3, both of which are upregulated in HPV-infected cells, we cloned their gene promoters into luciferase expression (pGL3-luciferase) vectors for transcription analysis. The SRSF1 promoter (previously assayed in a chloramphenicol acetyltransferase [CAT] construct) was cloned in the same vector in order to use as a known E2-regulated control. The promoter for SRSF7 (9G8) was also cloned and analyzed in order to compare an SRSF family member that we have shown is not upregulated during differentiation of HPV-infected cervical epithelial cells (24). A non-SR protein promoter control, the hTERT promoter whose expression is downregulated by HPV E2 (33), and a pGL3 empty vector with no promoter sequence was also used. U2OS subclones A4 and B1, stably express different amounts of HPV16 E2 protein: A4 expresses low levels, and B1 expresses high levels (Fig. 1A) (25, 29). Transient transfections of the different luciferase expression vectors were performed in U2OS, U2OSA4, and U2OSB1 cells. An EGFP expression vector was cotransfected in each transfection experiment in order to normalize transfection efficiency. Figure 1B shows quantification of luciferase expression from the different promoters indicated on the x axis. Control transfections of the promoterless pGL3 vector showed very low luciferase activity (pGL-3 empty). hTERT promoter activity was low in all the experiments. This is due to U2OS cells displaying low telomerase activity (34). Despite the low levels of reporter gene expression some decrease was detected in the U2OSB1 cell line that expresses higher levels of E2 as expected (33). Similar to what we reported previously, the SRSF1 promoter was transactivated by E2 protein in the U2OSA4 cell clone, which expresses low levels of HPV16 E2. However, the higher levels of E2 expressed by the U2OSB1 cell clone did not upregulate the SRSF1 promoter, confirming our previous data (25). The SRSF2 promoter also showed a small but statistically significant upregulation by HPV16 E2 protein. Under the control of the SRSF2 promoter, luciferase activity was increased 1.35-fold in U2OSA4 cells and 1.24-fold in U2OSB1 cells (P < 0.05). However, a greater increase in activity of the SRSF3 promoter than that observed for SRSF1 and SRSF2 in the presence of E2 was observed. The basal activity of the SRSF3 promoter in U2OS cells was lower than either the SRSF1 or 2 promoters and HPV E2 protein gave a greater transactivation response, showing a larger absolute difference in the fold change in luciferase activity. The luciferase activity was increased 2.93-fold in U2OSA4 cells and 2.69-fold in U2OSB1 cells (P < 0.001) (Fig. 1B). In contrast to the repressive effect seen on SRSF1 promoter activity, high levels of E2 expression did not abrogate the transactivation of the SRSF3 promoter. The SRSF7 promoter is not controlled by HPV16 infection (24) and showed no statistically significant change in activity in the presence of E2 demonstrating that the E2-mediated upregulation of SRSF1, -2, and -3 is specific (Fig. 1B).
FIG 1.
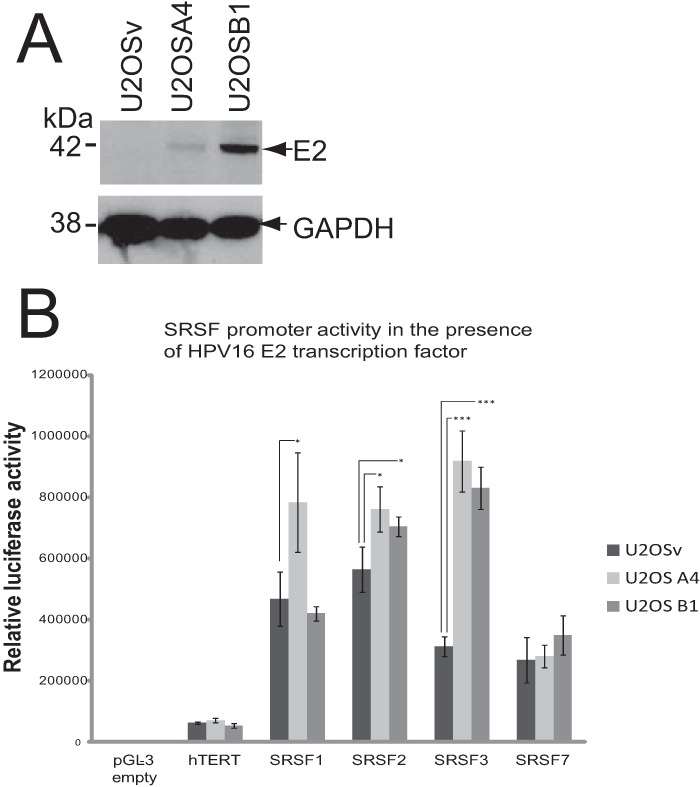
HPV16 E2 transactivates the promoters of SRSFs 1 to 3. (A) Western blot analysis of levels of E2 protein in two clones of U2OS cells (U2OSA4 and U2OSB1) stably transfected with an HPV16 E2 expression vector (29). U2OSv cells were transfected with empty vector. GAPDH is shown as a control for protein loading levels. (B) Luciferase transcription assays reveal E2 transactivation of the SRSF1, -2, and -3 promoters. A graph is shown of the luciferase activity in U2OSv (dark gray bars), U2OSA4 (light gray bars), and U2OSB1 (mid gray bars) cells transiently transfected with luciferase expression vectors under the control of the promoters, shown on the x axis, or the promoterless vector, pGL3, as a negative control. The means and standard deviations from three separate experiments are shown. Asterisks show statistically significant changes (P values) determined using a Student t test (two-tailed). *, P < 0.05; ***, P < 0.001.
Differentiation of HPV31-positive keratinocytes increases SRSF1, -2, and -3 levels.
W12 cells are cervical epithelial cells derived from a patient with an HPV16-positive low grade cervical lesion (35). Cells of the non-tumor subclone W12E (clone 20863), when maintained at low passage (P > 17), contain around 50 to 100 episomal copies of the HPV16 genome (26, 35). W12E cells can be induced to differentiate to keratinocytes that express markers of terminal differentiation (36), viral late mRNAs and late proteins (10) by culturing to high density in the presence of 1.88 mM Ca2+. Previously, we showed that a subset of SRSF proteins, including SRSF1, -2, and -3, were upregulated upon differentiation of W12E cells (24). To determine whether control of SRSF expression during epithelial differentiation was associated with other HR-HPV infections, we examined SR protein levels in HPV31-infected CIN612-9E cervical epithelial cells (28). CIN612-9E cells were established from an HPV31-infected low-grade cervical lesion and contain episomal HPV31 viral genome copies. CIN612-9E cells can also be induced to differentiate in culture using the same protocol as for W12 cells (by culturing to high density in the presence of 1.88 mM Ca2+) to express viral late mRNAs and synthesize virions (37, 38). Monoclonal antibodies against SRSF1, SRSF3, and SRSF7 perform well in Western blotting. However, to detect the remainder of the SR proteins, we used Mab104, which detects a phospho-epitope on most of the classical SR proteins (SRSF1, -2, 4, -5, and -6). Therefore, in Fig. 2A, detection of SRSF1 is shown both with Mab104 and the SRSF1-specific antibody Mab96. Semiquantitative Western blot analysis of protein titrations (5, 10, and 20 μg) showed that the levels of SRSF1, -2, and -3 were higher in differentiated compared to undifferentiated HPV31-positive cells (Fig. 2A), similar to what was found in W12E cells (24). In contrast to W12E cells, CIN612-9E cells expressed SRSF4 (SRp75), whose levels also increased upon differentiation. Levels of SRSF5 (SRp40) and SRSF6 (SRp55) increased slightly upon differentiation. The levels of SRSF7 did not change significantly. Cell differentiation was revealed by detection of increased levels of the epithelial differentiation marker involucrin in the differentiated keratinocytes (Fig. 2A).
FIG 2.

The transactivation domain of HPV31 E2 is required for the control of SRSF1, -2, and -3 and the production of L1 protein. (A) SRSF levels are increased in differentiated HPV31-infected CIN612-9E cells. Semiquantitative Western blot analysis was performed to determine the levels of SR proteins in undifferentiated (U) and differentiated (D) CIN612-9E cells. Protein extracts (5, 10, or 20 μg) were loaded as indicated above the blots. SRSF1, -3, and -7 were detected with specific monoclonal antibodies, as indicated on the right-hand sides of the blots. SRSF1, -2, -4, -5, and -6 were detected with Mab104, which detects phospho-epitopes of all of the classical SR proteins except SRSF9. Involucrin was detected as a control for differentiated epithelial cells. Involucrin is detected in the undifferentiated cells due to ca. 20% of these having undergone differentiation. GAPDH was detected as a protein loading control. A single gel was blotted and probed with antibodies against SRSF1 and involucrin. The same samples were electrophoresed on identical gels for probing with GAPDH/SRSF7 or with Mab104/SRSF3. This experiment was repeated three times, with very similar results from each Western blot. (B) Keratinocytes containing a transactivation-negative E2 point mutant HPV31 genome have reduced levels of SRSF1, -2, and -3. Western blot analysis of SRSF1, -2, and -3 levels in normal human foreskin keratinocytes (NFKs) stably transfected with wild-type E2 (E2wt) or point mutant E2:I73L HPV31 genomes (E2:IL-73). SRSF7 was detected as a control for an SR protein whose levels are not changed upon epithelial differentiation or significantly transactivated by HPV16E2. The levels of L1 protein in the two lines are also shown. The experiment was carried out three times using two different clones of each E2-expressing keratinocyte line. Very similar results were obtained in each experiment. (C) Western blot analysis of levels of E2 protein and differentiation status of NFKs stably transfected with wild-type E2 (E2wt) or point mutant E2:I73L HPV31 genomes (E2:IL-73). As a marker of differentiation, the levels of involucrin are shown in undifferentiated (U) and differentiated (D) CIN6129E keratinocytes for comparison. The experiment was carried out twice using two different clones of each E2-expressing keratinocyte line. Very similar results were obtained in each experiment. (D) Quantification of levels of the SR proteins shown in panel B. The graph shows the means and standard deviations from three separate experiments. Values were calculated relative to the GAPDH levels. Very similar data were obtained with two different clones of each keratinocyte line. (E) Western blot quantification of levels of L1 protein in differentiated NFKs stably maintaining wild-type HPV31 and in NFKs stably maintaining point mutant E2:I73L HPV31 genomes. The graph shows the means and standard deviations from three separate experiments. Values were calculated relative to the GAPDH levels. P values were calculated using a Student t test.
The E2 transactivation domain is required for control of SRSF1, -2, and -3 levels.
The data support the hypothesis that HR-HPV E2, which is expressed at highest levels in differentiating HPV-infected epithelial cells (5, 6), controls expression of SR proteins. To test this directly, we compared levels of SR proteins in normal foreskin keratinocytes (NFKs) stably transfected with wild-type HPV genomes or with genomes containing an inactivating point mutation in the transactivation domain of E2. HPV16 genomes containing such a mutant cannot be maintained episomally in keratinocytes (39). However, keratinocytes containing HPV31 genomes with this mutation are available (mutant E2:I73L) (30). If E2 transactivated the SRSF1-3 promoters, there would be reduced levels of the proteins in these keratinocytes. Western blot quantification indeed revealed decreased levels of SRSF1, -2, and -3 in the keratinocytes expressing the mutant E2 protein compared to keratinocytes expressing wild-type E2 (E2wt), but there was no change in the levels of SRSF7, as expected, since it is not regulated by E2 (Fig. 2B). There were similar viral genome copy numbers in E2wt and E2:IL-73 NFKs (data not shown). Figure 2C shows the levels of E2 protein in each NFK line. Compared to the levels of GAPDH, there were similar levels of E2 protein expressed in E2wt compared to E2:IL-73 cells (Fig. 2C, lanes 3 and 4). The NFK E2wt and E2:IL-73 protein extracts used in Fig. 2B were prepared from differentiated NFK clones because both expressed involucrin, a marker of epithelial differentiation (Fig. 2C, lanes 3 and 4). Both E2wt and E2:IL-73 NFKs clearly expressed more involucrin than undifferentiated HPV31-positive CIN6129E cells (Fig. 2C, lanes 2), and the levels were almost as high as fully differentiated CIN6129E cells (Fig. 2C, lanes 1). The levels of SRSF1 in the E2:I73L-expressing cells were 52% ± 3.2% of the wild type, while the levels of SRSF2 and -3 were 31% ± 6.5% and 29% ± 5.0% of the wild type, respectively (Fig. 2D). These data indicate that E2 transactivates the SRSF gene promoters to control expression of SRSF1, -2, and -3 proteins in infected epithelial cells. E2:I73L mutant HPV31 genomes express ca. 80% less late mRNA than wild-type genomes (30), but whether this leads to reduced L1 protein expression has not been tested. In accordance with the reported change in late mRNA production, Western blotting revealed that there was a reduction of around 75% in L1 protein expression from E2:I73L genomes compared to E2 wild-type genomes (P < 0.01) (Fig. 2E). Our data suggest that HR-HPV E2 upregulates the expression of SR proteins 1, 2, and 3 in infected, differentiated keratinocytes.
SRSF3 controls expression of the HPV16 L1 capsid protein in differentiated keratinocytes.
E2-mediated upregulation of cellular SR proteins 1, 2, and 3 in differentiated infected epithelial cells could indicate that increased levels of these RNA processing proteins are required for completion of the viral replication cycle. Therefore, we next determined which of the differentiation stage-specific controlled SR proteins controlled HPV capsid protein expression during the HPV life cycle in differentiating keratinocytes. For these experiments, we used both the HPV16-infected W12E life cycle model we had used previously (24) and a second model of the HPV16 infectious life cycle (NIKS16) to corroborate our data with W12 cells. NIKS16 cells are normal immortalized foreskin keratinocytes (NIKS) stably transfected with episomal HPV16 genomes (27). If used at low passage, NIKS16 clone 2L stably maintains episomal genomes. Like W12E and CIN612 9E cells, they can differentiate in monolayer culture and display a CIN1-like phenotype in organotypic raft culture (27). Differentiation of W12E, NIKS16, and the parental NIKS cells can be induced by culturing in 1.88 mM Ca2+ to high density (10, 26). At the end of the differentiation protocol between 75 and 85% of NIKS or NIKS16 cells expressed the differentiation marker involucrin similar to that observed with W12E cells (10). Using Mab104 that simultaneously detects SRSFs1, -2, -4, -5, and -6 and antibody 7B4 that detects specifically SRSF3, semiquantitative Western blotting of 5, 10, and 20 μg of HPV-negative NIKS protein extracts revealed very little change in levels of SR proteins upon differentiation, a finding similar to what we observed previously using HPV-negative HaCaT cells (25) (Fig. 3A). However, semiquantitative Western blotting of 5, 10, or 20 μg of HPV16-positive NIKS16 protein extract revealed a pattern of change in SRSF expression between undifferentiated and differentiated NIKS16 cells similar to that observed for W12E (24) and CIN612-9E cells (Fig. 2A). Figure 3B shows that levels of SRSFs1, -2, and -3 were higher in differentiated NIK16 cells. The levels of SRSF4 and -6 also appeared to increase slightly, while the SRSF5 and -7 levels (SRSF7 was detected with specific monoclonal antibody clone 98) did not change. Quantification of changes in SRSF3 levels between undifferentiated and differentiated HPV-negative NIKS cells revealed a reduction of 30% upon differentiation (Fig. 3C). In contrast, SRSF3 levels increased 4-fold upon differentiation of HPV16-positive NIKS16 cells similar to the increase in SRSF3 levels we quantified in W12 cells previously (24) (Fig. 3D). Immunohistochemistry staining of normal cervical epithelium revealed staining in the basal layer cells and some in the suprabasal layers (Fig. 3E). Immunohistochemistry staining of a representative low-grade cervical lesion revealed that whereas more cells in the basal epithelial layers stained positive for SRSF3 compared to cells in the upper layers, the intensity of staining of cells in the middle to upper layers was strong, indicating high levels of SRSF3 expression (Fig. 3F).
FIG 3.

SRSF3 levels increase upon differentiation of HPV16-infected cells (A) SRSF1, -2, and -3 levels do not alter upon differentiation of HPV-negative NIKS cells. Semiquantitative Western blot analysis of levels of SR proteins in undifferentiated (U) and differentiated (D) NIKS cells. Protein extracts (5, 10, or 20 μg) were loaded as indicated above the blots. SRSF3 was detected with the specific monoclonal antibody 7B4. SRSF1, -2, -4, -5, and -6 were detected with Mab104. SRSF7 was not tested in this experiment because its levels did not change upon HPV infection (24). Involucrin was detected as a control for differentiation of epithelial cells. GAPDH was detected as a protein loading control. (B) SRSF1, -2, and -3 levels are increased in differentiated NIKS16 cells. Semiquantitative Western blot analysis of the levels of SR proteins in undifferentiated (U) and differentiated (D) NIKS16 cells was performed. Protein extracts (5, 10, or 20 μg) were loaded as indicated above the blots. SRSF3 and -7 were detected with specific monoclonal antibodies. SRSF1, -2, -4, -5, and -6 were detected with Mab104. Involucrin was detected as a control for differentiated epithelial cells. GAPDH was detected as a protein loading control. The experiments in panels A and B were carried out at least three times, with very similar results obtained each time. (C) Quantification of SRSF3 levels in undifferentiated (U) and differentiated (D) HPV-negative NIKS cells. The graph shows the means and standard deviations from three separate experiments. Values were calculated relative to the GAPDH levels. (D) Quantification of SRSF3 levels in undifferentiated (U) and differentiated (D) NIKS16 cells. The graph shows the means and standard deviations from five separate experiments. Values were calculated relative to the GAPDH levels. (E) Immunohistochemical staining of a representative normal cervical epithelium (10 normal lesions were stained) with an antibody against SRSF3. Note that the strongly stained nuclei are present mainly in the lower epithelial layers. (F) Immunohistochemical staining of a representative low-grade cervical lesion (10 low-grade lesions were stained) with an antibody against SRSF3. Note the strongly stained nuclei in the middle to upper epithelial layers. The picture in panel E is taken at a lower magnification than that in panel F to show that the staining pattern is consistent over a wide area of tissue.
Having demonstrated that NIKS16 cells control expression of SRSFs1, -2, and -3 in a similar manner to W12E and CIN612-9E cells (24), these SR proteins were depleted using siRNA in NIKS16 keratinocytes. SRSFs 5 and 7 were also depleted as controls for SR proteins that are not regulated by E2 or HPV infection. Figure 4A shows that levels of each of the SR proteins were significantly reduced by their respective siRNAs. SRSF1, -2, and -5 were detected with Mab104 (top panel) that detects most of the classical SR proteins. The second panel from the top shows detection of SRSF1 using the specific antibody Mab96. A second panel showing SRSF1 and -2 detected together is included as the third panel down from the top because SRSF2 depletion is not clearly seen in the top panel. The bottom two panels show SRSF3 detected by specific antibody 7B4 and SRSF7 detected by specific antibody clone 98. Levels of L1 protein were assessed as a marker of late events in the virus life cycle in the SRSF-depleted cells by Western blotting with an HPV L1 antibody. L1 was expressed in differentiated (Fig. 4B, lane 2) but not in undifferentiated (Fig. 4B, lane 1) NIKS16 cells, as expected from data from raft cultures of this line (27). Differentiation was assessed by increased expression of the epithelial differentiation marker involucrin (Fig. 4B, compare lanes 1 and 2). There was no significant change in levels of L1 protein in differentiated NIKS16 cells when either SRSF1, -2, -5, or -7 were depleted. However, depletion of SRSF3 (Fig. 4B, lane 5) caused a significant decrease in L1 protein expression. Quantification of three separate experiments showed that loss of SRSF3 caused a 53% ±11% (P < 0.05) decrease in L1 protein expression in differentiated NIKS16 cells (Fig. 4C).
FIG 4.
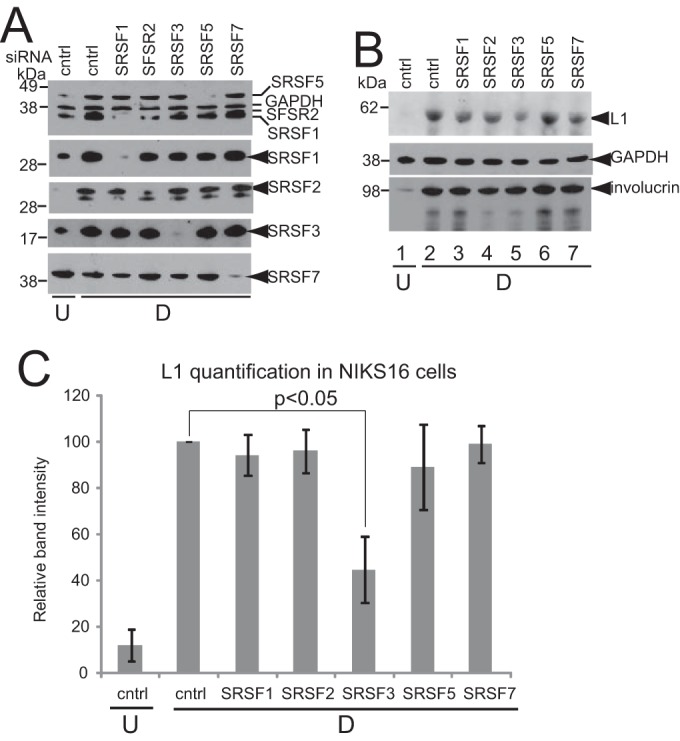
SRSF3 is required for L1 protein expression. (A) Western blot analysis of SR protein depletion for the experiment shown in panel B. SRSF1, -3, and -7 were detected with specific antibodies. SRSF1, -2, and -5 were detected with Mab104. The panel showing SRSF2 depletion was probed with Mab104. Only the area of the blot showing SRSF2 and SRSF1 bands is shown. SRSF2 is the upper band. U, undifferentiated NIKS16 cells; D, differentiated NIKS16 cells; Cntrl, cells transfected with a control siRNA. (B) SRSF3 depletion results in reduced L1 protein levels. Western blot analysis of L1 protein in differentiated keratinocytes (D) with the various SR proteins depleted was performed. Undifferentiated NIKS16 cell extracts (U) were analyzed as a negative control for L1 protein expression; Cntrl, cells transfected with a control siRNA. Involucrin was detected as a control for differentiated epithelial cells. GAPDH was detected as a protein loading control. (C) Quantification of levels of L1 protein in NIKS16 keratinocytes transfected with a control siRNA (cntrl) or siRNAs against each of the SR proteins shown on the x axis. U, undifferentiated keratinocytes; D, differentiated keratinocytes. The graph shows the means and standard deviations from three separate experiments. Values were calculated relative to the GAPDH levels. The P value was calculated using a Student t test.
Next, we confirmed the SRSF3 regulation of L1 expression in the W12E model of the HPV16 life cycle. SRSF1 was used as a control SR protein since this did not significantly affect the levels of HPV16 L1 protein in NIKS16 cells (Fig. 4B and C). SRSF1 and SRSF3 were successfully depleted using specific siRNAs in undifferentiated (W12U) and differentiated (W12D) W12E cells (Fig. 5A). Involucrin expression was higher in the differentiated cell population as expected (Fig. 5A, lanes 4 to 6). No L1 protein expression was detected in undifferentiated W12 cells, but high levels of L1 protein were detected in differentiated W12E cells (Fig. 5A, lane 4). Depletion of SRSF1 had a small reduction effect in L1 protein expression in the differentiated cells (Fig. 5A, lane 5) but, consistent with the data using NIKS16 cells, SRSF3 depletion reduced the L1 protein levels significantly (Fig. 5A, lane 6). Quantification of the data from three separate experiments showed that L1 protein levels were reduced by 55% ±11% (P < 0.05) in differentiated W12E cells (Fig. 5B).
FIG 5.

SRSF3 controls L1 protein levels in W12E cells. (A) Western blot analysis of SRSF1, -2, and -3 protein depletion by siRNA in undifferentiated (U) and differentiated (D) W12E cells. Much higher levels of SRSF1 and SRSF3 were detected in differentiated W12E cells, as expected (24). L1 protein was not detected in undifferentiated W12E cells even with SRSF1 or SRSF3 depletion. L1 protein was detected in differentiating W12E cells, but the levels were reduced in cells treated with siRNA against SRSF3. Cntrl, cells transfected with a control siRNA. Involucrin was detected as a control for differentiated epithelial cells. GAPDH was detected as a protein loading control. This experiment was carried out five times, and very similar data were obtained each time. (B) Western blot quantification of levels of L1 protein in differentiated W12E keratinocytes transfected with a control siRNA (cntrl) or siRNAs against SRSF1 or SRSF3. The graph shows the means and standard deviations from three separate experiments. Values were calculated relative to the GAPDH levels. The P value was calculated using a Student t test. (C) Undifferentiated NIKS16 cells were transfected with vector control (Cntrl, lane 2) or a plasmid encoding the SRSF3 protein (SRSF3 transfect, lane 3). Differentiated NIKS16 cell extract (DNIKS16, lane 1) was probed as a positive control for L1 expression. GAPDH was used as a loading control. Involucrin was detected as a marker of epithelial differentiation. The level of involucrin was so high in the differentiated NIK16 cell extracts compared to undifferentiated NIK16 cell extracts that the blot was bleached out in lane 1. The experiment was repeated twice, and very similar data were obtained each time.
If SRSF3 depletion in differentiated NIKS16 cells results in the loss of L1 expression, then the overexpression of SFSF3 in undifferentiated NIKS16 cells might activate L1 expression. To test this, we transfected undifferentiated NIKS16 cells with an expression construct for SRSF3 (pcDNA3.1SRSF3). Cells were maintained at low cell density in medium with low calcium concentrations over the course of the experiment to minimize differentiation (26). At 72 h after transfection, the cells were harvested, and the protein lysates were prepared. Western blot analysis revealed that cells transfected with vector alone did not express L1 protein (Fig. 5C, lane 2). In contrast, cells transfected with the SRSF3 expression construct showed some L1 expression (Fig. 5C, lane 3) but not to the level detected in fully differentiated NIKS16 cells (Fig. 5C, lane 1). The transfected NIKS16 cells had not been induced to differentiate because levels of involucrin differentiation marker were low in the transfected cells (Fig. 5C, lanes 2 and 3) compared to the fully differentiated NIKS16 cell control (Fig. 5C, lane 1). Taken together, the data reveal that SRSF3 is a key regulator of HPV16 L1 protein levels.
SRSF3 depletion results in loss of the E4^L1 spliced RNA.
SRSF3 can regulate mRNA expression at a number of different levels (40), and it is known to be a master regulator of alternative splicing (41). Alternative splicing is a key control mechanism of HPV gene expression. HPV L1 capsid protein is thought to be expressed from an E4^L1 spliced transcript (RNA B) but may also be expressed from an E1^L1 (RNA A) or the bicistronic L2L1 transcript (RNA C) (Fig. 5A) (10, 42). Therefore, we examined whether the levels of any of these mRNAs were affected by SRSF3 depletion during the HPV16 life cycle. We mock depleted or siRNA depleted SRSFs 1 and 3 in W12E cells and allowed the cells to differentiate. siRNA treatment successfully depleted levels of SRSF1 and -3 (Fig. 5B). We used primer pairs to PCR amplify across splice junctions E1^L1 and E4^L1 or across the junction of the open reading frames in the readthrough L2L1 mRNA. E1^L1 proved very difficult to detect, indicating that it is an mRNA present at very low levels in HPV16-positive keratinocytes. In viral late mRNAs, the end of the E4 open reading frame contains a splice donor site at nt 3632 that allows splicing to the L1 splice acceptor site at nt 5639. E4^L1 mRNAs were detected readily. Compared to the effect of SRSF1 depletion, which has only a small effect on L1 protein production, SRSF3 depletion caused a remarkable decrease in levels of late mRNA E4^L1 (RNA B) in differentiated W12E cells (Fig. 5C, lane 6). SRSF1 depletion did not alter production of this RNA (Fig. 5C, lane 4). In contrast, depletion of SRSF1 or SRSF3 appeared to increase the levels of the L2/L1 bicistronic mRNA (RNA C) (Fig. 5C, lower panel, lanes 4 and 6). The levels of GADPH did not change upon SRSF1 or SRSF3 depletion. qRT-PCR analysis using previously published probe/primer sets (43) confirmed that E4^L1 mRNA levels were reduced significantly upon SRSF3 depletion in differentiated cells (Fig. 5D), whereas the bicistronic L2L1 mRNA levels increased when either SRSF1 (2.0-fold) or SRSF3 (1.7-fold) were depleted (Fig. 5E). Taken together, these data suggest that SRSF3 can control production of L1 capsid protein via enhancing the expression of the E4^L1 mRNA.
DISCUSSION
Focusing on the paradigm SR protein SRSF1, we demonstrated previously that E2 bound the SRSF1 promoter and its transactivation domain was essential for the upregulation of SRSF1 transcription (25). Here, we have shown that E2 transcriptionally activates the genes encoding not only SRSF1 but also SRSF2 and -3. The SRSF3 promoter displayed the highest level of transactivation in our transcription assays. E2 regulated the SRSF3 promoter in a dose-dependent manner. This is from the SRSF1 promoter that we found was activated by low but repressed by high levels of E2 (25; the present study). We do not yet understand how E2 transactivates the SRSF promoters because, similar to what we have found for the SRSF1 promoter (24), the SFSR2 and -3 promoters do not contain a cognate E2 binding site. In silico analysis of the proximal promoter sequences of SRSF1, -2, and -3 revealed that they are each predicted to bind the E2-interacting factors Sp1 and cEBP (data not shown), but it remains to be tested whether E2 binds and transactivates via these proteins and whether other factors are also involved. It is even possible that the array of cellular transcription factors that control each promoter may be different and mediate different transcriptional responses to E2. SRSF7 was used in the transcription assays as a control promoter since it is not regulated by E2, but we did not clone and test E2 control of other SRSFs. Therefore, we cannot conclusively rule out the possibility that E2 controls the expression of additional SRSF proteins. However, our data clearly reveal that the cellular SRSF1, -2, and -3 genes are upregulated by HPV16 E2. This is the first reported cloning of these SRSF promoters.
Semiquantitative Western blotting experiments revealed a similar profile of changes in SRSF protein levels upon differentiation of HPV16-positive W12E cells (24), NIKS16 cells (Fig. 3A), and the HPV31-positive CIN612-9E cells. This allowed us to test the mechanism of E2 control of the SRSF promoters by comparing levels of the SRSF proteins in normal foreskin keratinocytes stably transfected with a wild-type HPV31 genome or a genome containing a single inactivating point mutation in the E2 transactivation domain (30). There was a significant decrease in expression of SRSFs 1 to 3 in keratinocytes expressing the point mutant E2. These data support our hypothesis that HR-HPV E2 controls the promoters of these SR proteins via its transactivation domain. SRSF3 levels showed the greatest change, with a 71% decrease detected in cells expressing the mutant E2 compared to wild-type E2. The reported increased levels of E2 in the middle to upper layers of HPV-infected epithelia (5, 6) fits well with the increase in SRSF1, -2, and -3 expression in differentiated compared to undifferentiated W12E cells (24), NIKS16 cells, and CIN612-9E cells (the present study). It will be of interest to determine whether other high-risk and low-risk HPV types control SRSFs during the infectious life cycle. Our data imply that E2 control of SRSF1, -2, and -3 is likely at the level of transcription initiation. However, because E2 has been shown to possess some properties of SR proteins (44–46), we cannot discount a direct E2-mediated regulation of HPV mRNA processing or indeed E2 control of alternatively spliced isoforms of SR protein mRNAs that are more stimulatory of L1 splicing.
Also, although E2 is a regulator of SRSFs, we cannot rule out the possibility that other HPV proteins could be involved in controlling SRSF expression. For example, E6 and E7 may activate cellular transcription factors and control SRSF levels indirectly in differentiated keratinocytes (47, 48).
Our data suggest that a specific subset of SR proteins (SRSFs 1 to 3) is regulated by E2, indicating that these proteins may have important functions during the HPV replicative life cycle. Reasoning that increased SR protein expression in differentiated keratinocytes might impact late events in the viral life cycle, we compared the effect of siRNA depletion of each of the E2-regulated SRSFs 1 to 3 with SRSF5 and -7 (the latter two of which are not controlled by E2 in the NIKS16 model) on capsid protein expression. Depletion of SR proteins 2, 5, and 7 had no significant effect on L1 protein expression. Depletion of SRSF1 caused a modest reduction in L1 protein expression in NIKS16 and W12 cells as predicted by a previous study (20). However, SRSF3 depletion caused a major reduction in L1 protein expression in the differentiated NIKS16 cells. SRSF3 control of L1 expression was confirmed in a second HPV16 life cycle model using W12E cells. Conversely, the overexpression of SRSF3 in undifferentiated NIKS16 cells resulted in some induction of L1 protein expression. We were unable to repeat the experiment in the W12 cell model due to problems inhibiting differentiation in these cells during the course of the overexpression experiment. Although further work is required to determine whether the effect of SRSF3 on L1 induction in undifferentiated epithelial cells is at the level of mRNA processing, our data indicate that SRSF3 is a key regulator of viral capsid protein expression.
Although we did not carry out SR protein depletion experiments in CIN612-9E cells, we observed a significant decrease in capsid protein production in human keratinocytes maintaining E2:I73L mutant genomes, with a concomitant 50% reduction in SRSF3 expression, compared to cells containing wild-type HPV31 genomes. This does not prove that SRSF proteins control L1 protein production in HPV31-positive keratinocytes, but it is correlative with the observations in the HPV16-positive lines. The ∼4-fold level of L1 protein depletion we observed (Fig. 2D) agrees with the 80% reduction observed previously in L1 mRNA production in foreskin keratinocytes expressing the HPV31 E2:I73L mutant genome (30). The greater effect of the E2 inactivating point mutation on L1 protein expression compared to SRSF3 depletion could be due to incomplete knock down of SRSF3 in our experiments. However, it may be due to other effects of E2 on HPV or cellular gene expression. E2 appears to be required for induction of HPV late mRNA expression by inhibiting the viral early polyadenylation signal through interaction with CPSF30 (11). Although in our study an HPV31 E2 transactivation domain point mutant was all that was required to reduce virus capsid protein expression, in the study by Johannson et al. both the E2 transactivation domain and the hinge region were required for control of the L1 and L2 mRNA levels (11). This suggests that there at least two E2-regulated mechanisms for controlling the viral late gene expression, and these are not mutually exclusive. Apart from these important mechanisms, E2 can bind other RNA processing factors, including the SR proteins SRSF1, -2, and -7 (49) and could modulate their splicing activities as well as or instead of controlling transcription of SRSF genes. Moreover, E2 possesses SR protein-like properties itself since it can bind RNA and affect splicing in vitro (44, 45). Importantly, a recent study revealed that in U2OS cells E2 can control human gene expression by inducing alternative splice isoforms of mRNAs that encode proteins involved in cancer formation and cell motility (46). Therefore, it is possible that E2 could control late mRNA expression directly. A complex combination of different E2-regulated mechanisms may be essential in differentiation stage-specific control of HPV gene expression.
Several previous studies have reported the effects of SR proteins on HPV gene expression. These reports have delineated ESEs and ESSs and their interacting proteins on viral RNAs. SRSF1 binds a downstream ESE in the E4 open reading frame to control the viral splice acceptor site 3258 (20). SRSF2 controls HPV16 oncoprotein expression by regulating mRNA stability (23). SRSF3 also controls HPV16 oncoprotein expression (19), but to a lesser extent than SRSF2 (23). SRSF9 can activate the E4 splice donor 3632 while inhibiting splice acceptor 3358 at the 5′ end of the E4 open reading frame (21). Of interest, another splicing control element in the E4 coding region (50) binds SRSF3 (19). Knocking down SRSF3 in U2OS osteosarcoma cells increased levels of both L1 and L2 mRNAs while mutating the SRSF3 binding site increased L1 mRNA production ∼2-fold (19). In contrast to that study, we found that SRSF3 depletion in undifferentiated epithelial cells did not induce L1 protein expression (Fig. 3E). Moreover, our data indicate that SRSF3 is required for the L1-encoding E4^L1 mRNA and L1 protein expression in differentiating keratinocytes. Although some of the effects we have observed are rather modest, the differences in our data compared to the previous studies can be attributed to our use of differentiating, HPV-infected nontumor epithelial cells (three different model systems) that better mimic the natural HPV life cycle. Further, wild-type HPV genomes are present in NIKS16 and W12E and CIN612-9E cells. In contrast, in the previous studies, subgenomic constructs with deletions in important cis-acting elements (e.g., the late negative regulatory region [51]) controlling gene expression were used (20, 21, 50). In our HPV-infected lines, viral transcription is driven from the natural viral promoters (10, 38) which are relatively weakly active. Expression of HPV late mRNAs from the subgenomic constructs is under the control of the strong CMV promoter. Of importance, RNA polymerase II elongation rate, determined by promoter strength, is a major determinant of the use of weak over strong splice sites regardless of ESE/ESS activity (52, 53). Finally, the exact levels of SR proteins in cells are known to be vital to their function in splicing (54), meaning the effects on HPV late gene expression of the SRSF levels found in undifferentiated tumor cells may be different from their effects in differentiated keratinocytes.
However, it is important to consider that because we have analyzed HPV gene expression, together with keratinocyte differentiation upon SRSF knockdown, some of the effects we observed could be an indirect result of SRSF-mediated control of other cellular factors or of epithelial differentiation itself. For example, in Fig. 6A we observed an increase in expression of the epithelial differentiation marker involucrin upon SRSF3 depletion, implying that SRSF3 may repress involucrin expression upon epithelial differentiation. Further work will be required to determine whether SRSF3 is controlling HPV late gene expression directly or whether it does so through other cellular pathways.
FIG 6.

SRSF3 controls levels of the E4^L1 mRNA. (A) Schematic diagram of the HPV16 genome. Open reading frames are represented with light gray boxes with the gene names beneath. Promoters are indicated with arrowheads. Polyadenylation sites are indicated with downward arrows [Poly(A)E/L]. The major late mRNA splicing events with the open reading frames (dark gray boxes), and introns (dark gray lines) are indicated below the genome map. Each mRNA is shown truncated at the 5′ end to indicate the presence of several possible initiation sites for the transcription of these mRNAs (10). The open reading frames in each mRNA are indicated on the right-hand side. (B) Western blot analysis of levels of SRSF1 and SRSF3 in NIKS16 cells following siRNA knockdown. siSRSF1, siRNA against SRSF1; siSRSF3, siRNA against SRSF3; siCntrl, control siRNA. GAPDH was detected as a protein loading control. (C) RT-PCR detection of HPV late mRNAs type B (upper gel) or type C (lower gel) in cDNA synthesized from RNA isolated from the same cell populations analyzed in panel B. Cntrl, control siRNA; SRSF1, siRNA against SRSF1; SRSF3, siRNA against SRSF3; RT, reverse transcriptase; M, 1-kb DNA marker ladder. GAPDH was amplified as an internal control for RNA concentrations. (D) Relative levels of E4^L1 spliced mRNA B compared to GAPDH mRNA determined by qPCR. The means and standard deviations from three separate experiments are shown. (E) Relative levels of unspliced L2L1 mRNAs compared to GAPDH mRNA, as determined by qPCR. The means and standard deviations from three separate experiments are shown. P values were calculated using a Student t test.
Immunohistochemical staining of HPV16-infected low-grade cervical lesions has revealed a distinct pattern of SRSF3 expression (24; the present study). SRSF3 is expressed in the majority of basal epithelial cells. Although fewer cells in the upper epithelial layers express SRSF3, those that do express the protein contain very densely stained nuclei (Fig. 3F). We propose that these cells are HPV infected and support late events in the viral replication cycle, including E2-mediated control of SRSF3 expression. In preparation for capsid protein production, viral late mRNAs may be produced in epithelial cells below the granular layer (8), colocating with high levels of E2 and SRSF3. SRSF3 appears to be required for expression of the E4^L1 late mRNA that encodes the L1 capsid protein. Conversely, SRSF1 and SRSF3 negatively regulate production of the L2L1 read through RNA that likely encodes L2. This could be one regulatory mechanism that ensures high levels of L1 and low levels of L2 protein production to provide the correct ratio of L1 to L2 protein for capsid formation. SRSF3 can cross-regulate SRSF2, SRSF5, and SRSF7 and has been proposed as a master regulator of RNA splicing (41). The cooperative activity of SR proteins imposes positive and negative combinatorial control on splicing (54). Therefore, in the future it will be important to examine the effect of siRNA depletion of combinations of E2-controlled SR proteins on the HPV gene expression program.
In conclusion, if HPV-regulated SR proteins are key to completion of the viral replication cycle, they may represent a useful target for antiviral therapy. Small-molecule inhibitors of SR protein kinases that modulate the functions of SR proteins are available. Since they have been shown to be effective in inhibiting replication of HIV, hepatitis C virus, and Sindbis virus (14), they may also be effective against HPV replication. However, it will also be important also to understand any potential effects of these therapies on the switch from the viral replicative cycle to persistent or latent infection that underlies tumor progression. Nevertheless, such targets may prove effective for future therapeutic study.
ACKNOWLEDGMENTS
This study was funded by Wellcome Trust grant Wtd004098. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
We thank Margaret Stanley (University of Cambridge) and Paul Lambert and Denis Lee (University of Wisconsin) for provision of the W12 cells. We are grateful to Craig Meyers (Penn State University) for the gift of CIN612-9E cells and to John Doorbar (University of Cambridge) for the NIKS16 cells. Iain Morgan (Virginia Commonwealth University) kindly provided the U2OS cells lines transfected with HPV16 E2. We are grateful to Lou Laimins (Northwestern University, Chicago, IL) for agreeing to provide the protein extracts of the wild-type and E2:I73L mutant HPV31-transfected keratinocytes. We declare no conflicts of interest.
Funding Statement
This work was funded by Wellcome Trust grant Wtd004098 to S. V. Graham. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.zur Hausen H. 2009. Papillomaviruses in the causation of human cancers: a brief historical account. Virology 384:260–265. doi: 10.1016/j.virol.2008.11.046. [DOI] [PubMed] [Google Scholar]
- 2.Graham SV. 2010. Human papillomavirus: gene expression, regulation and prospects for novel diagnostic methods and antiviral therapies. Future Microbiol 5:1493–1505. doi: 10.2217/fmb.10.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Groves IJ, Coleman N. 2015. Pathogenesis of human papillomavirus-associated mucosal disease. J Pathol 235:527–538. doi: 10.1002/path.4496. [DOI] [PubMed] [Google Scholar]
- 4.Doorbar J. 2005. The papillomavirus life cycle. J Clin Virol 32S:S7–S15. [DOI] [PubMed] [Google Scholar]
- 5.Xue Y, Bellanger S, Zhang W, Lim D, Low J, Lunny D, Thierry F. 2010. HPV16 E2 is an immediate early marker of viral infection, preceding E7 expression in precursor structures of cervical carcinoma. Cancer Res 70:5316–5325. doi: 10.1158/1538-7445.AM10-5316. [DOI] [PubMed] [Google Scholar]
- 6.Coupe VM, Gonzalez-Barreiro L, Gutierrez-Berzal J, Melian-Boveda AL, Lopez-Rodriguez O, Alba-Dominguez J, Alba-Losada J. 2012. Transcriptional analysis of human papillomavirus type 16 in histological sections of cervical dysplasia by in situ hybridization. J Clin Pathol 65:164–170. doi: 10.1136/jclinpath-2011-200330. [DOI] [PubMed] [Google Scholar]
- 7.McBride AA. 2013. The papillomavirus E2 proteins. Virology 445:57–79. doi: 10.1016/j.virol.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stoler MH, Rhodes CR, Whitbeck A, Wolinsky SM, Chow LT, Broker TR. 1992. Human papillomavirus type 16 and 18 gene expression in cervical neoplasia. Hum Pathol 23:117–128. doi: 10.1016/0046-8177(92)90232-R. [DOI] [PubMed] [Google Scholar]
- 9.Zhou J, Lui WJ, Peng SW, Sun XY, Frazer IH. 1999. Papillomavirus capsid protein expression levels depends on the match between codon usage and tRNA availability. J Virol 73:4972–4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Milligan SG, Veerapraditsin T, Ahamat B, Mole S, Graham SV. 2007. Analysis of novel human papillomavirus type 16 late mRNAs in differentiated W12 cervical epithelial cells. Virology 360:172–181. doi: 10.1016/j.virol.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johannson C, Somberg M, Li X, Winquist EB, Fay J, Ryan F, Pim D, Banks L, Schwartz S. 2012. HPV-16 E2 contributes to induction of HPV-16 late gene expression by inhibiting early polyadenylation. EMBO J 31:3212–3227. doi: 10.1038/emboj.2012.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johannson C, Schwartz S. 2013. Regulation of human papillomavirus gene expression by splicing and polyadenylation. Nat Rev Microbiol 11:239–251. doi: 10.1038/nrmicro2984. [DOI] [PubMed] [Google Scholar]
- 13.Singh RK, Cooper TA. 2012. Pre-mRNA splicing in disease and therapeutics. Trends Mol Med 18:472–482. doi: 10.1016/j.molmed.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hernandez-Lopez HR, Graham SV. 2012. Alternative splicing in tumour viruses: a therapeutic target? Biochem J 445:145–156. doi: 10.1042/BJ20120413. [DOI] [PubMed] [Google Scholar]
- 15.Busch A, Hertel KJ. 2012. Evolution of SR proteins and hnRNP splicing regulatory factors. Wiley Interdiscip Rev RNA 3:1–12. doi: 10.1002/wrna.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roca X, Krainer AR, Eperon IC. 2013. Pick one, but be quick: 5′ splice sites and the problem of too many choices. Genes Dev 27:129–144. doi: 10.1101/gad.209759.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Long JC, Caceres JF. 2009. The SR protein family of splicing factors: master regulators of gene expression. Biochem J 417:15–27. doi: 10.1042/BJ20081501. [DOI] [PubMed] [Google Scholar]
- 18.Kajitani N, Schwartz S. 2015. RNA binding proteins that control human papillomavirus gene expression. Biomolecules 5:758–774. doi: 10.3390/biom5020758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jia R, Liu X, Tao M, Kruhlak M, Guo M, Meyers C, Baker CC, Zheng ZM. 2009. Control of the papillomavirus early-to-late switch by differentially expressed SRp20. J Virol 83:167–180. doi: 10.1128/JVI.01719-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Somberg M, Schwartz S. 2010. Multiple ASF/SF2 sites in the human papillomavirus type 16 (HPV-16) E4-coding region promote splicing to the most commonly used 3′-splice site on the HPV-16 genome. J Virol 84:8219–8230. doi: 10.1128/JVI.00462-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Somberg M, Li X, Johannson C, Orru B, Chang R, Rush M, Fay J, Ryan F, Schwartz S. 2011. Serine/arginine-rich protein 30c activates human papillomavirus type 16 L1 mRNA expression via a bimodal mechanism. J Gen Virol 92:2411–2421. doi: 10.1099/vir.0.033183-0. [DOI] [PubMed] [Google Scholar]
- 22.Li X, Johansson C, Cardoso Palacios C, Mossberg A, Dhanjal S, Bergvall M, Schwartz S. 2013. Eight nucleotide substitutions inhibit splicing to HPV-16 3′-splice site SA3358 and reduce the efficiency by which HPV-16 increases the life span of primary human keratinocytes. PLoS One 8:e72776. doi: 10.1371/journal.pone.0072776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McFarlane M, MacDonald AI, Stevenson A, Graham SV. 2015. Human papillomavirus 16 oncoprotein expression is controlled by the cellular splicing factor SRSF2 (SC35). J Virol 89:5276–5287. doi: 10.1128/JVI.03434-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mole S, McFarlane M, Chuen-Im T, Milligan SG, Millan D, Graham SV. 2009. RNA splicing factors regulated by HPV16 during cervical tumour progression. J Pathol 219:383–391. doi: 10.1002/path.2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mole S, Milligan SG, Graham SV. 2009. Human papillomavirus type 16 E2 protein transcriptionally activates the promoter of a key cellular splicing factor, SF2/ASF. J Virol 83:357–367. doi: 10.1128/JVI.01414-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jeon S, Allen-Hoffman BL, Lambert PF. 1995. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J Virol 69:2989–2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weschler EI, Wang Q, Roberts I, Pagliarulo E, Jackson D, Untersperger C, Coleman N, Griffin H, Doorbar J. 2012. Reconstruction of human papillomavirus type 16-mediated early-stage neoplasia implicated E6/E7 deregulation and the loss of contact inhibition in neoplastic progression. J Virol 86:6358–6364. doi: 10.1128/JVI.07069-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hummel M, Hudson JB, Laimins LA. 1992. Differentiation-induced and constitutive transcription of human papillomavirus type 31b in cell lines containing viral episomes. J Virol 66:6070–6080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taylor ER, Boner W, Dornan ES, Corr EM, Morgan IM. 2003. UVB irradiation reduces the half life and transactivation potential of the human papillomavirus 16 E2 proteins. Oncogene 22:4469–4477. doi: 10.1038/sj.onc.1206746. [DOI] [PubMed] [Google Scholar]
- 30.Stubenrauch F, Colbert AME, Laimins LA. 1998. Transactivation by the E2 protein of oncogenic human papillomavirus type 31 is not essential for early and late viral functions. J Virol 72:8115–8123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McPhillips MG, Veerapraditsin T, Cumming SA, Karali D, Milligan SG, Boner W, Morgan IM, Graham SV. 2004. SF2/ASF binds the human papillomavirus type 16 late RNA control element and is regulated during epithelial differentiation. J Virol 78:10598–10605. doi: 10.1128/JVI.78.19.10598-10605.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fay J, Kelehan P, Lambkin H, Schwartz S. 2009. Increased expression of cellular RNA-binding proteins in HPV-induced neoplasia and cervical cancer. J Med Virol 81:897–907. doi: 10.1002/jmv.21406. [DOI] [PubMed] [Google Scholar]
- 33.Lee D, Kim H-Z, Jeong KW, Shim YS, Horikawa I, Barrett JC, Choe J. 2002. Human papillomavirus E2 down-regulates the human telomerase reverse transcriptase promoter. J Biol Chem 277:27748–27756. doi: 10.1074/jbc.M203706200. [DOI] [PubMed] [Google Scholar]
- 34.Frolkis M, Fischer MB, Wang Z, Lebkowski JS, Chiu C-P, Majumdar AS. 2003. Dendritic cells reconstituted with human telomerase gene induce potent cytotoxic T-cell response against different types of tumors. Cancer Gene Ther 10:239–249. doi: 10.1038/sj.cgt.7700563. [DOI] [PubMed] [Google Scholar]
- 35.Stanley MA, Browne HM, Appelby M, Minson AC. 1989. Properties of a non-tumorigenic human cervical keratinocyte cell line. Int J Cancer 43:672–676. doi: 10.1002/ijc.2910430422. [DOI] [PubMed] [Google Scholar]
- 36.Chuen-Im T, Zhang J, Milligan SG, McPhillips MG, Graham SV. 2008. The alternative splicing factor hnRNP A1 is up-regulated during virus-infected epithelial cell differentiation and binds the human papillomavirus type 16 late regulatory element. Virus Res 131:189–198. doi: 10.1016/j.virusres.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hummel M, Lim HB, Laimins LA. 1995. Human papillomavirus type 31b late gene expression is regulated through protein kinase C-mediated changes in RNA processing. J Virol 69:3381–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ozbun MA, Meyers C. 1997. Characterisation of late gene transcripts expressed during vegetative replication of human papillomavirus type 31b. J Virol 71:5161–5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gauson EJ, Donaldson MM, Dornan ES, Wang X, Bristol M, Bodily JM, Morgan IM. 2015. Evidence supporting a role for TopBP1 and Brd4 in the initiation but not continuation of human papillomavirus 16 E1/E2-mediated DNA replication. J Virol 89:4980–4990. doi: 10.1128/JVI.00335-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Corbo C, Orru S, Salvatore F. 2013. SRp20: An overview of its role in human disease. Biochem Biophys Res Commun 436:1–5. doi: 10.1016/j.bbrc.2013.05.027. [DOI] [PubMed] [Google Scholar]
- 41.Anko M-L, Muller-McNicoll M, Brandi H, Curk T, Gorup C, Henry I, Ule J, Neugebauer KM. 2012. The RNA-binding landscapes of two SR proteins reveal unique functions and binding to diverse RNA classes. Genome Biol 13:R17. doi: 10.1186/gb-2012-13-3-r17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang X, Meyers C, Wang H, Chow LT, Zheng ZM. 2011. Construction of a full transcription map of human papillomavirus type 18 during productive viral infection. J Virol 85:8080–8092. doi: 10.1128/JVI.00670-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmitt M, Dalstein V, Waterboer T, Clavel C, Gissman L, Pawlita M. 2010. Diagnosing cervical cancer and high grade precursors by HPV16 transcription patterns. Cancer Res 70:249–256. doi: 10.1158/0008-5472.CAN-09-2514. [DOI] [PubMed] [Google Scholar]
- 44.Lai M-C, Teh BH, Tarn W-Y. 1999. A human papillomavirus E2 transcriptional activator. J Biol Chem 274:11832–11841. doi: 10.1074/jbc.274.17.11832. [DOI] [PubMed] [Google Scholar]
- 45.Bodaghi S, Jia R, Zheng Z. 2009. Human papillomvirus type 16 E2 and E6 are RNA-binding proteins and inhibit in vitro splicing of pre-mRNA with suboptimal splice sites. Virology 386:32–43. doi: 10.1016/j.virol.2008.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gauson EJ, Windle B, Donaldson MM, Caffarel MM, Dornan ES, Coleman N, Henderson SC, Wang X, Morgan IM. 2014. Regulation of human genome expression and RNA splicing by human papillomavirus E2 protein. Virology 468-470:10–18. doi: 10.1016/j.virol.2014.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roman A, Munger K. 2013. The papillomavirus E7 proteins. Virology 445:138–168. doi: 10.1016/j.virol.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vande Pol SB, Klingelhutz AJ. 2013. Papillomavirus E6 proteins. Virology 445:115–137. doi: 10.1016/j.virol.2013.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Muller M, Demert C. 2012. The HPV E2-host protein-protein interactions: a complex hijacking of the cellular network. Open Virol J 6:173–189. doi: 10.2174/1874357901206010173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rush M, Zhao X, Schwartz S. 2005. A splicing enhancer in the E4 coding region of human papillomavirus type 16 is required for early mRNA splicing and polyadenylation as well as inhibition of premature late gene expression. J Virol 79:12002–12015. doi: 10.1128/JVI.79.18.12002-12015.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Graham SV. 2008. Papillomavirus 3′UTR regulatory elements. Front Biosci 13:5646–5663. [DOI] [PubMed] [Google Scholar]
- 52.Proudfoot N. 2000. Connecting transcription to messenger RNA processing. Trends Biochem Sci 25:290–293. doi: 10.1016/S0968-0004(00)01591-7. [DOI] [PubMed] [Google Scholar]
- 53.Moreno NN, Giono LE, Cambindo Botto AE, Munoz MJ, Kornblihtt AR. 2015. Chromatin, DNA structure and alternative splicing. FEBS Lett 589:3370–3378. doi: 10.1016/j.febslet.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 54.Bradley T, Cook ME, Blanchette M. 2015. SR proteins control a complex network of RNA-processing events. RNA 21:75–92. doi: 10.1261/rna.043893.113. [DOI] [PMC free article] [PubMed] [Google Scholar]