

Abstract
MicroRNAs (miRNAs) are critical regulators of gene expression. Amplification and overexpression of individual ‘oncomiRs’ or genetic loss of tumour suppressor miRNAs are associated with human cancer and are sufficient to drive tumorigenesis in mouse models. Furthermore, global miRNA depletion caused by genetic and epigenetic alterations in components of the miRNA biogenesis machinery is oncogenic. This, together with the recent identification of novel miRNA regulatory factors and pathways, highlights the importance of miRNA dysregulation in cancer.
MicroRNAs (miRNAs) repress gene expression by binding to complementary sequences in the 3′ untranslated region (3′ UTR) of mRNAs to target them for degradation and thereby prevent their translation1. Considering that more than 1,000 individual miRNA genes have been identified, that an individual miRNA can target hundreds or thousands of different mRNAs, and that an individual mRNA can be coordinately suppressed by multiple different miRNAs, the miRNA biogenesis pathway therefore has an important role in gene regulatory networks. Over the past decade, it has emerged that miRNAs have crucial roles in cancer. Propelled by the original publication that described the deletion of the miR-15 and miR-16 loci in the majority of samples from patients with B cell chronic lymphocytic leukaemia (B-CLL), a plethora of subsequent publications described altered miRNA expression in diverse types of cancer2,3. Functionally, it has been shown through both loss-of-function and gain-of-function experiments in human cancer cells, mouse xenografts, transgenic mouse models and knockout mouse models that miRNAs have key roles in cancer initiation, progression and metastasis4,5. The first example was provided by enforced expression of the miR-17~92 cluster, the so-called oncomiR-1, that acted with MYC to accelerate tumour development in a mouse model of B cell lymphoma6. Certain other miRNAs can function as tumour suppressors: for example, the let-7 family of miRNAs targets important oncogenes such as MYC, RAS family members (HRAS, KRAS and NRAS) and high-mobility group AT-hook 2 (HMGA2) to suppress tumour growth7–9. Therefore, cancer-associated changes in miRNA expression patterns are emerging as promising diagnostic markers that often correlate with disease progression and patient survival. This pathway might also represent a new therapeutic target for multiple types of cancer2. Mechanistically, miRNAs can control cell proliferation, differentiation, survival, metabolism, genome stability, inflammation, invasion and angiogenesis to affect tumour development.
Although individual miRNAs can have either oncogenic or tumour-suppressive function, several studies have shown that miRNA expression is globally suppressed in tumour cells compared with normal tissue, suggesting that miRNA biogenesis might be impaired in cancer10,11. Indeed, the expression levels of miRNAprocessing machinery components such as the ribonuclease III (RNase III) DROSHA and DICER1 are decreased in some cancers, such as lung cancer, ovarian cancer and neuroblastoma12–14. Additionally, low DROSHA or DICER1 expression levels are associated with advanced tumour stage and poor clinical outcome in patients with neuroblastoma and patients with ovarian cancer13,14. Support that this global suppression can have a causative role in cancer was initially provided by the demonstration that genetic deficiency of components of the miRNA biogenesis pathway can accelerate tumour growth in a mouse model of lung cancer15. Although this work provided proof-of-concept that the miRNA biogenesis pathway can have an important role in cancer progression, it is the recently reported mutations in and dysregulation of miRNA biogenesis pathway components that highlight the pathophysiological relevance of the miRNA biogenesis machinery in human tumours16–24. Moreover, the recent discovery of certain molecular and cellular mechanisms that control miRNA biogenesis provided compelling evidence that disruption of this pathway is crucially important for a wide variety of paediatric and adult cancers.
In this Review, we discuss what is known about dysregulation of the miRNA biogenesis pathway in cancer, summarize the growing evidence that germline mutations and somatic mutations in core components of the miRNA biogenesis machinery promote oncogenesis, and provide specific examples of how certain RNA-binding proteins and cell signalling pathways contribute to cancer through their control of miRNA expression. With these examples, we aim to highlight emerging themes and the relevance of the miRNA biogenesis pathway in cancer.
miRNAs and their biogenesis
miRNAs are a group of short non-coding RNAs that mediate post-transcriptional gene silencing. The first miRNA was reported in Caenorhabditis elegans in 1993 (REF. 25); however, the general regulatory function of miRNAs was not well appreciated until 2001 (REFS 26–28). Since then, thousands of miRNAs have been identified in various species29. Binding of the ~22-nucleo tide miRNA to target mRNA mediates mRNA degradation and blocks translation30. The majority of miRNA genes are transcribed by RNA polymerase II (Pol II) in the nucleus, and the primary miRNAs (pri-miRNAs) are capped, spliced and polyadenylated31. Approximately 30% of miRNAs are processed from introns of proteincoding genes, whereas most other miRNAs are expressed from dedicated miRNA gene loci. An individual primiRNA can either produce a single miRNA or contain clusters of two or more miRNAs that are processed from a common primary transcript. Nonetheless, these long pri-miRNAs are cleaved by Microprocessor, which comprises the double-stranded RNase III enzyme DROSHA and its essential cofactor, the double-stranded RNA (dsRNA)-binding protein DiGeorge syndrome critical region 8 (DGCR8)32,33. DROSHA contains two RNase III domains, each of which cleaves one strand of the dsRNA towards the base of stem–loop secondary structures contained within pri-miRNAs to liberate ~60–70-nucleotide hairpin-shaped precursor miRNAs (pre-miRNAs)32–35. Microprocessor recognizes the single-stranded RNA (ssRNA)–stem junction as well as the distance from the terminal loop region. It specifically cleaves the dsRNA ~11 bp from the junction with the flanking ssRNA to produce hairpin-shaped pre-miRNAs with an overhang at the 3′ end of either 2 nucleotides (group I miRNAs) or 1 nucleotide (group II miRNAs)36–39. Although the core components, DROSHA and DGCR8, are required for the biogenesis of almost all miRNAs in the cell, and Microprocessor activity can be reconstituted in vitro with recombinant DROSHA and DGCR8 proteins32,35, numerous accessory factors are known to have a role in pri-miRNA processing in cells (discussed in more detail below). The pre-miRNAs are then exported from the nucleus to the cytoplasm by exportin 5 (XPO5)40–42 and further processed by DICER1, an RNase III enzyme that measures from the 5′ and 3′ ends of the pre-miRNA43. DICER1 binding to the end of the pre-miRNA positions its two catalytic RNase III domains so that asymmetrical cleavage of the dsRNA stem, close to the terminal loop sequence, produces the mature ~22-nucleotide miRNA duplex with 2-nucleotide 3′ overhangs44. DICER1 associates with transactivation-responsive RNA-binding protein (TRBP; also known as TARBP2), which binds to dsRNA45. Although it is not required for pre-miRNA processing by DICER1, TRBP enhances the fidelity of DICER1-mediated cleavage of a subset of pre-miRNAs in a structure-dependent manner and alters miRNA guide-strand selection by triggering the formation of isomiRNAs, which are 1 nucleotide longer than the regular miRNAs46,47. TRBP also physically bridges DICER1 with the Argonaute proteins (AGO1, AGO2, AGO3 or AGO4) to participate in the assembly of the miRNAinduced silencing complex (miRISC)45. One strand of the mature miRNA (the guide strand) is bound by an Argonaute protein and retained in the miRISC to guide the complex, together with members of the GW182 family of proteins, to complementary target mRNAs for post-transcriptional gene silencing. This occurs in processing bodies (P-bodies), which are the cytoplasmic foci that are induced by mRNA silencing and decay but are not necessarily required for miRNA-mediated gene silencing48–50 (FIG. 1).
Figure 1. Overview of miRNA biogenesis pathway.
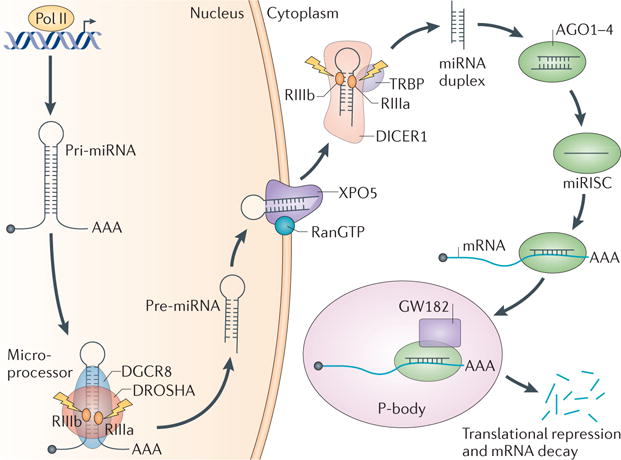
MicroRNA (miRNA) genes are transcribed as primary miRNAs (pri-miRNAs) by RNA polymerase II (Pol II) in the nucleus. The long pri-miRNAs are cleaved by Microprocessor, which includes DROSHA and DiGeorge syndrome critical region 8 (DGCR8), to produce the 60–70-nucleotide precursor miRNAs (pre-miRNAs). The pre-miRNAs are then exported from the nucleus to the cytoplasm by exportin 5 (XPO5) and further processed by DICER1, a ribonuclease III (RIII) enzyme that produces the mature miRNAs. One strand of the mature miRNA (the guide strand) is loaded into the miRNA-induced silencing complex (miRISC), which contains DICER1 and Argonaute (AGO) proteins, directs the miRISC to target mRNAs by sequence complementary binding and mediates gene suppression by targeted mRNA degradation and translational repression in processing bodies (P-bodies). TRBP, transactivation-responsive RNA-binding protein.
Pri-miRNA transcription in cancer
miRNA biogenesis initiates with the transcription of the pri-miRNA, and this step is dysregulated in multiple human cancers. A considerable number of human miRNA genes are located at fragile sites or in genomic regions that are deleted, amplified or translocated in cancer51. These genomic variations alter pri-miRNA transcription and miRNA expression, which leads to the aberrant expression of downstream target mRNAs that can promote cancer initiation and progression51,52. For example, the locus including miR-15 and miR-16 on chromosome 13q14 is frequently deleted in B-CLL, resulting in the loss or reduced expression of these two miRNAs in ~70% of B-CLLs3. miR-15 and miR-16 normally control apoptosis by targeting BCL-2 mRNAs53. In another example, a point mutation in the miR-128b (also known as miR-128-2) gene blocks the processing of pri-miR-128b and reduces the levels of mature miR-128b, thus leading to glucocorticoid resistance in acute lymphoblastic leukaemia (ALL) cells with the mixed-lineage leukaemia (MLL)–AF4 (also known as KMT2A–AFF1) translocation54.
In addition to genomic alterations, dysregulated miRNA expression can arise from alterations in tumour suppressor or oncogenic factors that function as transcriptional activators or repressors to control pri-miRNA transcription. For example, expression of the miR-34 family of miRNAs is driven by p53 and reflects the status of p53 in human cancers55–59. The miR-34a, miR-34b and miR-34c miRNAs repress growth-promoting genes and coordinate with other members of the p53 tumour-suppressive network to inhibit uncontrolled cell proliferation and to promote apoptosis55–59. In addition, the proto-oncoprotein MYC activates expression of oncogenic miRNAs, including the miR-17~92 cluster, in cancer60,61. These MYC-target miRNAs promote cancer progression by controlling the expression of E2F1, thrombospondin 1 (THBS1), connective tissue growth factor (CTGF) and other target mRNAs to regulate cell cycle progression and angiogenesis60,61. MYC can also contribute to the widespread repression of tumour-suppressive miRNAs in B cell lymphoma62. Expression of the miR-200 family (miR-200a, miR-200b and miR-200c) is frequently suppressed in human tumours. These miRNAs are known to directly target the mRNAs encoding the zinc-finger E-box-binding homeobox (ZEB) transcription factors, ZEB1 and ZEB2, which suppress the expression of epithelial genes to promote the epithelial–mesenchymal transition (EMT)63. Interestingly, ZEB1 and ZEB2 directly bind to a regulatory element at the miR-200 promoter to repress transcription of miR-200 as part of a negative regulatory feedback loop that promotes EMT64. Many other cancer-associated transcription factors also aberrantly regulate miRNA transcription in cancer. Therefore, transcriptional dysregulation — through either genetic loss of miRNA genes or aberrant transcription factor activity — is an important mechanism for altered miRNA expression in cancer.
Epigenetic modification of histone proteins and DNA controls local chromatin structure and has an important role in the regulation of both coding and non-coding gene expression. Indeed, epigenetic alteration is a common feature of cancer pathogenesis that drives the dysregulation of miRNA expression. The CpG islands at the gene promoters of tumour-suppressive miRNAs are frequently hypermethylated in cancer, thereby leading to the epigenetic silencing of these miRNAs. Treatment of cancer cells with DNA-demethylating agents can reactivate the expression of tumour-suppressive miRNAs, such as miR-148a, miR-34b, miR-34c and miR-9, that inhibit tumour growth and metastasis65. In addition to DNA methylation, histone modifications have important roles in chromatin remodelling and cooperate with DNA methylation to suppress miRNA expression in cancer66. Overall, epigenetic silencing is an important mechanism underlying miRNA repression in cancer.
Defective Microprocessor in cancer
The nascent pri-miRNA generated by Pol II forms a typical secondary structure consisting of a stem–loop hairpin flanked by ssRNA that is a substrate for cleavage by Microprocessor to generate pre-miRNA intermediates. A negative feedback mechanism involving the Microprocessor-mediated cleavage and destabilization of DGCR8 mRNA operates to help to control the relative DGCR8 expression level and to maintain the homeostatic control of miRNA biogenesis in cells67–69. The expression and function of the Microprocessor components are often dysregulated in cancer. For example, copy-number gain or overexpression of DROSHA occurs in more than 50% of advanced cervical squamous cell carcinomas70. In addition, DROSHA expression levels are upregulated in multiple types of cancer (TABLE 1). The increased expression of DROSHA alters the global miRNA expression profile and promotes cell proliferation, migration and invasion, which contributes to cancer progression70,71. Conversely, DROSHA expression levels have been shown to be downregulated in many other types of cancer. DROSHA downregulation results in decreased miRNA expression13 and is correlated with metastasis, invasion72 and poor patient survival13,14,73,74 (TABLE 1). Knockdown of DROSHA in lung adenocarcinoma cells results in increased proliferation and tumour growth in vitro and in vivo15, suggesting that DROSHA can function as a tumour suppressor to inhibit cancer progression in some contexts. Why DROSHA is upregulated in certain types of cancer but downregulated in others is not well understood, but one possibility is that different cancers have different genetic or epigenetic mechanisms controlling DROSHA expression, thus resulting in the abnormal expression of oncogenic or tumour-suppressive miRNAs in a given cancer type.
Table 1.
Dysregulation of miRNA biogenesis machinery in cancers
| Protein | Dysregulation | Cancer type | Clinical correlation | Refs |
|---|---|---|---|---|
| DROSHA | Upregulation | Cervical SCC | Altered miRNA profile; associated with neoplastic progression | 70,162 |
| Oesophageal cancer | Regulates cell proliferation; associated with poor patient survival | 71 | ||
| BCC | Not determined | 163 | ||
| SCC | Not determined | 163 | ||
| Triple-negative breast cancer | No clinical correlation | 164,165 | ||
| Smooth muscle tumours | Associated with tumour progression | 166 | ||
| Gastric cancer | Associated with pathological characteristics and patient survival | 167 | ||
| Serous ovarian carcinoma | Associated with advanced tumour stages | 168 | ||
| Non-small cell lung cancer | Associated with poor prognosis | 169 | ||
| Downregulation | Bladder cancer | Altered miRNA profile | 170 | |
| Ovarian cancer | Associated with poor patient survival | 14 | ||
| Endometrial cancer | Correlated with histological grade | 171 | ||
| Nasopharyngeal carcinoma | Correlated with shorter patient survival | 73 | ||
| Breast cancer | Not determined | 172 | ||
| Gallbladder adenocarcinoma | Correlated with metastasis, invasion and poor prognosis | 72 | ||
| Neuroblastoma | Correlated with global downregulation of miRNAs and poor outcome | 13 | ||
| Cutaneous melanoma | Associated with cancer progression and poor survival | 74 | ||
| DGCR8 | Upregulation | Oesophageal cancer | Associated with poor patient survival | 71 |
| Bladder cancer | Altered miRNA profile | 170 | ||
| SCC and BCC | Not determined | 173 | ||
| Prostate cancer | Associated with dysregulated miRNA | 174 | ||
| Colorectal carcinoma | Not associated with any clinical parameters | 175 | ||
| Ovarian cancer | Required for cell proliferation, migration and invasion | 176 | ||
| DICER1 | Upregulation | Smooth muscle tumours | Associated with high-grade disease and tumour progression | 166 |
| Gastric cancer | Correlated with gastric tumour subtype | 167 | ||
| Serous ovarian carcinoma | Associated with advanced tumour stages | 168 | ||
| Prostate cancer | Dysregulated miRNA expression; correlated with tumour stage | 174,177 | ||
| Oral cancer | Required for proliferation | 178 | ||
| Colorectal cancer | Correlated with tumour stage and associated with poor survival | 179–181 | ||
| Precursor lesions of lung adenocarcinoma | Associated with histological subtypes and stages | 182 | ||
| Cutaneous melanoma | Correlated with clinical stage | 183 | ||
| Downregulation | Triple-negative breast cancer | No clinical correlation | 165,184 | |
| Bladder cancer | Altered miRNA profile | 170,185 | ||
| BCC | Not determined | 163 | ||
| Ovarian cancer | Associated with advanced tumour stage and poor patient survival | 14,186, 187 | ||
| Endometrial cancer | No association with histological grade detected | 171 | ||
| Nasopharyngeal carcinoma | Correlated with shorter patient survival | 73 | ||
| Neuroblastoma | Associated with global downregulation of miRNAs and poor outcome | 13 | ||
| Breast cancer | Associated with cancer progression and recurrence | 172,188 | ||
| Gallbladder adenocarcinoma | Correlated with metastasis, invasion and poor prognosis | 72 | ||
| Non-small cell lung cancer | Low levels of DICER1 expression correlate with shortened survival | 12,169 | ||
| Hepatocellular carcinoma | Not associated with clinical characteristics | 189 | ||
| Chronic lymphocytic leukaemia | Associated with progression and prognosis | 190 | ||
| Colorectal cancer | Associated with tumour stage and shorter survival | 191 | ||
| PACT | Upregulation | AK, SCC and BCC | Not determined | 173 |
| XPO5 | Downregulation | Bladder cancer | Associated with altered miRNA profile | 170 |
| AGO1 | Upregulation | AK, SCC and BCC | Not determined | 173 |
| Serous ovarian carcinoma | Associated with advanced tumour stages | 168 | ||
| AGO2 | Upregulation | AK, SCC and BCC | Not determined | 173 |
| Serous ovarian carcinoma | Correlated with advanced tumour stages and associated with shorter survival | 168 |
AGO, Argonaute; AK, actinic keratoses; BCC, basal cell carcinoma; DGCR8, DiGeorge syndrome critical region 8; miRNA, microRNA; PACT, interferon-inducible double-stranded RNA-dependent protein kinase activator A; SCC, squamous cell carcinoma; XPO5, exportin 5.
Mutational analysis revealed that DROSHA is frequently mutated in Wilms tumour samples21–24 (FIG. 2; see Supplementary information S1 (table)). More than 70% of the DROSHA mutations occur at E1147, a metalbinding residue in the RNase IIIb domain. The recurrent somatic missense mutation E1147K interferes with metal binding and therefore affects the function of DROSHA in the processing of pri-miRNAs through a dominantnegative mechanism21–24. As a result, mature miRNAs are globally downregulated in DROSHA-mutated Wilms tumours21–24. Several missense mutations and a splicesite mutation of the DROSHA gene have been found in ovarian cancer; however, these mutations do not affect DROSHA expression levels. Therefore, it remains to be characterized whether the functions of DROSHA are affected by these mutations14. In addition, DROSHA was found to be alternatively spliced in melanoma and teratocarcinoma cells75. The splice variants encode carboxy-terminal-truncated DROSHA proteins that partially lack the RNase IIIb domain and the dsRNAbinding domain (dsRBD). These truncated proteins fail to interact with DGCR8 and are deficient in pri-miRNA processing in vitro. However, the splice variants have little effect on mature miRNA expression, which might be due to the relatively low expression level of the splice variants in the cells75.
Figure 2. Mutation of the miRNA biogenesis pathway in cancer.
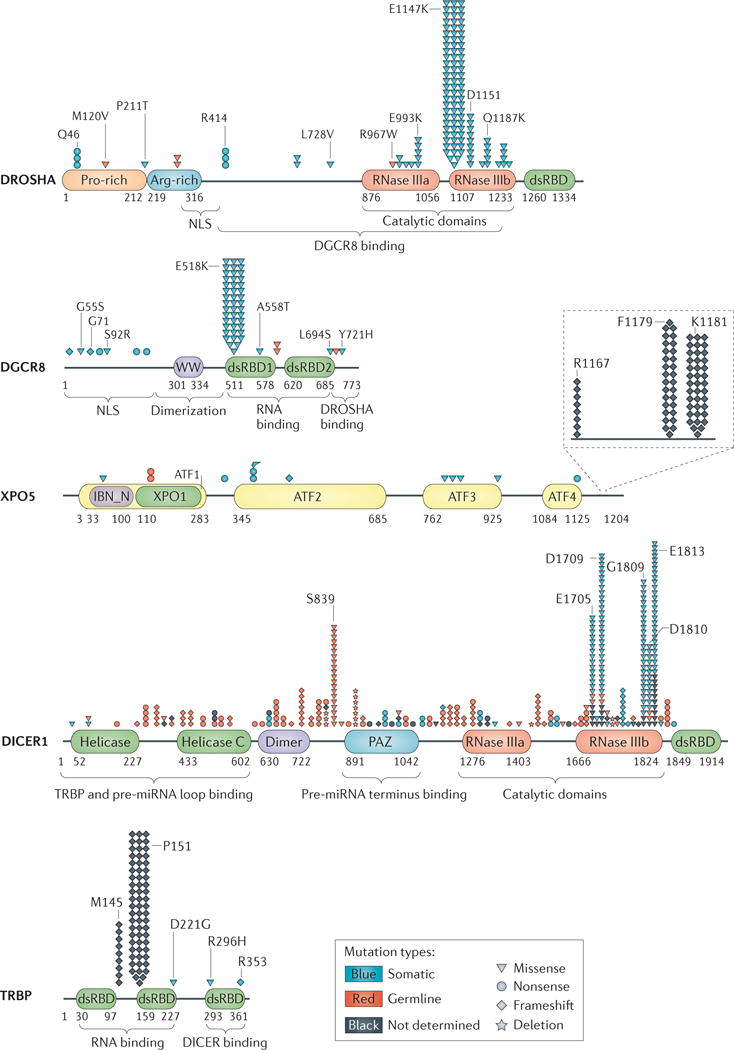
Mutations of the microRNA (miRNA) biogenesis pathway genes identified in cancer are summarized and represented by their relative locations in the protein and the type of mutation. The detailed mutational information (mutation locations, mutation types and tumour types) is provided in Supplementary information S1 (table). ATF, armadillo-type fold; DGCR8, DiGeorge syndrome critical region 8; Dimer, dimerization domain; dsRBD, double-stranded RNA-binding domain; IBN_N, importin-β amino-terminal domain; NLS, nuclear localization signal; PAZ, Piwi–Argonaute–Zwille domain; RNase, ribonuclease; pre-miRNA, precursor miRNA; TRBP, transactivation-responsive RNA-binding protein; WW, WW domain (also known as WWP-repeating motif); XPO1, exportin 1/importin β-like domain; XPO5, exportin 5.
DGCR8 expression is also dysregulated in cancer (TABLE 1). In addition, mutations of DGCR8 were reported in Wilms tumours: a recurrent mutation (E518K) in dsRBD1 results in the reduced expression of crucial miRNAs in the tumours22–24 (FIG. 2; see Supplementary information S1 (table)). Similar to knockdown of DROSHA, knockdown of DGCR8 also promotes cellular transformation and tumour growth15, further confirming the important role of Microprocessor in cancer.
Pre-miRNA export in cancer
Pre-miRNAs are exported into the cytoplasm to be processed into mature miRNAs. The export of pre-miRNAs is mediated by XPO5 and its cofactor, RanGTP41. Three recurrent heterozygous XPO5-inactivating mutations were identified in sporadic colon, gastric and endometrial tumours with microsatellite instability76 (FIG. 2; see Supplementary information S1 (table)). These XPO5 mutations impair pre-miRNA export and result in an accumulation of pre-miRNAs in the nucleus, leading to defects in miRNA biogenesis. In addition, genetic and epigenetic association studies revealed that XPO5 genetic variation and expression level are associated with the risk of breast cancer77. Therefore, XPO5 dysregulation contributes to miRNA processing defects and tumorigenesis.
Pre-miRNA processing in cancer
DICER1 mutations
After being exported to the cytoplasm, pre-miRNAs are then processed by DICER1 to form ~22-nucleotide mature miRNAs78. DICER1 is a large multi-domain nuclease that contains two helicase domains, a dimerization domain, a Piwi–Argonaute–Zwille (PAZ) domain, two RNase III domains (RNase IIIa and RNase IIIb) and a dsRBD (FIG. 2; see Supplementary information S1 (table)). In addition to its function in pre-miRNA cleavage, DICER1 is required for the assembly of the minimal miRISC that executes miRNA function in repressing target gene expression48. Depletion of DICER1 in cancer cells or mouse models promotes cell growth and tumorigenesis, indicating the important function of DICER1 in oncogenesis15,79. Furthermore, Dicer is considered a haploinsufficient tumour suppressor gene, as loss of a single Dicer1 allele reduces survival in a mouse model of lung cancer79.
Heterozygous germline DICER1 mutations were first identified to be responsible for pleuropulmonary blastoma (PPB), a rare paediatric lung tumour that arises during fetal lung development and is often part of an inherited cancer syndrome (Online Mendelian Inheritance in Man (OMIM) #601200)16. Germline frameshift or nonsense mutations mainly affect DICER1 upstream of the region encoding RNase III domains (FIG. 2), resulting in truncated DICER1 proteins lacking the C-terminal catalytic domains. DICER1 loss of heterozygosity (LOH) is almost never observed in human tumours, and homozygous Dicer1 loss is generally selected against in mouse cancer models79. Although more than 50% of heterozygous germline DICER1 mutation carriers are clinically unaffected, the tumours that develop in PPB patients are typically associated with another important group of DICER1 mutations: recurrent somatic mutations in the RNase IIIb domain18,80. The mutation hot spots of the RNase IIIb domain occur in the metal-binding residues (E1705, D1709, G1809, D1810 and E1813)18 (FIG. 2); this domain is responsible for the cleavage of the 3′ end of the miRNAs derived from the 5′ side of the pre-miRNA hairpin called 5p miRNAs. These mutations do not change DICER1 protein expression but instead cause defects in the function of the RNase IIIb domain. As a result, the maturation of 5p miRNAs is specifically blocked, while the processing of 3p miRNAs (miRNAs derived from the 3′ side of the pre-miRNA hairpin) remains unaffected, leading to the global loss of 5p miRNAs in cancer17,18. Particularly, DICER1 RNase IIIb mutations strongly reduce the expression of the members of the let-7 tumour-suppressive miRNA family (that are all 5′ derived), which probably helps to explain the selective pressures that give rise to this specific mutation spectrum in cancers. Interestingly, modelling of PPB in mice supports the idea that Dicer1 deletion in the distal airway epithelium causes non-cellautonomous tumour initiation, whereby Dicer1 loss in the epithelium causes the underlying mesenchymal cells to be malignantly transformed81. DICER1 mutations are frequently found in different types of inherited tumours: PPB16,80–84, non-epithelial ovarian cancer18,84,85, Wilms tumour22,86,87, pituitary blastoma88, cystic nephroma89, rhabdomyosarcoma90 and others91 (see Supplementary information S1 (table)). As a result, patients harbouring these DICER1 mutations have reduced DICER1 expression and/or impaired DICER1 function, which cause the abnormal expression of miRNAs and contribute to the pathogenesis of cancer. As such, DICER1 mutation is considered a tumour predisposition syndrome known as DICER1 syndrome20. This topic has recently been reviewed in detail19.
In addition to genetic mutations of DICER1, DICER1 expression is often dysregulated in cancer. Similar to that of DROSHA, DICER1 expression can be increased or decreased in cancer, depending on the cancer type (TABLE 1). Many oncoproteins and dysregulated tumour suppressors regulate cancer progression by targeting DICER1 expression. For example, the p53 family member TAp63 directly binds to the promoters of DICER1 and miR-130b and drives their expression to suppress tumorigenesis and metastasis92. Overall, both genetic mutation and dysregulation of DICER1 can result in aberrant miRNA expression and tumorigenesis.
TRBP mutations
Impaired function of TRBP also contributes to miRNA dysregulation in cancer. Sequencing of the genes encoding the miRNA processing machinery revealed two frameshift mutations of TRBP in sporadic and hereditary carcinomas with microsatellite instability93,94 (FIG. 2; see Supplementary information S1 (table)). These mutations cause reduced TRBP and DICER1 expression as well as defective processing of pre-miRNAs. Re-introduction of wild-type TRBP in the mutated cell lines rescued TRBP and DICER1 expression, restored miRNA processing and suppressed cancer cell growth in vitro and in vivo93. Interestingly, the expression of TRBP is repressed in the cancer stem cell (CSC) population of Ewing sarcoma family tumour (ESFT), which results in the miRNA profile of ESFT CSCs that is required for CSCassociated self-renewal and tumour growth95. Therefore, TRBP-mediated miRNA processing has an important tumour-suppressive role in normal cells.
Other miRNA regulators in cancer
Aberrant expression of or mutations in the genes encoding key components of the miRNA biogenesis pathway contributes to the global repression of miRNAs in cancer. However, a widespread suppression of miRNA expression has been observed in cancers with normal expression of the miRNA biogenesis machinery. This suggests that other pathways regulating miRNA processing are dysregulated in cancer. We highlight below recent discoveries of selected cancer-relevant pathways involved in the regulation of miRNA biogenesis.
Regulators of Microprocessor
The original characterization of a large DROSHA-containing complex identified multiple classes of RNA-binding proteins, including the DEAD (Asp-Glu-Ala-Asp) box helicases DDX5 (also known as p68) and DDX17 (also known as p72), Ewing sarcoma family proteins and heterogeneous nuclear ribonucleoproteins (hnRNPs)32. These Microprocessorassociated proteins can directly affect Microprocessor activity, and alterations in this regulation can result in aberrant miRNA biogenesis in cancer96. Other factors might also regulate Microprocessor activity in cancer: for example, the tumour suppressor BRCA1 interacts with multiple Microprocessor regulators to facilitate miRNA biogenesis97. Moreover, RNA-binding proteins such as KH type-splicing regulatory protein (KSRP; also known as FUBP2)98, serine/arginine-rich splicing factor 1 (SRSF1)99, hnRNP A1 (REFS 100,101) and FUS (also known as TLS)102 bind to certain regions of primiRNAs (stem or terminal loop) and facilitate DROSHA recruitment and function (FIG. 3).
Figure 3. Dysregulated miRNA biogenesis in cancer.
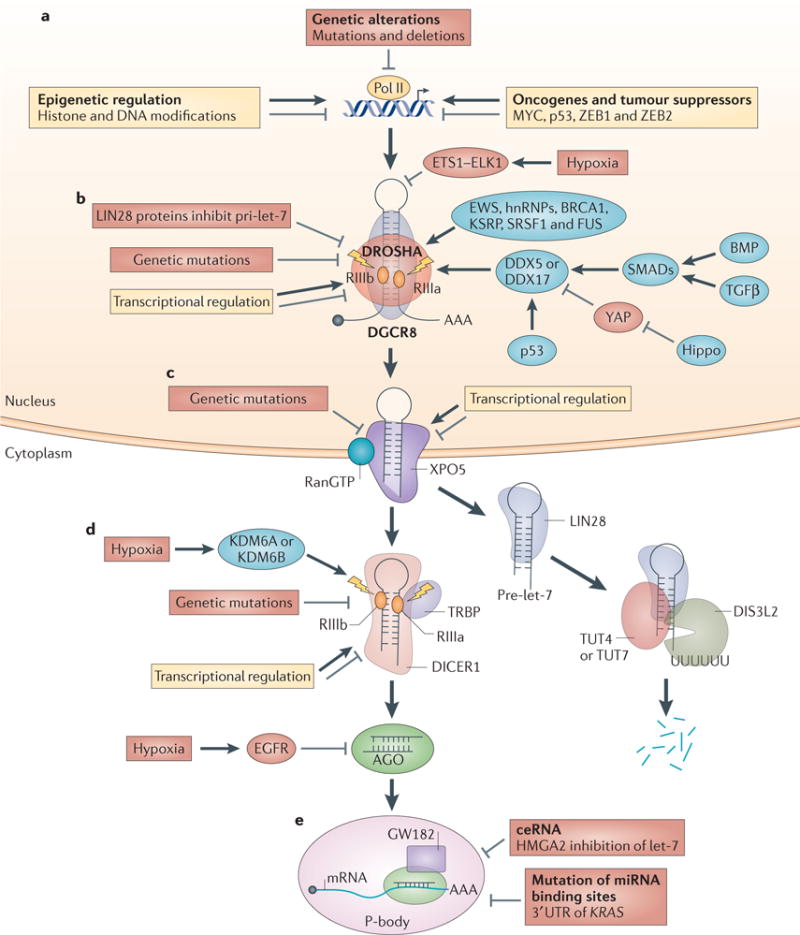
Aberrant microRNA (miRNA) biogenesis in cancer occurs at different steps during miRNA maturation. a | Genetic alterations, epigenetic modifications, oncogenes and tumour suppressors negatively or positively regulate primary miRNA (pri-miRNA) transcription in cancer. b | Pri-miRNA processing is regulated in the following ways: hypoxia, genetic mutations and transcriptional regulation control DROSHA and DiGeorge syndrome critical region 8 (DGCR8) expression in cancer; RNA-binding proteins such as DEAD box protein 5 (DDX5), DDX17 and BRCA1 modulate Microprocessor activity in cancer; cell signalling pathways such as Hippo and bone morphogenetic protein (BMP) regulate pri-miRNA processing; and LIN28 proteins selectively block the processing of pri-let-7. c | Genetic mutations in and transcriptional regulation of exportin 5 (XPO5) affect XPO5-mediated precursor miRNA (pre-miRNA) export in cancer. d | Pre-miRNA processing in cancer is regulated in the following ways: hypoxia, genetic mutations and transcriptional regulation modulate DICER1 expression and function to control pre-miRNA cleavage in cancer; LIN28 proteins selectively bind to pre-let-7 and recruit terminal uridylyltransferase 4 (TUT4), TUT7 and DIS3-like exonuclease 2 (DIS3L2) to degrade pre-let-7; and hypoxia-induced and epidermal growth factor receptor (EGFR)-induced phosphorylation of Y393 of Argonaute 2 (AGO2) inhibits pre-miRNA processing. e | miRNA function is regulated in the following ways: competing endogenous RNA (ceRNA) inhibits miRNA function in cancer (high-mobility group AT-hook 2 (HMGA2) blocks let-7 function), as do mutations of miRNA-binding sites in non-small cell lung cancer (mutation of let-7-binding site in the 3′ untranslated region (UTR) of KRAS mRNA). hnRNP, heterogeneous nuclear ribonucleoprotein; KDM6, lysine-specific demethylase 6; KSRP, KH-type splicing regulatory protein; Pol II, RNA polymerase II; RIII, ribonuclease III; SRSF1, serine/arginine-rich splicing factor 1; TGFβ, transforming growth factor-β; TRBP, transactivation-responsive RNA-binding protein; YAP, Yes-associated protein; ZEB, zinc-finger E-box-binding homeobox.
In addition to regulating Microprocessor activity, DDX5 and DDX17 function as bridging factors for important oncoproteins or tumour suppressors to regulate miRNA biogenesis in cancer. For example, the tumour suppressor protein p53 regulates miRNA biogenesis through association with DDX5 and DDX17. In response to DNA damage, the level of p53 expression increases, which enhances the expression levels of tumour-suppressive miRNAs including miR-34a, miR-16-1, miR-143 and miR-145 (REF. 103). In contrast to miR-34a, which is a transcriptional target of p53 (REF. 55), the other miRNAs are post-transcriptionally regulated by p53. Mediated by DDX5 and DDX17, p53 interacts with the DROSHA complex and promotes the processing of tumour-suppressive pri-miRNAs. Accordingly, miRNA processing is hindered in p53-mutant cells103. Given that p53 is frequently mutated in human cancer, dysregulation of miRNA biogenesis by p53 mutation might account for the widespread miRNA repression in cancer (FIG. 3).
Cell signalling control
Cell signalling pathways also modulate Microprocessor activity to dynamically control pri-miRNA processing and miRNA expression in cancer96 (FIG. 3). For example, SMADs — which transduce transforming growth factor-β (TGFβ) and bone morphogenetic protein (BMP) signalling — associate with DDX5 and promote miRNA processing by binding to a consensus sequence in the stem region of primiRNAs104,105. Moreover, the core biogenesis machinery components, including DROSHA, DGCR8, DICER1 and TRBP, are subject to post-translational control such as phosphorylation and/or acetylation (reviewed in REFS 106,107). The effect of these protein modifications, and their possible dysregulation in cancer, remains to be determined.
It was recently found that the Hippo pathway controls Microprocessor activity108. The Hippo pathway controls organ size by regulating cell proliferation and differentiation in response to cell density109. Given its key role in regulating organ size and cell proliferation, it is perhaps not surprising that the Hippo signalling pathway is frequently perturbed in a variety of human cancers109. miRNA biogenesis is activated by cell–cell contact and Hippo signalling108,110. Mechanistically, it was found that the Hippo downstream effector Yes-associated protein 1 (YAP1) post-transcriptionally regulates miRNA biogenesis by targeting DDX17. In in vitro cell culture systems, at low cell density, the growth-suppressive Hippo pathway is inactive, and nuclear YAP1 binds to and sequesters DDX17 to suppress pri-miRNA processing, whereas at high cell densities, the Hippo pathway is active, which leads to YAP1 phosphorylation and its retention in the cytoplasm. When YAP1 is cytoplasmic, DDX17 is able to bind to a specific sequence motif in pri-miRNA, associate with Microprocessor and enhance miRNA biogenesis. Accordingly, inactivation of the Hippo pathway or constitutive activation of YAP1, which occurs in cancer cells, results in widespread miRNA suppression both in human cancer cell lines and in mouse tumour models108. It will be interesting to explore whether Hippo signalling is responsible for the widespread repression of miRNA expression in cancer.
Stress response
Rapidly growing tumours often experience hypoxia owing to the limited oxygen supply in the tumour microenvironment. Interestingly, miRNA expression and function are dynamically regulated under stress conditions111. Oncogenic epidermal growth factor receptor (EGFR) signalling is activated by hypoxia to promote cell growth and oncogenesis112. Identification of the EGFR protein complex in serum-starved EGFtreated HeLa cells revealed that EGFR interacts with AGO2 (REF. 113). In response to hypoxia, EGFR induces the phosphorylation of AGO2 at Y393, which inhibits the interaction between DICER1 and AGO2 and blocks miRNA accumulation. Furthermore, EGFR-mediated AGO2-Y393 phosphorylation is required for cell survival and invasion under hypoxic conditions and is associated with poor survival rates in patients with breast cancer113. In addition, recent studies uncovered the important role of hypoxia in suppressing DROSHA and DICER1 expression in cancer cells, which results in aberrant miRNA biogenesis and promotes tumour progression114,115. These studies provide an interesting link between hypoxia and miRNA repression in cancer and uncover a novel oncogenic role of hypoxia in regulating miRNA biogenesis during tumorigenesis113–115 (FIG. 3).
LIN28-mediated blockade of let-7
The let-7 miRNA family members function as tumour suppressors in multiple cancer types by inhibiting expression of oncogenes and key regulators of mitogenic pathways116–118. In humans, there are 12 let-7 family members (let-7a-1, let-7a-2, let-7a-3; let-7b; let-7c; let-7d; let-7e; let-7f-1, let-7f-2; let-7g; let-7i; miR-98) located at 8 unlinked chromosomal loci. The let-7 miRNAs are downregulated in numerous cancer types, and low let-7 expression levels correlate with poor prognosis119–122. The expression of the let-7 miRNA family is coordinately regulated by the paralogous RNA-binding proteins LIN28A and LIN28B during early embryonic development123–126. Reactivation of this embryonic pathway in adult cells by expression of LIN28A and LIN28B is sufficient to promote cellular transformation and tumorigenesis in vitro and in vivo127–130. Of note, expression of LIN28B is sufficient to drive neuroblastoma, T cell lymphoma, intestinal adenocarcinoma, Wilms tumour (nephroblastoma) and hepatocellular carcinoma in mouse models128,130–133. LIN28 proteins block cell differentiation, promote cell proliferation and alter cellular metabolism to promote tumorigenesis134,135. The repression of the let-7 family in these contexts is crucial, as tumour formation is suppressed by enforced expression of let-7g, and genetic deletion of a let-7 locus (let7c2 and let7b) recapitulated the effects of LIN28B overexpression in the intestine127–129,133. Depletion of LIN28A or LIN28B in human cancer cell lines results in decreased cell proliferation, cell invasion and tumorigenicity129,136, and withdrawal of LIN28B expression can revert liver tumorigenesis in mice130. At least 15% of all human cancer samples investigated are characterized by reactivation of either LIN28A or LIN28B, with a corresponding reduction in let-7 levels129. Moreover, elevated LIN28A or LIN28B expression correlates with poor prognosis and decreased patient survival129,131,137–140. Considering also that LIN28A and LIN28B expression may characterize distinct tumorigenic subpopulations of cells within the tumour, known as tumour-initiating cells or CSCs141, these studies underscore the importance of the LIN28 proteins in promoting and characterizing various human malignancies and suggest that this pathway represents an important new target for effective cancer therapies.
Mechanistically, LIN28 proteins selectively bind to the terminal loop region of pre-let-7 through RNA–protein interactions through its cold-shock domain and tandem Cys-Cys-His-Cys (CCHC)-type zinc-fingers142,143. LIN28 proteins recruit two alternative 3′ terminal uridylyltransferases (TUTases), ZCCHC11 (also known as TUT4) and ZCCHC6 (also known as TUT7), to pre-let-7 RNA144–146. These TUTases are key mediators in the LIN28 blockade of let-7 biogenesis, in which they catalyse the addition of an oligouridine tail to pre-let-7. Uridylated pre-let-7 is resistant to DICER1 processing and is rapidly degraded to prevent let-7 biogenesis in LIN28Aor LIN28B-expressing cells125. The enzyme responsible for this decay pathway was recently identified as DIS3L2, a novel 3′–5′ exonuclease that selectively degrades 3′ oligouridylated (>12 uridines) RNA147–149 (FIG. 3). Intriguingly, DIS3L2 is a tumour suppressor gene that is deleted in Perlman syndrome, which is characterized by fetal overgrowth and cancer predisposition, as well as in ~30% of sporadic Wilms tumours analysed150. Considering the strong links between DROSHA and DICER1 mutations in Wilms tumours, the demonstrated ability of LIN28A and LIN28B to promote tumorigenesis as well as the tumour-suppressive role of DIS3L2, it is perhaps likely that loss of let-7 expression and/or function is a unifying driver of Wilms tumours and of other types of cancer. This let-7 loss might be accomplished by any of the aforementioned mechanisms as well as by the possible titration of let-7 function via the considerable overexpression of mRNAs containing let-7 binding sites, as was recently suggested for HMGA2 (REF. 151). Another possible mechanism involves mutations in the let-7 binding sites of key downstream targets, thus relieving these mRNAs from let-7 regulation. In support of this, a single-nucleotide polymorphism (SNP) in a let-7 binding site in the 3′ UTR of the KRAS mRNA has been genetically associated with an increased risk of cancer152 (FIG. 3).
Conclusions and perspectives
Discoveries over the past 15 years have provided substantial insights into the mechanisms controlling miRNA biogenesis. The identification and characterization of the core miRNA biogenesis machinery provided the framework for recent developments that uncovered cancer-causing mutations in miRNA biogenesis components as well as for the identification of cellular signalling and regulatory pathways that control different subsets of miRNAs. Although clear examples of individual miRNAs with oncogenic function have been described, the net effect of widespread miRNA depletion is to promote tumorigenesis. This was first demonstrated in human cancer cells and mouse models and is strongly supported by the mutations recently identified in core miRNA biogenesis genes.
Analogous to the defective differentiation phenotype of miRNA-deficient embryonic stem cells, it seems that also in the context of cancer the dominant function of miRNAs is to help to maintain differentiated cells in a particular cell state or lineage153,154. In this model, loss of miRNAs facilitates epigenetic reprogramming, loss of differentiated cell identity and adoption of an undifferentiated cancer phenotype. Indeed, DGCR8 depletion is sufficient to reprogramme human primary keratinocytes to induced pluripotent-like cells155. Furthermore, miRNA expression is globally elevated in confluent cells, which is consistent with their roles in suppressing cell proliferation and in coordinating the altered metabolic demands of less-proliferative cells and tissues108,110. Presumably this is how widespread miRNA depletion — through loss of components of the biogenesis machinery or loss of growth-suppressive signalling pathways (for example, the Hippo pathway) — contributes to rapid cancer cell proliferation and tumour growth. In this way, widespread loss of miRNAs functionally cooperates with other cancer hallmarks to regulate cancer progression156. Is loss of any particular miRNA or miRNA family responsible for these tumorigenic effects? One good candidate is the let-7 family. The let-7 family is required in adult fibroblasts to suppress the expression of a mid-gestation embryonic gene signature that is enriched with oncofetal genes157. Conversely, antagonizing let-7 with antisense oligonucleotides can enhance reprogramming to induced pluripotent stem cells, suggesting that let-7 has a dominant role in stem cell differentiation158. Indeed, re-introduction of let-7 into miRNA-deficient mouse embryonic stem cells rescued the stem cell differentiation phenotype158; similarly, restoration of let-7 expression was shown to effectively inhibit growth of lung and breast cancer cells, as well as in mouse models of hepatocellular carcinoma and Wilms tumours118,159,160. Thus, let-7 emerges as a key regulator in stem cell biology and tumorigenesis and, as outlined in this Review, there are multiple mechanisms by which cancer cells inactivate this miRNA ‘guardian’ of differentiation, proliferation and metabolic reprogramming.
Future work promises to illuminate the most relevant miRNAs in the context of different cancer types and will probably uncover additional pathways that control the expression of individual miRNAs or of miRNA subsets. Studies in this area will be facilitated by the recent advances in genome engineering using CRISPR–Cas9 (clustered regularly interspaced short palindromic repeat–CRISPR-associated protein 9) technology, in mouse modelling and in the use of organoid culture systems to model cancer161, as well as by the application of high-throughput sequencing technologies that will uncover cancer-causing mutations in patients and that can be applied in the laboratory to examine the effects of possible regulators on global miRNA expression profiles21. With this powerful toolkit in hand, the next several years promise exciting discoveries that will help to unlock the secrets of miRNA dysregulation in cancer. Understanding the molecular and cellular pathways controlling miRNA biogenesis and how these mechanisms go awry in cancer will identify promising therapeutic targets that might be readily manipulated by small pharmacological agents to allow restoration of miRNA expression profiles and to bypass the challenges associated with delivering synthetic miRNA mimics or antagomiRs.
Supplementary Material
Acknowledgments
S.L. is a Damon Runyon-Sohn Pediatric Cancer Research Fellow supported by the Damon Runyon Cancer Research Foundation (DRSG-7-13). R.I.G. is supported by grants from the US National Cancer Institute (NCI) (R01CA163467) and the American Cancer Society (121635-RSG-11-175-01-RMC).
Glossary
- 3′ untranslated region
(3′ UTR). The non-coding region of mRNA between the translation termination codon and the poly(A) tail. The 3′ UTR often contains regulatory elements, such as miRNA binding sites, for post-transcriptional regulation of gene expression
- Ribonuclease III
(RNase III). Enzymes that can specifically recognize and cleave double-stranded RNA with their ribonuclease III domains
- Germline mutations
Heritable gene mutations that occur in germline tissues
- Somatic mutations
Gene mutations that occur in non-germline tissues that are not inherited
- Post-transcriptional gene silencing
A gene-silencing effect that controls gene expression after transcription, often mediated by small non-coding RNAs such as small interfering RNAs (siRNAs) and microRNAs (miRNAs)
- Epithelial–mesenchymal transition
(EMT). A process that occurs during development or cancer progression in which the epithelial cells lose their cell polarity and cell–cell adhesion to become mesenchymal cells with migratory and invasive characteristics
- CpG islands
Genetic regions with high CpG content, often located at the gene promoter, that have important functions in regulating gene expression
- Microsatellite
Short (2–5 bp) tandem repeat of DNA that can be used as a genetic marker
- Loss of heterozygosity
(LOH). Deletion or mutation of the normal allele of a gene, of which the other allele is already deleted or inactivated, resulting in loss of both alleles of the gene
- Cold-shock domain
A protein domain of ~70 amino acids that is often found in DNA- or RNA-binding proteins and that functions to protect cells during cold temperatures
- Cys-Cys-His-Cys (CCHC)-type zinc-fingers
Protein domains that are found in RNA-binding proteins or single-stranded DNA-binding proteins
- Terminal uridylyltransferases
(TUTases). Enzymes that catalyse the addition of one or more uridine monophosphate (UMP) molecules to the 3′ end of RNA
- Oncofetal genes
Genes that are typically highly expressed during fetal development and repressed in adult life, and reactivated in cancers
Footnotes
Competing interests statement
The authors declare no competing interests.
References
- 1.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nature Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 3.Calin GA, et al. Frequent deletions and downregulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2002;99:15524–15529. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Di Leva G, Croce CM. Roles of small RNAs in tumor formation. Trends Mol Med. 2010;16:257–267. doi: 10.1016/j.molmed.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mendell JT, Olson EN. MicroRNAs in stress signaling and human disease. Cell. 2012;148:1172–1187. doi: 10.1016/j.cell.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He L, et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. This paper was the first to reveal that genes in the miR-17~92 cluster function as potential human oncogenes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim HH, et al. HuR recruits let-7/RISC to repress c-Myc expression. Genes Dev. 2009;23:1743–1748. doi: 10.1101/gad.1812509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson SM, et al. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635–647. doi: 10.1016/j.cell.2005.01.014. This paper was the first to show that members of the let-7 family of miRNAs function as tumour suppressors by targeting RAS. [DOI] [PubMed] [Google Scholar]
- 9.Kumar MS, et al. Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proc Natl Acad Sci USA. 2008;105:3903–3908. doi: 10.1073/pnas.0712321105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu J, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. This paper was the first to report that miRNAs are globally downregulated in cancers. [DOI] [PubMed] [Google Scholar]
- 11.Thomson JM, et al. Extensive post-transcriptional regulation of microRNAs and its implications for cancer. Genes Dev. 2006;20:2202–2207. doi: 10.1101/gad.1444406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karube Y, et al. Reduced expression of Dicer associated with poor prognosis in lung cancer patients. Cancer Sci. 2005;96:111–115. doi: 10.1111/j.1349-7006.2005.00015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin RJ, et al. microRNA signature and expression of Dicer and Drosha can predict prognosis and delineate risk groups in neuroblastoma. Cancer Res. 2010;70:7841–7850. doi: 10.1158/0008-5472.CAN-10-0970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Merritt WM, et al. Dicer, Drosha, and outcomes in patients with ovarian cancer. N Engl J Med. 2008;359:2641–2650. doi: 10.1056/NEJMoa0803785. This paper reveals that the expression levels of DICER1 and DROSHA are associated with clinical outcomes in patients with ovarian cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nature Genet. 2007;39:673–677. doi: 10.1038/ng2003. This paper shows that impaired miRNA biogenesis promotes oncogenesis. [DOI] [PubMed] [Google Scholar]
- 16.Hill DA, et al. DICER1 mutations in familial pleuropulmonary blastoma. Science. 2009;325:965. doi: 10.1126/science.1174334. This study was the first to identify the germline mutations of DICER1 in patients with familial PPB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anglesio MS, et al. Cancer-associated somatic DICER1 hotspot mutations cause defective miRNA processing and reverse-strand expression bias to predominantly mature 3p strands through loss of 5p strand cleavage. J Pathol. 2013;229:400–409. doi: 10.1002/path.4135. [DOI] [PubMed] [Google Scholar]
- 18.Heravi-Moussavi A, et al. Recurrent somatic DICER1 mutations in nonepithelial ovarian cancers. N Engl J Med. 2012;366:234–242. doi: 10.1056/NEJMoa1102903. This study identified the recurrent somatic mutations encoding the RNase IIIb catalytic domain of DICER1 that affect the processing of 5′ derived miRNAs. [DOI] [PubMed] [Google Scholar]
- 19.Foulkes WD, Priest JR, Duchaine TF. DICER1: mutations, microRNAs and mechanisms. Nature Rev Cancer. 2014;14:662–672. doi: 10.1038/nrc3802. [DOI] [PubMed] [Google Scholar]
- 20.Slade I, et al. DICER1 syndrome: clarifying the diagnosis, clinical features and management implications of a pleiotropic tumour predisposition syndrome. J Med Genet. 2011;48:273–278. doi: 10.1136/jmg.2010.083790. [DOI] [PubMed] [Google Scholar]
- 21.Rakheja D, et al. Somatic mutations in DROSHA and DICER1 impair microRNA biogenesis through distinct mechanisms in Wilms tumours. Nature Commun. 2014;2:4802. doi: 10.1038/ncomms5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Torrezan GT, et al. Recurrent somatic mutation in DROSHA induces microRNA profile changes in Wilms tumour. Nature Commun. 2014;5:4039. doi: 10.1038/ncomms5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wegert J, et al. Mutations in the SIX1/2 pathway and the DROSHA/DGCR8 miRNA microprocessor complex underlie high-risk blastemal type Wilms tumors. Cancer Cell. 2015;27:298–311. doi: 10.1016/j.ccell.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 24.Walz AL, et al. Recurrent DGCR8, DROSHA, and SIX homeodomain mutations in favorable histology Wilms tumors. Cancer Cell. 2015;27:286–297. doi: 10.1016/j.ccell.2015.01.003. References 21–24 identified the recurrent somatic mutation of DROSHA and DGCR8 in Wilms tumours. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. This study was the first to identify miRNA. [DOI] [PubMed] [Google Scholar]
- 26.Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294:853–858. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- 27.Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 2001;294:858–862. doi: 10.1126/science.1065062. [DOI] [PubMed] [Google Scholar]
- 28.Lee RC, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science. 2001;294:862–864. doi: 10.1126/science.1065329. [DOI] [PubMed] [Google Scholar]
- 29.Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39:D152–D157. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nature Rev Genet. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 31.Lee Y, et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23:4051–4060. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gregory RI, et al. The Microprocessor complex mediates the genesis of microRNAs. Nature. 2004;432:235–240. doi: 10.1038/nature03120. [DOI] [PubMed] [Google Scholar]
- 33.Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ. Processing of primary microRNAs by the Microprocessor complex. Nature. 2004;432:231–235. doi: 10.1038/nature03049. [DOI] [PubMed] [Google Scholar]
- 34.Lee Y, et al. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- 35.Han J, et al. The Drosha–DGCR8 complex in primary microRNA processing. Genes Dev. 2004;18:3016–3027. doi: 10.1101/gad.1262504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han J, et al. Molecular basis for the recognition of primary microRNAs by the Drosha–DGCR8 complex. Cell. 2006;125:887–901. doi: 10.1016/j.cell.2006.03.043. [DOI] [PubMed] [Google Scholar]
- 37.Zeng Y, Yi R, Cullen BR. Recognition and cleavage of primary microRNA precursors by the nuclear processing enzyme Drosha. EMBO J. 2005;24:138–148. doi: 10.1038/sj.emboj.7600491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burke JM, Kelenis DP, Kincaid RP, Sullivan CS. A central role for the primary microRNA stem in guiding the position and efficiency of Drosha processing of a viral pri-miRNA. RNA. 2014;20:1068–1077. doi: 10.1261/rna.044537.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heo I, et al. Mono-uridylation of pre-microRNA as a key step in the biogenesis of group II let-7 microRNAs. Cell. 2012;151:521–532. doi: 10.1016/j.cell.2012.09.022. [DOI] [PubMed] [Google Scholar]
- 40.Yi R, Qin Y, Macara IG, Cullen BR. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003;17:3011–3016. doi: 10.1101/gad.1158803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lund E, Guttinger S, Calado A, Dahlberg JE, Kutay U. Nuclear export of microRNA precursors. Science. 2004;303:95–98. doi: 10.1126/science.1090599. [DOI] [PubMed] [Google Scholar]
- 42.Bohnsack MT, Czaplinski K, Gorlich D. Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. RNA. 2004;10:185–191. doi: 10.1261/rna.5167604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park JE, et al. Dicer recognizes the 5′ end of RNA for efficient and accurate processing. Nature. 2011;475:201–205. doi: 10.1038/nature10198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409:363–366. doi: 10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- 45.Chendrimada TP, et al. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature. 2005;436:740–744. doi: 10.1038/nature03868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee HY, Doudna JA. TRBP alters human precursor microRNA processing in vitro. RNA. 2012;18:2012–2019. doi: 10.1261/rna.035501.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim Y, et al. Deletion of human tarbp2 reveals cellular microRNA targets and cell-cycle function of TRBP. Cell Rep. 2014;9:1061–1074. doi: 10.1016/j.celrep.2014.09.039. [DOI] [PubMed] [Google Scholar]
- 48.Gregory RI, Chendrimada TP, Cooch N, Shiekhattar R. Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell. 2005;123:631–640. doi: 10.1016/j.cell.2005.10.022. [DOI] [PubMed] [Google Scholar]
- 49.Liu J, Valencia-Sanchez MA, Hannon GJ, Parker R. MicroRNA-dependent localization of targeted mRNAs to mammalian P-bodies. Nature Cell Biol. 2005;7:719–723. doi: 10.1038/ncb1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eulalio A, Behm-Ansmant I, Schweizer D, Izaurralde E. P-body formation is a consequence, not the cause, of RNA-mediated gene silencing. Mol Cell Biol. 2007;27:3970–3981. doi: 10.1128/MCB.00128-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Calin GA, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci USA. 2004;101:2999–3004. doi: 10.1073/pnas.0307323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang L, et al. microRNAs exhibit high frequency genomic alterations in human cancer. Proc Natl Acad Sci USA. 2006;103:9136–9141. doi: 10.1073/pnas.0508889103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cimmino A, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA. 2005;102:13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kotani A, et al. A novel mutation in the miR-128b gene reduces miRNA processing and leads to glucocorticoid resistance of MLL–AF4 acute lymphocytic leukemia cells. Cell Cycle. 2010;9:1037–1042. doi: 10.4161/cc.9.6.11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.He L, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–1134. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Raver-Shapira N, et al. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell. 2007;26:731–743. doi: 10.1016/j.molcel.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 57.Chang TC, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26:745–752. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bommer GT, et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr Biol. 2007;17:1298–1307. doi: 10.1016/j.cub.2007.06.068. [DOI] [PubMed] [Google Scholar]
- 59.Tarasov V, et al. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle. 2007;6:1586–1593. doi: 10.4161/cc.6.13.4436. [DOI] [PubMed] [Google Scholar]
- 60.O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 61.Dews M, et al. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nature Genet. 2006;38:1060–1065. doi: 10.1038/ng1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chang TC, et al. Widespread microRNA repression by Myc contributes to tumorigenesis. Nature Genet. 2008;40:43–50. doi: 10.1038/ng.2007.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gregory PA, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nature Cell Biol. 2008;10:593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 64.Bracken CP, et al. A double-negative feedback loop between ZEB1–SIP1 and the microRNA-200 family regulates epithelial–mesenchymal transition. Cancer Res. 2008;68:7846–7854. doi: 10.1158/0008-5472.CAN-08-1942. [DOI] [PubMed] [Google Scholar]
- 65.Lujambio A, et al. A microRNA DNA methylation signature for human cancer metastasis. Proc Natl Acad Sci USA. 2008;105:13556–13561. doi: 10.1073/pnas.0803055105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guil S, Esteller M. DNA methylomes, histone codes and miRNAs: tying it all together. Int J Biochem Cell Biol. 2009;41:87–95. doi: 10.1016/j.biocel.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 67.Han J, et al. Posttranscriptional crossregulation between Drosha and DGCR8. Cell. 2009;136:75–84. doi: 10.1016/j.cell.2008.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Triboulet R, Chang HM, Lapierre RJ, Gregory RI. Post-transcriptional control of DGCR8 expression by the Microprocessor. RNA. 2009;15:1005–1011. doi: 10.1261/rna.1591709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kadener S, et al. Genome-wide identification of targets of the Drosha–Pasha/DGCR8 complex. RNA. 2009;15:537–545. doi: 10.1261/rna.1319309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Muralidhar B, et al. Functional evidence that Drosha overexpression in cervical squamous cell carcinoma affects cell phenotype and microRNA profiles. J Pathol. 2011;224:496–507. doi: 10.1002/path.2898. [DOI] [PubMed] [Google Scholar]
- 71.Sugito N, et al. RNASEN regulates cell proliferation and affects survival in esophageal cancer patients. Clin Cancer Res. 2006;12:7322–7328. doi: 10.1158/1078-0432.CCR-06-0515. [DOI] [PubMed] [Google Scholar]
- 72.Shu GS, Yang ZL, Liu DC. Immunohistochemical study of Dicer and Drosha expression in the benign and malignant lesions of gallbladder and their clinicopathological significances. Pathol Res Pract. 2012;208:392–397. doi: 10.1016/j.prp.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 73.Guo X, et al. The microRNA-processing enzymes: Drosha and Dicer can predict prognosis of nasopharyngeal carcinoma. J Cancer Res Clin Oncol. 2012;138:49–56. doi: 10.1007/s00432-011-1058-1. [DOI] [PubMed] [Google Scholar]
- 74.Jafarnejad SM, Sjoestroem C, Martinka M, Li G. Expression of the RNase III enzyme DROSHA is reduced during progression of human cutaneous melanoma. Mod Pathol. 2013;26:902–910. doi: 10.1038/modpathol.2012.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Grund SE, Polycarpou-Schwarz M, Luo C, Eichmuller SB, Diederichs S. Rare Drosha splice variants are deficient in microRNA processing but do not affect general microRNA expression in cancer cells. Neoplasia. 2012;14:238–248. doi: 10.1593/neo.111586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Melo SA, et al. A genetic defect in exportin-5 traps precursor microRNAs in the nucleus of cancer cells. Cancer Cell. 2010;18:303–315. doi: 10.1016/j.ccr.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 77.Leaderer D, et al. Genetic and epigenetic association studies suggest a role of microRNA biogenesis gene exportin-5 (XPO5) in breast tumorigenesis. Int J Mol Epidemiol Genet. 2011;2:9–18. [PMC free article] [PubMed] [Google Scholar]
- 78.Hutvagner G, et al. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science. 2001;293:834–838. doi: 10.1126/science.1062961. [DOI] [PubMed] [Google Scholar]
- 79.Kumar MS, et al. Dicer1 functions as a haploinsufficient tumor suppressor. Genes Dev. 2009;23:2700–2704. doi: 10.1101/gad.1848209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pugh TJ, et al. Exome sequencing of pleuropulmonary blastoma reveals frequent biallelic loss of TP53 and two hits in DICER1 resulting in retention of 5p-derived miRNA hairpin loop sequences. Oncogene. 2014;33:5295–5302. doi: 10.1038/onc.2014.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wagh PK, et al. Cell- and developmental stage-specific Dicer1 ablation in the lung epithelium models cystic pleuropulmonary blastoma. J Pathol. 2014;236:41–52. doi: 10.1002/path.4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.de Kock L, et al. Germ-line and somatic DICER1 mutations in a pleuropulmonary blastoma. Pediatr Blood Cancer. 2013;60:2091–2092. doi: 10.1002/pbc.24692. [DOI] [PubMed] [Google Scholar]
- 83.Seki M, et al. Biallelic DICER1 mutations in sporadic pleuropulmonary blastoma. Cancer Res. 2014;74:2742–2749. doi: 10.1158/0008-5472.CAN-13-2470. [DOI] [PubMed] [Google Scholar]
- 84.Schultz KA, et al. Ovarian sex cord-stromal tumors, pleuropulmonary blastoma and DICER1 mutations: a report from the International Pleuropulmonary Blastoma Registry. Gynecol Oncol. 2011;122:246–250. doi: 10.1016/j.ygyno.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Witkowski L, et al. DICER1 hotspot mutations in non-epithelial gonadal tumours. Br J Cancer. 2013;109:2744–2750. doi: 10.1038/bjc.2013.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Foulkes WD, et al. Extending the phenotypes associated with DICER1 mutations. Hum Mutat. 2011;32:1381–1384. doi: 10.1002/humu.21600. [DOI] [PubMed] [Google Scholar]
- 87.Wu MK, et al. Biallelic DICER1 mutations occur in Wilms tumours. J Pathol. 2013;230:154–164. doi: 10.1002/path.4196. [DOI] [PubMed] [Google Scholar]
- 88.de Kock L, et al. Pituitary blastoma: a pathognomonic feature of germ-line DICER1 mutations. Acta Neuropathol. 2014;128:111–122. doi: 10.1007/s00401-014-1285-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Doros LA, et al. DICER1 mutations in childhood cystic nephroma and its relationship to DICER1-renal sarcoma. Mod Pathol. 2014;27:1267–1280. doi: 10.1038/modpathol.2013.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Doros L, et al. DICER1 mutations in embryonal rhabdomyosarcomas from children with and without familial PPB-tumor predisposition syndrome. Pediatr Blood Cancer. 2012;59:558–560. doi: 10.1002/pbc.24020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schultze-Florey RE, et al. DICER1 syndrome: a new cancer syndrome. Klin Padiatr. 2013;225:177–178. doi: 10.1055/s-0033-1337976. [DOI] [PubMed] [Google Scholar]
- 92.Su X, et al. TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature. 2010;467:986–990. doi: 10.1038/nature09459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Melo SA, et al. A TARBP2 mutation in human cancer impairs microRNA processing and DICER1 function. Nature Genet. 2009;41:365–370. doi: 10.1038/ng.317. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 94.Garre P, Perez-Segura P, Diaz-Rubio E, Caldes T, de la Hoya M. Reassessing the TARBP2 mutation rate in hereditary nonpolyposis colorectal cancer. Nature Genet. 2010;42:817–818. doi: 10.1038/ng1010-817. [DOI] [PubMed] [Google Scholar]
- 95.De Vito C, et al. A TARBP2-dependent miRNA expression profile underlies cancer stem cell properties and provides candidate therapeutic reagents in Ewing sarcoma. Cancer Cell. 2012;21:807–821. doi: 10.1016/j.ccr.2012.04.023. [DOI] [PubMed] [Google Scholar]
- 96.van Kouwenhove M, Kedde M, Agami R. MicroRNA regulation by RNA-binding proteins and its implications for cancer. Nature Rev Cancer. 2011;11:644–656. doi: 10.1038/nrc3107. [DOI] [PubMed] [Google Scholar]
- 97.Kawai S, Amano A. BRCA1 regulates microRNA biogenesis via the DROSHA microprocessor complex. J Cell Biol. 2012;197:201–208. doi: 10.1083/jcb.201110008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Trabucchi M, et al. The RNA-binding protein KSRP promotes the biogenesis of a subset of microRNAs. Nature. 2009;459:1010–1014. doi: 10.1038/nature08025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wu H, et al. A splicing-independent function of SF2/ASF in microRNA processing. Mol Cell. 2010;38:67–77. doi: 10.1016/j.molcel.2010.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Guil S, Caceres JF. The multifunctional RNA-binding protein hnRNP A1 is required for processing of miR-18a. Nature Struct Mol Biol. 2007;14:591–596. doi: 10.1038/nsmb1250. [DOI] [PubMed] [Google Scholar]
- 101.Michlewski G, Guil S, Semple CA, Caceres JF. Posttranscriptional regulation of miRNAs harboring conserved terminal loops. Mol Cell. 2008;32:383–393. doi: 10.1016/j.molcel.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Morlando M, et al. FUS stimulates microRNA biogenesis by facilitating co-transcriptional Drosha recruitment. EMBO J. 2012;31:4502–4510. doi: 10.1038/emboj.2012.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Suzuki HI, et al. Modulation of microRNA processing by p53. Nature. 2009;460:529–533. doi: 10.1038/nature08199. [DOI] [PubMed] [Google Scholar]
- 104.Davis BN, Hilyard AC, Lagna G, Hata A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature. 2008;454:56–61. doi: 10.1038/nature07086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Davis BN, Hilyard AC, Nguyen PH, Lagna G, Hata A. Smad proteins bind a conserved RNA sequence to promote microRNA maturation by Drosha. Mol Cell. 2010;39:373–384. doi: 10.1016/j.molcel.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ha M, Kim VN. Regulation of microRNA biogenesis. Nature Rev Mol Cell Biol. 2014;15:509–524. doi: 10.1038/nrm3838. [DOI] [PubMed] [Google Scholar]
- 107.Drake M, et al. A requirement for ERK-dependent dicer phosphorylation in coordinating oocyte-to-embryo transition in C. elegans. Dev Cell. 2014;31:614–628. doi: 10.1016/j.devcel.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mori M, et al. Hippo signaling regulates Microprocessor and links cell-density-dependent miRNA biogenesis to cancer. Cell. 2014;156:893–906. doi: 10.1016/j.cell.2013.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nature Rev Cancer. 2013;13:246–257. doi: 10.1038/nrc3458. [DOI] [PubMed] [Google Scholar]
- 110.Hwang HW, Wentzel EA, Mendell JT. Cell–cell contact globally activates microRNA biogenesis. Proc Natl Acad Sci USA. 2009;106:7016–7021. doi: 10.1073/pnas.0811523106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Leung AK, Sharp PA. MicroRNA functions in stress responses. Mol Cell. 2010;40:205–215. doi: 10.1016/j.molcel.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Franovic A, et al. Translational up-regulation of the EGFR by tumor hypoxia provides a nonmutational explanation for its overexpression in human cancer. Proc Natl Acad Sci USA. 2007;104:13092–13097. doi: 10.1073/pnas.0702387104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shen J, et al. EGFR modulates microRNA maturation in response to hypoxia through phosphorylation of AGO2. Nature. 2013;497:383–387. doi: 10.1038/nature12080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rupaimoole R, et al. Hypoxia-mediated downregulation of miRNA biogenesis promotes tumour progression. Nature Commun. 2014;5:5202. doi: 10.1038/ncomms6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.van den Beucken T, et al. Hypoxia promotes stem cell phenotypes and poor prognosis through epigenetic regulation of DICER. Nature Commun. 2014;5:5203. doi: 10.1038/ncomms6203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Peter ME. Let-7 and miR-200 microRNAs: guardians against pluripotency and cancer progression. Cell Cycle. 2009;8:843–852. doi: 10.4161/cc.8.6.7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Barh D, Malhotra R, Ravi B, Sindhurani P. MicroRNA let-7: an emerging next-generation cancer therapeutic. Curr Oncol. 2010;17:70–80. doi: 10.3747/co.v17i1.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mayr C, Hemann MT, Bartel DP. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science. 2007;315:1576–1579. doi: 10.1126/science.1137999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Akao Y, Nakagawa Y, Naoe T. let-7 microRNA functions as a potential growth suppressor in human colon cancer cells. Biol Pharm Bull. 2006;29:903–906. doi: 10.1248/bpb.29.903. [DOI] [PubMed] [Google Scholar]
- 120.Boyerinas B, Park SM, Hau A, Murmann AE, Peter ME. The role of let-7 in cell differentiation and cancer. Endocr Relat Cancer. 2010;17:F19–F36. doi: 10.1677/ERC-09-0184. [DOI] [PubMed] [Google Scholar]
- 121.Bussing I, Slack FJ, Grosshans H. let-7 microRNAs in development, stem cells and cancer. Trends Mol Med. 2008;14:400–409. doi: 10.1016/j.molmed.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 122.Droge P, Davey CA. Do cells let-7 determine stemness? Cell Stem Cell. 2008;2:8–9. doi: 10.1016/j.stem.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 123.Rybak A, et al. A feedback loop comprising lin-28 and let-7 controls pre-let-7 maturation during neural stem-cell commitment. Nature Cell Biol. 2008;10:987–993. doi: 10.1038/ncb1759. [DOI] [PubMed] [Google Scholar]
- 124.Viswanathan SR, Daley GQ, Gregory RI. Selective blockade of microRNA processing by Lin28. Science. 2008;320:97–100. doi: 10.1126/science.1154040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Heo I, et al. Lin28 mediates the terminal uridylation of let-7 precursor MicroRNA. Mol Cell. 2008;32:276–284. doi: 10.1016/j.molcel.2008.09.014. References 124–126 reveal that LIN28A and LIN28B selectively inhibit let-7 miRNA biogenesis. [DOI] [PubMed] [Google Scholar]
- 126.Newman MA, Thomson JM, Hammond SM. Lin-28 interaction with the Let-7 precursor loop mediates regulated microRNA processing. RNA. 2008;14:1539–1549. doi: 10.1261/rna.1155108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Madison BB, et al. LIN28B promotes growth and tumorigenesis of the intestinal epithelium via Let-7. Genes Dev. 2013;27:2233–2245. doi: 10.1101/gad.224659.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Urbach A, et al. Lin28 sustains early renal progenitors and induces Wilms tumor. Genes Dev. 2014;28:971–982. doi: 10.1101/gad.237149.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Viswanathan SR, et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nature Genet. 2009;41:843–848. doi: 10.1038/ng.392. This paper reveals the roles of the LIN28–let-7 pathway in the regulation of oncogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Nguyen LH, et al. Lin28b is sufficient to drive liver cancer and necessary for its maintenance in murine models. Cancer Cell. 2014;26:248–261. doi: 10.1016/j.ccr.2014.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Molenaar JJ, et al. LIN28B induces neuroblastoma and enhances MYCN levels via let-7 suppression. Nature Genet. 2012;44:1199–1206. doi: 10.1038/ng.2436. [DOI] [PubMed] [Google Scholar]
- 132.Beachy SH, et al. Enforced expression of Lin28b leads to impaired T-cell development, release of inflammatory cytokines, and peripheral T-cell lymphoma. Blood. 2012;120:1048–1059. doi: 10.1182/blood-2012-01-401760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.King CE, et al. LIN28B fosters colon cancer migration, invasion and transformation through let-7-dependent and -independent mechanisms. Oncogene. 2011;30:4185–4193. doi: 10.1038/onc.2011.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Thornton JE, Gregory RI. How does Lin28 let-7 control development and disease? Trends Cell Biol. 2012;22:474–482. doi: 10.1016/j.tcb.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhu H, et al. The Lin28/let-7 axis regulates glucose metabolism. Cell. 2011;147:81–94. doi: 10.1016/j.cell.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-κB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139:693–706. doi: 10.1016/j.cell.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hamano R, et al. High expression of Lin28 is associated with tumour aggressiveness and poor prognosis of patients in oesophagus cancer. Br J Cancer. 2012;106:1415–1423. doi: 10.1038/bjc.2012.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Picard D, et al. Markers of survival and metastatic potential in childhood CNS primitive neuro-ectodermal brain tumours: an integrative genomic analysis. Lancet Oncol. 2012;13:838–848. doi: 10.1016/S1470-2045(12)70257-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Diskin SJ, et al. Common variation at 6q16 within HACE1 and LIN28B influences susceptibility to neuroblastoma. Nature Genet. 2012;44:1126–1130. doi: 10.1038/ng.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hovestadt V, et al. Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing. Nature. 2014;510:537–541. doi: 10.1038/nature13268. [DOI] [PubMed] [Google Scholar]
- 141.Zhang WC, et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell. 2012;148:259–272. doi: 10.1016/j.cell.2011.11.050. [DOI] [PubMed] [Google Scholar]
- 142.Piskounova E, et al. Determinants of microRNA processing inhibition by the developmentally regulated RNA-binding protein Lin28. J Biol Chem. 2008;283:21310–21314. doi: 10.1074/jbc.C800108200. [DOI] [PubMed] [Google Scholar]
- 143.Nam Y, Chen C, Gregory RI, Chou JJ, Sliz P. Molecular basis for interaction of let-7 microRNAs with Lin28. Cell. 2011;147:1080–1091. doi: 10.1016/j.cell.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hagan JP, Piskounova E, Gregory RI. Lin28 recruits the TUTase Zcchc11 to inhibit let-7 maturation in mouse embryonic stem cells. Nature Struct Mol Biol. 2009;16:1021–1025. doi: 10.1038/nsmb.1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Heo I, et al. TUT4 in concert with Lin28 suppresses microRNA biogenesis through pre-microRNA uridylation. Cell. 2009;138:696–708. doi: 10.1016/j.cell.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 146.Thornton JE, Chang HM, Piskounova E, Gregory RI. Lin28-mediated control of let-7 microRNA expression by alternative TUTases Zcchc11 (TUT4) and Zcchc6 (TUT7) RNA. 2012;18:1875–1885. doi: 10.1261/rna.034538.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chang HM, Triboulet R, Thornton JE, Gregory RI. A role for the Perlman syndrome exonuclease Dis3l2 in the Lin28–let-7 pathway. Nature. 2013;497:244–248. doi: 10.1038/nature12119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Faehnle CR, Walleshauser J, Joshua-Tor L. Mechanism of Dis3l2 substrate recognition in the Lin28–let-7 pathway. Nature. 2014;514:252–256. doi: 10.1038/nature13553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ustianenko D, et al. Mammalian DIS3L2 exoribonuclease targets the uridylated precursors of let-7 miRNAs. RNA. 2013;19:1632–1638. doi: 10.1261/rna.040055.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Astuti D, et al. Germline mutations in DIS3L2 cause the Perlman syndrome of overgrowth and Wilms tumor susceptibility. Nature Genet. 2012;44:277–284. doi: 10.1038/ng.1071. [DOI] [PubMed] [Google Scholar]
- 151.Kumar MS, et al. HMGA2 functions as a competing endogenous RNA to promote lung cancer progression. Nature. 2014;505:212–217. doi: 10.1038/nature12785. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 152.Chin LJ, et al. A SNP in a let-7 microRNA complementary site in the KRAS 3′ untranslated region increases non-small cell lung cancer risk. Cancer Res. 2008;68:8535–8540. doi: 10.1158/0008-5472.CAN-08-2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kanellopoulou C, et al. Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev. 2005;19:489–501. doi: 10.1101/gad.1248505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wang Y, Medvid R, Melton C, Jaenisch R, Blelloch R. DGCR8 is essential for microRNA biogenesis and silencing of embryonic stem cell self-renewal. Nature Genet. 2007;39:380–385. doi: 10.1038/ng1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Chakravarti D, et al. Induced multipotency in adult keratinocytes through down-regulation of ΔNp63 or DGCR8. Proc Natl Acad Sci USA. 2014;111:E572–E581. doi: 10.1073/pnas.1319743111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 157.Gurtan AM, et al. Let-7 represses Nr6a1 and a mid-gestation developmental program in adult fibroblasts. Genes Dev. 2013;27:941–954. doi: 10.1101/gad.215376.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Melton C, Judson RL, Blelloch R. Opposing microRNA families regulate self-renewal in mouse embryonic stem cells. Nature. 2010;463:621–626. doi: 10.1038/nature08725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Trang P, et al. Regression of murine lung tumors by the let-7 microRNA. Oncogene. 2009;29:1580–1587. doi: 10.1038/onc.2009.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Yu F, et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131:1109–1123. doi: 10.1016/j.cell.2007.10.054. [DOI] [PubMed] [Google Scholar]
- 161.Li X, et al. Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nature Med. 2014;20:769–777. doi: 10.1038/nm.3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Muralidhar B, et al. Global microRNA profiles in cervical squamous cell carcinoma depend on Drosha expression levels. J Pathol. 2007;212:368–377. doi: 10.1002/path.2179. [DOI] [PubMed] [Google Scholar]
- 163.Sand M, et al. Expression levels of the microRNA processing enzymes Drosha and dicer in epithelial skin cancer. Cancer Invest. 2010;28:649–653. doi: 10.3109/07357901003630918. [DOI] [PubMed] [Google Scholar]
- 164.Passon N, et al. Expression of Dicer and Drosha in triple-negative breast cancer. J Clin Pathol. 2012;65:320–326. doi: 10.1136/jclinpath-2011-200496. [DOI] [PubMed] [Google Scholar]
- 165.Avery-Kiejda KA, Braye SG, Forbes JF, Scott RJ. The expression of Dicer and Drosha in matched normal tissues, tumours and lymph node metastases in triple negative breast cancer. BMC Cancer. 2014;14:253. doi: 10.1186/1471-2407-14-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Papachristou DJ, et al. Immunohistochemical analysis of the endoribonucleases Drosha, Dicer and Ago2 in smooth muscle tumours of soft tissues. Histopathology. 2012;60:E28–E36. doi: 10.1111/j.1365-2559.2012.04192.x. [DOI] [PubMed] [Google Scholar]
- 167.Tchernitsa O, et al. Systematic evaluation of the miRNA-ome and its downstream effects on mRNA expression identifies gastric cancer progression. J Pathol. 2010;222:310–319. doi: 10.1002/path.2759. [DOI] [PubMed] [Google Scholar]
- 168.Vaksman O, Hetland TE, Trope CG, Reich R, Davidson B. Argonaute, Dicer, and Drosha are up-regulated along tumor progression in serous ovarian carcinoma. Hum Pathol. 2012;43:2062–2069. doi: 10.1016/j.humpath.2012.02.016. [DOI] [PubMed] [Google Scholar]
- 169.Diaz-Garcia CV, et al. DICER1, DROSHA and miRNAs in patients with non-small cell lung cancer: implications for outcomes and histologic classification. Carcinogenesis. 2013;34:1031–1038. doi: 10.1093/carcin/bgt022. [DOI] [PubMed] [Google Scholar]
- 170.Catto JW, et al. Distinct microRNA alterations characterize high- and low-grade bladder cancer. Cancer Res. 2009;69:8472–8481. doi: 10.1158/0008-5472.CAN-09-0744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Torres A, et al. Major regulators of microRNAs biogenesis Dicer and Drosha are down-regulated in endometrial cancer. Tumour Biol. 2011;32:769–776. doi: 10.1007/s13277-011-0179-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Yan M, et al. Dysregulated expression of dicer and drosha in breast cancer. Pathol Oncol Res. 2012;18:343–348. doi: 10.1007/s12253-011-9450-3. [DOI] [PubMed] [Google Scholar]
- 173.Sand M, et al. Expression levels of the microRNA maturing microprocessor complex component DGCR8 and the RNA-induced silencing complex (RISC) components argonaute-1, argonaute-2, PACT, TARBP1, and TARBP2 in epithelial skin cancer. Mol Carcinog. 2011;51:916–922. doi: 10.1002/mc.20861. [DOI] [PubMed] [Google Scholar]
- 174.Ambs S, et al. Genomic profiling of microRNA and messenger RNA reveals deregulated microRNA expression in prostate cancer. Cancer Res. 2008;68:6162–6170. doi: 10.1158/0008-5472.CAN-08-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kim B, et al. An essential microRNA maturing microprocessor complex component DGCR8 is up-regulated in colorectal carcinomas. Clin Exp Med. 2013;14:331–336. doi: 10.1007/s10238-013-0243-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Guo Y, et al. Silencing the double-stranded RNA binding protein DGCR8 inhibits ovarian cancer cell proliferation, migration, and invasion. Pharm Res. 2013;32:769–778. doi: 10.1007/s11095-013-1219-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Chiosea S, et al. Up-regulation of dicer, a component of the microRNA machinery, in prostate adenocarcinoma. Am J Pathol. 2006;169:1812–1820. doi: 10.2353/ajpath.2006.060480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Jakymiw A, et al. Overexpression of dicer as a result of reduced let-7 microRNA levels contributes to increased cell proliferation of oral cancer cells. Genes Chromosomes Cancer. 2010;49:549–559. doi: 10.1002/gcc.20765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Faber C, Horst D, Hlubek F, Kirchner T. Overexpression of Dicer predicts poor survival in colorectal cancer. Eur J Cancer. 2011;47:1414–1419. doi: 10.1016/j.ejca.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 180.Stratmann J, et al. Dicer and miRNA in relation to clinicopathological variables in colorectal cancer patients. BMC Cancer. 2011;11:345. doi: 10.1186/1471-2407-11-345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Papachristou DJ, et al. Expression of the ribonucleases Drosha, Dicer, and Ago2 in colorectal carcinomas. Virchows Arch. 2011;459:431–440. doi: 10.1007/s00428-011-1119-5. [DOI] [PubMed] [Google Scholar]
- 182.Chiosea S, et al. Overexpression of Dicer in precursor lesions of lung adenocarcinoma. Cancer Res. 2007;67:2345–2350. doi: 10.1158/0008-5472.CAN-06-3533. [DOI] [PubMed] [Google Scholar]
- 183.Ma Z, et al. Up-regulated Dicer expression in patients with cutaneous melanoma. PLoS ONE. 2011;6:e20494. doi: 10.1371/journal.pone.0020494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Dedes KJ, et al. Down-regulation of the miRNA master regulators Drosha and Dicer is associated with specific subgroups of breast cancer. Eur J Cancer. 2011;47:138–150. doi: 10.1016/j.ejca.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 185.Wu D, et al. Downregulation of Dicer, a component of the microRNA machinery, in bladder cancer. Mol Med Rep. 2012;5:695–699. doi: 10.3892/mmr.2011.711. [DOI] [PubMed] [Google Scholar]
- 186.Pampalakis G, Diamandis EP, Katsaros D, Sotiropoulou G. Down-regulation of dicer expression in ovarian cancer tissues. Clin Biochem. 2010;43:324–327. doi: 10.1016/j.clinbiochem.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 187.Faggad A, et al. Prognostic significance of Dicer expression in ovarian cancer — link to global microRNA changes and oestrogen receptor expression. J Pathol. 2010;220:382–391. doi: 10.1002/path.2658. [DOI] [PubMed] [Google Scholar]
- 188.Khoshnaw SM, et al. Loss of Dicer expression is associated with breast cancer progression and recurrence. Breast Cancer Res Treat. 2012;135:403–413. doi: 10.1007/s10549-012-2169-3. [DOI] [PubMed] [Google Scholar]
- 189.Wu JF, et al. Down-regulation of Dicer in hepatocellular carcinoma. Med Oncol. 2011;28:804–809. doi: 10.1007/s12032-010-9520-5. [DOI] [PubMed] [Google Scholar]
- 190.Zhu DX, et al. Downregulated Dicer expression predicts poor prognosis in chronic lymphocytic leukemia. Cancer Sci. 2012;103:875–881. doi: 10.1111/j.1349-7006.2012.02234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Faggad A, et al. Down-regulation of the microRNA processing enzyme Dicer is a prognostic factor in human colorectal cancer. Histopathology. 2012;61:552–561. doi: 10.1111/j.1365-2559.2011.04110.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.