

Abstract
A highly modular approach to N-substituted 4-(1-fluorovinyl)triazoles is described. In situ desilylation and Cu-catalyzed ligation reaction of TMS-protected α-fluoropropargyl benzothiazole sulfone with aryl, alkyl, and metallocenyl azides furnished second-generation Julia-Kocienski reagents in good to excellent yields. Condensation reactions of these reagents with aldehydes can be tuned to yield E or Z-alkenes selectively. Under mild conditions with DBU as base, reactions of aldehydes furnished E-alkenes as the major isomer. On the other hand, in condensations with LHMDS as base and in appropriate solvents, both aldehydes and ketones reacted to yield fluoroalkenes with Z-selectivity. Stereochemical assignment to E/Z olefins obtained in the reaction of a ketone with two Julia reagents was performed via X-ray crystallographic analysis and comparisons of NMR data. The method allows efficient and ready diversification of N1-substituent and substituents at the double bond.
Graphical abstract
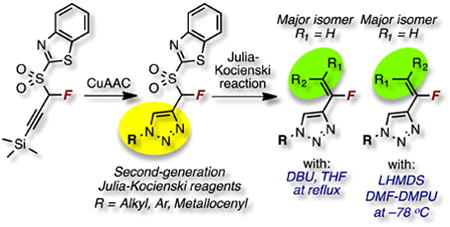
Introduction
The high modularity and 100% atom economy of the Huisgen ligation make this an attractive approach to the triazole moiety.1 As a class both 1,4- and 1,5-disubstituted 1,2,3-triazoles are highly valuable in a number of areas, from materials and polymer chemistry, to pharmaceutics and medicine.2 The complexation of the triazole ring has also been exploited in supramolecular chemistry.3 Discovery of the Cu-catalyzed variant of the Huisgen ligation enabled facile and highly regioselective access to 4-substituted 1,2,3-triazoles.4,5 This prompted resurgence of the azide-alkyne cycloaddition, leading to plethora of 1,2,3-triazole-derived structures. Figure 1 shows some representative examples of 1,4-disubstituted 1,2,3-triazole-containing biologically active compounds,6,7 as well as new macromolecules with valuable mechanical and/or thermal properties.2d–f,8
Figure 1. Examples of 1,4-disubstituted 1,2,3-triazoles as biologically active compounds and as new macromolecules.

In the area of fluoroorganic chemistry,9 we10,11 and others12 have been involved in modular synthesis of variously functionalized fluoroolefins10,13 using Julia-Kocienski approach.14 The effect of fluorine atom incorporation on pharmaceuticals is well established.15 For example, Z- and E-fluorovinyl groups are hydrolytically stable isosteres of trans and of cis peptide bonds, respectively.16 Given the high modularity of the azide-alkyne cycloadditions (CuAAC) as well as the Julia-Kocienski olefination, we became interested in a facile approach to merging of the pharmacologically interesting triazole and fluorovinyl moieties. We recently described an approach to 4-vinyl and 4-(α-fluorovinyl)-triazoles via the use of a building block containing a triple bond and an olefination handle. Although synthesis of N-fluorovinyl triazoles17 has been reported, our preliminary communication11a was, to the best of our knowledge, the first report on the synthesis of 4-(α-fluorovinyl)-triazoles.
In our preliminary communication11a we demonstrated that regioselectively difunctionalized triazoles, with readily varied N1 and vinyl substituents could be accessed by a combination of the CuAAC and the Julia-Kocienski reactions. In that work we showed that triazole-substituted Julia-Kocienski reagents can be first obtained via a Cu-catalyzed azide-alkyne click ligation of propargyl and α-fluoropropargyl benzothiazole sulfones with one aryl and two alkyl azides. The resulting reagents then underwent olefination reactions with aldehydes and a ketone. Herein, we describe the broader generality of the azide-alkyne ligation reactions of α-fluoropropargyl benzothiazole sulfone with a larger series of azides, and the reactivity of these second-generation Julia-Kocienski reagents in olefinations with aldehydes and ketones. Notably, evaluation of olefination conditions showed that selectivity could be tuned to yield E- or Z-fluorovinyl triazoles as the major isomer in the Julia-Kocienski reactions. This adds to the overall modularity of this approach.
Results and Discussion
In order to explore the generality of azide-alkyne ligation reactions leading to second-generation Julia-Kocienski reagents, TMS-protected α-fluoropropargyl benzothiazole sulfone 111a,c was deprotected in situ and subjected to ligation with a series of azides, following our published protocol11a (Scheme 1). It should be noted that the use of 1-phenyl-1H-tetrazol-5-yl propargyl sulfone (propargyl PT-sulfone) was also considered for the synthesis of comparable second-generation Julia-Kocienski reagents. The heteroaryl moiety can influence the selectivity in Julia-Kocienski olefinations,14 e.g. olefinations of n-alkanals with alkyl PT-sulfones proceed with superior selectivity as compared to benzothiazole-derived sulfones.18 However, it has been reported that TMS-protected propargyl PT-sulfone is unstable.19 This sulfone on reaction with benzaldehyde gave the enyne product in a low yield, plausibly due to its instability.20 Because our protocol requires initial fluorination of a TMS-protected propargyl heteroaryl sulfone, followed by CuAAC reactions, we chose the more stable benzothiazolyl derivative (Scheme 1).
Scheme 1. Synthesis of fluoro(1-substituted-1H-1,2,3-triazol-4-yl)methyl 1,3-benzothiazol-2-yl sulfones 2-10 via CuAAC reaction.

Briefly, in situ alkyne deprotection with AgBF4, followed by Cu(CH3CN)4PF6 mediated reaction with nine azides furnished corresponding triazoles in 66–93% yield (2-10, Scheme 1). Azidobenzene and 1-azido-4-nitrobenzene gave triazole products 2 and 6 in 79% and 82% yield, respectively, whereas electron-rich aromatic azides gave triazoles 3–5 in slightly higher 90–93% yields. 1-Azidoadamantane and 1-azidodecane gave products 7 and 8 in 85% and 81% yield, respectively, and 3-(azidopropyl)benzene gave triazole 9 in a slightly lower 68% yield. Azidoferrocene reacted as well, to give product 10 in a respectable 66% yield. Reactions were typically complete in 1.5–4 h, except in the case of 2-azidonaphthalene and 1-azido-4-nitrobenzene, where longer reaction times were required (19 h and 50 h, respectively).
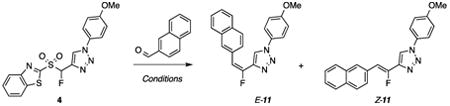
Screening of olefination conditions was then performed using N-p-methoxyphenyl triazolyl Julia-Kocienski reagent 4 and 2-naphthaldehyde (Table 1).
Table 1. Screening of olefination conditions.
| |||||
|---|---|---|---|---|---|
|
| |||||
| entry | base (molar equiv) | solvent | Additive (molar equiv) | T (°C); time | 11, yield,a (%) E/Zb |
| 1 | LHMDS (2.4) | DMF-DMPUc | -- | –78; 3 min | 76%, 7/93 |
| 2 | LHMDS (2.4) | THF | MgBr2 (1.8) | –78; 51 h | NRd |
| 3 | DBU (4.0) | DMF | -- | –55; 3.5 h | NA,e 22/78 |
| 4 | DBU (4.0) | DMF-DMPUc | -- | –78; 24 h | Inc,f 27/73 |
| 5 | DBU (4.0) | THF | -- | –78; 50 h | Inc,f 63/37 |
| 6 | DBU (4.0) | THF | -- | rt; 10 h | 77%, 77/23 |
| 7 | DBU (4.0) | THF | -- | 50; 6 h | NA,e 77/23 |
| 8 | DBU (4.0) | THF | -- | reflux; 2.5 h | 74%, 78/22 |
| 9 | DBU (4.0) | THF | MgBr2 (1.8) | reflux; 40 min | NA,e 69/31 |
| 10 | DBU (4.0) | CHCl3 | -- | reflux; 4.5 h | NA,e 75/25 |
Yields are of isolated and purified products.
E/Z olefin ratios in the crude reaction mixtures were determined by 19F NMR prior to isolation.
DMF-DMPU 1:1 v/v.
Formation of products was not observed.
Products were not isolated.
Incomplete reaction and products were not isolated.
In our prior communication we have demonstrated that condensations of 4 and 9 with some aldehydes proceeded with Z-selectivity, in low-temperature LHMDS-mediated condensations in DMF-DMPU.11a In the present case as well, 2-naphthaldehyde reacted with 4 to furnish fluorovinyl triazole (E/Z)-11 with high Z-selectivity (Table 1, entry 1, E/Z 7/93). However, when reaction was performed with LHMDS in THF and in the presence of MgBr2, no product formation was observed even after 51 h (entry 2). We were now curious to assess whether condensations would proceed under milder conditions. Indeed, the use of DBU as base in DMF at –55 °C gave (E/Z)-11, but with a lower Z-selectivity than the reaction with LHMDS (compare entry 3 to entry 1). A DBU-mediated reaction in DMF-DMPU at –78 °C gave selectivity comparable to entry 3, but the reaction was incomplete after 24 h (entry 4). We were pleased that these reactions could be tuned to E-selectivity upon changing the solvent to THF. However, this reaction was incomplete even after 50 h (entry 5, E/Z 63/37). When the reaction was performed at rt, E-selectivity improved and the reaction was complete within 10 h (entry 6, E/Z 77/23, 77% yield). Further increase in temperature to 50 °C gave the same E/Z ratio, but in a shorter reaction time (entry 7). At reflux, reaction was complete in 2.5 h (entry 8, E/Z 78/22, 74% yield). In the presence of MgBr2, the E/Z ratio dropped to 69/31 (entry 9). Reaction in CHCl3 at reflux gave a similar E/Z ratio 75/25 as compared to the reaction in THF, but a longer reaction time was required (compare entry 10 to entry 8).
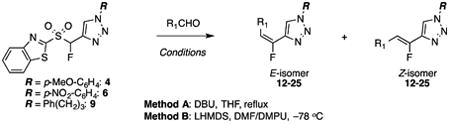
Having determined conditions that allowed olefinations to be tuned towards Z- or E-selectivity, we next assessed the scope of condensations with regard to the carbonyl compound and the structure of the Julia-Kocienski reagent. For this, we performed condensation reactions with three Julia-Kocienski reagents 4, 6, and 9 under two sets of conditions, i.e. with DBU in refluxing THF (Method A, Table 2), and with LHMDS in DMF-DMPU at –78 °C (Method B, Table 2).
Table 2. Tunable selectivity in condensations of sulfones 4, 6, and 9 with aldehydes.
| ||||
|---|---|---|---|---|
|
| ||||
| product, yield,a % E/Z ratiob | ||||
| entry | sulfone | R1-CHO | method A | method B |
| 1 | 4 |
|
12, 80%, 81:19 | 12, 72%, 14:86c |
| 2 | 6 | 13, 82%, 87:13 | 13, 62%, 19:81 | |
| 3 | 9 | 14, 75%, 76:24 | 14, 90%, 7:93c | |
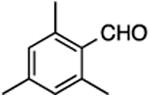
| 4 | 9 |
|
--d | 15, 47%, 26:74c |
| 5 | 4 |
|
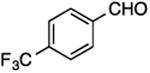
16, 52%, 64:36 | 16, 77%, 30:70 |
| 6 | 6 | 17, 87%, 60:40 | 17, 73%, 15:85 | |
| 7 | 9 | 18, 80%, 63:37 | 18, 72%, 20:80 | |
| 8 | 9 |
|
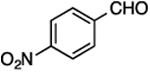
19, 85%, 57:43 | 19, 76%, 38:62c |
| 9 | 4 |
|
20, 61%, 57:43 | 20, 68%, 34:66c |
| 10 | 6 | 21, 33%, 58:42 | 21, 61%, 32:68 | |
| 11 | 9 | 22, 56%, 61:39 | 22, 63%, 36:64c | |
| 12 | 4 |
|
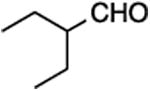
23, 47%, 54:46 | 23, 59%, 15:85c |
| 13 | 6 | 24, 3%, 54:46 | 24, 68%, 12:88 | |
| 14 | 9 | 25, 5%, 61:39 | 25, 57%, 19:81c | |
Yields are of isolated and purified products.
E/Z olefin ratios in the crude reaction mixtures were determined by 19F NMR prior to isolation.
Data reported in ref 11a.
Reaction was not performed.
Under DBU-mediated conditions, all reactions proceeded with E-selectivity. Selectivity appears to depend on the aldehyde, and to a lesser extent on the triazole reagent. Good E-selectivity was observed with electron-rich aromatic aldehydes (entry 8 in Table 1 and entries 1–3 in Table 2), whereas E-selectivity was moderate to poor with increasing electron deficiency in the aldehydes, and with alkanals (entries 5–14, Table 2). Alpha braching in the aldehyde did not alter the E/Z ratio (compare entry 9 to 12). In the case of aromatic aldehydes, yields were in the range of 74–87% (entry 8 in Table 1, and entries 1–3 and 6–8 in Table 2), except for the reaction of p-trifluoromethylbenzaldehyde with sulfone 4, where product 16 was obtained in moderate 52% yield (entry 5). n-Octanal gave products in moderate (entries 9 and 11) to poor yield (entry 10). 2-Ethylbutanal reacted with sulfone 4 to give product in 47% yield (entry 12), whereas reactions with sulfones 6 and 9 gave only traces of products (entries 13, 14). It is plausible that in the case of enolizable aldehydes, yields were low due to a competing aldol reaction. Consistent with this, yields were lower with 2-ethylbutanal compared to n-octanal, where steric hindrance likely results in slower addition of the Julia reagent to the aldehyde and subsequent spirocyclization.
LHMDS-mediated condensations proceeded with complementary Z-selectivity (Table 2). As with the DBU-mediated reactions, this selectivity depended on the structure of the aldehyde, and to a much lesser extent on that of the sulfone. Selectivity was good to excellent with electron-rich aldehydes and the sterically hindered 2-ethylbutanal, and moderate to good with electron-deficient aromatic aldehydes and n-octanal. These condensations were very fast and complete disappearance of the aldehyde was observed within 5 minutes at –78 °C.
Next, reactivity of triazole-derived Julia-Kocienski reagents with ketones was tested as well. For this, N-p-methoxyphenyl (4), N-(3-phenylpropyl) (9), and N-ferrocenyl (10) derived reagents were reacted with three ketones. Table 3 shows yields, E/Z ratios, and 19F chemical shifts of the products.
Table 3. Condensations of sulfones 4, 9, and 10 with ketones.
| ||||
|---|---|---|---|---|
|
| ||||
| entry | sulfone | R1R2CO | product, yield,a % isomer ratiob | 19F NMR δ (ppm)c |
| 1 | 4 |
|
26, 87%, NAd | –121.99 ppmd |
| 2 | 9 |
|
27, 58%, NA | –121.52 ppm |
| 3 | 4 |
|
28, 64%,e 28:72 | –116.46 ppm (minor)f –116.57 ppm (major)f |
| 4 | 9 |
|
29, 77%,e 20:80 | –115.56 ppm (minor E-29)g –116.33 ppm (major Z-29)g |
| 5 | 10 |
|
30, 58%, one isomer onlyh | –123.76 ppm |
Yields are of isolated and purified products.
Olefin isomer ratios in the crude reaction mixtures were determined by 19F NMR prior to isolation.
Referenced to CFCl3 as internal standard; 282 MHz, CDCl3 solvent.
Data reported in ref 11a.
Combined yield of E and Z-isomers.
Stereochemistry of the isomers was assigned by comparison to compound 29 (see text).
Stereochemistry of the major isomer was assigned by X-ray crystallography (see text).
Stereochemistry was not assigned.
All reactions proceeded under LHMDS-mediated conditions, in THF at 0 °C, to give fluorovinyl triazoles 26–30 in 58–87% yields (Table 3). Initially, condensations were performed using conditions that were employed with aldehydes, i.e. with LHMDS in DMF-DMPU at –78 °C, but incomplete conversions of the starting ketone were observed. Complete conversions were achieved upon warming of the reaction mixture to room temperature, however, yields were lower in DMF-DMPU. For example, addition of LHMDS to a solution of acetophenone and 9 in DMF-DMPU at –78 °C with subsequent warming to room temperature gave (E/Z)-29 in 63% yield. In comparison, a 77% yield was obtained when the reaction was performed in THF at 0 °C (entry 4). Stereoselectivity, on the other hand, was similar in both reactions.
In order to assess the stereoselectivity of the condensations with acetophenone, the E/Z isomers produced in the reaction of N-(3-phenylpropyl) derived Julia-Kocienski reagent 9 (Table 3, entry 4) with acetophenone were chromatographically separated and the major isomer was crystallized. Analysis by X-ray diffraction21 showed the stereochemistry of the major isomer to be Z-29 (Figure 2). The X-ray structure also confirmed regioselectivity of the azide-alkyne cycloaddition reaction.
Figure 2. Crystal structure of major isomer of fluorovinyl triazole 29 (C, black; H, grey; F, green; N, blue).
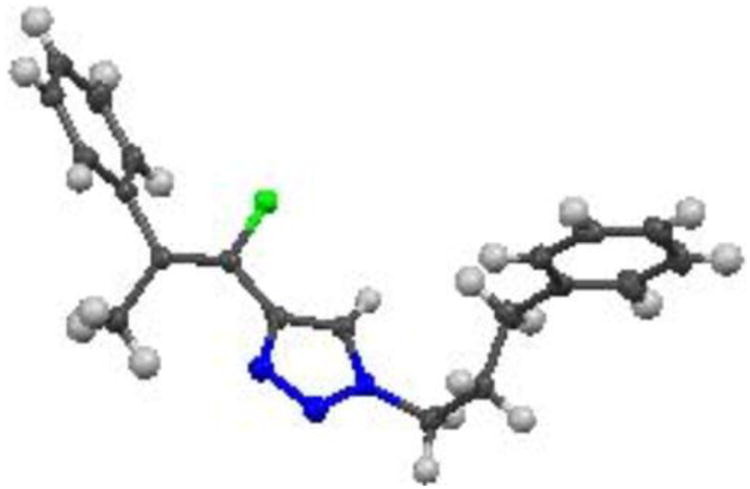
The Z-stereoselectivity observed in the reaction of sulfone 9 with acetophenone is consistent with that observed in LHMDS-mediated reactions of aldehydes (Table 2, Method B). We assessed the stereochemistry in E-28 and Z-28, formed by reaction of sulfone 4 with acetophenone (Table 3, entry 3), by comparison to NMR characteristics of E- and Z-29 (Table 4).
Table 4. Chemical shifts of the H-5 proton resonance in the major and minor isomers of compounds 28 and 29.
| compound | triazolyl H-5 resonancea | Δδ ppmb |
|---|---|---|
| Major isomer of 28 | 8.04 ppm |
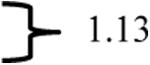
|
| Minor isomer of 28 | 6.91 ppm | |
| Z-29 | 7.65 ppm |
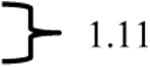
|
| E-29 | 6.54 ppm |
Obtained at 500 MHz in CDCl3;
δ ppm of major isomer – δ ppm of minor isomer.
In comparing the 1H NMR spectra of E- and Z-29, a distinctive 1.11 ppm difference was observed in the chemical shifts of the H-5 triazolyl proton singlets, and the triazolyl proton resonance in Z-29 (the major isomer) appeared most downfield in the spectrum (Table 4). The 19F resonance of the major isomer of Z-29 is more upfield shifted as compared to the E-isomer (see data in Table 3). In comparing the NMR data of E/Z-isomers of compound 29 to those of 28, a similar 1.13 ppm difference was observed between the chemical shifts of the triazolyl H-5 proton singlets in the major and the minor isomers of olefins 28. Also, the 1H NMR of the major isomer showed an aromatic singlet as the most downfield shifted resonance. Furthermore, in the 19F NMR, the major isomer in olefin mixture 28 produced the more upfield resonance. Due to the close structural similarities of (E/Z)-28 and (E/Z)-29 and the parallels in the NMR spectra, the major isomer of 28 was assigned a Z-geometry and the minor isomer E. The condensation product from indanone (30) is structurally very different to postulate the olefin stereochemistry by such comparisons. Moreover, because formation of only one isomer was detected, comparison of chemical shifts was not possible.
Conclusions
In conclusion, a highly modular approach to N-substituted 4-(1-fluorovinyl)triazoles has been developed, where N1-substituent as well as substituents at the double bond can be readily varied. Cu-catalyzed azide-alkyne ligation reactions of TMS-protected α-fluoropropargyl benzothiazole sulfone, a Julia-Kocienski reagent, proceed with aryl, alkyl, or metallocenyl azides, to give triazole-derived second-generation Julia-Kocienski reagents in good to excellent yields. Condensation reactions of triazole-derived Julia-Kocienski reagents with aldehydes are tunable and proceed with either E-selectivity under mild DBU-mediated conditions in THF at reflux, or with Z-selectivity in low temperature LHMDS-mediated reactions in DMF-DMPU. Ketones react in the presence of LHMDS in THF, to give tetrasubstituted olefins. In the cases studied, where both isomers were formed in reactions with ketones, E- and Z-fluorovinyl triazoles were separable under chromatographic conditions. The method offers high flexibility for diversification of N1 and vinyl substituents.
Experimental Section
General Experimental Considerations
THF was distilled over LiAlH4 and then over sodium, toluene was distilled over sodium, and CH2Cl2 was distilled over CaCl2. DMF and DMPU were obtained from commercial sources and were used without further purification. For reactions performed in a nitrogen atmosphere, glassware was dried with heat gun under vacuum. LDA (2.0 M solution in heptane/THF/EtPh) and LHMDS (1.0 M in THF) were obtained from commercial sources. All other reagents were obtained from commercial sources and were used as received. 1-Azidoadamantane and N-fluorobenzenesulfonimide (NFSI) are commercially available. Synthesis of 2-[1-fluoro-3-(trimethylsilyl)prop-2-ynylsulfonyl]benzo[d]thiazole (1) was first reported in our preliminary communication,11a we then reported an improved method.11c Syntheses of 2-{fluoro[1-(4-methoxyphenyl)-1H-1,2,3-triazol-4-yl]methylsulfonyl}benzo[d]-thiazole (4), 2-[(1-decyl-1H-1,2,3-triazol-4-yl)fluoromethylsulfonyl]benzo[d]thiazole (8) and 2-{fluoro[1-(3-phenylpropyl)-1H-1,2,3-triazol-4-yl]methylsulfonyl}benzo[d]-thiazole (9) have been reported in our preliminary communication.11a 1-Azido-4-phenoxybenzene,22a1-azido-4-nitrobenzene, 22a azidobenzene,22,23 2-azidonaphthalene,23 azidoferrocene,24 1-azido-4-methoxybenzene,11a,22 3-(azidopropyl)benzene,11a and 1-azidodecane11a were synthesized via literature methods. Thin layer chromatography was performed on glass-backed silica gel plates (250 μm). Column chromatographic purifications were performed on 200–300 mesh silica gel. 1H NMR spectra were recorded at 500 MHz and were referenced to residual protio solvent. 13C NMR spectra were recorded at 125 MHz and were referenced to the carbon resonance of the deuterated solvent. 19F NMR spectra were recorded at 282 MHz with CFCl3 as the internal standard. Chemical shifts (δ) are reported in parts per million and coupling constants (J) are in hertz (Hz). HRMS data were obtained using a TOF analyzer, the ionization modes are specified under each compound heading.
Synthesis of Triazoles
General procedure
To a stirring solution of azide (1 molar equiv) in 4:1 (v/v) CH2Cl2/MeOH (28.0 mL/mmol of azide), 2-[1-fluoro-3-(trimethylsilyl)prop-2-ynylsulfonyl]benzo[d]thiazole 1 (1.10 to 1.50 molar equiv), Cu(CH3CN)4PF6 (0.20 molar equiv) and AgBF4 (0.20 molar equiv) were sequentially added. The stirring was continued at room temperature until TLC showed disappearance of the azide. The solvents were evaporated under reduced pressure and the crude reaction mixture was purified by column chromatography on silica gel. The quantities of azides and fluoropropargyl sulfone 1, reaction times, eluting solvents for chromatography, product yields, Rf values, and spectroscopic characterization data are provided under the individual compound headings.
2-{[Fluoro(1-phenyl-1H-1,2,3-triazol-4-yl)methyl]sulfonyl}benzo[d]thiazole (2).
![2-{[Fluoro(1-phenyl-1H-1,2,3-triazol-4-yl)methyl]sulfonyl}benzo[d]thiazole (2)](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/e1bb/4859785/b8cb73efd92f/nihms779409f1.jpg)
Prepared from azidobenzene (60.0 mg, 0.500 mmol) and sulfone 1 (206 mg, 0.630 mmol, 1.26 molar equiv), in a reaction time of 4 h. Chromatography was performed with 20% EtOAc in hexanes and compound 2 was obtained as an off-white solid (148 mg, 79%). Rf (40% EtOAc in hexanes) = 0.45. 1H NMR (500 MHz, CDCl3): δ 8.54 (d, 1H, Ar-H, J = 1.4 Hz), 8.32 (d, 1H, Ar-H, J = 8.3 Hz), 8.07 (d, 1H, Ar-H, J = 8.3 Hz), 7.78 (d, 2H, Ar-H, J = 7.4 Hz), 7.71 (t, 1H, Ar-H, J = 7.5 Hz), 7.66 (t, 1H, Ar-H, J = 7.6 Hz), 7.58 (t, 2H, Ar-H, J = 7.6 Hz), 7.52 (t, 1H, Ar-H, J = 7.4 Hz), 7.02 (d, 1H, CHF, 2JFH = 46.5 Hz). 13C NMR (125 MHz, CDCl3): δ 162.0, 153.1, 137.8, 136.6, 136.0 (d, 2JCF = 24.3 Hz), 130.2 129.9, 128.9, 128.3, 126.2, 124.0, 122.6, 121.2, 96.0 (d, 1JCF = 219.7 Hz). 19F NMR (282 MHz, CDCl3): δ –165.6 (d, 2JFH = 45.8 Hz). HRMS (ESI) calcd. for C16H12FN4O2S2 [M + H]+ 375.380, found 375.379.
2-{[Fluoro(1-(naphthalen-2-yl)-1H-1,2,3-triazol-4-yl)methyl]sulfonyl}benzo[d]thiazole (3).
![2-{[Fluoro(1-(naphthalen-2-yl)-1H-1,2,3-triazol-4-yl)methyl]sulfonyl}benzo[d]thiazole (3)](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/e1bb/4859785/e3ae70324c6c/nihms779409f2.jpg)
Prepared from 2-azidonaphthalene (85.0 mg, 0.500 mmol) and sulfone 1 (206 mg, 0.630 mmol, 1.26 molar equiv) in a reaction time of 19 h. Chromatography was performed with 20% EtOAc in hexanes and compound 3 was obtained as an off-white solid (194 mg, 91%). Rf (40% EtOAc in hexanes) = 0.50. 1H NMR (500 MHz, CDCl3): δ 8.67 (d, 1H, Ar-H, J = 1.8 Hz), 8.32 (d, 1H, Ar-H, J = 8.3 Hz), 8.22 (d, 1H, Ar-H, J = 1.9 Hz), 8.06 (d, 1H, Ar-H, J = 8.3 Hz), 8.04 (d, 1H, Ar-H, J = 8.8 Hz), 7.96–7.93 (m, 2H, Ar-H), 7.90 (dd, 1H, Ar-H, J = 8.8, 2.3 Hz), 7.72–7.59 (m, 4H, Ar-H), 7.06 (d, 1H, CHF, 2JFH = 46.5 Hz). 13C NMR (125 MHz, CDCl3): δ 162.0, 153.1, 137.8, 136.1 (d, 2JCF = 24.3 Hz), 133.9, 133.4, 133.3, 130.5 128.9, 128.6, 128.2, 127.9, 127.7, 126.6, 126.2,124.2, 122.6, 119.4, 119.0, 96.1 (d, 1JCF = 220.2 Hz). 19F NMR (282 MHz, CDCl3): δ –165.5 (d, 2JFH = 45.8 Hz). HRMS (ESI) calcd. for C20H14FN4O2S2 [M + H]+ 425.0537, found 425.0541.
2-{Fluoro[1-(4-phenoxyphenyl)-1H-1,2,3-triazol-4-yl)methyl]sulfonyl}benzo[d]thiazole (5).
![2-{Fluoro[1-(4-phenoxyphenyl)-1H-1,2,3-triazol-4-yl)methyl]sulfonyl}benzo[d]thiazole (5)](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/e1bb/4859785/8274813ae911/nihms779409f3.jpg)
Prepared from 1-azido-4-phenoxybenzene (20.0 mg, 0.095 mmol) and sulfone 1 (total 46.6 mg, 0.143 mmol, 1.50 molar equiv; 0.095 mmol were added initially and 0.048 mmol after 45 min) in a reaction time of 2 h 45 min. Chromatography was performed with CH2Cl2 and compound 5 was obtained as a white solid (39.7 mg, 90%). Rf (40% EtOAc in hexanes) = 0.52. 1H NMR (500 MHz, CDCl3): δ 8.48 (d, 1H, Ar-H, J = 1.9 Hz), 8.33 (d, 1H, Ar-H, J = 8.3 Hz), 8.07 (d, 1H, Ar-H, J = 7.8 Hz), 7.72–7.65 (m, 4H, Ar-H), 7.41 (t, 2H, Ar-H, J = 8.1 Hz), 7.21 (t, 1H, Ar-H, J = 7.4 Hz), 7.16 (d, 2H, Ar-H, J = 8.8 Hz), 7.09 (d, 2H, Ar-H, J = 7.8 Hz), 7.01 (d, 1H, CHF, 2JFH = 46.5 Hz). 13C NMR (125 MHz, CDCl3): δ 161.9, 158.9, 156.2, 153.1 137.8, 135.9 (d, 2JCF = 23.8 Hz), 131.6, 130.3 128.9, 128.2, 126.2, 124.6, 124.1, 122.9, 122.6, 119.9, 119.4, 96.0 (d, 1JCF = 220.2 Hz). 19F NMR (282 MHz, CDCl3): δ –165.7 (d, 2JFH = 45.8 Hz). HRMS (ESI) calcd. for C22H16FN4O3S2 [M + H]+ 467.0642, found 467.0646.
2-{[Fluoro(1-(4-nitrophenyl)-1H-1,2,3-triazol-4-yl)methyl]sulfonyl}benzo[d]thiazole (6).
![2-{[Fluoro(1-(4-nitrophenyl)-1H-1,2,3-triazol-4-yl)methyl]sulfonyl}benzo[d]thiazole (6)](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/e1bb/4859785/c904c932422e/nihms779409f4.jpg)
Prepared from 1-azido-4-nitrobenzene (425 mg, 2.59 mmol) and sulfone 1 (932 mg, 2.85 mmol, 1.10 molar equiv) in a reaction time of 50 h. Chromatography was performed with 40% EtOAc in hexanes and compound 6 was obtained as a yellow solid (889.8 mg, 82%). Rf (40% EtOAc in hexanes) = 0.47. 1H NMR (500 MHz, DMSO-d6): δ 9.57 (s, 1H, Ar-H), 8.47 (d, 2H, Ar-H, J = 8.8 Hz), 8.41 (d, 1H, Ar-H, J = 8.3 Hz), 8.36 (d, 1H, Ar-H, J = 7.4 Hz), 8.30 (d, 2H, Ar-H, J = 8.8 Hz), 7.81–7.76 (m, 2H, Ar-H), 7.72 (d, 1H, CHF, 2JFH = 43.3 Hz). 13C NMR (125 MHz, DMSO-d6): δ 161.8, 152.2, 147.2, 140.2, 137.3, 136.4 (d, 2JCF = 23.8 Hz), 128.8, 128.3, 126.1, 125.5, 125.3, 123.6, 121.3, 95.9 (d, 1JCF = 217.4 Hz). 19F NMR (282 MHz, DMSO-d6): δ –172.0 (d, 2JFH = 42.7 Hz). HRMS (ESI) calcd. for C16H11FN5O4S2 [M + H]+ 420.0231, found 420.0223.
2-{[(1-(Adamantan-1-yl)-1H-1,2,3-triazol-4-yl)fluoromethyl]sulfonyl}benzo[d]thiazole (7).
![2-{[(1-(Adamantan-1-yl)-1H-1,2,3-triazol-4-yl)fluoromethyl]sulfonyl}benzo[d]thiazole (7)](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/e1bb/4859785/59d533179b68/nihms779409f5.jpg)
Prepared from 1-azidoadamantane (177 mg, 1.00 mmol) and sulfone 1 (409 mg, 1.25 mmol, 1.25 molar equiv) in a reaction time of 4 h. Chromatography was peroformed with CH2Cl2 and compound 7 was isolated as a white solid (367 mg 85%). Rf (40% EtOAc in hexanes) = 0.53. 1H NMR (500 MHz, CDCl3): δ 8.26 (d, 1H, Ar-H, J = 8.3 Hz), 8.12 (s, 1H, Ar-H), 8.02 (d, 1H, Ar-H, J = 7.8 Hz), 7.66–7.59 (m, 2H, Ar-H), 6.93 (d, 1H, CHF, 2JFH = 46.5 Hz), 2.25–2.23 (m, 9H), 1.80-1.74 (m, 6H). 13C NMR (125 MHz, CDCl3): δ 162.2, 153.0, 137.7, 134.1 (d, 2JCF = 21.5 Hz), 128.7, 128.1, 126.1, 122.5, 122.3, 96.4 (d, 1JCF = 219.2 Hz), 61.0, 43.0, 35.9, 29.6. 19F NMR (282 MHz, CDCl3): δ –163.8 (d, 2JFH = 45.8 Hz). HRMS (ESI) calcd. for C20H22FN4O2S2 [M+H]+ 433.1163, found 433.1169.
2-{[(1-Ferrocenyl-1H-1,2,3-triazol-4-yl)fluoromethyl]sulfonyl}benzo[d]thiazole (10).
![2-{[(1-Ferrocenyl-1H-1,2,3-triazol-4-yl)fluoromethyl]sulfonyl}benzo[d]thiazole (10)](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/e1bb/4859785/09a824e3cba2/nihms779409f6.jpg)
Prepared from azidoferrocene (160 mg, 0.710 mmol) and sulfone 1 (279 mg, 0.850 mmol, 1.20 molar equiv) in a reaction time of 4 h. Chromatography was performed with 20% EtOAc in hexanes and compound 10 was obtained as a yellowish solid (226 mg, 66%). Rf (40% EtOAc in hexanes) = 0.52. 1H NMR (500 MHz, CDCl3): δ 8.33–8.32 (m, 2H, Ar-H), 8.07 (d, 1H, Ar-H, J = 7.8 Hz), 7.72–7.63 (m, 2H, Ar-H), 6.97 (d, 1H, CHF, 2JFH = 46.5 Hz), 4.88–4.879 (m, 2H, Ar-H), 4.33 (t, 2H, Ar-H, J = 1.8 Hz), 4.25 (s, 5H, Ar-H). 13C NMR (125 MHz, CDCl3): δ 162.0, 153.1, 137.8, 135.2 (d, 2JCF = 24.3 Hz), 128.9, 128.2, 126.2, 125.5, 122.6, 96.1 (d, 1JCF = 219.7 Hz), 93.3, 70.6, 67.4, 62.8. 19F NMR (282 MHz, CDCl3): δ –165.7 (d, 2JFH = 45.8 Hz). HRMS (ESI) calcd. for C20H16FFeN4O2S2 [M+H]+ 483.0043, found 483.0048.
General procedures for condensations of sulfones 4, 6, and 9 with aldehydes
Method A: DBU-mediated condensations
To a stirring solution of the aldehyde (1 molar equiv) and sulfone (1.2–1.7 molar equiv, see specific compound headings for the stoichiometry) in THF (20 mL/mmol of aldehyde), DBU (4 molar equiv) in THF (8 mL/mmol of aldehyde) was added. The reaction mixture was stirred at reflux and monitored for disappearance of the aldehyde. If disappearance of sulfone was observed prior to consumption of the aldehyde, more sulfone was added to the reaction mixture and the reaction was continued at reflux. Upon complete consumption of the aldehyde, the solvent was removed under reduced pressure. The product E/Z ratio was determined by 19F NMR, prior to purification by column chromatography. The combined E/Z product mixture was purified by column chromatography over silica gel. The quantities of reactants and solvents, reaction times, eluting solvents for chromatography, product yields, Rf values, and spectroscopic data are provided under the individual compound headings.
Method B: LHMDS-mediated condensations
A stirring solution of aldehyde (1 molar equiv) and sulfone (1.2 molar equiv) in 1:1 (v/v) DMF-DMPU (15.2 mL/mmol of aldehyde) was cooled to –78 °C (dry ice/iPrOH), under a nitrogen atmosphere. LHMDS (1.0 M solution in THF, 2.40 molar equiv) was then added to the mixture. The reaction mixture was stirred at –78 °C for 5 min, saturated aq NH4Cl was added, and the mixture was poured into EtOAc. The organic layer was separated and the aqueous layer was extracted with EtOAc (3×). The combined organic layer was washed with water and brine, dried over anhydrous Na2SO4, and evaporated under reduced pressure. The product E/Z ratio was determined by 19F NMR, prior to purification by column chromatography. The combined E/Z product mixture was purified by column chromatography over silica gel. The quantities of reactants and solvents, eluting solvents for chromatography, product yields, Rf values, and spectroscopic data are provided under the individual compound headings.
(E/Z)-4-[1-Fluoro-2-(naphthalen-2-yl)vinyl]-1-(4-methoxyphenyl)-1H-1,2,3-triazole (11).
![(E/Z)-4-[1-Fluoro-2-(naphthalen-2-yl)vinyl]-1-(4-methoxyphenyl)-1H-1,2,3-triazole (11)](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/e1bb/4859785/58312db3cf96/nihms779409f7.jpg)
Prepared by method B using 2-naphthaldehyde (20.0 mg, 0.128 mmol), sulfone 4 (62.3 mg, 0.154 mmol, 1.2 molar equiv), LHMDS (0.307 mL, 0.307 mmol, 2.4 molar equiv), in 1:1 (v/v) DMF-DMPU (2.0 mL). The E/Z ratio was determined to be 7/93. Chromatography was performed using 20% EtOAc in hexanes and E/Z-11 was obtained as a white solid (33.4 mg, 76%). Rf (20% EtOAc in hexanes) = 0.23. 1H NMR (500 MHz, CDCl3): δ 8.09 (s, 1H, Ar-H, Z-isomer), 8.05 (s, 1H, Ar-H, Z-isomer), 7.96 (s, 1H, Ar-H, E-isomer), 7.87–7.78 (m, 4H, Ar-H, both E and Z-isomers), 7.68 (d, 2H, Ar-H, J = 8.5 Hz, Z-isomer), 7.59 (d, 1H, Ar-H, J = 8.5 Hz, E-isomer), 7.55 (d, 2H, Ar-H, J = 8.6 Hz, E-isomer), 7.50–7.46 (m, 2H, Ar-H, both E and Z-isomers), 7.05 (d, 2H, Ar-H, J = 8.2 Hz, Z-isomer), 6.99 (d, 2H, Ar-H, J = 8.9 Hz, E-isomer), 6.98 (d, 1H, CHF, 3JFH = 41.2 Hz, Z-isomer), 6.78 (d, 1H, CHF, 3JFH = 22.3 Hz, E-isomer), 3.89 (s, 3H, CH3, Z-isomer), 3.85 (s, 3H, CH3, E-isomer). 19F NMR (282 MHz, CDCl3): δ –105.98 (d, 3JFH = 24.4 Hz, E-isomer), –118.47 (d, 3JFH = 39.7 Hz, Z-isomer). HRMS (ESI) calcd. for C21H17FN3O [M+H]+ 346.1350, found 346.1334.
(E/Z)-4-[1-Fluoro-2-(4-methoxyphenyl)vinyl]-1-(4-methoxyphenyl)-1H-1,2,3-triazole (12).
![(E/Z)-4-[1-Fluoro-2-(4-methoxyphenyl)vinyl]-1-(4-methoxyphenyl)-1H-1,2,3-triazole (12)](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/e1bb/4859785/f10ef4059f69/nihms779409f8.jpg)
Prepared by method A using 4-methoxybenzaldehyde (68.1 mg, 0.500 mmol), sulfone 4 (total 283.1 mg, 0.700 mmol, 1.4 molar equiv; 0.6 mmol was added first and 0.1 mmol was added after 4.5 h), and DBU (304.5 mg, 2.00 mmol, 4.0 molar equiv) in THF (14 mL), in a reaction time of 6.5 h. The E/Z ratio was determined to be 81/19. Chromatography was performed using 20% EtOAc in hexanes and E/Z-12 was obtained as a white solid (130.1 mg, 80%). Rf (20% EtOAc in hexanes) = 0.22. 1H NMR (500 MHz, CDCl3): δ 7.98 (s, 1H, Ar-H, Z-isomer), 7.81 (s, 1H, Ar-H, E-isomer), 7.66 (d, 2H, Ar-H, J = 9.2 Hz, Z-isomer), 7.61–7.57 (m, 2H, Ar-H, both E and Z-isomers), 7.46 (d, 2H, Ar-H, J = 8.8 Hz, E-isomer), 7.04 (d, 2H, Ar-H, J = 9.2 Hz, Z-isomer), 7.01 (d, 2H, Ar-H, J = 9.2 Hz, E-isomer), 6.92 (d, 2H, Ar-H, J = 8.8 Hz, Z-isomer), 6.87 (d, 2H, Ar-H, J = 8.8 Hz, E-isomer), 6.75 (d, 1H, CHF, 3JFH = 41.4 Hz, Z-isomer), 6.57 (d, 1H, CHF, 3JFH = 23.0 Hz, E-isomer), 3.88 (s, 3H, CH3, Z-isomer), 3.86 (s, 3H, CH3, E-isomer), 3.84 (s, 3H, CH3, Z-isomer), 3.81 (s, 3H, CH3, E-isomer). 19F NMR (282 MHz, CDCl3): δ –108.51 (d, 3JFH = 21.4 Hz, E-isomer), –121.54 (d, 3JFH = 42.7 Hz, Z-isomer). HRMS (ESI) calcd. for C18H17FN3O2 [M+H]+ 326.1299, found 326.1304.
(E/Z)-4-[1-Fluoro-2-(4-methoxyphenyl)vinyl]-1-(4-nitrophenyl)-1H-1,2,3-triazole (13).
![(E/Z)-4-[1-Fluoro-2-(4-methoxyphenyl)vinyl]-1-(4-nitrophenyl)-1H-1,2,3-triazole (13)](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/e1bb/4859785/47569a33ceec/nihms779409f9.jpg)
Prepared by method A using 4-methoxybenzaldehyde (20.5 mg, 0.150 mmol), sulfone 6 (75.5 mg, 0.180 mmol, 1.2 molar equiv), and DBU (91.3 mg, 0.600 mmol, 4.0 molar equiv), in THF (4.3 mL), in a reaction time of 3 h. The E/Z ratio was determined to be 87/13. Chromatography was performed using CH2Cl2 and E/Z-13 was obtained as a pale yellow solid (41.9 mg, 82%). Rf (20% EtOAc in hexanes) = 0.29. 1H NMR (500 MHz, DMSO-d6): δ 9.34 (s, 1H, Ar-H, Z-isomer), 9.31 (s, 1H, Ar-H, E-isomer), 8.49–8.46 (m, Ar-H, 2H, both E and Z-isomers), 8.30–8.27 (m, Ar-H, 2H, both E and Z-isomers), 7.64 (d, 2H, Ar-H, J = 8.8 Hz, Z-isomer), 7.46 (d, 2H, Ar-H, J = 8.8, E-isomer), 7.01 (d, 2H, Ar-H, J = 8.7 Hz, Z-isomer), 6.90 (d, 2H, Ar-H, J = 8.7 Hz, E-isomer), 6.783 (d, 1H, CHF, 3JFH = 24.0 Hz, E-isomer), 6.778 (d, 1H, CHF, 3JFH = 40.1 Hz, Z-isomer), 3.80 (s, 3H, Z-isomer), 3.75 (s, 3H, E-isomer). 19F NMR (282 MHz, CDCl3): δ –109.57 (d, 3JFH = 21.4 Hz, E-isomer), –122.23 (d, 3JFH = 39.7 Hz, Z-isomer). HRMS (ESI) calcd. for C17H14FN4O3 [M +H]+ 341.1044, found 341.1043.
(E/Z)-4-[1-Fluoro-2-(4-methoxyphenyl)vinyl]-1-(3-phenylpropyl)-1H-1,2,3-triazole (14).
![(E/Z)-4-[1-Fluoro-2-(4-methoxyphenyl)vinyl]-1-(3-phenylpropyl)-1H-1,2,3-triazole (14)](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/e1bb/4859785/e48b80716996/nihms779409f10.jpg)
Prepared by method A using 4-methoxybenzaldehyde (20 mg, 0.15 mmol), sulfone 9 (total 106.5 mg, 0.26 mmol, 1.7 molar equiv; 0.21 mmol was added first and 0.05 mmol was added after 8 h), and DBU (91.3 mg, 0.60 mmol, 4.0 molar equiv), in THF (4.2 mL), in a reaction time of 20 h. The E/Z ratio was determined to be 76/24. Chromatography was performed using 20% EtOAc in hexanes and E/Z-14 was obtained as a white solid (38.2 mg, 75%). Rf (20% EtOAc in hexanes) = 0.29. 1H NMR (500 MHz, CDCl3): δ 7.60 (s, 1H, Ar-H, Z-isomer), 7.58 (d, 2H, Ar-H, J = 8.8 Hz, Z-isomer), 7.39 (d, 2H, Ar-H, J = 8.8 Hz, E-isomer), 7.38 (s, 1H, E-isomer), 7.33–7.18 (m, Ar-H, 3H, E-isomer and 5H Z-isomer), 7.14 (d, 2H, Ar-H, J = 7.8 Hz, E-isomer), 6.91 (d, 2H, Ar-H, J = 8.8 Hz, Z-isomer), 6.85 (d, 2H, Ar-H, J = 8.3 Hz, E-isomer), 6.67 (d, 1H, CHF, 3JFH = 41.4 Hz, Z-isomer), 6.52 (d, 1H, CHF, 3JFH = 22.6 Hz, E-isomer), 4.39 (t, 2H, CH2, J = 6.9 Hz, Z-isomer), 4.32 (t, 2H, CH2, J = 6.9 Hz, E-isomer), 3.83 (s, 3H, CH3, Z-isomer), 3.80 (s, 3H, CH3, E-isomer), 2.69 (t, 2H, CH2, J = 7.4 Hz, Z-isomer), 2.63 (t, 2H, CH2, J = 7.4 Hz, E-isomer), 2.29 (quint, 2H, CH2, J = 7.2 Hz, Z-isomer), 2.23 (quint, 2H, CH2, J = 7.4 Hz, E-isomer). 19F NMR (282 MHz, CDCl3): δ –108.20 (d, 3JFH = 21.4 Hz, E-isomer), –121.30 (d, 3JFH = 42.7 Hz, Z-isomer). HRMS (ESI) calcd. for C20H21FN3O [M + H]+ 338.1663, found 338.1667.
(E/Z)-4-(1-Fluoro-2-mesitylvinyl)-1-(3-phenylpropyl)-1H-1,2,3-triazole (15).
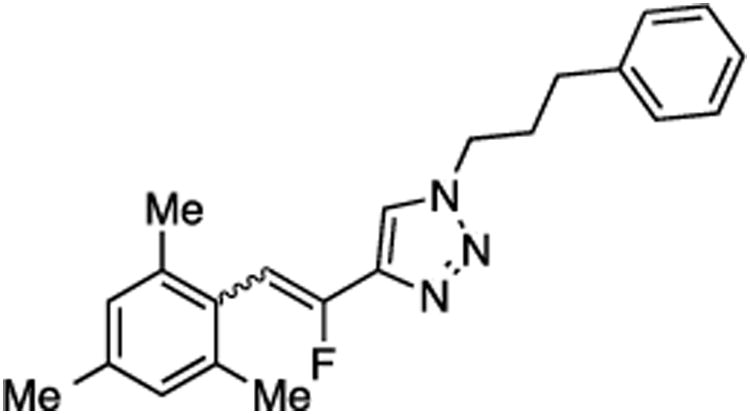
Prepared by method B using 2,4,6-trimethylbenzaldehyde (15.0 mg, 0.10 mmol), sulfone 9 (50.4 mg, 0.12 mmol, 1.2 molar equiv), and LHMDS (0.240 mL, 0.242 mmol, 2.4 molar equiv), in 1:1 (v/v) DMF-DMPU (1.52 mL). The E/Z ratio was determined to be 26/74. Chromatography was performed using 20% EtOAc in hexanes and E/Z-15 was obtained as a white solid (16.7 mg, 47%). Rf (20% EtOAc in hexanes) = 0.45. 1H NMR (500 MHz, CDCl3): δ 7.61 (s, 1H, Ar-H, Z-isomer), 7.33–7.19 (m, Ar-H, 3H E-isomer and 5H Z-isomer), 7.07 (d, 2H, Ar-H, J = 7.3 Hz, E-isomer), 6.92 (s, 2H, Ar-H, both E and Z-isomers), 6.76 (d, 1H, CHF, 3JFH = 41.5 Hz, Z-isomer), 6.59 (s, 1H, Ar-H, E-isomer), 6.36 (d, 1H, CHF, 3JFH = 18.5 Hz, E-isomer), 4.40 (t, 2H, CH2, J = 7.1 Hz, Z-isomer), 4.17 (t, 2H, CH2, J = 6.8 Hz, E-isomer), 2.71 (t, 2H, CH2, J = 7.3 Hz, Z-isomer), 2.49 (t, 2H, CH2, J = 7.3 Hz, E-isomer), 2.33–2.27 (m, 3H E-isomer and 11H Z-isomers), 2.15 (s, 6H, E-isomer, 2CH3), 2.10 (quint, 2H, E-isomer, J = 7.2 Hz). 19F NMR (282 MHz, CDCl3): δ –114.17 (d, 3JFH = 18.3 Hz, E-isomer), –115.50 (d, 3JFH = 42.7 Hz, Z-isomer). HRMS (ESI) calcd. for C22H25FN3 [M + H]+ 350.2027, found 350.2034.
(E/Z)-4-{1-Fluoro-2-[4-(trifluoromethyl)phenyl]vinyl}-1-(4-methoxyphenyl)-1H-1,2,3-triazole (16).
![(E/Z)-4-{1-Fluoro-2-[4-(trifluoromethyl)phenyl]vinyl}-1-(4-methoxyphenyl)-1H-1,2,3-triazole (16)](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/e1bb/4859785/26e911e88e01/nihms779409f12.jpg)
Prepared by method A using 4-(trifluoromethyl)benzaldehyde (20.0 mg, 0.115 mmol), sulfone 4 (55.8 mg, 0.138 mmol, 1.2 molar equiv), and DBU (70.0 mg, 0.460 mmol, 4.0 molar equiv), in THF (3.2 mL), in a reaction time of 4h. The E/Z ratio was determined to be 64/36. Chromatography was performed using 10% EtOAc in hexanes and E/Z-16 was obtained as a white solid (21.6 mg, 52%). Rf (10% EtOAc in hexanes) = 0.21. 1H NMR (500 MHz, CDCl3): δ 8.06 (s, 1H, Ar-H, Z-isomer), 7.94 (s, 1H, Ar-H, E-isomer), 7.75 (d, 2H, Ar-H, J = 8.3 Hz, Z-isomer), 7.68 (d, 2H, Ar-H, J = 7.8 Hz, E-isomer), 7.65 (d, 2H, Ar-H, J = 7.4 Hz, Z-isomer), 7.62–7.58 (m, Ar-H, 4H E-isomer and 2H Z-isomer), 7.05 (d, 2H, Ar-H, J = 8.8 Hz, Z-isomer), 7.03 (d, 2H, Ar-H, J = 9.2 Hz, E-isomer), 6.86 (d, 1H, CHF, 3JFH = 40.5 Hz, Z-isomer), 6.61 (d, 1H, CHF, 3JFH = 23.0 Hz, E-isomer), 3.89 (s, 3H, CH3, Z-isomer), 3.87 (s, 3H, CH3, E-isomer). 19F NMR (282 MHz, CDCl3): δ –63.17 (CF3, both E and Z-isomers), –103.52 (d, 3JFH = 21.4 Hz, E-isomer), –116.10 (d, 3JFH = 39.7 Hz, Z-isomer). HRMS (ESI) calcd. for C18H14F4N3O [M+H]+ 364.1068, found 364.1068.
(E/Z)-4-{1-Fluoro-2-[4-(trifluoromethyl)phenyl]vinyl}-1-(4-nitrophenyl)-1H-1,2,3-triazole (17).
![(E/Z)-4-{1-Fluoro-2-[4-(trifluoromethyl)phenyl]vinyl}-1-(4-nitrophenyl)-1H-1,2,3-triazole (17)](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/e1bb/4859785/b45814f11fb0/nihms779409f13.jpg)
Prepared by method A using 4-(trifluoromethyl)benzaldehyde (15.0 mg, 0.086 mmol), sulfone 6 (43.0 mg, 0.103 mmol, 1.2 molar equiv), and DBU (52.0 mL, 0.344 mmol, 4.0 molar equiv), in THF (2.5 mL), in a reaction time of 4h. The E/Z ratio was determined to be 60/40. Chromatography was performed using 50% CH2Cl2 in hexanes and E/Z-17 was obtained as a white solid (28.5 mg, 87%). Rf (10% EtOAc in hexanes) = 0.19. 1H NMR (500 MHz, CDCl3): δ 8.48–8.44 (m, Ar-H, 2H, both E and Z-isomers), 8.26 (s, 1H, Ar-H, Z-isomer), 8.17 (s, 1H, Ar-H, E-isomer), 8.03 (d, 2H, Ar-H, J = 8.8 Hz, Z-isomer), 7.97 (d, 2H, Ar-H, J = 8.8 Hz, E-isomer), 7.77 (d, 2H, Ar-H, J = 7.8 Hz, Z-isomer), 7.66–7.60 (m, Ar-H, 4H E-isomer and 2H Z-isomer), 6.92 (d, 1H, CHF, 3JFH = 40.1 Hz, Z-isomer), 6.69 (d, 1H, CHF, 3JFH = 23.0 Hz, E-isomer). 19F NMR (282 MHz, CDCl3): δ –63.20 (CF3, both E and Z-isomers), –104.80 (d, 3JFH = 21.4 Hz, E-isomer), –116.81 (d, 3JFH = 39.7 Hz, Z-isomer). HRMS (ESI) calcd. for C17H10F4N4NaO2 [M + Na]+ 401.0632, found 401.0616.
(E/Z)-4-{1-Fluoro-2-[4-(trifluoromethyl)phenyl]vinyl}-1-(3-phenylpropyl)-1H-1,2,3-triazole (18).
![(E/Z)-4-{1-Fluoro-2-[4-(trifluoromethyl)phenyl]vinyl}-1-(3-phenylpropyl)-1H-1,2,3-triazole (18)](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/e1bb/4859785/d41bfdbd1816/nihms779409f14.jpg)
Prepared by method A using 4-(trifluoromethyl)benzaldehyde (20 mg, 0.115 mmol), sulfone 9 (57.0 mg, 0.14 mmol, 1.2 molar equiv), and DBU (70.0 mg, 0.46 mmol, 4.0 molar equiv), in THF (3.2 mL), in a reaction time of 4 h. The E/Z ratio was determined to be 63/37. Chromatography was performed 50% CH2Cl2 in hexanes and E/Z-18 was obtained as a white solid (34.2 mg, 80%). Rf (10% EtOAc in hexanes) = 0.35. 1H NMR (500 MHz, CDCl3): δ 7.72 (d, 2H, Ar-H, J = 8.3 Hz, Z-isomer), 7.69 (s, 1H, Ar-H, Z-isomer), 7.62 (d, 2H, Ar-H, J = 7.4 Hz, both E and Z-isomers), 7.57 (d, 2H, Ar-H, J = 8.3 Hz, E-isomer), 7.53 (s, 1H, Ar-H, E-isomer), 7.34–7.21 (m, 3H, Ar-H, both E and Z-isomers), 7.19 (d, 2H, J =7.4 Hz, Z-isomer), 7.15 (d, 2H, Ar-H, J = 7.4 Hz, E-isomer), 6.78 (d, 1H, CHF, 3JFH = 40.5 Hz, Z-isomer), 6.55 (d, 1H, CHF, 3JFH = 23.0 Hz, E-isomer), 4.41 (t, 2H, J = 7.1 Hz, Z-isomer), 4.36 (t, 2H, CH2, J = 7.1 Hz, E-isomer), 2.69 (t, 2H, J = 7.4 Hz, Z-isomer), 2.64 (t, 2H, CH2, J = 7.4 Hz, E-isomer), 2.30 (quint, 2H, J = 7.4 Hz, Z-isomer), 2.25 (quint, 2H, CH2, J = 7.4 Hz, E-isomer). 19F NMR (282 MHz, CDCl3): δ –63.14 (s, CF3), –102.93 (d, 3JFH = 21.4 Hz, E-isomer), –115.81 (d, 3JFH = 39.7 Hz, Z-isomer). HRMS (ESI) calcd. for C20H18F4N3 [M + H]+ 376.1431, found 376.1427.
(E/Z)-4-[1-Fluoro-2-(4-nitrophenyl)vinyl]-1-(3-phenylpropyl)-1H-1,2,3-triazole (19).
![(E/Z)-4-[1-Fluoro-2-(4-nitrophenyl)vinyl]-1-(3-phenylpropyl)-1H-1,2,3-triazole (19)](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/e1bb/4859785/b40943dd81fb/nihms779409f15.jpg)
Prepared by method B using 4-nitrobenzaldehyde (75.6 mg, 0.50 mmol), sulfone 9 (249.9 mg, 0.60 mmol, 1.2 molar equiv), and LHMDS (1.20 mL, 1.20 mmol, 2.4 molar equiv), in 1:1 (v/v) DMF-DMPU (7.6 mL). The E/Z ratio was determined to be 38/62. Chromatography was performed using 40% EtOAc in hexanes and E/Z-19 was obtained as a white solid (133.9 mg, 76%). Rf (20% EtOAc in hexanes) = 0.12. 1H NMR (500 MHz, CDCl3): δ 8.22 (d, 2H, Ar-H, J = 8.8 Hz, Z-isomer), 8.17 (d, 2H, Ar-H, J = 8.8 Hz, E-isomer), 7.75 (d, 2H, Ar-H, J = 8.3 Hz, Z-isomer), 7.74 (d, 2H, Ar-H, J = 8.3 Hz, E-isomer), 7.72 (s, 1H, Ar-H, Z-isomer), 7.64 (s, 1H, Ar-H, E-isomer), 7.33–7.29 (m, 2H, Ar-H, both E and Z-isomers), 7.25–7.22 (m, 1H, Ar-H, both E and Z-isomers), 7.21–7.16 (m, 2H, Ar-H, both E and Z-isomers), 6.82 (d, 1H, CHF, 3JFH = 39.6 Hz, Z-isomer), 6.55 (d, 1H, CHF, 3JFH = 23.5 Hz, E-isomer), 4.42 (t, 2H, J = 7.1 Hz, Z-isomer), 4.38 (t, 2H, CH2, J = 7.4 Hz, E-isomer), 2.71–2.65 (m, 2H, both E and Z-isomers), 2.34–2.25 (m, 2H, both E and Z-isomers). 19F NMR (282 MHz, CDCl3): δ –100.64 (d, 3JFH = 21.4 Hz, E-isomer), –113.37 (d, 3JFH = 39.7 Hz, Z-isomer). HRMS (ESI) calcd. for C19H18FN4O2 [M + H]+ 353.1408, found 353.1411.
(E/Z)-4-(1-Fluoronon-1-en-1-yl)-1-(4-methoxyphenyl)-1H-1,2,3-triazole (20).
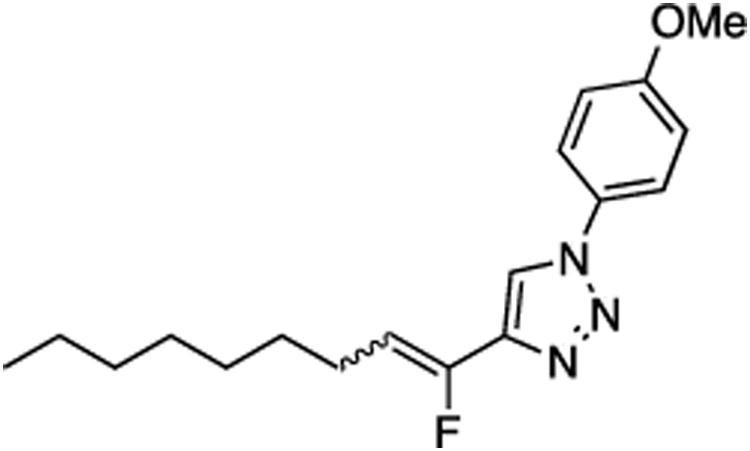
Prepared by method B using n-octanal (20.0 mg, 0.160 mmol), sulfone 4 (76.8 mg, 0.190 mmol, 1.2 molar equiv), and LHMDS (0.384 mL, 0.384 mmol, 2.4 molar equiv), in 1:1 (v/v) DMF-DMPU (2.4 mL). The E/Z ratio was determined to be 34/66. Chromatography was performed using 20% EtOAc in hexanes and E/Z-20 was obtained as a colorless semi solid (34.7 mg, 68%). Rf (20% EtOAc in hexanes) = 0.40. 1H NMR (500 MHz, CDCl3): δ 7.94 (s, 1H, Ar-H, E-isomer), 7.86 (s, 1H, Ar-H, Z-isomer), 7.65–7.62 (m, 2H, Ar-H, both E and Z-isomers), 7.04–7.02 (m, 2H, Ar-H, both E and Z-isomers), 5.85 (dt, 1H, CHF, 3JFH = 39.1, 3JHH = 7.8 Hz, Z-isomer), 5.54 (dt, 1H, CHF, 3JFH = 22.6, 3JHH = 8.3 Hz, E-isomer), 3.87 (s, 3H, both E and Z-isomers), 2.66 (q, 2H, J = 7.4 Hz, E-isomer), 2.30 (q, 2H, J = 7.2 Hz, Z-isomer), 1.50 (quint, 2H, J = 7.4 Hz, both E and Z-isomers), 1.40–1.26 (m, 8H, both E- and Z-isomers) 0.90–0.86 (m, 3H, both E and Z-isomers). 19F NMR (282 MHz, CDCl3): δ –115.56 (d, 3JFH = 21.4 Hz, E-isomer), –124.67 (d, 3JFH = 39.7 Hz, Z-isomer). HRMS (ESI) calcd. for C18H25FN3O [M+H]+ 318.1976, found 318.1983.
(E/Z)-4-(1-Fluoronon-1-en-1-yl)-1-(4-nitrophenyl)-1H-1,2,3-triazole (21).
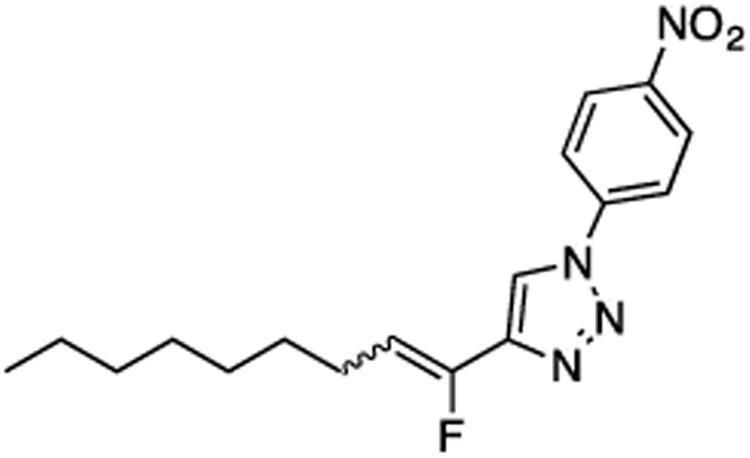
Prepared by method B using n-octanal (20.0 mg, 0.160 mmol), sulfone 6 (79.7 mg, 0.19 mmol, 1.2 molar equiv), and LHMDS (0.380 mL, 0.380 mmol, 2.4 molar equiv), in 1:1 (v/v) DMF-DMPU (2.2 mL). The E/Z ratio was determined to be 32/68. Chromatography was performed using 20% EtOAc in hexanes and E/Z-21 was obtained as a pale yellow solid (32.5 mg, 61%). Rf (20% EtOAc in hexanes) = 0.48. 1H NMR (500 MHz, CDCl3): δ 8.45–8.42 (m, 2H, Ar-H, both E and Z-isomers), 8.14 (s, 1H, Ar-H, E-isomer), 8.05 (s, 1H, Ar-H, Z-isomer), 8.01–7.97 (m, 2H, Ar-H, both E and Z-isomers), 5.93 (dt, 1H, CHF, 3JFH = 39.0 Hz, 3JHH = 7.8 Hz, Z-isomer), 5.62 (dt, 1H, CHF, 3JFH = 22.9 Hz, 3JHH = 8.3 Hz, E-isomer), 2.68 (q, 2H, CH2, J = 7.7 Hz, E-isomer), 2.32 (q, 2H, CH2, J = 7.5 Hz, Z-isomer), 1.50 (quint, 2H, CH2, J = 7.2 Hz, both E and Z-isomers), 1.41–1.29 (m, 8 H, both E and Z-isomers), 0.88 (app q, 3H, CH3, J = 6.8 Hz, both E and Z-isomers). 19F NMR (282 MHz, CDCl3): δ –116.23 (d, 3JFH = 21.4 Hz, E-isomer), –125.21 (d, 3JFH = 39.7 Hz, Z-isomer). HRMS (ESI) calcd. for C17H22FN4O2 [M +H]+ 333.1721, found 333.1719.
(E/Z)-4-(1-Fluoronon-1-en-1-yl)-1-(3-phenylpropyl)-1H-1,2,3-triazole (22).
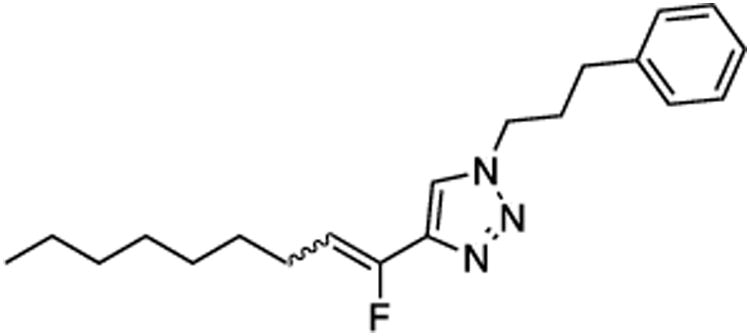
Prepared by method B using n-octanal (20.0 mg, 0.160 mmol), sulfone 9 (79.1 mg, 0.19 mmol, 1.2 molar equiv), and LHMDS (0.380 mL, 0.380 mmol, 2.4 molar equiv), in DMF-DMPU (v/v) (2.2 mL). The E/Z ratio was determined to be 36/64. Chromatography was performed using 20% EtOAc in hexanes and E/Z-22 was obtained as a pale yellow solid (32.9 mg, 63%). Rf (20% EtOAc in hexanes) = 0.2. 1H NMR (500 MHz, CDCl3): δ 7.56 (s, 1H, Ar-H, E-isomer), 7.48 (s, 1H, Ar-H, Z-isomer), 7.31 (t, 2H, Ar-H, J = 7.4 Hz, both E and Z-isomers), 7.22 (t, 1H, Ar-H, J = 7.1 Hz, both E and Z-isomers), 7.19–7.17 (m, 2H, Ar-H, both E and Z-isomers), 5.77 (dt, 1H, CHF, 3JFH = 39.1 Hz, 3JHH = 7.8 Hz, Z-isomer), 5.48 (dt, 1H, CHF, 3JFH = 22.6 Hz, 3JHH = 8.1 Hz, E-isomer), 4.38–4.34 (m, 2H, both E and Z-isomers), 2.67 (q, 2H, J = 7.1 Hz, both E and Z-isomers), 2.60 (q, 2H, J = 7.7 Hz, E-isomer), 2.30–2.23 (m, 2H E-isomer and 4H Z-isomer), 1.49–1.43 (m, 2H, both E and Z-isomers), 1.37–1.23 (m, 8 H, both E and Z-isomers), 0.90–0.86 (m, 3H, CH3, both E and Z-isomers). 19F NMR (282 MHz, CDCl3): δ –115.34 (d, 3JFH = 21.4 Hz, E-isomer), –124.51 (d, 3JFH = 36.6 Hz, Z-isomer). HRMS (ESI) calcd. for C20H28FN3Na [M+Na]+ 352.2159, found 352.2161.
(E/Z)-4-(3-Ethyl-1-fluoropent-1-en-1-yl)-1-(4-methoxyphenyl)-1H-1,2,3-triazole (23).
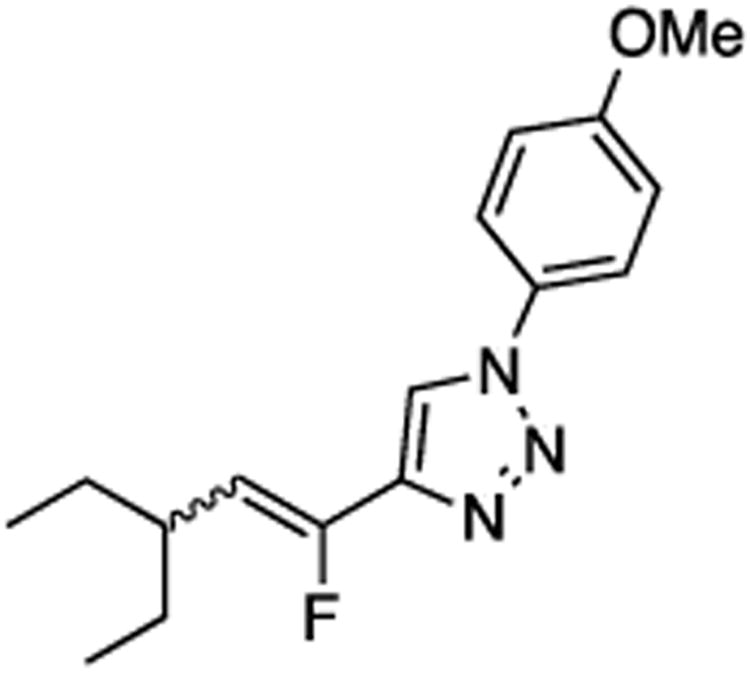
Prepared by method B using 2-ethylbutanal (15.0 mg, 0.15 mmol), sulfone 4 (72.8 mg, 0.18 mmol, 1.2 molar equiv), and LHMDS (0.360 mL, 0.360 mmol, 2.4 molar equiv), in DMF-DMPU (v/v) (2.2 mL). The E/Z ratio was determined to be 15/85. Chromatography was performed using 20% EtOAc in hexanes and E/Z-23 was obtained as a colorless semi solid (25.7 mg, 59%). Rf (20% EtOAc in hexanes) = 0.39. 1H NMR (500 MHz, CDCl3): δ 7.94 (s, 1H, Ar-H, E-isomer), 7.87 (s, 1H, Ar-H, Z-isomer), 7.66–7.62 (m, 2H, Ar-H, both E and Z-isomers), 7.03 (d, 2H, Ar-H, J = 8.8 Hz, both E and Z-isomers), 5.61 (dd, 1H, CHF, 3JFH = 39.1 Hz, 3JHH = 10.6 Hz, Z-isomer), 5.27 (dd, 1H, CHF, 3JFH = 24.0 Hz, 3JHH = 11.0 Hz, E-isomer), 3.87 (s, 3H, both E and Z-isomers), 3.31–3.23 (m, 1H, E-isomer), 2.57–2.49 (m, 1H, Z-isomer), 1.60–1.52 (m, 2H, both E and Z-isomers), 1.40–1.30 (m, 2H, both E and Z-isomers), 0.93 (t, 6H, J = 7.6 Hz, both E and Z-isomers). 19F NMR (282 MHz, CDCl3): δ –114.37 (d, 3JFH = 21.4 Hz, E-isomer), –124.68 (d, 3JFH = 39.7 Hz, Z-isomer). HRMS (ESI) calcd. for C16H21FN3O [M+H]+ 290.1663, found 290.1666.
(E/Z)-4-(3-Ethyl-1-fluoropent-1-en-1-yl)-1-(4-nitrophenyl)-1H-1,2,3-triazole (24).
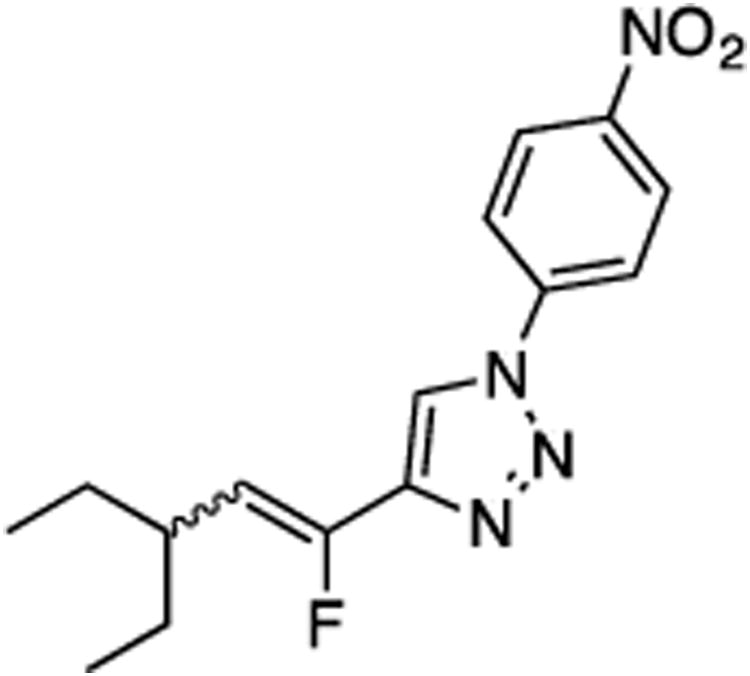
Prepared by method B using 2-ethylbutanal (15.0 mg, 0.15 mmol), sulfone 6 (75.5 mg, 0.18 mmol, 1.2 molar equiv), and LHMDS (0.360 mL, 0.360 mmol, 2.4 molar equiv), in DMF-DMPU (v/v) (2.0 mL). The E/Z ratio was determined to be 12/88. Chromatography was performed using 20% EtOAc in hexanes and compound E/Z-24 was obtained as a pale yellow solid (31.0 mg, 68%). Rf (20% EtOAc in hexanes) = 0.53. 1H NMR (500 MHz, CDCl3): δ 8.43 (d, 2H, Ar-H, J = 8.9 Hz, both E and Z-isomers), 8.14 (s, 1H, Ar-H, E-isomer), 8.05 (s, 1H, Ar-H, Z-isomer), 8.01–7.98 (m, 2H, Ar-H, both E and Z-isomers), 5.70 (dd, 1H, CHF, 3JFH = 39.4 Hz, 3JHH = 10.4 Hz, Z-isomer), 5.35 (dd, 1H, CHF, 3JFH = 24.0 Hz, 3JHH = 11.1 Hz, E-isomer), 3.34–3.24 (m, 1H, E-isomer), 2.59–2.51 (m, 1H, Z-isomer), 1.64–1.54 (m, 2H, both E and Z-isomers), 1.41–1.32 (m, 2H, both E and Z-isomers), 0.93 (t, 6H, J = 7.5 Hz, both E and Z-isomers). 19F NMR (282 MHz, CDCl3): δ –114.94 (d, 3JFH = 24.4 Hz, E-isomer), –125.07 (d, 3JFH = 39.7 Hz, Z-isomer). HRMS (ESI) calcd. for C15H18FN4O2 [M +H]+ 305.1408, found 305.1414.
(E/Z)-4-(3-Ethyl-1-fluoropent-1-en-1-yl)-1-(3-phenylpropyl)-1H-1,2,3-triazole (25).
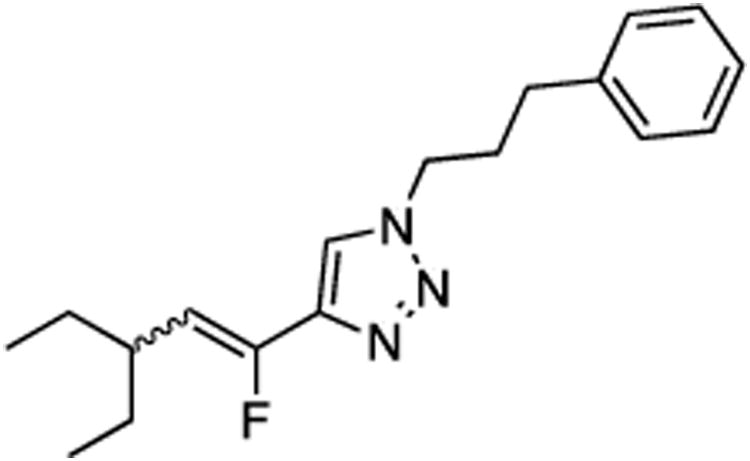
Prepared by method B using 2-ethylbutanal (20.0 mg, 0.20 mmol), sulfone 9 (100.0 mg, 0.24 mmol, 1.2 molar equiv), and LHMDS (0.480 mL, 0.480 mmol, 2.4 molar equiv), in DMF-DMPU (v/v) (3.0 mL). The E/Z ratio was determined to be 19/81. Chromatography was performed using 20% EtOAc in hexanes and compound E/Z-25 was obtained as a white solid (34.5 mg, 57%). Rf (20% EtOAc in hexanes) = 0.21. 1H NMR (500 MHz, CDCl3): δ 7.55 (s, 1H, Ar-H, E-isomer), 7.49 (s, 1H, Ar-H, Z-isomer), 7.31 (t, 2H, Ar-H, J =7.4 Hz, both E and Z-isomers), 7.22 (t, 1H, Ar-H, J = 7.4 Hz, both E and Z-isomers), 7.18 (d, 2H, Ar-H, J = 7.4 Hz, both E and Z-isomers), 5.53 (dd, 1H, CHF, 3JFH = 39.6 Hz, 3JHH = 10.1 Hz, Z-isomer), 5.21 (dd, 1H, CHF, 3JFH = 23.7 Hz, 3JHH = 11.3 Hz, E isomer), 4.36 (t, 2H, CH2, J = 7.1 Hz, both E and Z-isomers), 3.22–3.14 (m, 1H, CH, E-isomer), 2.67 (t, 2H, CH2, J = 7.4 Hz, both E and Z-isomers), 2.54–2.46 (m, 1H, CH, Z-isomer), 2.31–2.23 (m, 2H, CH2, both E and Z-isomers), 1.57–1.50 (m, 2H, CH2, both E and Z-isomers), 1.37–1.26 (m, 2H, CH2, both E and Z-isomers), 0.93–0.88 (m, 6H, 2CH3, both E and Z-isomers). 19F NMR (282 MHz, CDCl3): δ –113.99 (d, 3JFH = 24.4 Hz, E-isomer), –124.55 (d, 3JFH = 39.7 Hz, Z-isomer). HRMS (ESI) calcd. for C18H25FN3 [M + H]+ 302.2027, found 302.2031.
General procedure for condensations of sulfones 4, 9, and 10 with ketones
A stirring solution of the ketone (1 molar equiv) and sulfone (1.2-1.3 molar equiv) in THF (12 mL/mmol of ketone, except for compound 30, see below) was cooled to 0 °C under a nitrogen atmosphere. LHMDS (2.40 molar equiv in every case and 4.0 molar equiv for compound 30, see below) was added to the mixture. The reaction mixture was then stirred at 0 °C and was monitored for disappearance of the ketone. Saturated aq NH4Cl was added and the mixture was poured into EtOAc. The organic layer was separated and the aqueous layer was extracted with EtOAc (3×). The combined organic layer was washed with water and brine, dried over anhydrous Na2SO4, and evaporated under reduced pressure. The product E/Z ratio was determined by 19F NMR, prior to purification by column chromatography. The combined E/Z product mixture was isolated by column chromatography over silica gel. The quantities of reactants and solvents, reaction times, eluting solvents for chromatography, product yields, Rf values, and spectroscopic data are provided under the individual compound headings.
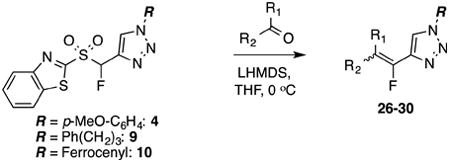
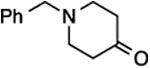
1-Benzyl-4-{fluoro[1-(4-methoxyphenyl)-1H-1,2,3-triazol-4-yl]methylene}piperidine (26)11a.
![1-Benzyl-4-{fluoro[1-(4-methoxyphenyl)-1H-1,2,3-triazol-4-yl]methylene}piperidine (26)11a](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/e1bb/4859785/082a28399e41/nihms779409f22.jpg)
Prepared from 1-benzylpiperidin-4-one (20.0 mg, 0.11 mmol), sulfone 4 (56.6 mg, 0.14 mmol, 1.3 molar equiv), and LHMDS (0.260 mL, 0.260 mmol, 2.4 molar equiv), in THF (1.3 mL), in a reaction time of 5 min. Chromatography was performed using 40% EtOAc in hexanes to obtain compound 26 as a semi solid (36.2 mg, 87%). Rf (30% EtOAc in hexanes) = 0.20. 1H NMR (500 MHz, CDCl3): δ 7.92 (s, 1H, Ar-H), 7.63 (d, 2H, Ar-H, J = 9.3 Hz), 7.36–7.32 (m, 4H, Ar-H), 7.28–7.25 (m, 1H, Ar-H), 7.02 (d, 2H, Ar-H, J = 8.3 Hz), 3.87 (s, 3H, CH3), 3.57 (s, 2H, CH2), 3.00 (t, 2H, CH2, J = 4.9 Hz), 2.59–2.54 (m, 6H, 3CH2). 13C NMR (125 MHz, CDCl3): δ 160.2, four resonances at 143.4, 143.2, 142.9, 141.6 (two doublets for 2C), 138.4, 130.4, 129.4 (2C), 128.4 (2C), 127.3, 122.4 (2C), 119.9, 118.9 (d, JCF = 15.1 Hz), 115.0 (2C), 63.1, 55.8, 54.3, 53.8, 27.0 (d, JCF = 4.6 Hz), 26.2 (d, JCF = 7.3 Hz). 19F NMR (282 MHz, CDCl3): δ –121.99 (s). HRMS (ESI) calcd. for C22H24FN4O [M +H]+ 379.1929, found 379.1924.
1-Benzyl-4-{fluoro[1-(3-phenylpropyl)-1H-1,2,3-triazol-4-yl]methylene}piperidine (27).
![1-Benzyl-4-{fluoro[1-(3-phenylpropyl)-1H-1,2,3-triazol-4-yl]methylene}piperidine (27)](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/e1bb/4859785/17249e79e522/nihms779409f23.jpg)
Prepared from 1-benzylpiperidin-4-one (20.0 mg, 0.11 mmol), sulfone 9 (52.8 mg, 0.13 mmol, 1.2 molar equiv), and LHMDS (0.264 mL, 0.264 mmol, 2.4 molar equiv), in THF (1.3 mL), in a reaction time of 2 h. Chromatography was performed using 20% EtOAc in hexanes to obtain compound 27 as an off-white semi solid (25.0 mg, 58%). Rf (20% EtOAc in hexanes) = 0.23. 1H NMR (500 MHz, CDCl3): δ 7.53 (s, 1H, Ar-H), 7.36–7.25 (m, 7H, Ar-H), 7.22 (t, 1H, Ar-H, J = 7.4 Hz), 7.18 (d, 2H, Ar-H, J = 7.8 Hz), 4.36 (t, 2H, CH2, J = 6.9 Hz), 3.55 (s, 2H, CH2), 2.94 (t, 2H, CH2, J = 4.8 Hz), 2.66 (t, 2H, CH2, J = 7.6 Hz), 2.56–2.52 (m, 6H), 2.26 (quint, 2H, CH2, J = 7.2 Hz). 13C NMR (125 MHz, CDCl3): δ 142.8 (d, JCF = 227.9 Hz), 142.7 (d, JCF = 40.3 Hz), 140.3 138.8, 129.4 (2C), 128.9 (2C), 128.7 (2C), 128.4 (2C), 127.2, 126.7, 121.6, 118.5 (d, JCF = 14.8 Hz), 63.2, 54.4, 53.9, 49.7, 32.8, 31.7, 27.2 (d, JCF = 4.6 Hz), 26.3 (d, JCF = 7.8 Hz). 19F NMR (282 MHz, CDCl3): δ –121.52 (s). HRMS (ESI) calcd. for C24H28FN4 [M + H]+ 391.2293, found 391.2298.
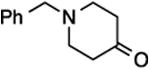
(E/Z)-4-(1-Fluoro-2-phenylprop-1-en-1-yl)-1-(4-methoxyphenyl)-1H-1,2,3-triazole (28).
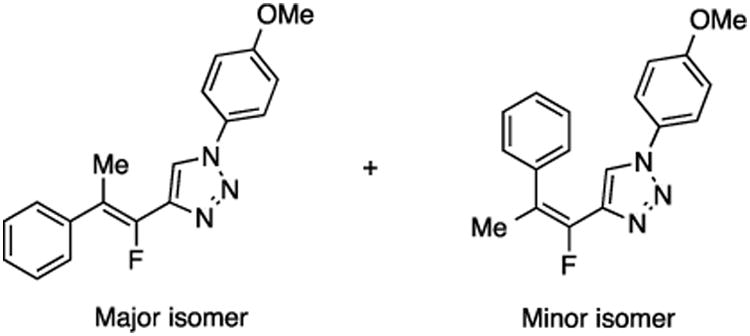
Prepared from acetophenone (20.0 mg, 0.17 mmol), sulfone 4 (80.9 mg, 0.20 mmol, 1.2 molar equiv), and LHMDS (0.410 mL, 0.410 mmol, 2.4 molar equiv), in THF (2.0 mL), in a reaction time of 15 min. The major/minor isomer ratio was determined to be 72:28. Chromatography was performed using 20% EtOAc in hexanes and compound E/Z-28 was obtained as a white solid (33.0 mg, 64%). Major isomer: Rf (20% EtOAc in hexanes) = 0.27. 1H NMR (500 MHz, CDCl3): δ 8.04 (s, 1H, Ar-H), 7.67 (d, 2H, Ar-H, J = 8.8 Hz), 7.50 (d, 2H, Ar-H, J = 7.8 Hz), 7.41 (t, 2H, Ar-H, J = 7.6 Hz), 7.31 (t, 1H, Ar-H, J = 7.4 Hz), 7.04 (d, 2H, Ar-H, J = 9.2 Hz), 3.87 (s, 3H, CH3), 2.57 (d, 3H, CH3, J = 3.7 Hz). 13C NMR (125 MHz, CDCl3): δ 160.2, four resonances at 145.8, 143.9, 143.7, 143.4 (two doublets for 2C), 138.6, 130.3, 128.5 (d, 2C, JCF = 4.1 Hz), 128.4 (2C), 127.5, 122.4 (2C), 120.5, 118.0 (d, JCF = 12.4 Hz), 115.1 (2C), 55.9, 17.5 (d, JCF = 3.7 Hz). 19F NMR (282 MHz, CDCl3): δ –116.57 (s). Minor isomer: Rf (20% EtOAc in hexanes) = 0.22. 1H NMR (500 MHz, CDCl3): δ 7.40–7.32 (m, 5H, Ar-H), 7.27–7.25 (m, 2H, Ar-H), 6.94 (d, 2H, Ar-H, J = 9.2 Hz), 6.91 (s, 1H, Ar-H), 3.83 (s, 3H, CH3), 2.21 (d, 3H, CH3, J = 3.7 Hz). 13C NMR (125 MHz, CDCl3): δ 160.1, 145.9 (d, JCF = 240.8 Hz), 141.5 (d, JCF = 35.0 Hz), 139.4 (d, JCF = 7.3 Hz), 130.2, 129.2 (2C), 128.8 (d, 2C, JCF = 2.7 Hz), 128.1, 122.3 (2C), 120.6 (d, JCF = 5.1 Hz), 119.7 (d, JCF = 19.2 Hz), 114.9 (2C), 55.8, 17.8 (d, JCF = 6.0 Hz). 19F NMR (282 MHz, CDCl3): δ –116.46 (s). HRMS (ESI) calcd. for C18H17FN3O [M + H]+ 310.1350, found 310.1353.
(E/Z)-4-(1-Fluoro-2-phenylprop-1-en-1-yl)-1-(3-phenylpropyl)-1H-1,2,3-triazole (29).
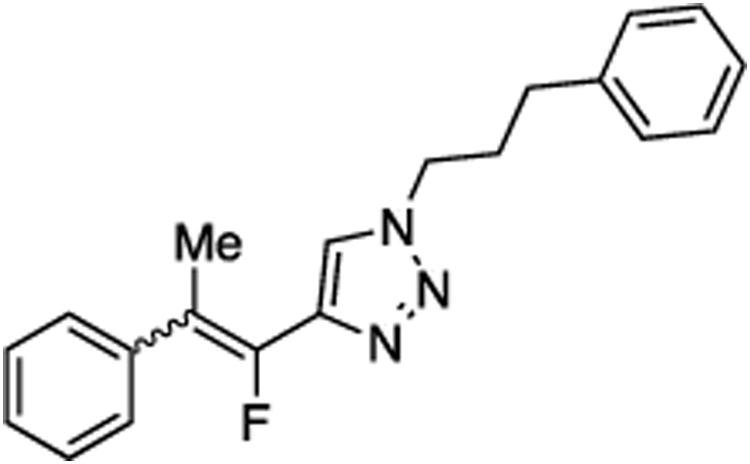
Prepared from acetophenone (20.0 mg, 0.17 mmol), sulfone 9 (83.2 mg, 0.20 mmol, 1.2 molar equiv), and LHMDS (0.410 mL, 0.410 mmol, 2.4 molar equiv), in THF (2.0 mL), in a reaction time of 30 min. The major/minor isomer ratio was determined to be 80:20. Chromatography was performed using 20% EtOAc in hexanes to obtain the major isomer Z-29 as a white solid (33.0 mg, 60%), and the minor isomer E-29 as a white solid (9.2 mg, 17%). Analysis of the major isomer by X-ray diffraction showed Z stereochemistry of the alkene (crystals were obtained by slow evaporation from a solution in methylene chloride).
Major isomer Z-29:
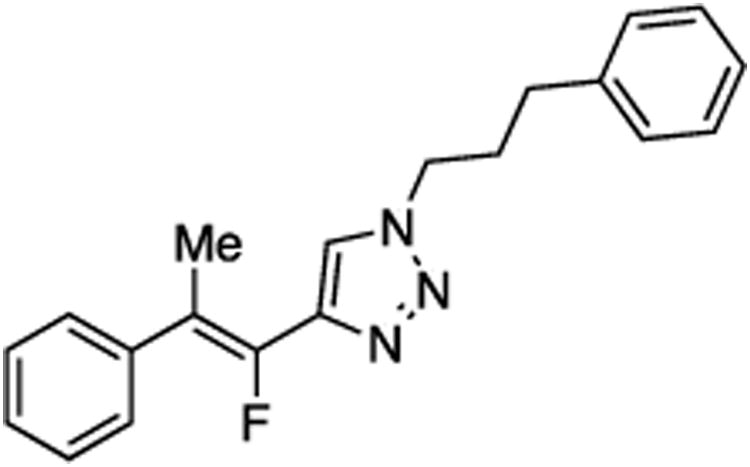
Rf (20% EtOAc in hexanes) = 0.20. 1H NMR (500 MHz, CDCl3): δ 7.65 (s, 1H, Ar-H), 7.48 (d, 2H, Ar-H, J = 7.4 Hz), 7.39 (t, 2H, Ar-H, J = 7.6 Hz), 7.33–7.28 (m, 3H, Ar-H), 7.24–7.19 (m, 3H, Ar-H), 4.40 (t, 2H, CH2, J = 7.1 Hz), 2.69 (t, 2H, CH2, J = 7.4 Hz), 2.52 (d, 3H, CH3, J = 3.2 Hz), 2.30 (quint, 2H, CH2, J = 7.2 Hz). 13C NMR (125 MHz, CDCl3): δ 145.1 (d, JCF = 233.9 Hz), 143.0 (d, JCF = 39.4 Hz), 140.2, 138.6, 128.9 (2C), 128.6 (2C), 128.4 (d, 2C, JCF = 4.1 Hz), 128.3 (2C), 127.5, 126.6, 122.3 (d, JCF = 2.3 Hz), 117.4 (d, JCF = 13.3 Hz), 49.7, 32.6, 31.8, 17.4 (d, JCF = 4.1 Hz). 19F NMR (282 MHz, CDCl3): δ –116.33 (s).
Minor isomer E-29:
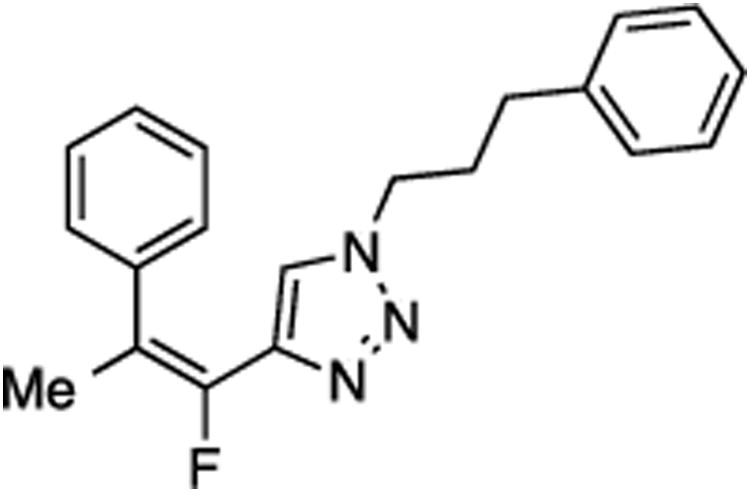
Rf (20% EtOAc in hexanes) = 0.10. 1H NMR (500 MHz, CDCl3): δ 7.36–7.31 (m, 3H, Ar-H), 7.28 (t, 2H, Ar-H, J = 7.4 Hz), 7.22–7.19 (m, 3H, Ar-H), 7.06 (d, 2H, Ar-H, J = 7.4 Hz), 6.54 (s, 1H, Ar-H), 4.15 (t, 2H, CH2, J = 6.7 Hz), 2.48 (t, 2H, CH2, J = 7.4 Hz), 2.18 (d, 3H, CH3, J = 4.1 Hz), 2.07 (quint, 2H, CH2, J = 7.2 Hz). 13C NMR (125 MHz, CDCl3): δ 146.1 (d, JCF = 241.2 Hz), 141.0 (d, JCF = 33.4 Hz), 140.2, 139.6 (d, JCF = 7.3 Hz) 129.1 (2C), 128.82 (2C), 128.77 (d, 2C, JCF = 2.7 Hz), 128.6 (2C), 127.9, 126.6, 122.6 (d, JCF = 5.5 Hz), 119.3 (d, JCF = 19.7 Hz), 49.4, 32.4, 31.6, 17.7 (d, JCF = 6.0 Hz). 19F NMR (282 MHz, CDCl3): δ –115.56 (s). HRMS (ESI) calcd. for C20H20FN3Na [M + Na]+ 344.1533, found 344.1536.
(E) or (Z)-4-[(2,3-Dihydro-1H-inden-1-ylidene)fluoromethyl]-1-ferrocenyl-1H-1,2,3-triazole (30).
![(E) or (Z)-4-[(2,3-Dihydro-1H-inden-1-ylidene)fluoromethyl]-1-ferrocenyl-1H-1,2,3-triazole (30)](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/e1bb/4859785/24953286ede1/nihms779409f28.jpg)
Prepared from 1-indanone (20.0 mg, 0.15 mmol), sulfone 10 (87.0 mg, 0.18 mmol, 1.2 molar equiv), and LHMDS (total 0.600 mL, 0.600 mmol, 4.0 molar equiv; 2.4 molar equiv was added first and 1.6 molar equiv was added after 2h), in THF (5.0 mL), in a reaction time of 4 h. Chromatography was performed using 20% EtOAc in hexanes, with stepwise increase to 40% EtOAc in hexanes, and compound 30 was obtained as a yellow-orange solid (35.1 mg, 58%). Rf (40% EtOAc in hexanes) = 0.48. Formation of only one isomer was detected by 1H and 19F NMR, and olefin stereochemistry was not determined. 1H NMR (500 MHz, CDCl3): δ 7.91 (d, 1H, Ar-H, J = 6.8 Hz), 7.90 (s, 1H), 7.32 (d, 1H, Ar-H, J = 6.4 Hz), 7.29–7.24 (m, 2H), 4.88 (t, 2H, J = 1.9 Hz), 4.30 (t, 2H, J = 1.9 Hz), 4.25 (s, 5H), 3.34 (td, 2H, J = 6.8; 2.4 Hz), 3.17 (br t, 2H, J = 7.3 Hz). 13C NMR (125 MHz, CDCl3): δ 146.8, 143.9 (d, 1JCF = 236.2 Hz), 143.6 (d, JCF = 38.0 Hz), 139.1 (d, JCF = 2.7 Hz), 128.4 (d, JCF = 2.3 Hz), 127.0, 126.0 (d, JCF = 14.2 Hz), 125.1, 124.0 (d, JCF = 11.0 Hz), 120.2, 93.7, 70.5, 67.0, 62.4, 31.3, 28.3 (d, JCF = 5.5Hz). 19F NMR (282 MHz, CDCl3): δ –123.76 (s). HRMS (ESI) calcd. for C22H19FFeN3 [M + H]+ 400.0907, found 400.0944.
Single-Crystal X-ray Diffraction for (Z)-29
The intensity data for (Z)-29 were measured on a KappaCCD diffractometer (graphite-monochromated Mo Kα radiation, λ = 0.71073 Å, ϕ-ω scans) at 100 (1) K. The data were not corrected for absorption. Details of the solution and refinements for C20H20FN3 (Z-29) are as follows. The crystals of (Z)-29, with approximate dimensions 0.10 × 0.28 × 0.30 mm, were monoclinic with space group P2/c. The final unit-cell constants of (Z)-29 were a = 18.306(4) b = 5.6030(11), c = 16.578(3) Å, β = 101.41(3)°, V = 1666.8(6) Å3, Z = 4, ρ = 1.281 g cm-1, μ = 0.085 mm-1, formula weight = 321.39. The structure of Z-29 was solved with SHELXS-97 and refined by full-matrix least squares on F2 with SHELXL-97. The hydrogen atoms were calculated with the riding model in the structure-factor calculations, but their parameters were not refined. The final discrepancy indices, 2.95 < θ < 27.47°, were R = 0.0610 (calculated on F for 2290 reflections) and Rw = 0.1395 (calculated on F2 for all 3804 reflections) with 219 parameters varied. The major peaks of the final difference map are – 0.29 and + 0.22 e Å3.
Supplementary Material
Acknowledgments
This work was supported by National Science Foundation Grant CHE-1058618 and a PSC CUNY award. Infrastructural support was provided by National Institutes of Health Grant 8G12MD007603 from the National Institute on Minority Health and Health Disparities. We thank Dr. Andrew Poss (Honeywell) for a sample of NFSI.
Footnotes
Supporting Information Available: Copies of 1H and 13C NMR spectra.
References
- 1.(a) Huisgen R. Angew Chem, Int Ed. 1963;2:565. [Google Scholar]; (b) Huisgen R. Angew Chem, Int Ed. 1963;2:633. [Google Scholar]
- 2.For reviews see: Kolb HC, Sharpless KB. Drug Discovery Today. 2003;8:1128. doi: 10.1016/s1359-6446(03)02933-7.Moses JE, Moorhouse AD. Chem Soc Rev. 2007;36:1249. doi: 10.1039/b613014n.Angell YL, Burgess K. Chem Soc Rev. 2007;36:1674. doi: 10.1039/b701444a.Lutz JF. Angew Chem, Int Ed. 2007;46:1018. doi: 10.1002/anie.200604050.Binder WH, Sachsenhofer R. Macromol Rapid Commun. 2007;28:15.Beghdadi S, Miladi IA, Addis D, Romdhane HB, Bernard J, Drockenmuller E. Polym Chem. 2012;3:1680.
- 3.Schulze B, Schubert US. Chem Soc Rev. 2014;43:2522. doi: 10.1039/c3cs60386e. [DOI] [PubMed] [Google Scholar]
- 4.For reviews see: Kolb HC, Finn MG, Sharpless KB. Angew Chem, Int Ed. 2001;40:2004. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5.Gil MV, Arévalo MJ, López Ó. Synthesis. 2007:1589.Tornøe CW, Meldal M. Chem Rev. 2008;108:2952. doi: 10.1021/cr0783479.
- 5.(a) Tornøe CW, Christensen C, Meldal M. J Org Chem. 2002;67:3057. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]; (b) Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem, Int Ed. 2002;41:2596. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 6.For reviews see: Agalave SG, Maujan SR, Pore VS. Chem Asian J. 2011;6:2696. doi: 10.1002/asia.201100432.Kharb R, Sharma PC, Yar MS. J Enzyme Inhib Med Chem. 2011;26:1. doi: 10.3109/14756360903524304.Shalini K, Kumar N, Drabu S, Sharma PK. Beilstein J Org Chem. 2011;7:668. doi: 10.3762/bjoc.7.79.Siddiqui N, Ahsan W, Alam MS, Ali R, Jain S, Azad B, Akhtar J. Int J Pharm Sci Rev Res. 2011;8:161.Zhou CH, Wang Y. Curr Med Chem. 2012;19:239. doi: 10.2174/092986712803414213.
- 7.(a) De Simone R, Chini MG, Bruno I, Riccio R, Mueller D, Werz O, Bifulco G. J Med Chem. 2011;54:1565. doi: 10.1021/jm101238d. [DOI] [PubMed] [Google Scholar]; (b) Guo L, Ye C, Chen W, Ye H, Zheng R, Li J, Yang H, Yu X, Zhang D. J Pharmacol Exp Ther. 2008;325:10. doi: 10.1124/jpet.107.131888. [DOI] [PubMed] [Google Scholar]; (c) Gupte A, Boshoff HI, Wilson DJ, Neres J, Labello NP, Somu RV, Xing C, Barry CE, III, Aldrich CC. J Med Chem. 2008;51:7495. doi: 10.1021/jm8008037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Zhu Y, Huang Y, Meng WD, Li H, Qing FL. Polymer. 2006;47:6272. [Google Scholar]; (b) Malkoch M, Vestberg R, Gupta N, Mespouille L, Dubois P, Mason AF, Hedrick JL, Liao Q, Frank CW, Kingsbury K, Hawker CJ. Chem Commun. 2006:2774. doi: 10.1039/b603438a. [DOI] [PubMed] [Google Scholar]
- 9.(a) Kirsch P. Modern Fluoroorganic Chemistry. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim: 2004. [Google Scholar]; (b) Laali KK, editor. Modern Organofluorine Chemistry–Synthetic Aspects. Vol. 2 Bentham Science Publishers; 2006. [Google Scholar]; (c) Bégué JP, Bonnet-Delpon D. Bioorganic and Medicinal Chemistry of Fluorine. John Wiley & Sons, Inc.; Hoboken, NJ: 2008. [Google Scholar]; (d) O'Hagan D. Chem Soc Rev. 2008;37:308. [Google Scholar]; (e) Liang T, Neumann CN, Ritter T. Angew Chem, Int Ed. 2013;52:8214. doi: 10.1002/anie.201206566. [DOI] [PubMed] [Google Scholar]
- 10.For earlier references see review on Julia-Kocienski fluoroolefination: Zajc B, Kumar R. Synthesis. 2010:1822. doi: 10.1055/s-0029-1218789.
- 11.For recent examples see: Kumar R, Pradhan P, Zajc B. Chem Commun. 2011;47:3891. doi: 10.1039/c0cc05083k.Mandal SK, Ghosh AK, Kumar R, Zajc B. Org Biomol Chem. 2012;10:3164. doi: 10.1039/c2ob07031f.Kumar R, Zajc B. J Org Chem. 2012;77:8417. doi: 10.1021/jo300971w.Chowdhury M, Mandal SK, Banerjee S, Zajc B. Molecules. 2014;19:4418. doi: 10.3390/molecules19044418.
- 12.See for example: Prakash GKS, Shakhmin A, Zibinsky M, Ledneczki I, Chacko S, Olah GA. J Fluorine Chem. 2010;131:1192.Allendörfer N, Es-Sayed M, Nieger M, Bräse S. Synthesis. 2010:3439.Calata C, Pfund E, Lequeux T. Tetrahedron. 2011;67:1398.Jacobsen CB, Nielsen M, Worgull D, Zweifel T, Fisker E, Jørgensen KA. J Am Chem Soc. 2011;133:7398. doi: 10.1021/ja110624k.Larnaud F, Malassis J, Pfund E, Linclau B, Lequeux T. Org Lett. 2013;15:2450. doi: 10.1021/ol400917j.Larnaud F, Pfund E, Linclau B, Lequeux T. Tetrahedron. 2014;70:5632.Cao CR, Ou S, Jiang M, Liu JT. Org Biomol Chem. 2014;12:467. doi: 10.1039/c3ob42093k.
- 13.(a) Landelle G, Bergeron M, Turcotte-Savard MO, Paquin JP. Chem Soc Rev. 2011;40:2867. doi: 10.1039/c0cs00201a. [DOI] [PubMed] [Google Scholar]; (b) Yanai H, Taguchi T. Eur J Org Chem. 2011:5939. [Google Scholar]
- 14.For reviews see: Blakemore PR. J Chem Soc, Perkin Trans I. 2002:2563.Plesniak K, Zarecki A, Wicha J. Top Curr Chem. 2007;275:163. doi: 10.1007/128_049.Aïssa C. Eur J Org Chem. 2009:1831.Blakemore PR. Olefination of carbonyl compounds by main-group element mediators. In: Knochel P, Molander G, editors. Comprehensive Organic Synthesis. 2nd. Elsevier Ltd; Oxford: 2013.
- 15.(a) Bégué JP, Bonnet-Delpon D. J Fluorine Chem. 2006;127:992. [Google Scholar]; (b) Müller K, Faeh C, Diederich F. Science. 2007;317:1881. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]; (c) Wang J, Sánchez-Roselló M, Aceña JL, del Pozo C, Sorochinsky AE, Fustero S, Soloshonok VA, Liu H. Chem Rev. 2014;114:2432. doi: 10.1021/cr4002879. [DOI] [PubMed] [Google Scholar]
- 16.(a) Allmendinger T, Furet P, Hungerbühler E. Tetrahedron Lett. 1990;31:7297. [Google Scholar]; (b) Allmendinger T, Furet P, Hungerbühler E. Tetrahedron Lett. 1990;31:7301. [Google Scholar]; (c) Welch JT, editor. Selective Fluorination in Organic and Bioorganic Chemistry. American Chemical Society; Washington, DC: 1991. [Google Scholar]; (d) Zhao K, Lim DS, Funaki T, Welch JT. Bioorg Med Chem. 2003;11:207. doi: 10.1016/s0968-0896(02)00384-x. [DOI] [PubMed] [Google Scholar]; (e) Choudhary A, Raines RT. ChemBioChem. 2011;12:1801. doi: 10.1002/cbic.201100272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiong Y, Zhang X, Huang T, Cao S. J Org Chem. 2014;79:6395. doi: 10.1021/jo5005845. [DOI] [PubMed] [Google Scholar]
- 18.Blakemore PR, Cole WJ, Kocienski PJ, Morley A. Synlett. 1998:26. [Google Scholar]
- 19.Bonini C, Chiummiento L, Videtta V. Synlett. 2005:3067. [Google Scholar]
- 20.Bonini C, Chiummiento L, Videtta V. Synlett. 2006:2079. [Google Scholar]
- 21.X-ray crystallographic data for Z-29 have been deposited at the Cambridge Crystallographic Data Centre and allocated the deposition number CCDC 1028722. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif
- 22.(a) Grimes KD, Gupte A, Aldrich CC. Synthesis. 2010:1441. doi: 10.1055/s-0029-1218683. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tao CZ, Cui X, Li J, Liu AX, Liu L, Guo QX. Tetrahedron Lett. 2007;48:3525. [Google Scholar]
- 23.Ouchi A, Awen BZS, Hatsuda R, Ogura R, Ishii T, Araki Y, Ito O. J Phys Chem A. 2004;108:9584. [Google Scholar]
- 24.Tennyson AG, Khramov DM, Varnado CD, Jr, Creswell PT, Kamplain JW, Lynch VM, Bielawski CW. Organometallics. 2009;28:5142. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.