

Abstract
Cas9 is an RNA-guided DNA endonuclease that targets foreign DNA for destruction as part of a bacterial adaptive immune system mediated by CRISPR (clustered regularly interspaced short palindromic repeats)1,2. Together with single-guide RNAs (sgRNA)3, Cas9 also functions as a powerful genome engineering tool in plants and animals4–6, and efforts are underway to increase the efficiency and specificity of DNA targeting for potential therapeutic applications7,8. Studies of off-target effects have shown that DNA binding is far more promiscuous than DNA cleavage9–11, yet the molecular cues that govern strand scission have not been elucidated. Here we show that the conformational state of the HNH nuclease domain directly controls DNA cleavage activity. Using intramolecular Förster resonance energy transfer (FRET) experiments to detect relative orientations of the Cas9 catalytic domains when associated with on- and off-target DNA, we find that DNA cleavage efficiencies scale with the extent to which the HNH domain samples an activated conformation. We furthermore uncover a surprising mode of allosteric communication that ensures concerted firing of both Cas9 nuclease domains. Our results highlight a proofreading mechanism beyond initial PAM recognition12 and RNA–DNA base-pairing3 that serves as a final specificity checkpoint before DNA double-strand break formation.
Cas9 is a large, multi-domain protein that undergoes RNA-induced conformational changes to reach a DNA binding-competent state13. Crystal structures of apo13, sgRNA-bound14, and sgRNA/DNA-bound15,16 Cas9 from Streptococcus pyogenes (Fig. 1a, b) have revealed distinct conformational states of the protein but failed to explain its DNA cleavage mechanism because in each structure, the HNH domain active site is positioned at least 30 Å away from the DNA cleavage site15,16. Furthermore, available structures could not explain why DNA cleavage is precluded at stably bound off-target sites with incomplete complementarity. We hypothesized that functionally important HNH conformational dynamics could influence the cleavage specificity of the Cas9-guide RNA enzyme complex. To test this possibility, we developed a FRET-based approach to investigate Cas9structural changes in response to binding sgRNA and DNA ligands.
Figure 1. Full-length sgRNA drives inward lobe closure of Cas9.
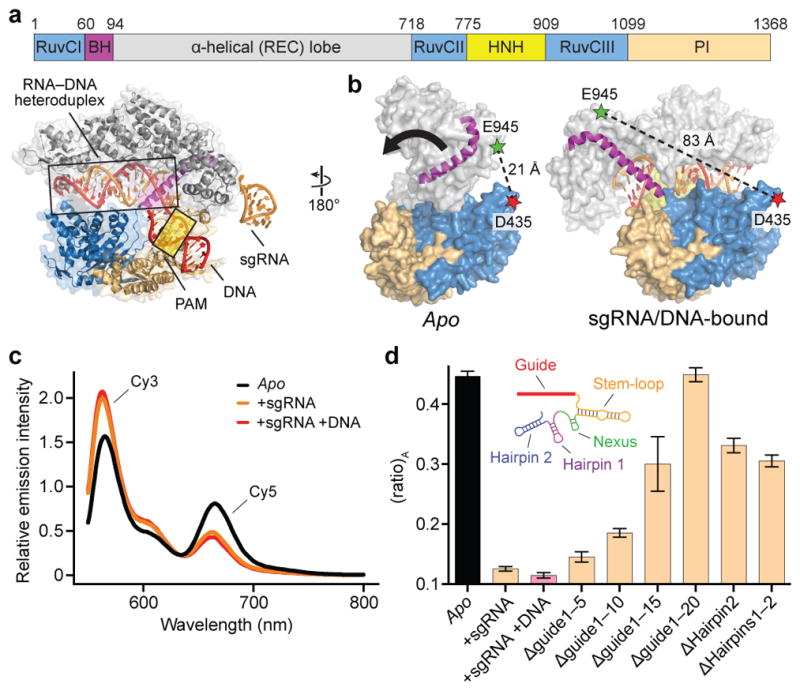
a, Domain organization of S. pyogenes Cas9 (top) and X-ray crystal structure of sgRNA/DNA-bound Cas9 (PDB ID 4UN3, ref. 16) (bottom), with HNH domain omitted for clarity. BH, bridge helix; REC, recognition; PI, PAM-interacting. b, Design of Cas9hinge FRET construct. Measured distances between D435 and E945 in apo (PDB ID 4CMP, ref. 13) and sgRNA/DNA-bound Cas9 structures are indicated. Inward lobe closure is exemplified by movement of the BH (arrow). Regions of the PI domain, sgRNA, and DNA are omitted for clarity. c, Fluorescence emission spectra for Cas9hinge in the presence of the indicated substrates. d, (ratio)A data for Cas9hinge. Inset: schematic of full-length sgRNA coloured by motif29. Error bars represent the standard deviation; n=3.
We generated a FRET construct to monitor Cas9 structural rearrangements upon sgRNA binding13 (Fig. 1b). Starting with a cysteine-free Cas9 variant, we introduced cysteine residues at positions D435 and E945 near the hinge region and labeled these residues with Cy3- and Cy5-maleimide dyes, generating Cas9hinge. Control labeling reactions with cysteine-free Cas9 confirmed the specificity of conjugation, and doubly-labeled Cas9 was fully functional for DNA cleavage (Extended Data Fig. 1a–c). Measurements from available structures revealed an expected distance change of ~60 Å upon sgRNA and DNA binding (Extended Data Table 1), and indeed, when Cy3 of sgRNA-bound Cas9hinge was excited at 530 nm, we observed a substantial decrease in energy transfer compared to apo-Cas9hinge, as evidenced by a relative increase in donor (Cy3) fluorescence relative to acceptor (Cy5) fluorescence (Fig. 1c); the observed change scaled with the molar ratio of sgRNA to Cas9. A mixture of donor-only and acceptor-only labeled Cas9hinge showed no evidence of energy transfer, and an sgRNA specific to Neisseria meningitidis Cas9 (ref. 17), which significantly impairs S. pyogenes Cas9 binding (data not shown), elicited a negligible change (Extended Data Fig. 2a–c). We conclude that the change in fluorescence intensities resulted from an sgRNA-induced, intramolecular conformational change in Cas9hinge.
Cas9hinge exhibited an ~70% decrease in energy transfer upon sgRNA binding as determined by (ratio)A, whereby the acceptor fluorescence intensity via energy transfer is normalized to that via direct excitation18,19 (Methods and Extended Data Fig. 2d). Target DNA binding induced little further change in FRET (Fig. 1c, d), consistent with available structural data (Extended Data Table 1). To identify the molecular determinants that trigger conformational rearrangement of Cas9, we tested truncated variants of the sgRNA (Extended Data Table 2) and found that the 20-nt target recognition sequence has a critical role in controlling the Cas9 conformational state (Fig. 1d). An sgRNA lacking the entire guide segment (Δguide1-20) generated a (ratio)A value indistinguishable from apo-Cas9hinge while being >95% bound under our experimental conditions14, whereas sgRNAs containing part of the 20-nt guide segment partially restored the change in (ratio)A. sgRNA variants lacking one or both hairpins at the 3′ end (Δhairpins1-2) also generated intermediate (ratio)A values (Fig. 1d) while retaining sub-nanomolar binding affinity to Cas9 (ref. 20), and similar data were obtained with catalytically dead (i.e. D10A/H840A) dCas9hinge (Extended Data Fig. 2e). We conclude that motifs at both ends of the sgRNA are required to stabilize a closed state of Cas9, but that in the case of Δhairpins1-2, a fully closed state is not required for rapid cleavage kinetics20. We propose that intermediate (ratio)A changes reflect stable sgRNA-Cas9hinge complexes interconverting between open and closed conformers.
We next focused on the HNH nuclease domain. Since existing crystal structures exhibit inactive HNH domain conformations15,16, we built a model for the putative activated state by docking a homologous HNH–dsDNA crystal structure21 onto the sgRNA/DNA-bound Cas9 structure (Extended Data Fig. 3a–d). We selected two pairs of positions (S355–S867 and S867–N1054) whose inter-residue distances, according to our model, would change substantially upon target DNA binding (Fig. 2a, Extended Data Fig. 3e, and Extended Data Table 1). Cas9 labeled with Cy3 and Cy5 at these sites (Cas9HNH-1 and Cas9HNH-2) retained nearly wild-type DNA cleavage activity (Extended Data Fig. 1c).
Figure 2. FRET experiments reveal an activated conformation of the HNH nuclease domain.

a, Design of Cas9HNH-1 FRET construct. Measured distances between S355 and S867 in the sgRNA/DNA-bound Cas9 structure16 and a model of the HNH domain docked at the cleavage site are indicated, as are putative conformational changes of the HNH domain (arrow). The model was generated using an HNH homolog structure (PDB ID 2QNC, ref. 21). b, Fluorescence emission spectra for dCas9HNH-1 in the presence of the indicated substrates. Inset: (ratio)A values; mut, mutation. c, (ratio)A data for dCas9HNH-1. Mismatches were introduced sequentially from the PAM-distal end of the target. d, Cleavage rate constants using wild-type Cas9. ND, cleavage not detected. e, (ratio)A data for catalytically active Cas9HNH-1 and Cas9HNH-2. Error bars in b–e represent the standard deviation; n=3.
We observed a substantial FRET increase for catalytically inactive dCas9HNH-1 upon target DNA binding relative to sgRNA alone (Fig. 2b), and control experiments with non-target DNA or off-target DNA substrates containing either PAM or seed mutations failed to generate this change (Fig. 2b and Extended Data Table 2). We next monitored FRET with off-target DNA substrates containing mutations distal from the PAM, which retain high-affinity Cas9 binding12,22. Remarkably, the observed (ratio)A values decreased as the number of mismatches increased (Fig. 2c), and these changes were not attributable to decreasing occupancy of the sgRNA/DNA-bound complex: direct binding assays indicate ≥89% of the dCas9HNH-1 population should be bound to all tested DNA substrates, and increasing the concentration of dsDNA had no discernible effect on (ratio)A (Extended Data Fig. 4a, b). Our results show that the HNH domain samples a conformational equilibrium with on-target DNA that is distinct from partially matching off-target DNA, and suggest that the high FRET state corresponds to an active HNH conformation at the cleavage site.
We suspected that altered conformational states of the HNH domain could explain which off-target DNA substrates are cleaved by Cas9. Substrates with ≥4-bp mismatches that displayed a low (ratio)A value were cleaved slowly if at all (Fig. 2d and Extended Data Fig. 4c), as observed previously22,23, indicating that the inability to access the high FRET state associated with an activated HNH conformation precludes cleavage. Interestingly, substrates with only 1–3 bp mismatches at the distal end of the target sequence were cleaved at near wild-type rates despite having diminished (ratio)A values relative to the on-target. This suggests that rapidly interconverting conformational states, one of which is the activated state, may still enable rapid cleavage. Truncated sgRNAs with shorter regions of target complementarity that exhibit enhanced fidelity in genome editing experiments24 may similarly facilitate efficient on-target cleavage without stabilizing an activated HNH conformation. Single-molecule experiments will be necessary to reveal these putative dynamics, which are unavoidably averaged in our ensemble measurements.
We observed a similar pattern of (ratio)A changes using catalytically active Cas9HNH, and the opposite trend of (ratio)A changes was observed with Cas9HNH-2, a construct designed to undergo a high-to-low FRET efficiency transition upon on-target DNA binding (Fig. 2e and Extended Data Fig. 3e, 4d). These data suggest that positioning of the HNH domain is largely unaffected by actual strand scission, but instead reflects a conformational equilibrium that is particularly sensitive to RNA-DNA heteroduplex formation at the distal end of the target. These observations underscore the importance of RNA-DNA complementarity throughout the target region, rather than only the seed sequence closest to the PAM, in controlling Cas9 cleavage specificity.
The HNH and RuvC nuclease domains cleave target and non-target strands 3 bp upstream of the PAM, respectively3,25. For partially unwound off-target substrates with mismatches >10 bp further upstream, target strand cleavage is precluded by conformational control of the HNH domain. However, the mechanism by which RuvC domain-catalyzed non-target strand cleavage is avoided remains unknown. We hypothesized that this activity would be sensitive to HNH domain conformational changes. We first separately measured HNH and RuvC domain cleavage rates for a panel of partially mismatched substrates and found that both strands were consistently cleaved in synchrony (Fig. 3a, b and Extended Data Fig. 5a, b). We next used shorter DNA substrates with or without internal mismatches, such that Cas9-mediated DNA unwinding up to the site of an sgRNA-DNA mismatch would theoretically present identical substrates to the RuvC domain active site (Fig. 3a). After separately measuring non-target strand cleavage kinetics and Cas9HNH-1 FRET, we observed a tight correlation between RuvC domain cleavage activity and the presence of an activated HNH conformational state (Fig. 3c and Extended Data Fig. 5c–e). This finding provides strong evidence that HNH conformational dynamics exert allosteric control over the RuvC nuclease domain. Furthermore, the RuvC domain could still effectively cleave the non-target strand of a bubbled substrate that induced an activated HNH conformation, but whose target strand could not be cleaved by the HNH domain due to mismatches in the seed (Fig. 3d, e). Together, these data argue that HNH conformational changes, but not HNH nuclease function, trigger RuvC domain nuclease activity.
Figure 3. RuvC nuclease activity is allosterically controlled by HNH conformational changes.
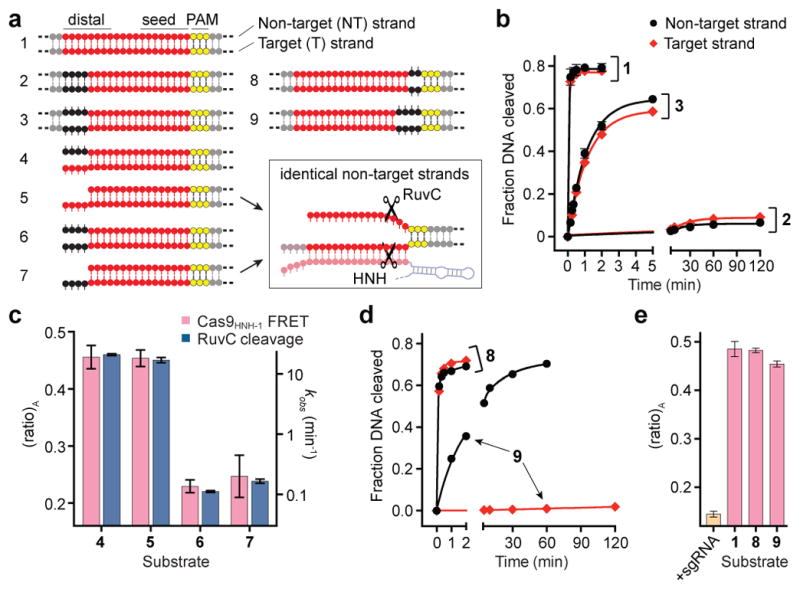
a, Tested DNA substrates, with on-target (1) at top. Matched and mismatched positions of DNA target strand sequences relative to the sgRNA are coloured red and black, respectively, with the PAM in yellow. Some substrates contain internal mismatches between the two DNA strands; dashed lines indicate additional flanking sequence. Schematic at bottom right depicts identical non-target strand substrates presented to the RuvC nuclease domain in substrates 5 and 7. b, Non-target (black) and target (red) strand cleavage time courses for the indicated DNA substrates using wild-type Cas9. Exponential fits are shown as solid lines. c, (ratio)A data for Cas9HNH-1 (red bars, left y-axis) and non-target strand cleavage kinetics of the RuvC domain (blue bars, right y-axis) for the indicated DNA substrates. d, Non-target and target strand cleavage time courses for the indicated DNA substrates using wild-type Cas9. Exponential fits are shown as solid lines. e, (ratio)A data for Cas9HNH-1. Error bars in b–e represent the standard deviation; n=3.
We wondered how Cas9 achieves this functional coupling. The HNH domain is inserted between RuvC domain motifs II and III, but linkers connecting both domains are consistently disordered in available crystal structures and there are relatively few inter-domain contacts13,15,16 (Extended Data Fig. 6a). We purified an HNH deletion construct, ΔHNH–Cas9 (Extended Data Fig. 6a–c), that retained nearly wild-type DNA binding activity while being defective in non-target strand cleavage by the RuvC domain (Fig. 4a, b and Extended Data Fig. 6d). Thus, the HNH domain is required for RuvC nuclease domain activation but is dispensable for RNA-guided DNA targeting.
Figure 4. Mechanism of communication between the HNH and RuvC nuclease domains to achieve concerted DNA cleavage.

a, Target DNA binding assay with dCas9 and ΔHNH–Cas9, resolved by native PAGE (top); for gel source data, see Supplementary Figure 1. Quantified data are below; binding fits are shown as solid lines. b, Target DNA cleavage assay with dCas9, wild-type (WT) Cas9, and ΔHNH–Cas9, resolved by denaturing PAGE. S, substrate; NT, cleaved non-target strand; T, cleaved target strand. c, Zoom-in view of the sgRNA/DNA-bound Cas9 structure16 (top) highlights two α-helices connecting the HNH domain C-terminus and RuvC III N-terminus. Bottom shows sequence alignment30 of this region, and residues mutated to proline or alanine are indicated (arrows). d, Target DNA cleavage assay with the indicated Cas9 variants, resolved by denaturing PAGE. e, Target (red) and non-target (black) strand cleavage time courses with the indicated Cas9 variants (for WT-Cas9 data, see Fig. 3b). Exponential fits are shown as solid lines. Error bars in a and e represent the standard deviation; n=5 or 3, respectively. f, Model for conformational control of target cleavage by CRISPR-Cas9.
Finally, we sought to identify the basis of allostery between the HNH and RuvC domains. We hypothesized that two α-helices connecting the HNH and RuvC III motifs (residues S909–N940), previously shown to adopt an extended conformation and proposed to assist the HNH domain in approaching the cleavage site15, was instead acting as a signal transducer (Extended Data Fig. 7a). We introduced a series of proline residues to specifically disrupt this α-helix and found that target strand cleavage kinetics by the HNH domain were minimally affected (Fig. 4c–e and Extended Data Fig. 7b, c). In stark contrast, RuvC domain nuclease activity was almost completely blocked with an E923P/T924P–Cas9 mutant, and this effect could be reversed with the corresponding alanine mutations (Fig. 4d, e and Extended Data Fig. 7c). The finding that this effect was not confined to highly conserved residues supports the idea that disruption of the helix-forming propensity of this region, and not specific point mutations, disabled the RuvC domain. We conclude that an intact, extended α-helix acts as an allosteric switch to communicate the HNH conformational change to the RuvC domain and activate it for cleavage. Understanding the mechanism of activation will likely require additional structures of Cas9 in a pre-cleavage state, with the intact non-target strand substrate bound in the RuvC active site.
Our data support a model in which Cas9 endonuclease employs multiple levels of regulation to ensure accurate target DNA cleavage (Fig. 4f and Supplementary Video 1). After identification of potential targets via PAM binding and directional DNA unwinding dependent on sgRNA-DNA complementarity, recognition of on-target DNA drives a conformational change in the HNH nuclease domain that enables productive engagement with the scissile phosphate. Importantly, this same structural transition triggers RuvC domain catalytic activity, ensuring concerted cleavage of both DNA strands. Partially complementary off-target DNA sequences may stably bind Cas9, but by failing to drive HNH conformational changes, avoid cleavage. This proofreading mechanism is strikingly similar to the R-loop locking mechanism employed by the RNA-guided targeting complex (Cascade) from Type I CRISPR-Cas systems, in which RNA-DNA heteroduplex formation at the PAM-distal end of the target exerts allosteric control over Cascade conformational rearrangements near the PAM-proximal end that are required for subsequent target cleavage22,26. Beyond providing fundamental insights into the mechanism of DNA interrogation by Cas9, our findings have important implications for the use of Cas9 as a genome engineering technology. For example, our data can explain why little cleavage occurs at off-target DNA sequences identified in ChIP-seq experiments9–11, and suggest that DNA nicking by the native Cas9 enzyme is disfavored in cells due to concerted cutting by the HNH and RuvC nuclease domains. Finally, our findings demonstrate an exciting opportunity to use protein conformational changes that report on target DNA recognition for fluorescence-based readout of DNA binding in cells.
METHODS
Cas9 and nucleic acid preparation
Streptococcus pyogenes Cas9 was cloned into a custom pET-based expression vector encoding an N-terminal His10-tag followed by maltose-binding protein (MBP) and a TEV protease cleavage site. Point mutations were introduced using site-directed mutagenesis or around-the-horn PCR and verified by DNA sequencing. dCas9 refers to catalytically inactive (dead) Cas9 containing D10A and H840A mutations. ΔHNH-Cas9 contains a deletion of residues T769–K918 and replacement with a GGSGGS linker. The HNH domain for add-back experiments (Extended Data Fig. 6d) encodes residues N776–G907. All Cas9 variants were purified as described13.
Single-guide RNA (sgRNA) templates were PCR amplified and cloned into EcoRI and BamHI sites in pUC19, and encode full-length crRNA and tracrRNA sequences connected via a GAAA tetraloop (Extended Data Table 2). sgRNAs were transcribed in vitro as described27 and purified using 5–10% denaturing polyacrylamide gel electrophoresis (PAGE).
DNA substrates (Extended Data Table 2) were prepared from commercially synthesized oligonucleotides (Integrated DNA Technologies). DNA duplexes without internal mismatches were prepared and purified by native PAGE as described13. DNA duplexes containing internal mismatches or overhangs were prepared by mixing a 5X molar excess of one strand with its complementary strand in hybridization buffer (20 mM Tris-Cl pH 7.5, 100 mM KCl, 5 mM MgCl2), heating at 95 °C for 1–2 min, and slow-cooling on the benchtop. For FRET experiments, the non-target strand was in excess over the target strand; for biochemical cleavage experiments, the non-radiolabeled strand was in excess over the radiolabeled strand.
Preparation of dye-labeled Cas9
Labeling reactions were conducted in Cas9 gel filtration buffer (20 mM Tris-Cl pH 7.5, 200 mM KCl, 5% glycerol, 1 mM TCEP) and contained 10 μM Cas9 and 200 μM Cy3- and Cy5-maleimide (GE Healthcare). Dyes were initially dissolved in anhydrous DMSO before being mixed with Cas9, and the final DMSO concentration did not exceed 5%. Reactions were incubated in the dark for 2 h at room temperature followed by overnight incubation at 4 °C. Reactions were quenched by adding 10 mM DTT, and labeled Cas9 was separated from free dye by size exclusion chromatography on a Superdex 200 10/300 column. Samples were then concentrated, snap frozen in liquid nitrogen, and stored at −80 °C. Control labeling reactions contained either cysteine-free Cas9 or only one of the two dyes.
FRET experiments
All fluorescence measurements were conducted at room temperature in reaction buffer (20 mM Tris-Cl pH 7.5, 100 mM KCl, 5 mM MgCl2, 5% glycerol, 1 mM DTT), supplemented with 50 μg/mL heparin to reduce non-specific DNA binding12. 60 μL reactions with Cas9hinge (C80S/D435C/C574S/E945C-Cas9 labeled with Cy3/Cy5) and dCas9hinge (Cas9hinge with additional nuclease-inactivating D10A/H840A mutations) contained either 50 nM or 100 nM Cas9, and when present, a 10X and 4X molar excess of sgRNA and target DNA, respectively. 60 μL reactions with Cas9HNH-1 (C80S/S355C/C574S/S867C-Cas9 labeled with Cy3/Cy5), dCas9HNH-1 (Cas9HNH-1 with additional nuclease-inactivating D10A/H840A mutations), and Cas9HNH-2 (C80S/C574S/S867C/N1054C-Cas9 labeled with Cy3/Cy5) contained 50 nM Cas9, and when present, 200 nM sgRNA and DNA unless otherwise indicated.
We observed substantial aggregation of apo-Cas9 upon 10 min incubation at 37 °C, as indicated by apparent intermolecular FRET with a single-cysteine Cas9 (C80S/C574S/S867C) that had been labeled with a mixture of Cy3- and Cy5-maleimide (data not shown). This aggregation could be completely avoided by incubating reactions for 10 min at room temperature instead, centrifuging reactions for 5 min at 16,000 × g and 4 °C, and using the supernatant for subsequent fluorescence measurements. This binding protocol was used for all reported FRET data. Reactions were kept at room temperature for ~10–100 min before acquisition of fluorescence spectra, and this variable time delay had no effect on the resulting data, even for reactions with catalytically active Cas9 (data not shown).
Fluorescence measurements were collected with a 3 mm path-length quartz cuvette (Hellma Analytics) and a FluoroMax-3 (HORIBA Jobin Yvon), using 5 nm slit widths and 0.2 s integration time. For each sample, two fluorescence emission spectra were recorded: 1) the sample was excited at 530 nm and emitted light was collected from 550–800 nm in 1 nm increments, and 2) the sample was excited at 630 nm and emitted light was collected from 650–800 nm in 1 nm increments. Data processing was conducted using FluorEssence software (HORIBA Jobin Yvon). Experiments were replicated at least three times, and the presented data are representative results unless stated otherwise.
FRET analysis
The distance between donor and acceptor dyes can be directly calculated from the FRET efficiency, but accurately relating these variables requires knowledge of numerous complex parameters18. Our labeling strategy resulted in a heterogeneous mixture of unlabeled, singly- and doubly-labeled species, further complicating the analysis. We therefore report (ratio)A as defined by Clegg and colleagues18,19, whereby acceptor (Cy5) fluorescence via energy transfer is normalized against acceptor fluorescence via direct excitation (Extended Data Fig. 2d), without pursuing a more rigorous calculation of exact distances. (ratio)A is directly proportional to FRET efficiency, and changes in (ratio)A across different experimental conditions serve as a proxy for conformational changes.
For each FRET construct, a donor-only (Cy3-labeled) sample was prepared and its emission spectrum in the apo state after 530 nm excitation collected. This spectrum was normalized to and subtracted from each experimental emission spectrum to generate an extracted fluorescence spectrum for the acceptor via energy transfer. The integrated area under this curve from 650–800 nm was calculated and divided by the integrated area under the curve of a spectrum resulting from direct acceptor excitation at 630 nm, resulting in (ratio)A, the enhancement of acceptor fluorescence due to FRET. Raw fluorescence emission spectra presented in the Figures and Extended Data Figures were normalized and smoothed using the Savistsky-Golay method, and all data analysis was conducted using Prism (GraphPad Software).
DNA binding and cleavage assays
Biochemical assays were conducted essentially as described13. Binding reactions utilized <0.1 nM 5′-[32P]-DNA duplex substrates radiolabeled on both strands and a constant excess of 100 nM sgRNA in the presence of increasing concentrations of dCas9 or ΔHNH-Cas9. Cas9 and sgRNA were pre-incubated at 37 °C for 10 min in reaction buffer supplemented with 50 μg/mL heparin before being incubated with DNA for ~30 min at room temperature. Reactions were resolved by 5% native PAGE (0.5X TBE, 5 mM MgCl2) at 4 °C and visualized by phosphorimaging (GE Healthcare).
DNA cleavage experiments presented in Figures 2d, 4b, d and Extended Data Figures 1c, 6d used 5′-[32P]-DNA duplex substrates radiolabeled on both strands; all other cleavage experiments used DNA duplex substrates with a single 5′-[32P]-radiolabeled strand that had been annealed to a 5X molar excess of unlabeled complementary strand. Cas9 and sgRNA were pre-incubated at 37 °C for 10 min in reaction buffer before adding DNA. Cleavage reactions were performed at room temperature and contained 1 nM DNA and 100 nM Cas9–sgRNA complex. Aliquots were removed at various time points and quenched by mixing with an equal volume of formamide gel loading buffer supplemented with 50 mM EDTA. Cleavage products were resolved by 10% denaturing PAGE and visualized by phosphorimaging (GE Healthcare). Reported pseudo-first order rate constants (kobs) represent the population-weighted average from double-exponential fits (target strand cleavage data for proline mutants, Fig. 4e and Extended Data Fig. 7b, c) or the result from single-exponential fits (all other data). In some cases where the observed fraction of cleaved DNA was <0.1 after 2 h, the exponential fit plateau was fixed at 0.75 to avoid overestimating the rate constant.
Experiments were replicated at least three times, and presented data are representative results unless stated otherwise.
Cas9 structural analysis
All structure figures were generated using Pymol (Schrödinger). Cas9 molecules from distinct crystal structures were aligned using the RuvC and PI domains (RMSD ~0.5–0.7). To generate the modeled docked state for the HNH domain (Fig. 2a and Extended Data Fig. 3), nucleotides 12–13 of chain D of PDB ID 2QNC (endo VII-DNA structure) were first aligned to nucleotides 11–12 of chain C of PDB ID 4UN3 (sgRNA/DNA-bound Cas9 structure). A copy of the Cas9 HNH domain from PDB ID 4UN3 was then aligned to chain A of PDB ID 2QNC. Conservation rendering was done using a multiple sequence alignment of 250 Cas9 homologs and the ConSurf server28.
Extended Data
Extended Data Figure 1. Biochemical preparation and DNA cleavage activity of dye-labeled Cas9.

a, Size exclusion chromatograms of Cy3/Cy5-labeling reactions with cysteine-free Cas9 (C80S/C574S) or the two double-cysteine Cas9 variants used to generate Cas9hinge and Cas9HNH-1. Reactions contained 10 μM Cas9 and 200 μM Cy3- and Cy5-maleimide, and were separated on a Superdex 200 10/300 column (GE Healthcare). Cysteine-free Cas9 was unreactive. b, Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis of unlabeled and dye-labeled Cas9 variants. The gel was scanned for Cy3 and Cy5 fluorescence (right) before being stained with Coommassie blue (left). For gel source data, see Supplementary Figure 1. c, Representative radiolabeled DNA cleavage assay with wild-type (WT) Cas9 and doubly-labeled Cas9 variants used in this study, resolved by denaturing PAGE (left); quantified data and exponential fits are shown on the right. S, substrate; NT, cleaved non-target strand; T, cleaved target strand. Error bars represent the standard deviation from three experiments.
Extended Data Figure 2. Fluorescence control experiments with Cas9hinge and dCas9hinge, and representative analysis of fluorescence emission spectra to calculate (ratio)A.

a, Fluorescence emission spectra of 50 nM Cas9hinge in the presence of increasing concentrations of full-length sgRNA. Protein and sgRNA concentrations were calculated under non-denaturing conditions using theoretical extinction coefficients. b, Fluorescence emission spectra of: (1) Cy3-labeled Cas9hinge, (2) Cy5-labeled Cas9hinge, and (3) an equal mixture of Cy3-Cas9hinge and Cy5-Cas9hinge upon excitation at 530 nm. The minor fluorescence peak for Cy5 in the mixed sample results from residual absorbance of Cy5-Cas9hinge at 530 nm and not from intermolecular FRET (compare spectra 3 to 4, which is a sum of spectra 1 and 2). c, Fluorescence emission spectra of Cas9hinge in the presence of sgRNA substrates specific to Streptococcus pyogenes (Spy) or Neisseria meningitidis (Nme) Cas9. d, Determination of the (ratio)A parameter, which is proportional to FRET efficiency. Shown for apo-Cas9hinge are: (1) an emission spectrum of Cy3/Cy5-Cas9hinge upon excitation of the donor at 530 nm; (2) an emission spectrum of donor only Cy3-Cas9hinge upon excitation of the donor at 530 nm, normalized to 1; (3) the extracted fluorescence of the acceptor via energy transfer, obtained by subtracting 2 from 1; and (4) an emission spectrum of Cy3/Cy5-Cas9hinge upon direct excitation of the acceptor at 630 nm. (ratio)A is calculated by dividing the integrated intensity (650–800 nm) of 4 by the integrated intensity of 2. e, (ratio)A data for dCas9hinge in the presence of the same sgRNA substrates tested with nuclease-active Cas9hinge in Figure 1e. Error bars represent the standard deviation from three experiments.
Extended Data Figure 3. Modeling of the HNH domain docked at the cleavage site, and design of the Cas9HNH-2 FRET construct.

a, The scissile phosphate and flanking nucleotides of a DNA substrate co-crystallized with the phage T4 endonuclease VII (endo VII; PDB ID 2QNC; left) was aligned with the scissile phosphate and flanking nucleotides of the DNA target strand in the sgRNA/DNA-bound Cas9 crystal structure (PDB ID 4UN3; middle). Structural alignment of the Cas9 HNH domain with endo VII (middle) results in a model for how the Cas9 HNH domain docks at the cleavage site (right). Catalytic residues are labeled, target strands are shown in red and pink, and a magnesium ion is depicted as a blue sphere. b, A conservation rendering of the sgRNA/DNA-bound Cas9 crystal structure, generated using ConSurf, shows that the most highly conserved patches of the HNH domain, including the active site, are solvent exposed in the observed conformation. The HNH domain is omitted from the view on the left for clarity. c, Zoom-in view of the HNH domain in its observed conformation (left) and the model for the docked state (right), colored as in b. The DNA target strand fits snugly in a groove on the HNH domain in the model, with the most highly conserved patches located in the immediate vicinity of the scissile phosphate. DNA and sgRNA are colored red and orange, respectively. d, The conformational flexibility of the HNH domain in available Cas9 crystal structures is revealed by structural alignment of the nuclease lobe (RuvC and PI domains) from two sgRNA/DNA-bound structures (PDB IDs 4UN3 and 4OO8) and the sgRNA-bound structure (PDB ID 4ZT0). The modeled docked state from a is shown. e, Design of Cas9HNH-2 FRET construct. Measured distances between ~N1054 and S867 in the sgRNA/DNA-bound Cas9 structure and a model of the HNH domain docked at the cleavage site are indicated. Putative conformational changes of the HNH domain are shown with a black arrow.
Extended Data Figure 4. Evidence that variable (ratio)A values for dCas9HNH-1 reflect distinct conformational states/dynamics, and FRET data for Cas9HNH-2.
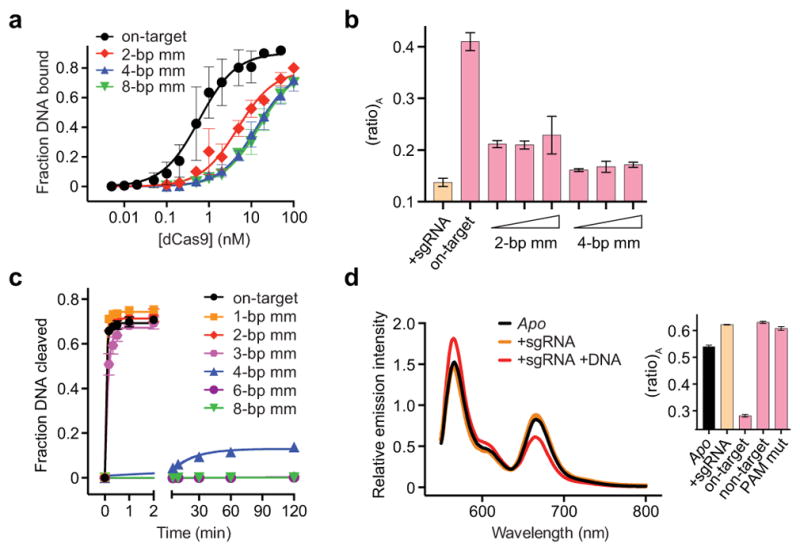
a, DNA binding assay with dCas9 and either on-target DNA or off-target DNAs containing 2, 4, or 8-bp mismatches at the PAM-distal end. Binding fits are shown as solid lines and yield equilibrium dissociation constants (Kd) of 0.80, 6.7, 19, and 20 nM, respectively. Given these values, 99%, 96%, 89%, and 89% of dCas9 should be bound to DNA under the conditions used for FRET experiments in Figure 2c (50 nM dCas9HNH-1, 200 nM DNA). b, (ratio)A data for 50 nM dCas9HNH-1 in the presence of 1 μM sgRNA and either 200 nM, 400 nM, or 1 μM off-target DNAs containing 2- or 4-bp mismatches. Data for sgRNA only and on-target DNA are shown for comparison. c, DNA cleavage time courses for the indicated DNA substrates using wild-type Cas9. Exponential fits are shown as solid lines, and extracted rate constants are shown in Figure 2d. d, Fluorescence emission spectra of Cas9HNH-2 in the presence of the indicated substrates. The inset shows (ratio)A values; mut, mutation. Error bars in a and b–d represent the standard deviation from three-to-five and three experiments, respectively.
Extended Data Figure 5. Additional experimental support for dependence of RuvC nuclease activity on HNH conformational changes.

a, Panel of DNA substrates tested in b, with on-target (1) at top. Matched and mismatched positions of DNA target strand sequences relative to the sgRNA are colored red and black, respectively, with the PAM in yellow. Some substrates contain internal mismatches between the two DNA strands; dashed lines indicate additional flanking sequence. b, Kinetics of non-target (black) and target (red) strand cleavage for the indicated DNA substrates. c, Panel of DNA substrates tested in d and e, depicted as in a. d, (ratio)A data for Cas9HNH-1 in the presence of the indicated DNA substrates. e, Non-target strand cleavage kinetics of the RuvC domain for the indicated DNA substrates. Error bars in b, d, e represent the standard deviation from three experiments.
Extended Data Figure 6. Design, purification, and DNA cleavage activity of ΔHNH-Cas9.

a, Domain organization of WT- and ΔHNH-Cas9 (top), showing the residues that were replaced with a GGS2 linker to generate ΔHNH-Cas9. Zoom-in view of connections between the HNH domain and RuvC II and III motifs in the apo (left) and sgRNA/DNA-bound (right) Cas9 crystal structures, as well as in the ΔHNH-Cas9 construct. Disordered linkers and the introduced GGS2 linker are shown as dashed lines. b, Size exclusion chromatograms of WT- and ΔHNH-Cas9 using a Superdex 200 16/60 column (GE Healthcare). c, SDS-PAGE analysis of dCas9 (D10A/H840A), WT-Cas9, ΔHNH-Cas9, and the purified HNH domain (residues 776–907). Expected molecular weights are 159 kDa, 159 kDa, 142 kDa, and 16 kDa, respectively. For gel source data, see Supplementary Figure 1. c, Representative radiolabeled DNA cleavage assay with WT-Cas9, ΔHNH-Cas9, ΔHNH-Cas9 in the presence of excess HNH domain, and HNH domain alone, resolved by denaturing PAGE.
Extended Data Figure 7. Structural analysis and perturbation of the HNH–RuvC III linker.
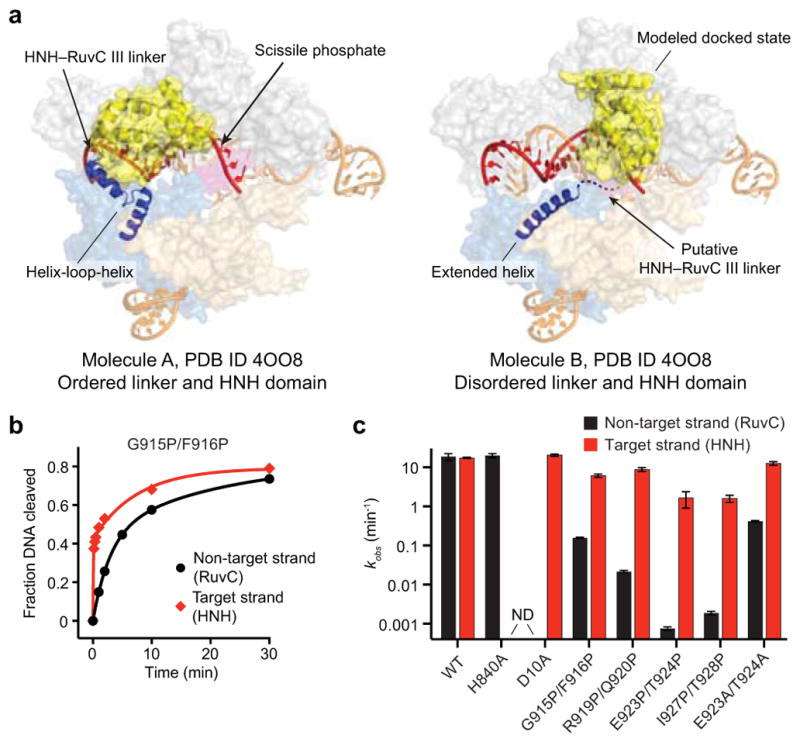
a, Molecules A (left) and B (right) of the sgRNA/DNA-bound Cas9 crystal structure (PDB ID 4OO8). Molecule A has an ordered HNH domain and HNH–RuvC III linker, whereas these are both disordered in molecule B; the missing density for the HNH domain is replaced with the modeled docked state (right). Another prominent difference is the N-terminal region of the RuvC III motif (blue helices), which rearranges from a helix-loop-helix in molecule A into an extended helix in molecule B. Proline pairs were inserted to prevent formation of this extended helix. b, Target (red) and non-target (black) strand cleavage time courses with the indicated Cas9 variant. Exponential fits are shown as solid lines. c, Kinetics of target (red) and non-target (black) strand cleavage for the indicated DNA substrates. ND, cleavage not detected. Error bars in b and c represent the standard deviation from three experiments.
Extended Data Table 1.
Measured distances between residues labeled with FRET pairs
| Inter-residue distance *
|
|||
|---|---|---|---|
| Structure used | D435–E945 | S355–S867 | S867–N1054 |
| Apo (4CMP) | 21 Å | 79 Å | 6 Å |
| sgRNA-bound (4ZT0) | 78 Å | 81 Å | 7 Å |
| sgRNA/DNA-bound (4OO8 mol A) | 77 Å † | 61 Å | 34 Å ‡ |
| sgRNA/DNA-bound (4UN3) | 83 Å | 59 Å | 28 Å § |
| sgRNA/DNA-bound, HNH docked state || | 21 Å | 57 Å § | |
Distances were measured between Cα atoms of the indicated residues, except where indicated, for the denoted structures (PDB IDs in parentheses).
E945 is disordered in the structure; an average of measured distances to T941 and I950 is reported.
N1054 is disordered in the structure; an average of measured distances to T1048 and I1063 is reported.
N1054 is disordered in the structure; an average of measured distances to I1050 and K1059 is reported.
The docked state for the HNH domain was generated using PDB IDs 4UN3 and 2QNC.
Extended Data Table 2.
RNA and DNA substrates used in this study

|
sgRNA guide sequences and matching DNA target strand sequences are shown in red. PAM sites (5′-NGG-3′) are highlighted in yellow on the non-target strand. Internal mismatches in select DNA substrates are denoted by misaligned text on the non-target strand.
All sgRNA constructs contain remnants of the BamHI sequence on the 3′ end resulting from run-off in vitro transcription.
sgRNA specific to Neisseria meningitidis (Nme) Cas9 contains an additional 3′ extension, which does not affect activity (data not shown), for purposes unrelated to this study.
Δguide1–20 sgRNA contains an extraneous 5′-G from in vitro transcription.
Supplementary Material
Acknowledgments
We thank David Taylor and Janice Chen for helpful discussions, Mitchell O’Connell, Linlin Ma, Nan Ma, and Kaihong Zhou for technical assistance, and members of the Doudna laboratory for critical reading of the manuscript. S.H.S. acknowledges support from the National Science Foundation and National Defense Science & Engineering Graduate Research Fellowship programs. J.A.D. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
S.H.S. and J.A.D. are inventors on a related patent application.
Methods, along with any additional Extended Data display items and Source Data, are available in the online version of the paper; references unique to these sections appear only in the online paper.
AUTHOR CONTRIBUTIONS
S.H.S. designed and conducted all experiments. B.L. and M.K. assisted with protein purification, dye labeling, cleavage assays, and fluorescence experiments. All authors discussed the data; S.H.S. and J.A.D. wrote the manuscript.
References
- 1.van der Oost J, Westra ER, Jackson RN, Wiedenheft B. Unravelling the structural and mechanistic basis of CRISPR-Cas systems. Nat Rev Microbiol. 2014;12:479–492. doi: 10.1038/nrmicro3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barrangou R, Marraffini LA. CRISPR-Cas systems: Prokaryotes upgrade to adaptive immunity. Mol Cell. 2014;54:234–244. doi: 10.1016/j.molcel.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jinek M, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 6.Sternberg SH, Doudna JA. Expanding the Biologist’s Toolkit with CRISPR-Cas9. Mol Cell. 2015;58:568–574. doi: 10.1016/j.molcel.2015.02.032. [DOI] [PubMed] [Google Scholar]
- 7.Wu X, Kriz AJ, Sharp PA. Target specificity of the CRISPR-Cas9 system. Quant Biol. 2014;2:59–70. doi: 10.1007/s40484-014-0030-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gori JL, et al. Delivery and Specificity of CRISPR-Cas9 Genome Editing Technologies for Human Gene Therapy. Hum Gene Ther. 2015;26:443–451. doi: 10.1089/hum.2015.074. [DOI] [PubMed] [Google Scholar]
- 9.Wu X, et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat Biotechnol. 2014;32:670–676. doi: 10.1038/nbt.2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsai SQ, et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol. 2015;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ran FA, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014;507:62–67. doi: 10.1038/nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jinek M, et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science. 2014;343:1247997. doi: 10.1126/science.1247997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang F, Zhou K, Ma L, Gressel S, Doudna JA. A Cas9-guide RNA complex preorganized for target DNA recognition. Science. 2015;348:1477–1481. doi: 10.1126/science.aab1452. [DOI] [PubMed] [Google Scholar]
- 15.Nishimasu H, et al. Crystal structure of cas9 in complex with guide RNA and target DNA. Cell. 2014;156:935–949. doi: 10.1016/j.cell.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anders C, Niewoehner O, Duerst A, Jinek M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature. 2014;513:569–573. doi: 10.1038/nature13579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hou Z, et al. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc Natl Acad Sci USA. 2013;110:15644–15649. doi: 10.1073/pnas.1313587110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Majumdar ZK, Hickerson R, Noller HF, Clegg RM. Measurements of internal distance changes of the 30S ribosome using FRET with multiple donor-acceptor pairs: quantitative spectroscopic methods. J Mol Biol. 2005;351:1123–1145. doi: 10.1016/j.jmb.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 19.Clegg RM. Fluorescence resonance energy transfer and nucleic acids. Meth Enzymol. 1992;211:353–388. doi: 10.1016/0076-6879(92)11020-j. [DOI] [PubMed] [Google Scholar]
- 20.Wright AV, et al. Rational design of a split-Cas9 enzyme complex. Proc Natl Acad Sci USA. 2015;112:2984–2989. doi: 10.1073/pnas.1501698112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Biertümpfel C, Yang W, Suck D. Crystal structure of T4 endonuclease VII resolving a Holliday junction. Nature. 2007;449:616–620. doi: 10.1038/nature06152. [DOI] [PubMed] [Google Scholar]
- 22.Szczelkun MD, et al. Direct observation of R-loop formation by single RNA-guided Cas9 and Cascade effector complexes. Proc Natl Acad Sci USA. 2014;111:9798–9803. doi: 10.1073/pnas.1402597111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cencic R, et al. Protospacer adjacent motif (PAM)-distal sequences engage CRISPR Cas9 DNA target cleavage. PLoS ONE. 2014;9:e109213. doi: 10.1371/journal.pone.0109213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32:279–284. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci USA. 2012;109:E2579–86. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rutkauskas M, et al. Directional R-Loop Formation by the CRISPR-Cas Surveillance Complex Cascade Provides Efficient Off-Target Site Rejection. Cell Rep. 2015;10:1534–1543. doi: 10.1016/j.celrep.2015.01.067. [DOI] [PubMed] [Google Scholar]
- 27.Sternberg SH, Haurwitz RE, Doudna JA. Mechanism of substrate selection by a highly specific CRISPR endoribonuclease. RNA. 2012;18:661–672. doi: 10.1261/rna.030882.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010;38:W529–33. doi: 10.1093/nar/gkq399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Briner AE, et al. Guide RNA functional modules direct cas9 activity and orthogonality. Mol Cell. 2014;56:333–339. doi: 10.1016/j.molcel.2014.09.019. [DOI] [PubMed] [Google Scholar]
- 30.Robert X, Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42:W320–4. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.