

Abstract
We investigated the anticancer potential of a new synthetic compound, 7-(3-fluorophenyl)-4-methylpyrido-[2,3-d]pyrimidin-5(8H)-one (MT3-037). We found that MT3-037 effectively decreased the cancer cell viability by inducing apoptosis. MT3-037 treatments led to cell cycle arrest at M phase, with a marked increase in both expression of cyclin B1 and cyclin-dependent kinase 1 (CDK1) as well as in CDK1 kinase activity. Key proteins that regulate mitotic spindle dynamics, including survivin, Aurora A/B kinases, and polo-like kinase 1 (PLK1), were activated in MT3-037-treated cells. MT3-037-induced apoptosis was accompanied by activation of a pro-apoptotic factor, FADD, and the inactivation of apoptosis inhibitors, Bcl-2 and Bcl-xL, resulting in the cleavage/activation of caspases. The activation of c-Jun N-terminal kinase (JNK) was associated with MT3-037-induced CDK1 and Aurora A/B activation and apoptosis. Immunofluorescence staining of tubulin indicated that MT3-037 altered tubulin networks in cancer cells. Moreover, an in vitro tubulin polymerization assay revealed that MT3-037 inhibited the tubulin polymerization by direct binding to tubulin. Molecular docking studies and binding site completion assays revealed that MT3-037 binds to the colchicine-binding site. Furthermore, MT3-037 significantly inhibited the tumor growth in both MDAMB-468 and Erlotinib-resistant MDA-MB-468 xenograft mouse models. In addition, MT3-037 inhibited the angiogenesis and disrupted the tube formation by human endothelial cells. Our study demonstrates that MT3-037 is a potential tubulin-disrupting agent for antitumor therapy.
Keywords: Microtubule, CDK1, JNK, apoptosis, angiogenesis
Introduction
The central constituent of the cytoskeleton, microtubules, are composed of α- and β-tubulin. With highly dynamic structures, microtubules are involved in a variety of fundamental cell functions, including maintenance of cell shape, transport of vesicles and proteins, regulation of cell movement, and coordination of cell division [1,2]. Because of their importance in the formation of the mitotic spindle and in regulating the mitotic apparatus, disruptions of microtubule dynamics can induce cell cycle arrest at G2/M phase, resulting in the formation of abnormal mitotic spindles and triggering apoptosis [3,4]. Therefore, impairing mitotic-spindle microtubules has been one of the most successful strategies for anticancer therapy [4-6].
Agents that affect microtubule functions can be divided into two groups based on their effects on microtubule assembly. The first group is the microtubule-stabilizing agents, such as taxanes (paclitaxel, docetaxel, laulimalide, and epothilones). These compounds stabilize microtubules, enhance microtubule polymerization, and induce the formation of microtubule bundles in cells [6,7]. The second group is microtubule-destabilizing agents, such as colchicine and the vinca alkaloids (vincristine, vinblastine) which inhibit microtubule polymerization and decrease the length of microtubules in cells [6,7]. Both types of microtubule agents alter microtubule dynamics via direct binding to tubulin. Several specific binding sites for these agents have been identified, including taxane, laulimalide, peloruside A, colchicine, and vinca alkaloids [7]. The strong ability of these agents, such as paclitaxel and vinblastine, to disrupt microtubules, inhibit proliferation, and induce programmed cell death have made them very effective in clinical therapy for cancers [4-6].
Chromosome segregation is a precisely regulated process directed by the mitotic spindle, a highly dynamic microtubule-based structure. The assembly and regulation of mitotic spindle rely on the coordination of many mitotic proteins, including survivin, Aurora kinases, and PLK1 [8-10]. CDK1 is essential for cells to enter into mitosis, and its activation requires the formation of a complex with cyclin B1 and removal of inhibitory phosphorylation [11,12]. CDK1 coordinates with mitotic kinases in a feedback activation loop to ensure proper mitotic progression. Its activation results in nuclear lamina disassembly and attachment of cytosolic microtubules to condensing chromatin [13,14]. CDK1 also acts as a pro-apoptotic mediator. Many tubulin-interfering agents, including paclitaxel and vinca alkaloids, induce apoptosis through the activation of CDK1 [15]. Therefore, CDK1 is a logical target for anticancer chemotherapy [16].
In recent years, we have designed and synthesized several series of antimitotic agents, including 2-phenylquinolin-4-one, 2-arylquinolin-4-one, 2-arylnaphthyridin-4-one, and 2-arylquinazoline-4-one [17-20]. Most of these compounds exhibited potent antitumor activity and interfered with microtubule dynamics [17-20]. The 4-pyrimidin-5-one series is a new synthesized antimitotic agent with novel scaffold structure. In primary screening of cell viability, 7-(3-fluorophenyl)-4-methylpyrido-[2,3-d] pyrimidin-5(8H)-one (MT3-037) showed the most potent anticancer activity. In this study, we further investigated the anticancer mechanism of MT3-037. We found that MT3-037 inhibits the cancer cell viability by interfering with tubulin dynamics, deregulating cell cycle progression, and inducing apoptotic signaling cascades. MT3-037 also showed the promising antitumor and anti-angiogenic activities.
Materials and methods
Compounds
MT3-037 was synthesized from commercially available 2’-acetonaphthone (Sigma-Aldrich, St. Louis, MO, USA) in our laboratory at the Graduate Institute of Pharmaceutical Chemistry, College of Pharmacy, China Medical University, as described in Supplementary Figure 1. The spectral data for MT3-037 were in full agreement with the assigned structure, and its purity was >95% as assessed with high-performance liquid chromatography. MT3-037 was dissolved in dimethyl sulfoxide (DMSO) and stored at -20°C. The final concentration of DMSO in the treatments was <0.2%.
Antibodies and reagents
Antibodies against caspase-3, caspase-8, caspase-9, poly(ADP-ribose) polymerase (PARP), survivin, cyclin B1, cyclin E, phospho-CDK1 (Tyr15), phospho-Aurora A (Thr288)/Aurora B (Thr232)/Aurora C (Thr198), Aurora A/AIK, Aurora B/AIM1, phospho-c-Jun (Ser73), and phospho-histone H3 (Ser10) were obtained from Cell Signaling Technology (Beverly, MA, USA). Antibodies against Bcl-2, Bcl-xL, fas-associated protein with death domain (FADD), tumor necrosis factor receptor type 1-associated death domain protein (TRADD), receptor-interacting protein kinase 1 (RIP), CDK1, and c-Jun N-terminal kinase (JNK) were purchased from BD Biosciences (Pharmingen, San Diego, CA). Antibodies against α-tubulin, histone H3, and c-Jun were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibody against phospho-CDK1 (Thr161) was purchased from GeneTex (Irvine, CA, USA). Antibodies against b-actin, mitotic protein monoclonal 2 (MPM-2), PLK1, and phospho-JNK1/2 (Thr183/Tyr185) were purchased from EMD Millipore (Billerica, MA, USA). Roscovitine was purchased from Cayman Chemical (Ann Arbor, MI, USA). Z-VAD-FMK and podophyllotoxin were obtained from Enzo Life Sciences (Farmingdale, NY, USA). RPMI-1640 medium, DMEM/F-12, MEM, Medium 199, non-essential amino acid mix, sodium pyruvate, fetal bovine serum (FBS), and penicillin/streptomycin were obtained from Thermo Fisher Scientific (Waltham, MA, USA). Other chemicals were purchased from Sigma-Aldrich.
Cell lines and cell culture
The human acute promyelocytic leukemia cell line (HL-60), human chronic myelogenous leukemia cell line (K-562), human T-cell acute lymphoblastic leukemia cell lines (CCRF-CEM and MOLT-4), human lung adenocarcinoma cell line (A549), human hepatocellular carcinoma cell lines (Hep3B and HepG2), human lung fibroblast cell line (MRC-5), human skin fibroblast cell line (Detroit 551), and human umbilical vein endothelial cell line (HUVEC) were purchased from the Bioresource Collection and Research Center of the Food Industry Research and Development Institute (Hsinchu, Taiwan). The human lung adenocarcinoma cell line (NCI-H522) and human breast cancer cell line (MDA-MB-468) were purchased from the American Type Culture Collection (Manassas, VA, USA). HL-60, K-562, CCRF-CEM, MOLT-4, and NCI-H522 cells were cultured in RPMI-1640 medium supplemented with 10% (v/v) FBS, penicillin (100 U/ml), streptomycin (100 μg/ml), and 2 mM l-glutamine. A549, Hep3B, HepG2, and MDA-MB-468 cells were maintained in DMEM/F-12 supplemented with 10% FBS, penicillin (100 U/ml), and streptomycin (100 μg/ml). MRC-5 and Detroit 551 cells were maintained in MEM supplemented with 10% FBS, 2 mM l-glutamine, 0.1 mM non-essential amino acid mix, 1 mM sodium pyruvate, penicillin (100 U/ml), and streptomycin (100 μg/ml). Erlotinib-resistant MDA-MB-468 cells were established by treatment with gradually increasing concentrations of the drug over one month and then maintained in the presence of 1 μM Erlotinib. HUVECs were cultured in Medium 199 supplemented with heparin (25 U/ml; Sigma-Aldrich), endothelial cell growth supplement (30 μg/ml; EMD Millipore), 10% FBS, penicillin (100 U/ml), and streptomycin (100 μg/ml). All cells were maintained in a humidified incubator at 37°C in 5% CO2.
Cell viability assay
Cell viability was evaluated by the reduction of MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide). Cells were cultured in 96-well plates and treated with the compound. After treatment, MTT solution (1 mg/ml) was added to each well and plates were incubated for another 2 h. The resulting blue formazan product was dissolved in DMSO and detected spectrophotometrically by absorbance at 570 (for MTT) and 630 (for background).
Cell morphology observations and staining with 4’,6’-diamino-2-phenylindole (DAPI)
For morphology observations, MT3-037-treated cells were visualized and photographed using a phase-contrast microscope equipped with a digital camera (Leica Microsystems, Wetzlar, Germany). To visualize nuclear morphological changes, cells were stained with DAPI (Invitrogen, Carlsbad, CA, USA) and observed by fluorescence microscopy (Zeiss, Jena, Germany).
Flow cytometry
After treatments, cells were harvested and stained with annexin V and propidium iodide using the Annexin V: FITC Apoptosis Detection Kit II (BD Biosciences) and subjected to flow cytometry (FACSCalibur; Becton Dickinson, Mountain View, CA, USA). The percentages of apoptotic cells were quantified with CellQuest software (Becton Dickinson). The cell cycle distribution of treated cells was determined by staining DNA with propidium iodide followed by flow cytometry. For MPM-2 mitotic phosphoprotein expression, paraformaldehyde-fixed cells were labeled with anti-MPM-2 antibody followed by incubation with fluorescein isothiocyanate (FITC)-conjugated anti-mouse IgG. Cells were then stained with propidium iodide and analyzed with flow cytometry.
CDK1 kinase activity assay
CDK1 activity was determined using CDK1 Kinase Assay Kit (MBL International, Nagoya, Japan) according to the manufacturer’s protocols.
Immunofluorescence staining
Cells were grown on sterile coverslips embedded in a 6-well plate. After treatments, cells were fixed with 3.7% (w/v) paraformaldehyde and permeabilized with 0.2% (v/v) Triton X-100. After blocking with 2% (w/v) bovine serum albumin in phosphate-buffered saline (PBS), tubulin was detected using anti-α-tubulin antibody followed by reaction with FITC-conjugated secondary antibody (Invitrogen). Coverslips were mounted onto glass slides with Prolong Gold Antifade Reagent (Invitrogen), and fluorescence images were taken on a Leica Microsystems TCS SP2 Confocal Spectral microscope.
Preparation of polymerized tubulin fractions from cells
After treatments, cells were lysed in Triton lysis buffer (0.5% (v/v) Triton X-100, 25 mM Tris-HCl (pH 7.5), 100 mM NaCl, 2.5 mM EGTA, 2.5 mM EDTA, 5 mM NaF, 1 mM Na3VO4, 20 mM sodium β-glycerophosphate, 1 mM phenylmethanesulfonyl fluoride (PMSF), and 5 μg/ml each of aprotinin, leupeptin, and pepstatin A). The lysates were centrifuged at 12,000 × g for 10 min at room temperature. Each supernatant constituted the unpolymerized tubulin fraction, and the pellet was the polymerized tubulin fraction which was resuspended in Triton lysis buffer and sonicated. Lysates were subjected to western blotting.
In vitro tubulin polymerization assay
The effect of MT3-037 on tubulin polymerization was determined using the Tubulin Polymerization Assay kit (BK006P, Cytoskeleton, Denver, CO, USA). Briefly, 300 μg of pure tubulin (> 99% purity) was suspended in 100 ml G-PEM buffer (80 mM piperazine-1,4-bis(2-ethanesulfonic acid), 2 mM MgCl2, 0.5 mM EGTA, 1 mM GTP, pH 6.9, and 5% (v/v) glycerol). MT3-037, paclitaxel, colchicine, or vehicle was added to the tubulin suspensions, and then the suspensions were transferred to a pre-warmed 96-well plate. The tubulin polymerization reaction was carried out at 37°C, and dynamic changes were measured at 340 nm every 30 sec for 30 min on a microplate reader (BioTek, Gen5, Winooski, VT, USA).
For the colchicine competitive binding assay, pure tubulin was incubated with various concentrations of MT3-037, podophyllotoxin, or vinblastine at 37°C for 1 h followed by addition of 10 μM colchicine. Changes in absorbance were measured at 340 nm.
Molecular modeling
Molecular flexible docking analysis was performed using Dock 5.1.1 software [21]. Kollman partial charges were applied to the atoms of protein models for the force field calculation in the Dock software. Energy-optimized three-dimensional coordination of small molecules was generated using Marvin 5.2.2 software (2009, http://www.chemaxon.com) [22] and Balloon 0.6 software [23]. Additionally, Gasteiger partial charges for ligands were calculated with OpenBabel 2.2.3 software [24]. The parameters for Dock were set to iteratively generate 2000 orientations and 100 conformers in the binding pocket with “anchor size” of 1. The docked conformers were subsequently scored and ranked with HotLig [25] to predict the protein-ligand binding position and molecular interactions. The Figures for structural models were rendered using Chimera 1.5.3 [26] and Ligplot 4.4 software [27].
Western blot analysis
Cells were harvested, washed, and lysed in PBS containing proteinase inhibitors (1 mM PMSF and 5 μg/ml each of leupeptin, aprotinin, and pepstatin A) and phosphatase inhibitors (1 mM Na3VO4, 5 mM NaF) and then sonicated. Protein concentrations were estimated using the Bio-Rad Protein Assay kit (Hercules, CA, USA). Samples were resolved by SDS-PAGE and transferred to the polyvinylidene difluoride membranes (EMD Millipore). Each membrane was blocked in 5% (w/v) non-fat milk in Tris-buffered saline with 0.1% (v/v) Tween-20 for 1 h followed by incubation with specific primary antibodies at 4°C overnight. Each membrane was then incubated with the appropriate horseradish peroxidase-conjugated secondary antibody at room temperature for 1 h. Protein signals were detected by the Immobilon Western Chemiluminescent HRP Substrate (EMD Millipore) and visualized using the LAS-4000 imaging system (Fuji Photo Film Co., Tokyo, Japan).
Xenograft mouse model
Female nu/nu mice (5 weeks old) were from National Laboratory Animal Center, Taipei, Taiwan. Mice were maintained under the procedures and guidelines from the Institutional Animal Care and Use Committee of the National Health Research Institutes, Taipei, Taiwan. All experiments were supervised under the Institutional Animal Care and Use Committee, China Medical University, Taichung, Taiwan. MDA-MB-468 or Erlotinib-resistant MDA-MB-468 breast cancer cells (5 × 106 cells per mouse) were suspended in 0.1 ml of Matrigel solution (50% (v/v) Matrigel in PBS) and inoculated into the mammary fat pads of the mice. When the tumor masses reached 100 mm3, the tumor-bearing mice were randomly divided into groups for treatments with vehicle (5% DMSO and 5% Cremophor in PBS) or MT3-037. MT3-037 was administered intravenously at 1 mg/kg once every day for 32 days. Tumor size and mouse body weight were measured once every 4 days, and tumor volume (mm3) was calculated using the equation: length × (width)2 × 0.5. At the end of experiments, mice were sacrificed and tumor nodules were dissected and weighed.
In vivo matrigel plug angiogenesis assay
Female nu/nu mice (5 weeks old) were injected subcutaneously in the left flank with 0.5 ml of growth factor-reduced Matrigel (BD Biosciences) containing recombinant human vascular endothelial growth factor (50 ng/ml; Prospec, East Brunswick, NJ, USA), basic fibroblast growth factor (150 ng/ml; Prospec), and heparin (100 U/ml) with vehicle (PBS) or MT3-037. After 7 days, the mice were euthanized by CO2 inhalation for Matrigel plug removal. The hemoglobin content in the Matrigel plug was measured using Drabkin’s reagent (Sigma-Aldrich) for quantification of blood vessel formation.
Tube formation assay
The MT3-037-treated HUVECs were seeded onto Matrigel-coated 48-well culture plates. After 6 h of seeding, capillary-like structures were stained with calcein-AM (Invitrogen) and photographed using fluorescence microscopy.
Statistical analysis
All data are expressed as the mean ± standard error (SEM) of three independent experiments unless stated otherwise. The data were analyzed with Student’s t-tests. P values < 0.05 were considered significant.
Results
Effects of MT3-037 on viability of human cancer cell lines
MT3-037 was synthesized in our laboratory, and its structure and synthesis scheme are shown in Figure 1A and Supplementary Figure 1. First, the effects of MT3-037 on viability of various human cancer cell lines were investigated with the MTT assay. MT3-037 reduced the cell viability in a time-dependent manner in multiple cancer cell lines, including leukemia (HL-60, K-562, CCRF-CEM, and MOLT-4), lung (A549 and NCI-H522), liver (Hep3B and HepG2), and breast cancer cells (MDA-MB-468 and Erlotinib-resistant MDA-MB-468). The IC50 values for cell viability were between 1.8 and 5.9 μM after 72 h of incubation (Figure 1B). No effect on the cell viability of normal lung (MRC-5) and skin (Detroit 551) fibroblasts were found after 72 h-treatment of 30 μM MT3-037 (Figure 1C). The examination of cellular morphology revealed that MT3-037 caused MOLT-4, A549, and Hep3B cells to become more rounded, with cytoplasmic membrane blebbing and cell shrinkage (Figure 1D). Meanwhile, the morphological observation showed that both MRC-5 and Detroit 551 fibroblasts grew normally at 30 μM MT3-037 (Data not shown). However, both cell types showed apoptosis-like morphology at a higher MT3-037 concentration (>100 μM) (Data not shown). These data suggested that non-tumor cells were much less sensitive to MT3-037. Nuclear staining of MOLT4, A549 and Hep3B cell lines with DAPI revealed that MT3-037 treatments resulted in an increase in the number of multinucleated cells and in condensed chromosomes (Figure 1E), suggesting that MT3-037 blocked the cell division and induced apoptosis. The micronuclei were also observed in cancer cells after 24 h of treatment with MT3-037, occurring in MOLT-4 was around 5% and 2% in A549 and Hep3B cells (Data not shown). To confirm that MT3-037 induces apoptosis, cells were stained with annexin V/propidium iodide and analyzed by flow cytometry. As shown in Figure 1F, MT3-037 significantly increased the annexin V-positive cell population, indicating the induction of apoptosis.
Figure 1.

MT3-037 inhibits the cell viability and induces apoptosis in several cancer cell lines. (A) Structure of MT3-037. (B, C) IC50 values for MT3-037 calculated from viability of different human cancer cell lines (B) and normal human cell lines (C). Cells were incubated with different concentrations of MT3-037. After 72 h incubation, cell viability was assessed by MTT assay, and IC50 values were calculated. (D, E) Changes in cell morphology in response to MT3-037. MOLT-4, A549, and Hep3B cells were incubated with vehicle (DMSO, CTL) or 5 μM MT3-037 for 24 h. (D) Cell morphology was assessed by phase-contrast microscopy. (E) Nuclei were stained with DAPI and visualized by florescence microscopy. Chromatin condensation (yellow arrows), multiple nuclei (red arrows), and apoptotic bodies (white arrowheads) are indicated. Magnification, 400 ×. (F) MT3-037 induces apoptosis. MOLT-4, A549, and Hep3B cells were treated with 5 μM MT3-037 for 24 h. Cells were collected, and the proportion of annexin V-positive (apoptotic) and propidium iodide-positive cells (dead) was determined by flow cytometry. A representative graph from three independent experiments is shown. All values are the mean ± SEM from three independent experiments. **P < 0.01, compared with control (untreated).
MT3-037 causes cell cycle arrest at M phase via activation of CDK1
We next investigated the effects of MT3-037 on cell cycle progression. MT3-037 caused a concentration- and time-dependent accumulation of MOLT-4 cells at the G2/M boundary and polyploidy with the concomitant reduction in cells in the G1 and S phases (Figure 2A). G2/M arrest and polyploidy was also observed in MT3-037-treated A549 and Hep3B cells (Supplementary Figure 2A). The population of MOLT-4 cells in G2/M increased significantly after 4 h of MT3-037 treatment, and the number of apoptotic cells (sub-G1) increased after 12 h of treatment (Figure 2A, right). These results indicated that MT3-037 induced cell cycle arrest at G2/M followed by cell death.
Figure 2.

CDK1 plays a critical role in the MT3-037-induced mitotic arrest. A. MT3-037 causes the cell cycle arrest. MOLT-4 cells were treated with vehicle (DMSO, CTL) or different concentrations of MT3-037 for 24 h (left) or 5 μM MT3-037 for the indicated times (right), and the DNA content/cell cycle distribution was assessed by flow cytometry. B. Expression of mitotic proteins in MT3-037-treated MOLT-4 cells. Cells were harvested after 24 h incubation with the indicated concentrations of MT3-037. MPM-2- and propidium iodide-stained cells were examined by flow cytometry. C. MT3-037 alters the expression and phosphorylation (p) of G2/M-phase regulators. MOLT-4 cells were treated with 5 μM MT3-037 for the indicated times, cells were harvested, and proteins were detected by western blotting. D. MT3-037 increases the CDK1 kinase activity. Cells were treated with 5 μM MT3-037 for the indicated times and then harvested for measurement of CDK1 kinase activity using CDK1 Kinase Assay Kit (MBL International). **P < 0.01, compared with control (untreated). E. Roscovitine prevents the MT3-037-induced altered expression of M-phase regulators. Cells were pretreated with 20 μM roscovitine (Rosco) for 1 h prior to 18 h incubation with 5 μM MT3-037, and proteins were analyzed by western blotting. F. Roscovitine attenuates the MT3-037-induced M-phase arrest. MOLT-4 cells were pretreated with 20 μM roscovitine for 1 h prior to 18 h incubation with 5 μM MT3-037, and then cells were collected for MPM-2 staining.
MT3-037-treated cells were then examined with an antibody against the mitotic phosphoprotein MPM-2 and flow cytometry to narrow the MT3-037-induced cell cycle arrest to G2 or M phase. The anti-MPM-2 antibody recognizes a group of proteins whose epitopes are exclusively phosphorylated during mitosis, particularly starting from early prophase to metaphase [28]. Cells with 4 N DNA content that were MPM-2-positive were considered arrested in M phase. In response to MT3-037, MPM-2 staining was elevated in a concentration-dependent manner, indicating an increase in the M-phase population (Figure 2B and Supplementary Figure 2B). These results suggested that the MT3-037-treated cells were blocked in M-phase.
We then investigated the relationship between MT3-037-induced M-phase arrest and the potential altered expression of regulatory proteins involved in the G2/M phase transition, which is driven by the activation of the cyclin B1-CDK1 complex [12,13]. Data showed that MT3-037 upregulated cyclin B1 and CDK1 expression and CDK1 (Thr161) phosphorylation in a time-dependent manner in MOLT-4 (Figure 2C), A549, and Hep3B cells (data not shown). Cyclin E were downregulated by MT3-037 (Figure 2C). The phosphorylation of Thr161 is required for CDK1 activation [29]. CDK1 activity significantly increased after 4 h of treatment with MT3-037 and remained elevated for 24 h (Figure 2D). The levels of CDK1 and cyclin B1 mRNA were elevated by MT3-037 (Supplementary Figure 3). We also examined the changes in the interaction between CDK1 and cyclin B1 and between CDK1 and MPM-2 in response to MT3-037 by co-immunoprecipitation and found that MT3-037 increased the association of these proteins (Supplementary Figure 4A). However, as we also found a significant increase in CDK1, cyclin B1, and MPM-2 levels in the MT3-037-treated samples (Supplementary Figure 4A), it is possible that the enhanced association followed by CDK1 activation is due, at least in part, to increase availability of the proteins. Furthermore, the addition of the CDK inhibitor, roscovitine, prevented the upregulation of MPM-2 and cyclin B1 and the activation of CDK1 (Figure 2E and Supplementary Figure 4B). In addition, the MT3-037-induced increase in the proportion of cells in M phase was reversed by roscovitine (Figure 2F). Taken together, these results suggested that the MT3-037-induced cell cycle arrest at M-phase is mediated by the CDK1 activation and leads to apoptosis.
MT3-037 activates the mitotic kinases, survivin, aurora A/B, and PLK1
Several key mitotic kinases, namely survivin, Aurora A/B, and PLK1, are critical for chromosome movement and segregation [9,21,30-32]. In MOLT-4 cells, MT3-037 stimulated the phosphorylation of Aurora A and B and upregulated survivin and PLK1 in a time-dependent manner (Figure 3A). The MT3-037-mediated induction of survivin, Aurora A/B, and PLK1 was also inhibited by roscovitine (Figure 3B), suggesting that MT3-037 activates the mitotic kinases through CDK1.
Figure 3.
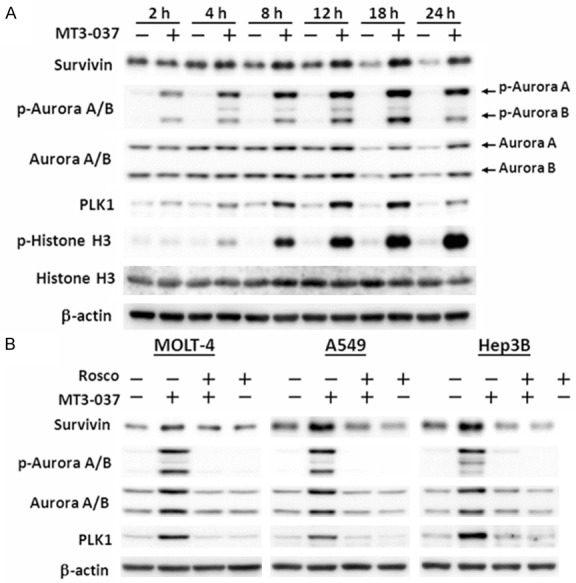
MT3-037 activates mitotic spindle kinases. A. MOLT-4 cells were incubated with 5 μM MT3-037 for the indicated times. Cells were harvested and proteins examined by western blotting. B. MOLT-4, A549, and Hep3B cells were pretreated with 20 μM roscovitine (Rosco) for 1 h, incubated with 5 μM MT3-037 for 18 h, and harvested for western blotting.
MT3-037 activates death receptor and mitochondrial apoptosis pathways
The relationship between MT3-037-induced M-phase arrest and apoptosis was further examined. As shown in Figure 4A, MT3-037 treatment of MOLT-4 cells resulted in a time-dependent cleavage activation of caspase-8, -9, and -3. Caspase activation was detected after 12 h incubation with MT3-037 and reached a maximum at 18 h. PARP was also activated following MT3-037 treatment (Figure 4A). FADD, a key mediator of the death receptor apoptotic pathway, was phosphorylated and activated by MT3-037. In contrast, Bcl-2 and Bcl-xL were phosphorylated and inactivated by MT3-037 in MOLT-4 cells (Figure 4A). These findings suggested the involvements of both death receptor (caspase-8-mediated) and mitochondrial (caspase-9-mediated) apoptotic pathways in MT3-037-induced apoptosis. Z-VAD-FMK, a pan-caspase inhibitor, effectively blocked the inhibitory effect of MT3-037 on cell viability in MOLT-4, A549, and Hep3B cells (Figure 4B), indicating that the inhibition of viability was mainly due to the induction of apoptosis. The addition of roscovitine also attenuated the inhibitory effects of MT3-037 on cell viability (Figure 4C), suggesting that CDK1 activation is also involved in the MT3-037-induced apoptosis. Furthermore, roscovitine prevented the MT3-037-induced phosphorylation of FADD, Bcl-2, and Bcl-xL (Figure 4D).
Figure 4.

MT3-037 activates the apoptotic pathways. (A) MOLT-4 cells were treated with 5 μM MT3-37 for the indicated times, and cell lysates were prepared and analyzed by western blotting. (B, C) MOLT-4, A549, and Hep3B cells were pretreated with 30 μM Z-VAD-FMK (Z-VAD, B) or 20 μM roscovitine (Rosco, C) for 1 h and then incubated with 5 μM MT3-037 for 24 h. Cell viability was analyzed by MTT assay. The values presented are the mean ± SEM from three independent experiments. **P < 0.01. (D) MOLT-4, A549, and Hep3B cells were treated with 20 μM roscovitine prior to 18 h-treatment with 5 μM MT3-037 and then examined by western blotting.
Involvement of JNK in regulating CDK1, mitotic arrest, and apoptosis
The mitogen-activated protein kinase pathways, such as JNK signaling cascade, play an important role in the induction of cell cycle arrest and apoptosis by tubulin-interfering agents [33-36]. Three specific mitogen-activated protein kinase inhibitors were applied to examine the importance of these kinases in the MT3-037-induced inhibition of cell viability. SP600125, a JNK inhibitor, significantly prevented the MT3-037-induced inhibition of cell viability in MOLT-4, A549, and Hep3B cells (Figure 5A). Both ERK (PD98059) and p38 MAPK (SB203580) inhibitors failed to reverse the effect of MT3-037 on viability (Figure 5A), suggesting that JNK may play a role in the MT3-037-induced apoptosis. Western blot analysis revealed that JNK phosphorylation/activation occurred at 2 h after MT3-037 treatment and persisted up to 24 h (Figure 5B). The phosphorylation/activation of JNK direct downstream effector c-Jun was also induced by MT3-037 in MOLT-4 cells (Figure 5B), indicating an increase in JNK kinase activity. These results suggested that JNK might be another target of MT3-037. The JNK inhibitors also attenuated the MT3-037-activated expression of Aurora A/B and PLK1 (Figure 5C) prevented the phosphorylation/activation of FADD and the phosphorylation/inactivation of Bcl-2 and Bcl-xL (Figure 5D). The MT3-037-induced caspase activation was also attenuated by SP600125 (data not shown). Furthermore, MT3-037-induced CDK1, JNK, and c-Jun activation as well as increase of cyclin B1 and MPM-2 were impaired by SP600125 (Figure 5E). Interestingly, the addition of roscovitine prevented the JNK phsophorylation (Figure 5F). Additionally, MT3-037 induced the formation of a JNK-CDK1 complex, and this complex could be disrupted by either SP600125 or roscovitine (Supplementary Figure 5).
Figure 5.
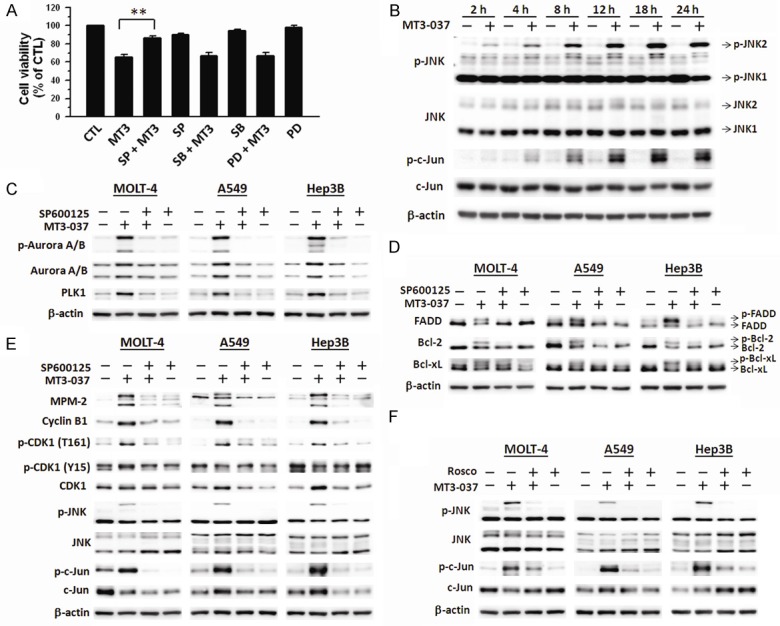
Involvement of JNK in the MT3-037-induced CDK1 activation and mitotic effects. (A) MOLT-4 cells were pretreated with 30 μM SP600125 (SP), 30 μM SB203580 (SB), or 30 μM PD98059 (PD) for 1 h followed by incubation with 5 μM MT3-037 for 24 h, and cell viability was assessed by MTT assay. The values presented are the mean ± SEM from three independent experiments. **P < 0.01. (B) MOLT-4 cells were treated with 5 μM MT3-037 for the indicated times, and protein expression was analyzed by western blotting. (C-F) MOLT-4 cells were pretreated with 30 μM SP600125 (SP) (C-E) or 20 μM roscovitine (Rosco) (F) for 1 h prior to 5 μM MT3-037 treatment for 18 h. Cells were collected for western blot analysis.
MT3-037 inhibits tubulin polymerization
Because MT3-037 arrested cells in M-phase and induced apoptosis, its effects on microtubule organization were evaluated in MOLT-4, A549, and Hep3B cells. Immunofluorescence staining of α-tubulin (Figure 6A) showed that the vehicle-treated cells displayed a clear and organized microtubule distribution. After MT3-037 treatment, the α-tubulin appeared dispersed and unpolymerized, similar to the effect of colchicine which destabilizes microtubules, and the α-tubulin immunofluorescence was significantly reduced in the MT3-037-treated cells compared with the vehicle-treated cells. However, paclitaxel, a microtubule-stabilizing compound, caused tubulin aggregation, condensation, and formation of extensive microtubule bundles (Figure 6A).
Figure 6.
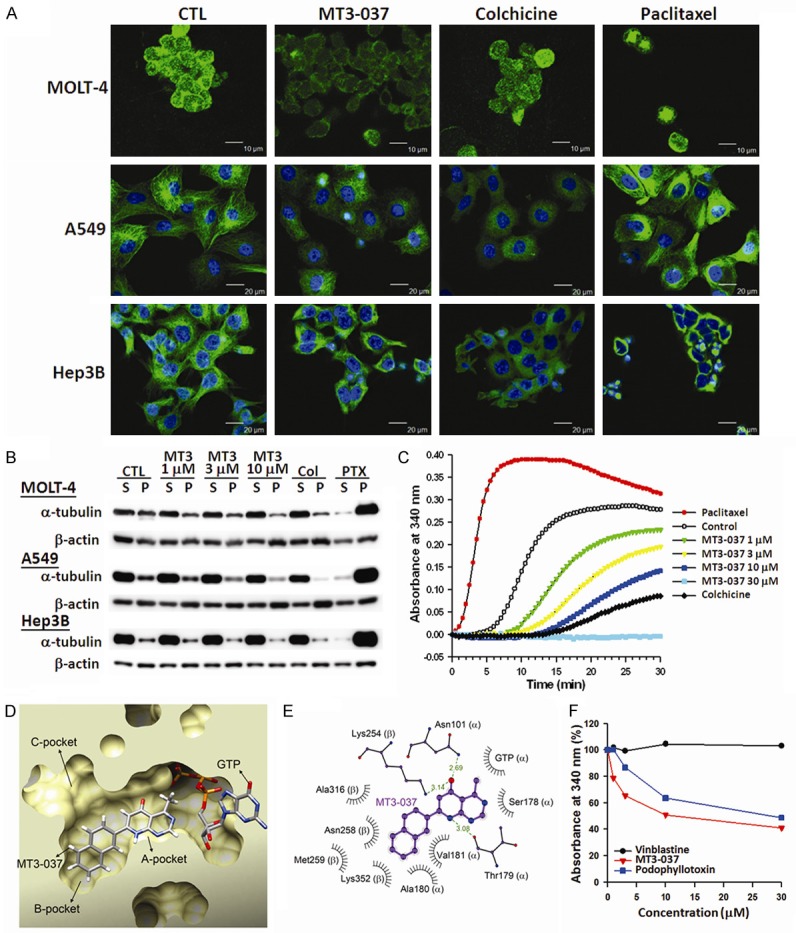
Effects of MT3-037 on the microtubule networks. A. MT3-037 affects the organization of cellular microtubules. MOLT-4, A549, and Hep3B cells were treated with vehicle (DMSO, CTL) 5 μM MT3-037, 5 μM colchicine, or 2 μM paclitaxel for 6 h. Cells were fixed, microtubules were stained with anti-α-tubulin (green, all three rows), and nuclei were counterstained with DAPI (blue, middle and bottom rows). Microtubules were observed by confocal microscopy at 400 × magnification. B. Western blotting of polymerized tubulin in MT3-037-treated cells. MOLT-4, A549, and Hep3B cells were treated with vehicle (DMSO, CTL) or different concentrations of MT3-037, 5 μM colchicine (Col), or 2 μM paclitaxel (PTX) for 18 h. Soluble (S) and pelleted (P) tubulin were collected and examined by western blotting. C. MT3-037 inhibits the microtubule assembly in vitro. Purified tubulin was polymerized in the absence (DMSO, CTL) or presence of MT3-037 (1, 3, 10, 30 μM), colchicine (10 μM), or paclitaxel (1 μM). The results presented are the representative of triplicate experiments. D. The HotLig scoring prediction showed that MT3-037 could fit into the A and B pockets of the colchicine-binding site in tubulin without displacing the bound GTP. E. Schematic diagram of potential molecular interactions between MT3-037 (purple) and tubulin. Hydrogen bonds are shown as green dashed lines, and bond lengths are indicated. MT3-037 might interact with tubulin through hydrogen bonding with residues Asn101 and Thr179 of α-tubulin and Lys254 of β-tubulin. Residues Ser178, Ala180, and Val181 of α-tubulin and Asn258, Met259, Ala316, and Lys352 of β-tubulin might contribute to the binding of MT3-037 through hydrophobic interactions. F. Competitive binding of MT3-037 to the colchicine-binding site of tubulin. Pure tubulin was preincubated with the indicated concentrations of MT3-037, vinblastine, or podophyllotoxin for 1 h, then colchicine was added for another 30-min incubation. The results presented are the representative of triplicate experiments.
To further examine whether MT3-037 affects the tubulin polymerization, cells were exposed to MT3-037, colchicine, or paclitaxel followed by the assessment of polymerized and unpolymerized tubulin. The MT3-037-treated cells showed a concentration-dependent reduction in the proportion of polymerized α-tubulin, similar to the colchicine-treated cells, whereas paclitaxel caused a significant increase in polymerized α-tubulin (Figure 6B). These results indicated that MT3-037 destabilized microtubules. A cell-free tubulin polymerization assay was performed to examine whether MT3-037 acts directly on tubulin polymerization. The half-maximal polymerization inhibitory concentration (IC50) of MT3-037 was 12.3 ± 0.5 μM. In contrast to the dramatic enhancement of tubulin polymerization by paclitaxel, MT3-037 suppressed tubulin polymerization in a concentration-dependent manner, similar to the colchicine-induced tubulin depolymerization (Figure 6C).
Docking site predictions for the known three-dimensional structure of tubulin also suggested that MT3-037 could bind at the colchicine-binding site (Figure 6D and 6E). We further assessed the ability of MT3-037 to compete with colchicine for binding to tubulin using a competitive binding assay. As shown in Figure 6F, vinblastine did not affect the binding to tubulin. Similar to podophyllotoxin, a microtubule-interfering agent that binds to the colchicine-binding site [37], MT3-037 reduced the absorbance of colchicine-tubulin complexes in a concentration-dependent manner, suggesting that MT3-037 competes with colchicine in binding to tubulin (Figure 6F).
In vivo antitumor activity of MT3-037
The in vivo antitumor activity of MT3-037 was investigated in nude mice inoculated with MDA-MB-468 or Erlotinib-resistant MDA-MB-468 cells. Tumor-bearing mice were intravenously injected with 1 mg/kg MT3-037. Treatment with MT3-037 significantly reduced tumor size and weight in both xenograft models (Figure 7A and 7B) without affecting body weight (Figure 7C). Angiogenesis is a critical event in tumor growth and metastasis. Angiogenesis requires microtubules [38], and numerous microtubule-targeting compounds can disrupt angiogenesis [4,39]. We next investigated the in vivo anti-angiogenic potency of MT3-037. MT3-037 inhibited blood vessel formation in a dose-dependent manner as shown by Matrigel assay (Figure 7D). The analysis of in vitro capillary-like tube formation in HUVECs also showed that MT3-037 could inhibit the tubular morphogenesis (Figure 7E). These data suggested that MT3-037 has both potent antitumor and anti-angiogenesis activities.
Figure 7.
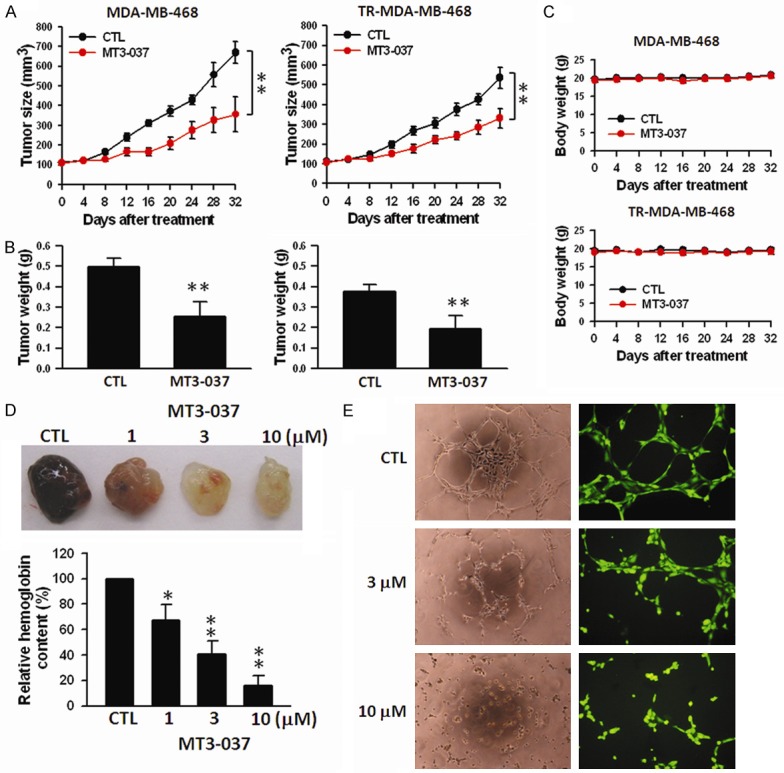
In vivo antitumor activity of MT3-037. (A) Antitumor activity of MT3-037 in breast cancer xenograft mouse models. MDA-MB-468 and Erlotinib-resistant (TR) MDA-MB-468 cells were used to inoculate nu/nu mice. Tumor-bearing mice were given vehicle (5% DMSO and 5% Cremophor in PBS) or MT3-037 (1 mg/kg per day) by intravenous injection. During treatment period, tumor volume (A) and mouse body weight (C) were measured once every 4 days. Data are expressed as the mean of tumor volume (mm3) ± SEM from five mice. **P < 0.01. (B) At the end of experiments, mice were sacrificed and tumor nodules dissected and weighed. Data are expressed as the mean of tumor weight (g) ± SEM from five mice. **P < 0.01. (D) Matrigel containing growth factors (human vascular endothelial and basic fibroblast) and heparin and with/without MT3-037 were injected into nu/nu mice. After one week, mice were sacrificed and the Matrigel plugs were excised. The hemoglobin content in the Matrigel was determined by the Drabkin method. The values presented are the mean ± SEM from three independent experiments. *P < 0.05; **P < 0.01, compared with control. (E) MT3-037-treated HUVEC were seeded on a Matrigel-coated culture plate. After 6 h incubation, tube formation was observed by phase-contrast microscopy (left, 200 × magnification), or cells were stained with calcein-AM and observed with fluorescence microscopy (right, 400 × magnification).
Discussion
The perturbation of microtubule assembly or stability can impair cell motility and causes cell cycle arrest and/or programmed cell death [4,5]. It is consistent with our observation that the MT3-037-induced changes in microtubule dynamics led to M-phase arrest. Mitotic arrest induced by the anti-microtubule agents is associated with the upregulation of cyclin B1/CDK1 activity in a variety of cells lines [40]. The phosphorylation of Thr161 is required for CDK1 activation, which is regulated by cyclins, upstream kinases (Cdk-activating kinase, Wee1, and Myt1), and phosphatases (Cdc25) [29]. An inappropriate accumulation of cyclin B1 and the activation of CDK1 were associated with the initiation of apoptosis in our study. Several microtubule-interfering agents exert the proapoptotic effects by increasing CDK1 activity [15] and disabling the anti-apoptotic function of Bcl-2 family proteins [41]. Similarly, MT3-037 caused a sustained upregulation of cyclin B1 and CDK1 as well CDK1 activity (Thr161 phosphorylation and kinase activity), which coincided with an increase in MPM-2 level. The MT3-037-induced M-phase arrest was modulated by the CDK1 inhibitor, roscovitine, demonstrating the importance of CDK1 activation in the MT3-037-induced mitotic block.
Cells treated with agents that induce mitotic arrest often show an abnormal morphology characteristic of mitotic catastrophe, including enlarged, irregular nuclei, multipolar mitosis, or multiple nuclei [42]. Survivin, Aurora A/B, and PLK1 play the multiple key roles in mitotic progression, including facilitating chromosome movement and spindle assembly [9,10,30-32,43]. The disruption of mitosis and activation of spindle assembly checkpoint are necessary to trigger apoptosis [44-46]. Despite the fact that Aurora A and B have distinct subcellular localization and functions, MT3-037 activated both proteins as well as PLK1, an important downstream effector of Aurora A [47]. PLK1 upregulation leads to mitotic arrest [48]. The MT3-037-treated cells exhibited irregular nuclei and multiple nuclei, indicating that MT3-037 impacts chromosome segregation and spindle assembly, likely through the activation of survivin-Aurora A/B-PLK1 axis. Survivin, Aurora A/B, and PLK1 are substrates of CDK1 in mitosis [49], which is consistent with our observation that CDK1 appears to activate these proteins in response to MT3-037. Survivin connects to the spindle dysfunction with apoptosis induction [50,51]. The activation of these mitotic-regulating proteins preceded caspase activation, suggesting that their activation might be required for the mitotic arrest and induction of apoptosis in response to MT3-037. Our results suggest that MT3-037 causes a mitotic defect that can lead to cell death, suggesting the induction of mitotic catastrophe.
MT3-037-induced apoptosis also appears to be mediated through the activation of caspase-8, -9, and -3 and PARP. Similar to other microtubule-disrupting agents [33,52], MT3-037 induced the Bcl-2 and Bcl-xL phosphorylation/inactivation, which is known to induce apoptosis [53]. The FADD phosphorylation is also critical for apoptosis induction [54], and we found that FADD was phosphorylated in the MT3-037-treated cells. These data indicated that both death receptor and mitochondrial apoptotic pathways are involved in the MT3-037-induced apoptosis.
Anti-microtubule agents increase JNK activity in a wide range of cancer cells, and the inhibition of JNK impedes apoptosis induced by paclitaxel, vinblastine, nocodazole, and colchicine [55,56]. Similarly, a JNK-specific inhibitor, SP600125, effectively abolished the MT3-037-induced reduction in cell viability. Therefore, the activation of JNK may be the primary stress signal resulting from the anti-microtubule agent-induced microtubule damage. Previous studies have shown that a sustained JNK activation is linked to apoptosis induction, whereas a transient JNK activation supports cell survival [34,35]. MT3-037 led to a sustained activation of JNK. Furthermore, the MT3-037-induced mitotic arrest and apoptosis-related events were prevented by the JNK inhibitors, demonstrating that JNK plays a critical role in MT3-037-induced apoptosis. The JNK activation is an upstream signal for Bcl-2 and Bcl-xL phosphorylation [52], and we found that FADD was also phosphorylated and activated through JNK in our present study, further suggesting that JNK may be the primary target for MT3-037. In addition, JNK contributes to cell cycle control by regulating histone H3, Aurora B, and Cdc25C [57-59]. Consistent with this, the JNK inhibitor also prevented the MT3-037-induced CDK1 activity as well as the phosphorylation of histone H3 and Aurora B, indicating that JNK may also regulate CDK1. Interestingly, the MT3-037-induced JNK activity could be reversed by a CDK1 inhibitor. Upon MT3-037 stimulation, we found that JNK physically associated with CDK1, suggesting that CDK1 and JNK may activate each other in a feedback loop, and coordinately contribute to the MT3-037-induced apoptosis.
MT3-037 displayed a broad-spectrum inhibition of cell viability on multiple cancer cells, including tyrosine kinase inhibitor-resistant breast cancer cells (Erlotinib-resistant MDA-MB-468). MT3-037 significantly attenuated the growth of MDA-MB-468 and Erlotinib-resistant MDA-MB-46 tumors. Our in vivo angiogenesis and in vitro tube formation assays showed that MT3-037 also decreased the vessel formation and disrupted the critical endothelial cell functions, including attachment, alignment, and motility, suggesting that its anti-angiogenesis via modulation of endothelial motility is involved in tumor growth inhibition.
Figure 8 presents a model of the cellular events underlying the anticancer activity of MT3-037 based on our results. MT3-037 interacts with microtubules and interferes with microtubule dynamics, which in turn causes the activation of cyclin B1, CDK1 and JNK, with a subsequent activation of the mitotic spindle-response axis involving survivin, Aurora A/B, and PLK1, which lead to mitotic block. When the spindle motility is hindered, the pro-apoptotic FADD and anti-apoptotic Bcl-2 and Bcl-xL undergo the phosphorylation and functional changes, which enhances the pro-apoptotic processes including the cleavage activation of caspases and PARP, eventually leading to cancer cell death (Figure 8).
Figure 8.
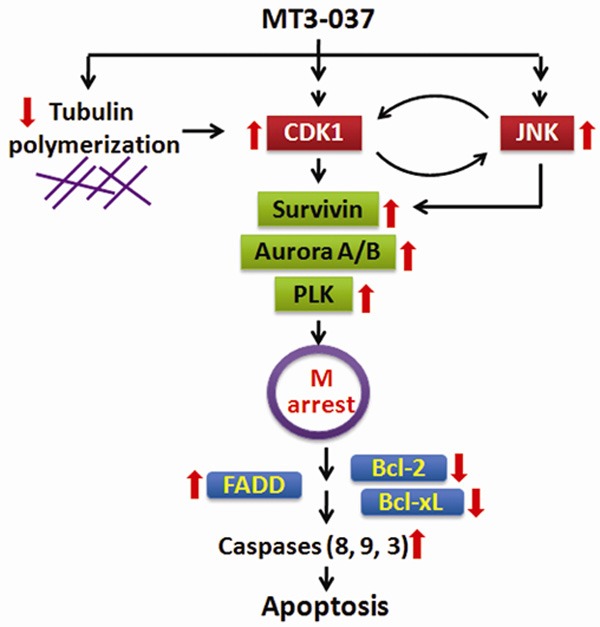
Model for key events leading to MT3-037-induced apoptosis in cancer cells.
MT3-037 was synthesized with a totally new scaffold structure. Unlike other natural occurring micrortubule-interfering agents with complicated structures, MT3-037 has a simple chemical structure, exhibiting a high antimitotic activity. The high synthetic yield and easy accessibility of starting materials for MT3-037 make it practical and promising in terms of its manufacture. Its broad-spectrum inhibitory effects against cancer cells and strong antitumor and anti-angiogenic activities make MT3-037 a promising potential antitumor drug candidate for further development.
Acknowledgements
This work was supported by the Ministry of Science and Technology of the Republic of China (grant numbers MOST 104-2314-B-039-034 to L.-C. Chang), National Science Council of the Republic of China (grant numbers NSC102-2325-B-039-005 and NSC102-2320-B-039-012 to S.-C. Kuo, and NSC101-2321-B-039-004 to Y.-L. Yu), the National Health Research Institute of the Republic of China (grant number NHRI-EX102-10245BI to Y.-L. Yu), and China Medical University Hospital (grant number DMR-104-097 and DMR-104-108 to L.-C. Chang).
Disclosure of conflict of interest
None.
Abbreviations
- CDK1
cyclin-dependent kinase 1
- DAPI
4’,6’-diamino-2-phenylindole
- FADD
fas-associated protein with death domain
- FBS
fetal bovine serum
- FITC
fluorescein isothiocyanate
- HUVEC
human umbilical vein endothelial cells
- JNK
c-Jun N-terminal kinase
- MPM-2
mitotic protein monoclonal 2
- PARP
poly(ADP-ribose) polymerase
- PLK1
polo-like kinase 1
- RIP
receptor-interacting protein kinase 1
- TRADD
tumor necrosis factor receptor type 1-associated death domain protein
Supporting Information
References
- 1.Desai A, Mitchison TJ. Microtubule polymerization dynamics. Annu Rev Cell Dev Biol. 1997;13:83–117. doi: 10.1146/annurev.cellbio.13.1.83. [DOI] [PubMed] [Google Scholar]
- 2.Wade RH. On and around microtubules: an overview. Mol Biotechnol. 2009;43:177–191. doi: 10.1007/s12033-009-9193-5. [DOI] [PubMed] [Google Scholar]
- 3.Esteve MA, Carre M, Braguer D. Microtubules in apoptosis induction: are they necessary? Curr Cancer Drug Target. 2007;7:7713–7729. doi: 10.2174/156800907783220480. [DOI] [PubMed] [Google Scholar]
- 4.Pasquier E, Kavallaris M. Microtubules: a dynamic target in cancer therapy. IUBMB Life. 2008;60:165–170. doi: 10.1002/iub.25. [DOI] [PubMed] [Google Scholar]
- 5.Wilson L, Jordan MA. New microtubule/tubulin-targeted anticancer drugs and novel chemotherapeutic strategies. J Chemother Suppl. 2004;4:83–85. doi: 10.1179/joc.2004.16.Supplement-1.83. [DOI] [PubMed] [Google Scholar]
- 6.Dumontet C, Jordan MA. Microtubulebinding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov. 2012;9:790–803. doi: 10.1038/nrd3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perez EA. Microtubule inhibitors: Differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol Cancer Ther. 2009;8:2086–2095. doi: 10.1158/1535-7163.MCT-09-0366. [DOI] [PubMed] [Google Scholar]
- 8.Nigg EA. Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol. 2001;2:21–32. doi: 10.1038/35048096. [DOI] [PubMed] [Google Scholar]
- 9.Salaun P, Rannou Y, Prigent C. CDK1, Plks, Auroras, and Neks: the mitotic bodyguards. Adv Exp Med Biol. 2008;617:41–56. doi: 10.1007/978-0-387-69080-3_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Musacchio A. Spindle assembly checkpoint: the third decade. Phil Trans R Soc B. 2011;366:3595–3604. doi: 10.1098/rstb.2011.0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elledge SJ. Cell cycle checkpoints: preventing an identity crisis. Science. 1996;274:1664–1672. doi: 10.1126/science.274.5293.1664. [DOI] [PubMed] [Google Scholar]
- 12.Vermeulen K, Berneman ZN, Van Bockstaele DR. Cell cycle and apoptosis. Cell Prolif. 2003;36:165–175. doi: 10.1046/j.1365-2184.2003.00267.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lindqvist A, van Zon W, Karlsson Rosenthal C, Wolthuis RM. Cyclin B1-CDK1 activation continues after centrosome separation to control mitotic progress. PLoS Biol. 2007;5:e123. doi: 10.1371/journal.pbio.0050123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lindqvist A, Rodriguez-Bravo V, Medema RH. The decision to enter mitosis: feedback and redundancy in the mitotic entry network. J Cell Biol. 2009;185:193–202. doi: 10.1083/jcb.200812045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ibrado AM, Kim CN, Bhalla K. Temporal relationship of CDK1 activation and mitotic arrest to cytosolic accumulation of cytochrome c and caspase-3 activity during Taxol-induced apoptosis of human AML HL-60 cells. Leukemia. 1998;12:1930–1936. doi: 10.1038/sj.leu.2401218. [DOI] [PubMed] [Google Scholar]
- 16.Zhao XF, Gartenhaus RB. Phospho-p70S6K and cdc2/cdk1 as therapeutic targets for diffuse large B-cell lymphoma. Expert Opin Ther Targets. 2009;13:1085–1093. doi: 10.1517/14728220903103833. [DOI] [PubMed] [Google Scholar]
- 17.Li L, Wang HK, Kuo SC, Wu TS, Lendnicer D, Lin CM, Hamel , Ernest , Lee KH. Antitumor agents. 150. 2’,3’,4’,5’,5,6,7-substituted 2-phenyl-4-quinolones and related compounds: their synthesis, cytotoxicity, and inhibition of tubulin polymerization. J Med Chem. 1994;37:1126–1135. doi: 10.1021/jm00034a010. [DOI] [PubMed] [Google Scholar]
- 18.Chen K, Kuo SC, Hsieh MC, Mauger A, Lin CM, Hamel E, Lee KH. Antitumoer agents. 174. 2’,3’,4’,5,6,7-Substituted 2-phenyl-1,8-naphthyridin-4-ones: their synthesis, cytotoxicity, and inhibition of tubulin polymerization. J Med Chem. 1997;40:2266–2275. doi: 10.1021/jm960858s. [DOI] [PubMed] [Google Scholar]
- 19.Hour MJ, Huang LJ, Kuo SC, Xia Y, Bastow K, Nakanishi Y, Hamel E, Lee KH. 6-Alkylamino- and 2,3-dihydro-3‘-methoxy-2-phenyl-4-quinazolinones and related Compounds: their Synthesis, cytotoxicity, and inhibition of tubulin Polymerization. J Med Chem. 2000;43:4479–4487. doi: 10.1021/jm000151c. [DOI] [PubMed] [Google Scholar]
- 20.Liu CY, Cheng YY, Chang LC, Huang LJ, Chou LC, Huang CH, Tsai MT, Liao CC, Hsu MH, Lin HY, Wu TS, Wen YF, Zhao Y, Kuo SC, Lee KH. Design and synthesis of new 2-arylnaphthyridin-4-ones as potent antitumor agents targeting tumorigenic cell lines. Eur J Med Chem. 2015;90:775–787. doi: 10.1016/j.ejmech.2014.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moustakas DT, Lang PT, Pegg S, Pettersen E, Kuntz ID, Brooijmans N, Rizzo RC. Development and validation of a modular, extensible docking program: DOCK 5. J Comput Aided Mol Des. 2006;20:601–619. doi: 10.1007/s10822-006-9060-4. [DOI] [PubMed] [Google Scholar]
- 22. Marvin 5.2.2, 2009, Chem Axon (http://www.chemaxon.com)
- 23.Vainio MJ, Johnson MS. Generating conformer ensembles using a multiobjective genetic algorithm. J Chem Inf Model. 2007;47:2462–2474. doi: 10.1021/ci6005646. [DOI] [PubMed] [Google Scholar]
- 24.Guha R, Howard MT, Hutchison GR, Murray-Rust P, Rzepa H, Steinbeck C, Wegner J, Willighagen EL. The blue obelisk-interoperability in chemical informatics. J Chem Inf Model. 2006;46:991–998. doi: 10.1021/ci050400b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang SH, Wu YT, Kuo SC, Yu J. HotLig: A molecular surface-directed approach to scoring protein-ligand interactions. J Chem Inf Model. 2013;53:2181–2195. doi: 10.1021/ci400302d. [DOI] [PubMed] [Google Scholar]
- 26.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 27.Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Prot Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 28.Davis FM, Tsao TY, Fowler SK, Rao PN. Monoclonal antibodies to mitotic cells. Proc Natl Acad Sci U S A. 1983;80:2926–2930. doi: 10.1073/pnas.80.10.2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lolli G, Johnson LN. CAK-Cyclin-dependent activation kinase: a key kinase in cell cycle control and a target for drugs. Cell Cycle. 2005;4:572–557. [PubMed] [Google Scholar]
- 30.Beardmore VA, Ahonen LJ, Gorbsky GJ, Kallio MJ. Survivin dynamics increases at centromeres during G2/M phase transition and is regulated by microtubule-attachment and Aurora B kinase activity. J Cell Sci. 2004;117:4033–4042. doi: 10.1242/jcs.01242. [DOI] [PubMed] [Google Scholar]
- 31.Vagnarelli P, Earnshaw WC. Chromosomal passengers: the four-dimensional regulation of mitotic events. Chromosoma. 2004;113:211–222. doi: 10.1007/s00412-004-0307-3. [DOI] [PubMed] [Google Scholar]
- 32.Mita AC, Mita MM, Nawrocki ST, Giles FJ. Survivin: Key regulator of mitosis and apoptosis and novel target for cancer therapeutics. Clin Cancer Res. 2008;14:5000–5005. doi: 10.1158/1078-0432.CCR-08-0746. [DOI] [PubMed] [Google Scholar]
- 33.Fan M, Du L, Stone AA, Gilbert KM, Chambers TC. Modulation of mitogen-activated protein kinases and phosphorylation of Bcl-2 by vinblastine represent persistent forms of normal fluctuation at G2-M1. Cancer Res. 2000;60:6403–6407. [PubMed] [Google Scholar]
- 34.Iordanov MS, Wong J, Newton DL, Rybak SM, Bright RK, Flavell RA, Davis RJ, Magun BE. Differential requirement for the stress-activated protein kinase/c-Jun NH(2)-terminal kinase in RNA damage- induced apoptosis in primary and in immortalized fibroblasts. Mol Cell Biol Res Commun. 2000;4:122–128. doi: 10.1006/mcbr.2000.0266. [DOI] [PubMed] [Google Scholar]
- 35.Fan M, Goodwin ME, Birrer MJ, Chambers TC. The c-Jun NH(2)-terminal protein kinase/AP-1 pathway is required for efficient apoptosis induced by vinblastine. Cancer Res. 2001;61:4450–4458. [PubMed] [Google Scholar]
- 36.Bacus SS, Gudkov AV, Lowe M, Lyass L, Yung Y, Komarov AP, Yarden Y, Seger R. Taxolinduced apoptosis depends on MAP kinase pathways (ERK and p38) and is independent of p53. Oncogene. 2011;20:147–155. doi: 10.1038/sj.onc.1204062. [DOI] [PubMed] [Google Scholar]
- 37.Cortese F, Bhattacharyya B, Wolff J. Podophyllotoxin as a probe for the colchicines binding of tubulin. J Biol Chem. 1977;252:1134–1140. [PubMed] [Google Scholar]
- 38.Bayless KJ, Johnson GA. Role of the cytoskeleton in formation and maintenance of angiogenic sprouts. J Vas Res. 2011;48:369–385. doi: 10.1159/000324751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jordan MA, Kamath K. How do microtubule-targeted drugs work? An overview. Curr Cancer Drug Targets. 2007;7:730–742. doi: 10.2174/156800907783220417. [DOI] [PubMed] [Google Scholar]
- 40.King RW, Jackson PK, Kirschner MW. Mitosis in transition. Cell. 1994;79:563–571. doi: 10.1016/0092-8674(94)90542-8. [DOI] [PubMed] [Google Scholar]
- 41.Debatin KM, Poncet D, Kroemer G. Chemotherapy: targeting the mitochondrial cell death pathway. Oncogene. 2002;21:8786–8803. doi: 10.1038/sj.onc.1206039. [DOI] [PubMed] [Google Scholar]
- 42.Vakifahmetoglu H, Olsson M, Zhivotovsky B. Death through a tragedy: mitotic catastrophe. Cell Death Diff. 2008;15:1153–1162. doi: 10.1038/cdd.2008.47. [DOI] [PubMed] [Google Scholar]
- 43.Barr AR, Gergely F. Aurora-A: the maker and breaker of spindle poles. J Cell Sci. 2007;120:2987–2996. doi: 10.1242/jcs.013136. [DOI] [PubMed] [Google Scholar]
- 44.Wignall SM, Gray NS, Chang YT, Juarez L, Jacob R, Burlingame A, Schultz PG, Heald R. Identification of a novel protein regulating microtubule stability through a chemical approach. J Chem Biol. 2004;11:135–146. [PubMed] [Google Scholar]
- 45.Gabrielli B, Chau YQ, Giles N, Harding A, Stevens F, Beamish H. Caffeine promotes apoptosis in mitotic spindle checkpoint-arrested cells. J Biol Chem. 2007;282:6954–6964. doi: 10.1074/jbc.M610104200. [DOI] [PubMed] [Google Scholar]
- 46.Wu YC, Yen WY, Yih LH. Requirement of a functional spindle checkpoint for arsenite-induced apoptosis. J Cell Biochem. 2008;105:678–687. doi: 10.1002/jcb.21861. [DOI] [PubMed] [Google Scholar]
- 47.Macurek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, Clouin C, Taylor SS, Yaffe MB, Medema RH. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature. 2008;455:119–123. doi: 10.1038/nature07185. [DOI] [PubMed] [Google Scholar]
- 48.Tang J, Eirkson RL, Liu X. Ecotpoic expression of Plk1 leads to activation of the spindle checkpoint. Cell Cycle. 2006;21:2484–2498. doi: 10.4161/cc.5.21.3411. [DOI] [PubMed] [Google Scholar]
- 49.O’Connor DS, Grossman D, Plescia J, Li F, Zhang H, Villa A, Tognin S, Marchisio PC, Altieri DC. Regulation of apoptosis at cell division by p34cdc2 phosphorylation of survivin. Proc Natl Acad Sci U S A. 2000;97:13103–13107. doi: 10.1073/pnas.240390697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li F, Ackermann EJ, Bennett CF, Rothermel AL, Plescia J, Tognin S, Villa A, Marchisio PC, Altieri DC. Pleiotropic cell-division defects and apoptosis induced by interference with survivin function. Nat Cell Biol. 1999;1:461–466. doi: 10.1038/70242. [DOI] [PubMed] [Google Scholar]
- 51.Lens SM, Wolthuis RM, Klompmaker R, Kauw J, Agami R, Brummelkamp T, Kops G, Medema RH. Survivin is required for a sustained spindle checkpoint arrest in response to lack of tension. EMBO J. 2003;22:2934–2947. doi: 10.1093/emboj/cdg307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yamamoto K, Ichijo H, Korsmeyer SJ. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol Cell Biol. 1999;19:8469–8478. doi: 10.1128/mcb.19.12.8469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kutuk O, Letai A. Regulation of Bcl-2 family proteins by posttranslational modifications. Curr Mol Med. 2008;8:102–118. doi: 10.2174/156652408783769599. [DOI] [PubMed] [Google Scholar]
- 54.Shimada K, Matsuyoshi S, Nakamura M, Ishida E, Kishi M, Konishi N. Phosphorylation of FADD is critical for sensitivity to anticancer drug-induced apoptosis. Carcinogenesis. 2004;25:1089–1097. doi: 10.1093/carcin/bgh130. [DOI] [PubMed] [Google Scholar]
- 55.Wang TH, Wang HS, Ichijo H, Giannakakou P, Foster JS, Fojo T, Wimalasena J. Microtubule-interfering agents activate c-Jun N-terminal kinase/stress-activated protein kinase through both Ras and apoptosis signalregulating kinase pathways. J Biol Chem. 1998;273:4928–4936. doi: 10.1074/jbc.273.9.4928. [DOI] [PubMed] [Google Scholar]
- 56.Chauhan D, Li G, Hideshima T, Podar K, Mitsiades C, Mitsiades N, Munshi N, Kharbanda S, Anderson KC. JNKdependent release of mitochondrial protein, Smac, during apoptosis in multiple myeloma (MM) cells. J Biol Chem. 2003;278:17593–17596. doi: 10.1074/jbc.C300076200. [DOI] [PubMed] [Google Scholar]
- 57.Lee K, Song K. Basal c-Jun N-terminal kinases promote mitotic progression through histone H3 phosphorylation. Cell Cycle. 2008;7:216–221. doi: 10.4161/cc.7.2.5155. [DOI] [PubMed] [Google Scholar]
- 58.Oktay K, Buyuk E, Oktem O, Oktay M, Giancotti FG. The c-Jun N-terminal kinase JNK functions upstream of Aurora B to promote entry into mitosis. Cell Cycle. 2008;7:533–541. doi: 10.4161/cc.7.4.5660. [DOI] [PubMed] [Google Scholar]
- 59.Gutierrez GJ, Tsuji T, Cross JV, Davis RJ, Templeton DJ, Jiang W, Ronai ZA. JNKmediated phosphorylation of Cdc25C regulates cell cycle entry and G(2)/M DNA damage checkpoint. J Biol Chem. 2010;285:14217–14228. doi: 10.1074/jbc.M110.121848. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.