

Abstract
A polymorphic mutation in the acetaldehyde dehydrogenase 2 (ALDH2) gene has been epidemiologically linked to the high susceptibility to esophageal carcinogenesis for individuals with alcohol use disorders. Mice subjected to alcohol drinking show increased oxidative stress and DNA adduct formation in esophageal epithelia where Aldh2 loss augments alcohol-induced genotoxic effects; however, it remains elusive as to how esophageal epithelial cells with dysfunctional Aldh2 cope with oxidative stress related to alcohol metabolism. Here, we investigated the role of autophagy in murine esophageal epithelial cells (keratinocytes) exposed to ethanol and acetaldehyde. We find that ethanol and acetaldehyde trigger oxidative stress via mitochondrial superoxide in esophageal keratinocytes. Aldh2-deficient cells appeared to be highly susceptible to ethanol- or acetaldehyde-mediated toxicity. Alcohol dehydrogenase-mediated acetaldehyde production was implicated in ethanol-induced cell injury in Aldh2 deficient cells as ethanol-induced oxidative stress and cell death was partially inhibited by 4-methylpyrazole. Acetaldehyde activated autophagy flux in esophageal keratinocytes where Aldh2 deficiency increased dependence on autophagy to cope with ethanol-induced acetaldehyde-mediated oxidative stress. Pharmacological inhibition of autophagy flux by chloroquine stabilized p62/SQSTM1, and increased basal and acetaldehyde-mediate oxidative stress in Aldh2 deficient cells as documented in monolayer culture as well as single-cell derived three-dimensional esophageal organoids, recapitulating a physiological esophageal epithelial proliferation-differentiation gradient. Our innovative approach indicates, for the first time, that autophagy may provide cytoprotection to esophageal epithelial cells responding to oxidative stress that is induced by ethanol and its major metabolite acetaldehyde. Defining autophagymediated cytoprotection against alcohol-induced genotoxicity in the context of Aldh2 deficiency, our study provides mechanistic insights into the tumor suppressor functions of ALDH2 and autophagy in alcohol-related esophageal carcinogenesis.
Keywords: Autophagy, ALDH2, acetaldehyde, alcohol, tobacco, esophageal squamous cell carcinoma, reactive oxygen species
Introduction
Alcohol consumption is a major environmental risk factor for esophageal squamous cell carcinoma (ESCC), one of the deadliest forms of all human squamous cell carcinomas common worldwide [1-3]. Acetaldehyde is a potent carcinogen produced as a result of alcohol metabolism [4]. Specifically, acetaldehyde is generated via ethanol oxidation through the action of alcohol dehydrogenase (ADH) 1B and/or cytochrome P450 2E1 (CYP2E1) enzymes present in the adult stomach and liver, the primary sites of first-pass metabolism. ADH1B is also expressed in esophageal epithelia; however, the role of esophageal ADH1B in acetaldehyde production and carcinogenesis remains elusive [5-7]. Additionally, mutagenic levels of acetaldehyde can be produced by oral microbiota in the saliva of individuals who consume alcohol beverages [8,9]. Acetaldehyde facilitates formation of carcinogenic DNA adducts such as N2-ethylidene-2’-deoxyguanosine [10-12], causing acetaldehyde-mediated DNA damage lesions in oral-esophageal mucosa [13]. The mitochondrial enzyme acetaldehyde dehydrogenase 2 (ALDH2) functions to detoxify acetaldehyde via an oxidation reaction that produces acetate and NADH, the latter of which facilitates ATP generation via mitochondrial respiration. Reactive oxygen species (ROS) are generated as byproducts of respiration [14]. ROS are important for cell signaling; however, ROS levels must be tightly regulated to prevent deleterious effects, including damage to intracellular components and cell death [15]. Thus, increased intake or reduced detoxification of acetaldehyde may promote oxidative stress and DNA damage.
Acetaldehyde accumulation due to reduced ALDH2 activity has been implicated in alcohol-related carcinogenesis [1,16,17]. At least 8% of the entire world population, including ~40% of East Asians (Japanese, Chinese and Korean), is estimated to carry a functional ALDH2 single nucleotide polymorphism (SNP) that delays acetaldehyde clearance [18]. This SNP is commonly found amongst individuals [18] who show significantly elevated blood, salivary and expiratory acetaldehyde levels following alcohol consumption [19-21]. Interestingly, acetaldehyde may have organ-specific roles in carcinogenesis since the ALDH2 SNP is more strongly associated with ESCC and head and neck cancers than other cancers arising in sites of alcohol absorption and metabolism such as the stomach, intestine and liver [22]. Alcohol drinking induces esophageal ALDH2 expression and Aldh2 loss increases oxidative stress in murine esophageal epithelia [23]. Nevertheless, long-term alcohol drinking alone fails to induce neoplastic changes in esophageal epithelia in mice [24]. Moreover, Aldh2-/- mice subjected to alcohol drinking show no histological neoplastic change in esophageal mucosa [13], suggesting that fail-safe and cytoprotective mechanisms may exist to avoid malignant transformation in esophageal epithelial cells (keratinocytes) exposed to oxidative stress induced by alcohol or acetaldehyde.
Autophagy (macroautophagy) is a cellular homeostatic and adaptive mechanism activated in response to physiologic stressors [25]. Autophagy is mediated by autophagic vesicles (AVs) that engulf dysfunctional intracellular components such as damaged mitochondria. Biogenesis of AVs involves multiple autophagy-related gene products including microtubule-associated protein 1 light chain 3 (LC3). In response to oxidative stress, LC3 undergoes proteolytic cleavage (LC3-I) [26] then lipidation (LC3-II), facilitating the protein’s incorporation into AVs. Subsequent AV-lysosome fusion allows degradation of autophagic cargo via lysosomal hydrolases. Autophagy may act as a tumor suppressor as autophagy-deficient mice are tumor prone to increased oxidative stress [27,28]. Autophagy has also been implicated in alcohol-related human diseases such as cardiomyopathies and hepatic steatosis [29,30]. The role of autophagy in esophageal keratinocytes exposed to ethanol or acetaldehyde remains unknown. Herein, we investigate the influence of Aldh2 upon autophagy-mediated cytoprotection in non-transformed murine esophageal keratinocytes.
Materials and methods
Primary esophageal cell culture, treatment, retrovirus-mediated gene transduction and live cell imaging
Primary murine esophageal keratinocytes (passages 2-5) isolated from Aldh2-/- and control Aldh2+/+ mice were grown and subjected to treatment with 100% ethanol (Decon Labs, King of Prussia, PA) or ≥99.5% acetaldehyde (Sigma-Aldrich, St. Louis, MO) as described previously [23,31]. To suppress alcohol dehydrogenase (ADH), cells were treated with 2 mM 4-methylpyrazole (Sigma-Aldrich). Subconfluent cells were subjected to live cell imaging or other analyses with or without additional retrovirus-mediated transduction of pBABE-puro mCherry-EGFP-LC3B [32] (a gift from Jayanta Debnath; Addgene, Cambridge, MA; plasmid # 22418) as described previously [33]. Cells expressing mCherry-EGFP-LC3 were imaged using a Leica DM IRB inverted microscope (Leica Microsystems, Buffalo Grove, IL) and the Find Maxima feature in ImageJ (National Institutes of Health) to determine mCherry-EGFP-LC3 puncta. At least 50 cells were counted for each condition.
Esophageal 3D organoid culture
Using 24-well plates, 5,000 cells were seeded per well in 50 µl Matrigel. After solidification, 500 µl of DMEM/F12 supplemented with 1X Glutamax, 1X HEPES, 1X N2 Supplement, 1X B27 Supplement, 0.1 mM N-acetyl-L-cysteine (Sigma-Aldrich), 50 ng/ml mouse recombinant epidermal growth factor (R&D Systems, Minneapolis, MN), 2.0% Noggin/R-Spondin-conditioned media and 10 µM Y27632 (Tocris Biosciences, Bristol, UK) was added and replenished every other day. Organoids were grown for 7 days in growth media before addition of acetaldehyde and/or Chloroquine (Sigma-Aldrich) for 2 days. Organoids were recovered by digesting MatrigelTM (BD Biosciences, San Jose, CA) with Dispase I (BD Biosciences, San Jose, CA; 1 U/ml) and fixed overnight in 4.0% paraformaldehyde. Specimens were embedded in 2.0% Bacto-Agar: 2.5% gelatin prior to paraffin embedding.
Aldh2-knockout (Aldh2-/-) mouse tissues
Paraffin embedded esophageal tissues from Aldh2-/- and control Aldh2+/+ mice treated with or without 10% ethanol in drinking water for 8 weeks (all male, beginning at the age of 6 weeks) were described previously [23].
Immunoblot analysis
Western blotting was performed as described previously [34]. In brief, 50 μg of denatured protein was fractionated on a NuPAGE Bis-Tris 4-12% gel (Life Technologies, Grand Island, NY). Following electrotransfer, Immobilon-P membranes (EMD Millipore, Billerica, MA) were blocked with Dulbecco’s Phosphate-Buffered Saline (DPBS) containing 0.1% Tween-20 and 5% milk, followed by overnight incubation with the following primary antibodies: rabbit-anti p53 (1:1000, Vector Laboratories, Burlingame, CA), rabbit-anti phospho-H2A.X Serine139 (p-H2A.XSer139; 1:1000, Cell Signaling Technology, Danvers, MA), rabbit anti-LC3B (1:1000, Cell Signaling Technology), mouse-anti-p62 (1:1000, Sigma-Aldrich) and mouse anti-actin (1:10000, Sigma-Aldrich) at 4°C. Secondary antibodies (Sigma-Aldrich) were used at 1:10000. Targeted proteins were visualized using a chemiluminescence detection system (Amersham ECL or ECL Prime; GE Healthcare Life Sciences; Buckinghamshire, UK) and exposed to Blue Lite Autorad film (ISC-BioExpress, Kaysville, UT).
Flow cytometry
Flow cytometry was performed to determine AVs, ROS and apoptosis as described previously [34] using FACSCalibur or LSR II cytometers (BD Biosciences, Franklin Lakes, NJ) and FlowJo software (Tree Star, Ashland, OR) for cells suspended in DPBS containing 1% bovine serum albumin (Sigma-Aldrich). AVs were determined with Cyto-ID® fluorescent dye (Enzo Life Sciences, Farmingdale, NY) by staining cells at 1:1000 at 37°C for 30 min in 1:1 mixture of DPBS containing 1% bovine serum albumin (Sigma-Aldrich) and full keratinocyte SFM medium (Life Technologies). ROS were determined with 2’,7’-dichlorodihydrofluorescein diacetate (DCF) and MitoSOX™ red mitochondrial superoxide indicator dyes (Life Technologies) as described previously [34,35]. Apoptosis was determined using the Annexin-V-FLUOS kit (Roche, Basel, Switzerland) according the manufacturer’s instructions. Viability of cells was determined by DAPI (4’,6-diamidino-2-phenylindole) (Life Technologies) staining.
Histology and immunohistochemistry
Hematoxylin and eosin (H&E) staining and immunohistochemistry (IHC) were performed and evaluated as described previously [36]. Sections were incubated with anti-cleaved LC3 polyclonal antibody (1:250; Abgent, San Diego, CA), or anti-phospho-Histone p-H2A.XSer139 (20E3) monoclonal antibody (1:250; Cell Signaling Technology, Danvers, MA) overnight at 4°C. A pathologist (AKS) blind to molecular data scored cleaved LC3 and phospho-Histone p-H2A.XSer139 stained specimens based on intensity and distribution. Intensity of cleaved LC3 staining was evaluated using the following criteria: Score 1, marginal to mild stain affecting the nuclei of basal and parabasal layers; Score 2, moderate to intense nuclear staining and occasional cytosolic stain in most layers, including superficial layers in which nuclear staining was either absent or less intense than in basal and parabasal layers; Score 3, very intense nuclear stain in all layers together with obvious cytosolic stain with or without puncta. Cleaved LC3 labeling index is reported as percent of stained cells in all epithelial layers. Cleaved LC3 score was calculated by multiplying the values of intensity and the label index. Intensity of p-H2A.XSer139 was evaluated using the following criteria: Score 2, strong positive with brown staining completely obscuring nucleus; Score 1, weak positive with any lesser degree of brown staining appreciable in cell nucleus; Score 0, absent with no appreciable staining in cell nucleus. p-H2A.XSer139 labeling index is reported as percent of stained cells in the basal layer. p-H2A.XSer139 score was calculated by multiplying the values of intensity and the labeling index.
Statistical analyses
Data are presented as mean ± standard error. Student’s t test was used to compare two groups. P<0.05 was considered significant.
Results
Aldh2 influences cytotoxicity and oxidative stress in esophageal epithelial cells exposed to ethanol and acetaldehyde
Since esophageal mucosa is directly exposed to ethanol upon alcohol consumption, we first asked whether esophageal keratinocytes have the capacity to generate acetaldehyde in response to ethanol exposure. To this end, primary esophageal keratinocytes isolated from Aldh2+/+ and Aldh2-/- mice [31] were exposed to 1.5% ethanol and evaluated for cell viability and oxidative stress. Within 48 hours, ethanol induced massive (>40%) cell death as determined by DAPI exclusion analysis in Aldh2-/-, but not Aldh2+/+ cells (Figure 1A). Moreover, flow cytometry for DCF, a general indicator of ROS, revealed that Aldh2-/- cells exhibit increased oxidative stress under both basal conditions and in response to ethanol exposure when compared to their Aldh2+/+ counterparts (Figure 1B). These data suggest that ethanol may be directly metabolized by esophageal keratinocytes to promote acetaldehyde-mediated oxidative stress and cell death in the absence of Aldh2. To determine how ethanol is metabolized to cause cytotoxicity and oxidative stress in esophageal keratinocytes, we utilized the pharmacological ADH inhibitor 4-methylpyrazole (4 MP) to prevent ADH-mediated oxidation of ethanol and generation of acetaldehyde. 4MP did not significantly impact cell viability or oxidative stress in Aldh2+/+ cells responding to ethanol (Figure 1A, 1B). By contrast, ethanol-induced ROS and cell death were suppressed in Aldh2-/- cells (Figure 1A, 1B), suggesting that ADH may play a role in ethanol metabolism by esophageal keratinocytes.
Figure 1.

ALDH2 level determines ROS generation and cytotoxicity in esophageal epithelial cells exposed to ethanol. A. Aldh2 +/+ and Aldh2 -/- cells were cultured in the presence of 1% ethanol with and without 4MP for 48 hours and then stained with DAPI to assess cell viability by flow cytometry. *p<0.05 (n=3) compared with Aldh2 -/- non-treatment, #p<0.05 (n=3) compared with non Aldh2 -/- treated with EtOH. B. Aldh2 +/+ and Aldh2 -/- cells were cultured in the presence of 1.5% ethanol with and without 4MP for 48 hours and then stained with DCF to assess ROS by flow cytometry. *p<0.05 (n=3) compared with non-treatment, #p<0.05 (n=3) compared with non Aldh2 +/+ non-treatment. †p<0.05 (n=3) compared with Aldh2 +/+ treated with EtOH.
We next assessed how esophageal keratinocytes respond to acetaldehyde exposure in primary culture. While acetaldehyde-mediated cytotoxicity was anticipated, caspase 3 cleavage was undetectable by immunoblot analysis in Aldh2+/+ cells treated with acetaldehyde up to a concentration of 0.5 mM for 72 hours (Figure 2A). While exposure to either 1.0 mM or 2.0 mM acetaldehyde promoted caspase 3 cleavage in Aldh2+/+ cells, the extent of this induction was minimal with 1.0 mM acetaldehyde (Figure 2A). Annexin-V/PI staining further revealed significant induction of apoptosis in Aldh2+/+ cells 72 hours following exposure to 1 mM acetaldehyde (Figure 2B). By contrast, Aldh2-/- cells exhibited higher apoptosis rates under basal conditions and in response to acetaldehyde, as exposure to 1 mM acetaldehyde induced apoptosis in 50% of Aldh2-/- cells within 72 hours (Figure 2B). Acetaldehyde-mediated cytotoxicity in Aldh2-/- cells was indeed associated with elevated basal and induced levels of hydrogen peroxide and mitochondrial super oxide, as evaluated by DCF and mitoSOX assays, respectively. (Figure 2C-E). Of note, the population doubling time of Aldh2-/- cells (50 hours) was longer than that of than Aldh2+/+ cells (35 hours), suggesting that Aldh2 may influence cell proliferation as well as apoptosis by modulating the level of acetaldehyde generated under cell culture conditions [37].
Figure 2.
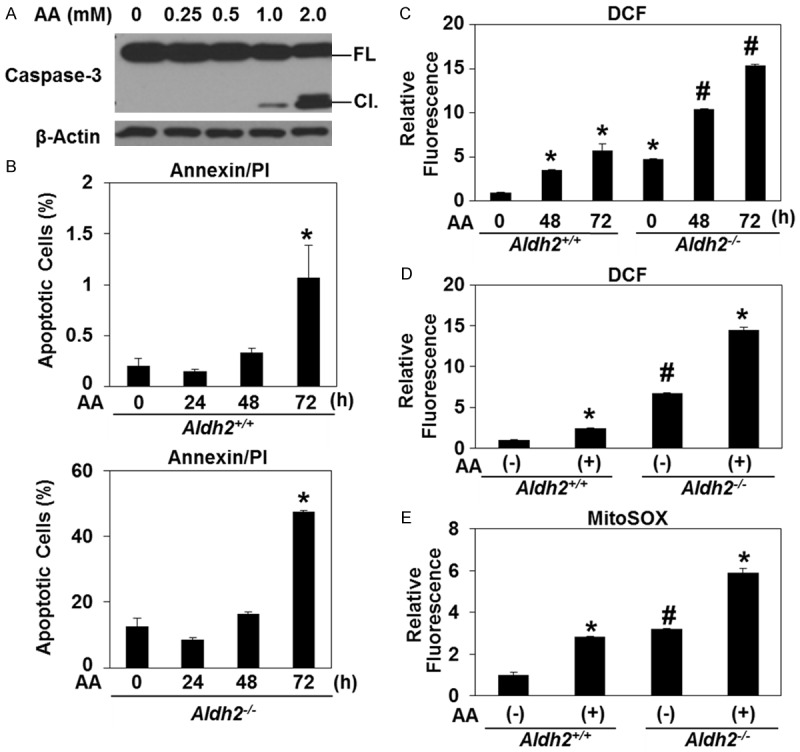
ALDH2 level determines ROS generation and cytotoxicity in esophageal epithelial cells exposed to acetaldehyde. A. Immunoblot analysis was performed to assess Caspase-3 with β-Actin as a loading control in Aldh2 +/+ cells treated with acetaldehyde. B. Aldh2 +/+ and Aldh2 -/- cells were cultured in the presence of 1 mM of acetaldehyde for indicated time periods and then stained with Annexin-V and PI to assess apoptotic cells by flow cytometry. *p<0.05 (n=3) compared with 0 h. C. Aldh2 +/+ and Aldh2 -/- cells were cultured in the presence of 1 mM of acetaldehyde for indicated time periods and then stained with DCF to assess ROS by flow cytometry. *p<0.05 (n=3) compared with non-treatment, #p<0.05 (n=3) compared with WT non-treatment. D. Aldh2 +/+ and Aldh2 -/- cells were cultured in the presence of 1 mM of acetaldehyde for 48 hours and then stained with DCF to assess ROS by flow cytometry. *p<0.05 (n=3) compared with non-treatment, #p<0.05 (n=3) compared with WT non-treatment. E. Aldh2 +/+ and Aldh2 -/- cells were cultured in the presence of 1 mM of acetaldehyde for 48 hours and then stained with MitoSOX to assess ROS by flow cytometry. *p<0.05 (n=3) compared with non-treatment, #p<0.05 (n=3) compared with WT non-treatment.
Autophagy is activated as a cytoprotective mechanism in response to oxidative stress induced by ethanol and acetaldehyde
While excessive ethanol and acetaldehyde may trigger apoptotic cell death, cells may cope with oxidative stress via autophagy. We hypothesized that autophagy may be activated to modulate ROS in esophageal keratinocytes exposed to ethanol and acetaldehyde. Western blotting in Aldh2-/- cells treated with 1 mM acetaldehyde suggested stress-mediated autophagy activation as LC3 protein lipidation (LC3-II), was induced following p53 stabilization (Figure 3A). To further evaluate autophagy, we carried out flow cytometry to for the AV-identifying fluorescent dye Cyto-ID. In Aldh2+/+ cells, Cyto-ID was induced by both 1.5% ethanol (EtOH) and sublethal concentrations of acetaldehyde (≤1 mM) in a time-dependent manner (Figure 3B, 3C). Moreover, Aldh2-/- cells displayed enhanced basal AV content that was robustly induced in response to exposure to ethanol or acetaldehyde (Figure 3B, 3C). Increased AV content may be reflective of enhanced autophagic or a distal defect in AV clearance. To evaluate autophagy flux, we further used chloroquine (CQ), a pharmacological autophagy inhibitor that prevents AV-lysosome fusion. Inhibition of AV-lysosome fusion is expected to stabilize LC3-II and p62/SQSTM (p62), an autophagy cargo identifying protein that is degraded upon AV-lysosome fusion. As expected, CQ increased expression of LC3-II as well as p62 in both Aldh2 +/+ and Aldh2 -/- cells under basal conditions (Figure 3D). CQ treatment further augmented expression of LC3-II in both acetaldehyde-treated Aldh2 +/+ and Aldh2 -/- cells (Figure 3D). Moreover, while p62 expression was decreased in Aldh2 -/- cells following acetaldehyde exposure, this effect was blocked via co-treatment with CQ (Figure 3D). These data indicate that autophagic flux is activated by acetaldehyde especially in Aldh2 -/- cells. In Aldh2+/+ cells, CQ-mediated suppression of autophagy flux also increased both basal and acetaldehyde-induced ROS as determined by DCF assays (Figure 3E), suggesting that autophagy may be activated to limit oxidative stress. Interestingly Aldh2-/- cells exhibited a basal ROS level that was significantly higher than Aldh2+/+ cells and that was further augmented upon co-treatment with CQ; however, acetaldehyde-mediated ROS production was only modestly influenced by CQ in Aldh2-/- cells (Figure 3E). These findings suggest that while autophagy flux may be present to decrease oxidative stress in Aldh2-/- cells under basal conditions, autophagy flux may rather be decreased or stalled in the presence of excessive ROS as found 72 hours following acetaldehyde treatment.
Figure 3.
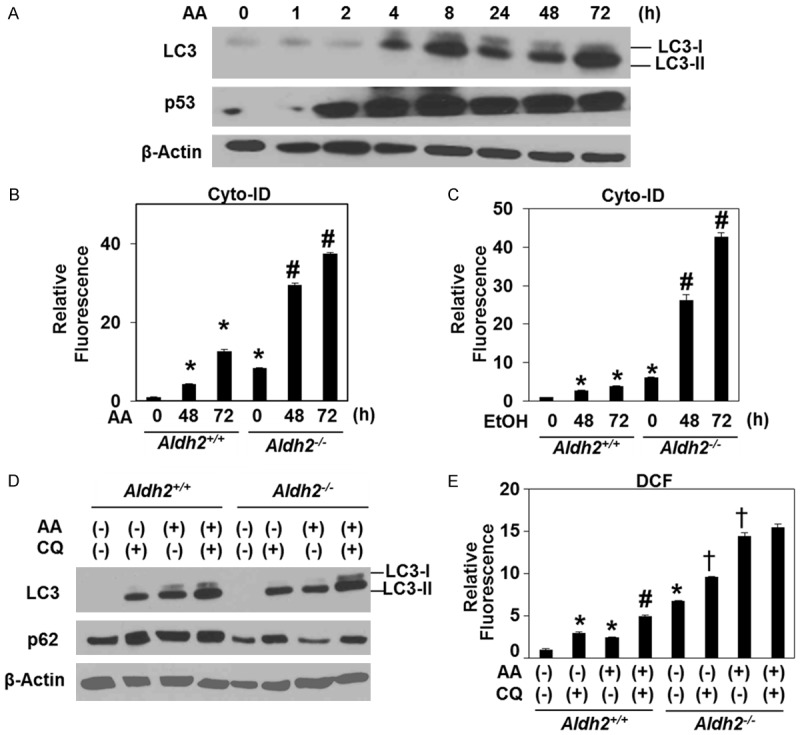
Autophagy is activated as a cytoprotective mechanism from oxidative stress induced by ethanol and acetaldehyde. A. Immunoblot analysis was performed to assess LC3 and p53 with β-Actin as a loading control in Aldh2 -/- treated with acetaldehyde. B. Aldh2 +/+ and Aldh2 -/- cells were treated with 1 mM acetaldehyde for indicated time periods. Cells were subjected to flow cytometry for Cyto-ID to determine relative AV levels. *p<0.05 (n=3) compared with Aldh2 +/+ 0 h, #p<0.05 (n=3) compared with Aldh2 -/- 0 h. C. Aldh2 +/+ and Aldh2 -/- cells were treated with 1% EtOH for indicated time periods. Cells were subjected to flow cytometry for Cyto-ID to determine relative AV levels. *p<0.05 (n=3) compared with Aldh2 +/+ 0 h #p<0.05 (n=3) compared with Aldh2 -/- 0 h. D. Immunoblot analysis was performed to assess LC3 and p62 with β-Actin as a loading control in Aldh2 +/+ and Aldh2-/- cells treated with 1 mM acetaldehyde with and without 1 µg/ml CQ for 48 hours. E. Aldh2 +/+ and Aldh2 -/- cells were treated with 1 mM acetaldehyde with and without 1 µg/ml CQ treatment for 48 hours. Cells were subjected to flow cytometry for Cyto-ID to determine ROS levels. *p<0.05 (n=3) compared with Aldh2 +/+ non-treatment, #p<0.05 (n=3) compared with Aldh2-/- non-treatment. †p<0.05 (n=3) compared with Aldh2 +/+ treated with acetaldehyde alone.
Autophagy flux in esophageal keratinocytes declines in response to extended acetaldehyde exposure
To gain further insight into the dynamic nature of autophagic flux in esophageal keratinocytes, we next visualized autophagy flux in Aldh2+/+ and Aldh2-/- cells stably transduced with a retrovirus expressing a fusion protein comprising acid-stable mCherry and acid-sensitive enhanced green fluorescent (EGFP) proteins fused to LC3 (mCherry-EGFP-LC3B) [32]. This reporter detects AVs as fluorescent proteins-labeled LC3-positive puncta expressing either mCherry alone (red puncta) or both mCherry and EGFP concurrently (yellow puncta). The former represents AVs fused to lysosomes (autolysosomes) where EGFP is degraded in the acidic lysosomal environment, thereby indicating autophagic flux. The latter represents AVs that have yet undergo lysosome-mediated cargo degradation. When Aldh2-/- cells were placed in Hanks’ Balanced Salt Solution (HBSS) for nutrient deprivation, a large number of red puncta emerged within 8 hours, indicating activation of autophagic flux (Figure 4A). By contrast, CQ treatment resulted in accumulation of yellow puncta (Figure 4A), suggesting stalled basal autophagic flux due to inhibition of AV-lysosome fusion. With these conditions serving as controls, only a small number of LC3 red puncta were observed in Aldh2+/+ cells under basal conditions while Aldh2-/- cells displayed a significant increase in red puncta at baseline (Figure 4B, 4C), suggesting that basal autophagy level was higher in Aldh2-/- cells than in their Aldh2+/+ counterparts. Following exposure to 1 mM acetaldehyde, the number of red puncta in both Aldh2+/+ and Aldh2-/- cells were significantly increased within 4 hours (Figure 4B, 4C). Thus, autophagy flux is robustly activated in esophageal keratinocytes upon acute acetaldehyde exposure. Of note, acetaldehyde-mediated induction of red puncta was more pronounced in Aldh2-/- cells, suggesting that Aldh2-/- cells may have a greater dependence of autophagy to cope with acetaldehyde toxicity than Aldh2+/+ cells (Figure 4B, 4C). Moreover, the number of yellow puncta became especially apparent in Aldh2-/- cells at 48 hours after acetaldehyde stimulation with 53.3% of LC3 puncta identified as AVs (yellow puncta) (Figure 4B, 4C). On the contrary, only 16.9% of LC3 puncta were AVs in Aldh2+/+ cells at 48 hours following acetaldehyde exposure (Figure 4B, 4C), suggesting that in comparison to cells with intact ALDH2 expression, Aldh2-/- cells are more prone to autophagy stalling upon prolonged acetaldehyde exposure which may increase oxidative stress and apoptotic cell death at later time points.
Figure 4.
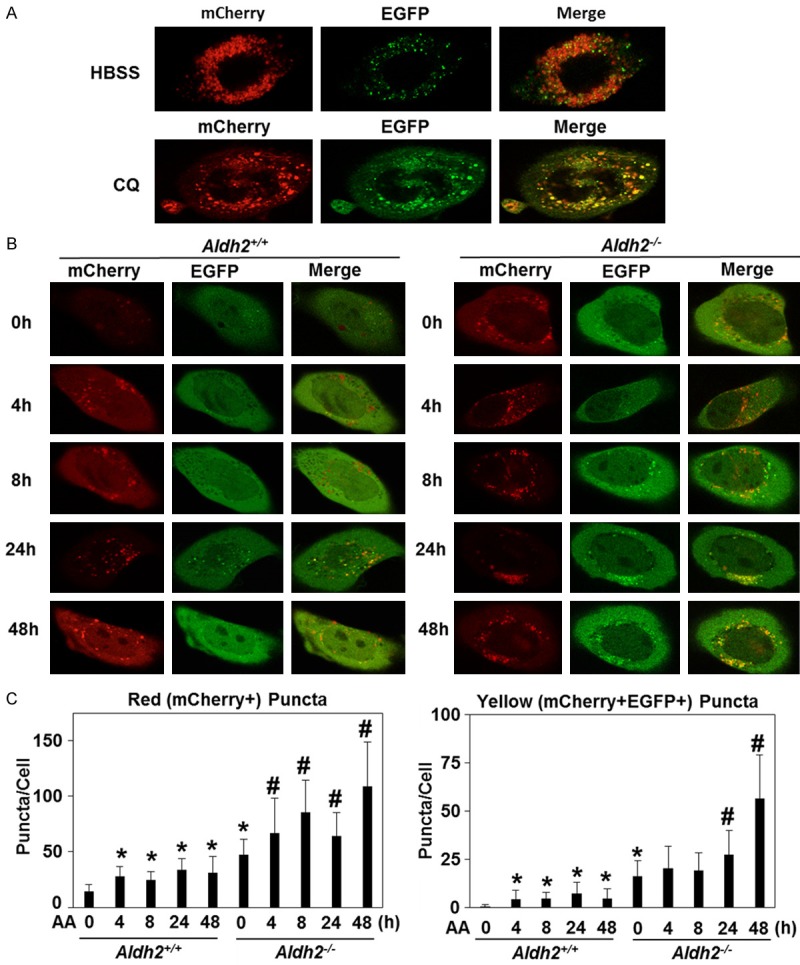
Autophagic flux is diminished upon extended exposure to acetaldehyde in Aldh2 -/- cells. A. Live cell imaging determined mCherry-EGFP-LC3B puncta in Aldh2 -/- cells treated with Hanks’ Balanced Salt Solution (HBSS) and CQ for 8 hours. Representative cell images. B. Live cell imaging determined mCherry-EGFP-LC3B puncta in Aldh2 +/+ and Aldh2 -/- cells treated with acetaldehyde for indicated time periods. Representative cell images. C. Histogram of average red (mCherry) puncta/cell and average yellow (co-localization of red and green puncta) puncta/cell in Aldh2 +/+ and Aldh2 -/- cells treated with acetaldehyde for indicated time periods. *p<0.05 compared with Aldh2 +/+ 0 h, #p<0.05 compared with non Aldh2 -/- 0 h. n=20-70 for all p-values.
Esophageal organoids reveal autophagy-mediated regulation of acetaldehyde-induced oxidative stress
We next employed a three dimensional organoid culture system to evaluate the influence of acetaldehyde treatment upon esophageal keratinocytes in a more physiologically relevant context. Acetaldehyde treatment significantly increased the expression of histone p-H2A.XSer139, an indicator of DNA damage that also serves as surrogate marker for oxidative stress, in esophageal organoids derived from both Aldh2+/+ and Aldh2-/- keratinocytes (Figure 5A, 5B). DNA damage level in Aldh2-/- organoids treated by acetaldehyde was significantly higher in Aldh2+/+ organoids treated by acetaldehyde. CQ treatment in addition to acetaldehyde treatment further increased DNA damage in Aldh2-/- organoids but not in Aldh2+/+ organoids (Figure 5A, 5B), suggesting that Aldh2-/- organoids depend on autophagy to decrease oxidative stress.
Figure 5.
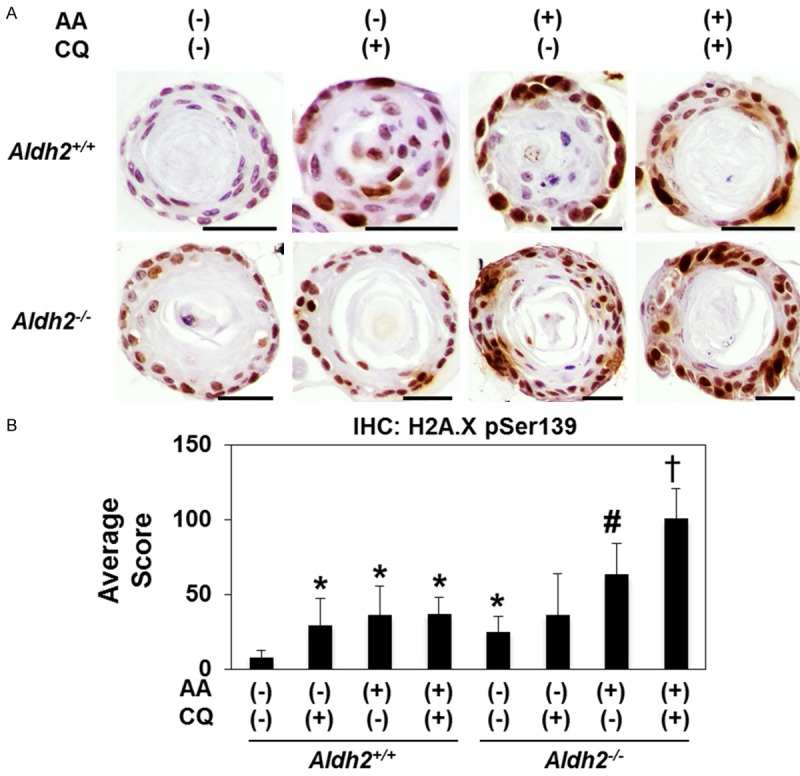
Esophageal organoids reveal autophagy-mediated regulation of oxidative stress induced by acetaldehyde Murine esophageal 3D organoids were grown ex vivo and treated from days 7-9 with 1 mM acetaldehyde and 1 μg/ml CQ as indicated and subjected IHC p-H2A.XSer139 (DNA damage). A. Representative images. Scale bars, 25 μm. B. p-H2A.XSer139 score in Aldh2 +/+ and Aldh2 -/- organoids treated with 1 mM acetaldehyde with and without CQ for 48 hours. *p<0.05 compared with Aldh2 +/+ non-treatment, #p<0.05 compared with Aldh2 -/- non-treatment, †p<0.05 compared with Aldh2 -/- treated with AA alone. n=10 for all p-values.
Alcohol drinking increases AV content in murine esophageal epithelia
Alcohol drinking induces DNA adduct formation, oxidative stress and Aldh2 upregulation in murine esophageal epithelia [23]. To determine the influence of alcohol drinking upon autophagy in esophageal epithelia in vivo, we evaluated expression of cleaved LC3 in Aldh2 +/+ mice provided ad libitum drinking water with or without alcohol (10% EtOH) for 8 weeks. A significant elevation of cleaved LC3 expression was found via IHC in esophageal epithelia of both Aldh2 +/+ and Aldh2 -/- mice exposed to alcohol as compared to animals given access to drinking water alone (Figure 6A, 6B). Despite a trend suggesting that Aldh2 -/- mice may exhibit increased AV content in response to alcohol drinking as compared to their Aldh2 +/+ counterparts, no significant difference was detected between genotypes with regard to LC3 expression (Figure 6A, 6B). These results indicate that alcohol drinking enhances AV content in murine esophageal epithelia.
Figure 6.
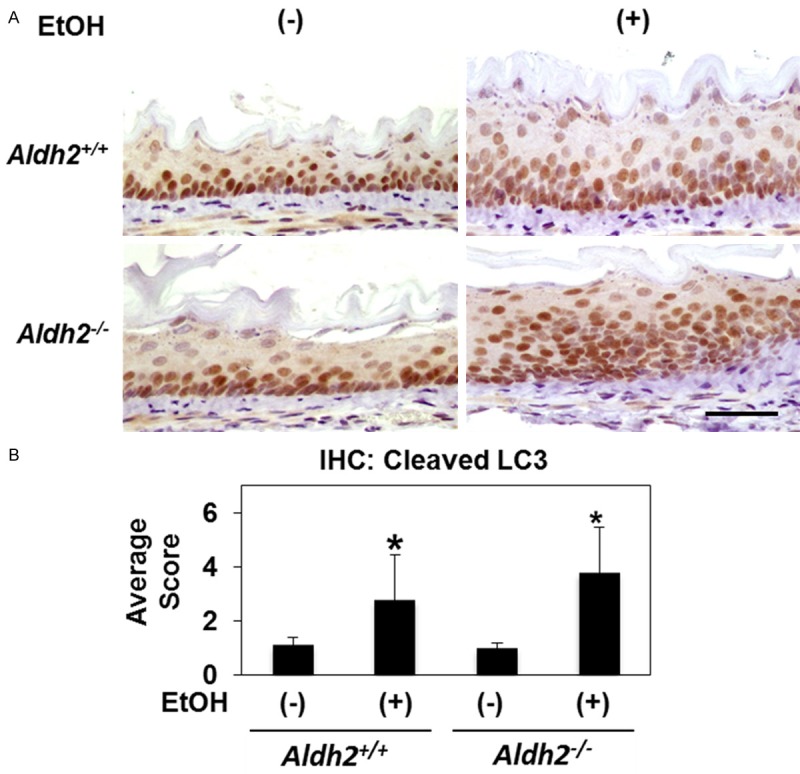
Alcohol drinking increases cleaved LC3 expression in murine esophageal epithelia Esophageal epithelia of Aldh2 +/+ and Aldh2 -/- mice provided with drinking water supplemented with or without 10% ethanol for 8 weeks were stained for cleaved LC3 by IHC. A. Representative images. Scale bar, 50 μm. B. Histogram representing average cleaved LC3 IHC score for indicated genotype and treatment group (n=5, each groups). *p<0.05 vs. Aldh2 +/+ and water.
In aggregate, these findings suggest that autophagy may provide cytoprotection to esophageal epithelial cells from oxidative stress induced by ethanol and its major metabolite acetaldehyde that is enhanced by ALDH2 dysfunction.
Discussion
In this study, we have for the first time demonstrated that esophageal keratinocytes undergo autophagy in response to ethanol or acetaldehyde exposure. Our flow cytometric and functional assays for ROS, AV content and autophagic flux coupled with esophageal 3D organoids revealed that Aldh2-/- cells display greater oxidative stress, more AV content as well as higher basal and inducible autophagic flux than Aldh2+/+ cells. Autophagy has been implicated in a variety of alcohol-related human pathologies. Autophagy contributes to loss of skeletal muscle mass (aka sarcopenia) in patients with alcoholic liver cirrhosis and hepatitis [38]. While autophagy is protective against ethanol-induced liver toxicity [39], alcohol-induced steatosis and liver injury are associated with decreased autophagic flux, which causes accumulation of cytoplasmic inclusions known as the Mallory-Denk bodies consisting of cytokeratins K8 and K18 as well as p62 [40]. Autophagy is also stalled in alcohol-induced cardiomyopathy to reduce cardiac muscle contractility [41]. Thus, alcohol-induced cellular dysfunction may occur when the level of oxidative stress and other abnormal substances exceeds cellular autophagic capability to remove them.
Autophagic flux was indeed decreased in acetaldehyde-treated Aldh2-/- cells as a function of time (Figure 4), providing an explanation for the higher vulnerability to alcohol-induced toxicity and oxidative stress in the presence of Aldh2 dysfunction. Autophagy defects cause accumulation of abnormal mitochondria and elevated ROS, culminating in DNA damage [27,42]. Acetaldehyde causes mitochondrial degeneration as well as DNA damage and mutations via DNA adduct formation [12,43]. Nevertheless, mice subjected to alcohol drinking alone display DNA damage, but not ESCC lesions with or without Aldh2 dysfunction [44,45]. While this may represent a species difference between human and mice with regard to disease susceptibility, autophagy may have a tumor suppressor role to maintain genetic stability and cellular homeostasis [46,47]. Thus, generation of genetically engineered mice with concurrent loss of autophagy (e.g. Atg7) and Aldh2 functions may be necessary to model alcohol-induced esophageal carcinogenesis in mice. Additionally, DNA damage normally triggers cell death or DNA repair functions; however, genetic alterations, such as mutations of the p53 tumor suppressor gene, promote cell survival, resulting in a further increase in abnormal cells with DNA lesions. p53 mutations are frequently found in ESCC precursor lesions. Acetaldehyde-mediated DNA damage activates the Fanconi anemia (FA) DNA repair pathway [48]. Patients with Fanconi anemia have an increased risk to develop young-onset ESCC and other squamous cell carcinomas [49-51]. Thus, these pathways may need to be impaired for survival and malignant transformation of esophageal cells with dysfunctional autophagy or Aldh2-mediated alcohol detoxification.
Decreased autophagy flux in Aldh2-/- cells was also associated with stabilization of p62 (Figure 4), a tumor promoting multifunctional scaffold protein. During autophagy, p62 directs autophagic cargo toward AVs where p62 is degraded along with autophagic substrates. Indeed, p62 accumulates in autophagy defective Atg7-/- mouse cells [52-54]. p62 also suppresses autophagy via physical interaction with the mammalian target of rapamycin complex 1 (mTORC1) [55], an inhibitor of autophagy. p62 interacts with tumor necrosis factor receptor family members to activate nuclear factor-κB signaling and expression of proinflammatory genes [56-60]. p62 stabilizes Nrf2 [42,61,62], another transcription factor mediating cellular antioxidant response [63]. While Nrf2 is required for squamous-cell differentiation in the esophagus [64], prolonged and enhanced Nrf2 activation impairs epithelial barrier function, promoting keratinocyte hyperproliferation and inflammation [65]. Thus, diminished autophagy flux in Aldh2-deficient cells under chronic exposure to ethanol or acetaldehyde may cooperate with p62 to alter esophageal cellular functions and tissue microenvironment which may then facilitate esophageal cell survival and malignant transformation.
The use of single cell-derived 3D esophageal organoids in this study represents a novel approach to elucidate esophageal epithelial function in a tissue-like context. By comparing Aldh2-/- to Aldh2+/+ organoids coupled with pharmacological inhibition of autophagy flux by chloroquine, autophagy-mediated inhibition of oxidative stress was found to be augmented in Aldh2-/- cells, recapitulating a similar finding in Aldh2-deficient murine esophagi [23]. However, we did not observe a statistically significant difference in cleaved LC3 expression between Aldh2+/+ and Aldh2-/- cells, organoids as well as murine esophageal tissues. Western blotting by an independent anti-LC3 antibody detecting the lipidated from of LC3 (LC3-II) also showed little difference in the LC3 expression levels between Aldh2-/- and Aldh2+/+ cells although either chloroquine or acetaldehyde, or a combination of the two, stabilized LC3-II, indicating the presence of basal and acetaldehyde-induced flux. Thus, LC3 did not appear to be as sensitive of an indicator of autophagic flux as the mCherry-EGFP-LC3B reporter. Thus, future studies may require development of functional assays using organoids as well as murine models, for example, mice carrying EGFP-LC3B reporter although EGFP-LC3B does not necessarily distinguish stalled autophagy, given the acid-sensitivity of EGFP. Alternatively, Cyto-ID fluorescent dye may be used to determine tissue autophagy flux along with chloroquine.
This study highlights ethanol metabolism in esophageal keratinocytes as suggested by aggravated oxidative stress and cytotoxicity in ethanol-exposed Aldh2-/- cells. Moreover, 4 MP alleviated partially ethanol-induced toxicity in Aldh2-/- cells, suggesting that acetaldehyde may be produced via multiple ethanol metabolizing pathways utilizing distinct enzymes including ADH1B and CYP2E1, the latter implicated in alcohol-related esophageal carcinogenesis [66]. Since ethanol impairs esophageal epithelial transport and barrier functions [67], acetaldehyde may be directly produced in oral-esophageal epithelia in long-term consumers of alcohol beverages, increasing the risk for carcinogenesis. Interestingly, Aldh2+/+ cells displayed limited lethality, oxidative stress and autophagy. Such cells may efficiently diminish acetaldehyde via Aldh2. It is also possible that acetaldehyde-mediated toxicity is alleviated by other cytoprotective mechanisms such as mitochondrial antioxidant pathways. Given their higher basal ROS level, Aldh2-/- cells may have more dysfunctional mitochondria. Besides acetaldehyde, Aldh2 detoxifies endogenous toxic aldehyde 4-hydroxy-2-nonenal (4-HNE) [68,69].
A series of studies led by Ren and others show protective myocardial protective roles of Aldh2 under a variety of conditions, including alcoholic cardiac contractile dysfunction [70], myocardial ischemia/reperfusion injury [68], diabetes-induced cardiac dysfunction [71] and doxorubicin cardiotoxicity [69], where 4-HNE has been implicated. As cigarette smoke extract induces 4-HNE [72], it is plausible that Aldh2 may have broader cytoprotective roles against tobacco smoke constituents containing nearly 80 chemical carcinogens including acetaldehyde [4]. Activators of ALDH2 such as Alda-1 [73,74] may not only confer cardioprotection, but reduce cancer risk.
In summary, our innovative approaches provided novel insights into the cytoprotective roles of Aldh2 and autophagy against acetaldehyde and oxidative stress, respectively, induced by ethanol or acetaldehyde, key human esophageal carcinogens.
Acknowledgements
We appreciate the support of Dr. Anil K. Rustgi and the members of his laboratory. We thank Ben Rhodes for technical support. We acknowledge the Flow Cytometry and Cell Sorting Resource Laboratory. PMC is currently in the Graduate program in Cell Biology, Physiology, and Metabolism at the University of Pennsylvania. This study was supported by the following NIH Grants: P01CA098101 (HN, KAW, KT, AKS) K26RR032714 (HN), P30 ES 013508 University of Pennsylvania Center of Excellence in Environmental Toxicology (HN), K01DK103953 (KAW), F32CA174176 (KAW), T32DK007066 (KAW), NIH/NIDDK P30-DK050306 Center of Molecular Studies in Digestive and Liver Diseases, The Molecular Pathology and Imaging, Molecular Biology/Gene Expression and Cell Culture Core Facilities. Additional supports were provided by Drexel University College of Medicine (EN) and Raptor Pharmaceuticals (HN, KT). KT is a recipient of the Japan Society for the Promotion of Science (JSPS) Postdoctoral Fellowship. This work was supported by the National Cancer Center and Development Fund (22-36 to M.M.); Grants-in-Aid for Scientific Research from the JSPS (24590917 to MM, 25460926 to SO) and the Japanese Society of Gastroenterology Research Foundation (SO). This research was supported by practical research for innovative cancer control from the Japan Agency for Medical Research and Development.
Disclosure of conflict of interest
None.
References
- 1.Pennathur A, Gibson MK, Jobe BA, Luketich JD. Oesophageal carcinoma. Lancet. 2013;381:400–412. doi: 10.1016/S0140-6736(12)60643-6. [DOI] [PubMed] [Google Scholar]
- 2.Rustgi AK, El-Serag HB. Esophageal carcinoma. N Engl J Med. 2014;371:2499–2509. doi: 10.1056/NEJMra1314530. [DOI] [PubMed] [Google Scholar]
- 3.Ohashi S, Miyamoto S, Kikuchi O, Goto T, Amanuma Y, Muto M. Recent Advances from Basic and Clinical Studies of Esophageal Squamous Cell Carcinoma. Gastroenterology. 2015;149:1700–15. doi: 10.1053/j.gastro.2015.08.054. [DOI] [PubMed] [Google Scholar]
- 4.Secretan B, Straif K, Baan R, Grosse Y, El Ghissassi F, Bouvard V, Benbrahim-Tallaa L, Guha N, Freeman C, Galichet L, Cogliano V. A review of human carcinogens--Part E: tobacco, areca nut, alcohol, coal smoke, and salted fish. Lancet Oncol. 2009;10:1033–1034. doi: 10.1016/s1470-2045(09)70326-2. [DOI] [PubMed] [Google Scholar]
- 5.Lee SP, Chiang CP, Lee SL, Hsia YJ, Chuang TL, Lin JC, Liang SC, Nieh S, Yin SJ. Immunochemical features in the classification of human alcohol dehydrogenase family. Alcohol. 2006;39:13–20. doi: 10.1016/j.alcohol.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 6.Yin SJ, Chou FJ, Chao SF, Tsai SF, Liao CS, Wang SL, Wu CW, Lee SC. Alcohol and aldehyde dehydrogenases in human esophagus: comparison with the stomach enzyme activities. Alcohol Clin Exp Res. 1993;17:376–381. doi: 10.1111/j.1530-0277.1993.tb00779.x. [DOI] [PubMed] [Google Scholar]
- 7.Boleda MD, Julia P, Moreno A, Pares X. Role of extrahepatic alcohol dehydrogenase in rat ethanol metabolism. Arch Biochem Biophys. 1989;274:74–81. doi: 10.1016/0003-9861(89)90416-5. [DOI] [PubMed] [Google Scholar]
- 8.Salaspuro V, Salaspuro M. Synergistic effect of alcohol drinking and smoking on in vivo acetaldehyde concentration in saliva. International journal of cancer. Int J Cancer. 2004;111:480–483. doi: 10.1002/ijc.20293. [DOI] [PubMed] [Google Scholar]
- 9.Marttila E, Uittamo J, Rusanen P, Lindqvist C, Salaspuro M, Rautemaa R. Acetaldehyde production and microbial colonization in oral squamous cell carcinoma and oral lichenoid disease. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013;116:61–68. doi: 10.1016/j.oooo.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 10.Hori K, Miyamoto S, Yukawa Y, Muto M, Chiba T, Matsuda T. Stability of acetaldehyde-derived DNA adduct in vitro. Biochem Biophys Res Commun. 2012;423:642–646. doi: 10.1016/j.bbrc.2012.05.158. [DOI] [PubMed] [Google Scholar]
- 11.Yukawa Y, Muto M, Hori K, Nagayoshi H, Yokoyama A, Chiba T, Matsuda T. Combination of ADH1B*2/ALDH2*2 polymorphisms alters acetaldehyde-derived DNA damage in the blood of Japanese alcoholics. Cancer Sci. 2012;103:1651–1655. doi: 10.1111/j.1349-7006.2012.02360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matsuda T, Matsumoto A, Uchida M, Kanaly RA, Misaki K, Shibutani S, Kawamoto T, Kitagawa K, Nakayama KI, Tomokuni K, Ichiba M. Increased formation of hepatic N2-ethylidene-2’-deoxyguanosine DNA adducts in aldehyde dehydrogenase 2-knockout mice treated with ethanol. Carcinogenesis. 2007;28:2363–2366. doi: 10.1093/carcin/bgm057. [DOI] [PubMed] [Google Scholar]
- 13.Yukawa Y, Ohashi S, Amanuma Y, Nakai Y, Tsurumaki M, Kikuchi O, Miyamoto S, Oyama T, Kawamoto T, Chiba T, Matsuda T, Muto M. Impairment of aldehyde dehydrogenase 2 increases accumulation of acetaldehyde-derived DNA damage in the esophagus after ethanol ingestion. Am J Cancer Res. 2014;4:279–284. [PMC free article] [PubMed] [Google Scholar]
- 14.Yokoyama A, Omori T, Tanaka Y, Yokoyama T, Sugiura H, Mizukami T, Matsushita S, Higuchi S, Maruyama K, Ishii H, Hibi T. p53 Protein accumulation, cancer multiplicity, and aldehyde dehydrogenase-2 genotype in Japanese alcoholic men with early esophageal squamous cell carcinoma. Cancer Lett. 2007;247:243–252. doi: 10.1016/j.canlet.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 15.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yokoyama A, Muramatsu T, Omori T, Yokoyama T, Matsushita S, Higuchi S, Maruyama K, Ishii H. Alcohol and aldehyde dehydrogenase gene polymorphisms and oropharyngolaryngeal, esophageal and stomach cancers in Japanese alcoholics. Carcinogenesis. 2001;22:433–439. doi: 10.1093/carcin/22.3.433. [DOI] [PubMed] [Google Scholar]
- 17.Yokoyama A, Omori T, Yokoyama T. Alcohol and aldehyde dehydrogenase polymorphisms and a new strategy for prevention and screening for cancer in the upper aerodigestive tract in East Asians. Keio J Med. 2010;59:115–130. doi: 10.2302/kjm.59.115. [DOI] [PubMed] [Google Scholar]
- 18.Brooks PJ, Enoch MA, Goldman D, Li TK, Yokoyama A. The alcohol flushing response: an unrecognized risk factor for esophageal cancer from alcohol consumption. PLoS Med. 2009;6:e50. doi: 10.1371/journal.pmed.1000050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yokoyama A, Tsutsumi E, Imazeki H, Suwa Y, Nakamura C, Yokoyama T. Polymorphisms of alcohol dehydrogenase-1B and aldehyde dehydrogenase-2 and the blood and salivary ethanol and acetaldehyde concentrations of Japanese alcoholic men. Alcohol Clin Exp Res. 2010;34:1246–1256. doi: 10.1111/j.1530-0277.2010.01202.x. [DOI] [PubMed] [Google Scholar]
- 20.Harada S, Agarwal DP, Goedde HW. Aldehyde dehydrogenase deficiency as cause of facial flushing reaction to alcohol in Japanese. Lancet. 1981;2:982. doi: 10.1016/s0140-6736(81)91172-7. [DOI] [PubMed] [Google Scholar]
- 21.Muto M, Nakane M, Hitomi Y, Yoshida S, Sasaki S, Ohtsu A, Yoshida S, Ebihara S, Esumi H. Association between aldehyde dehydrogenase gene polymorphisms and the phenomenon of field cancerization in patients with head and neck cancer. Carcinogenesis. 2002;23:1759–1765. doi: 10.1093/carcin/23.10.1759. [DOI] [PubMed] [Google Scholar]
- 22.Yokoyama A, Muramatsu T, Ohmori T, Yokoyama T, Okuyama K, Takahashi H, Hasegawa Y, Higuchi S, Maruyama K, Shirakura K, Ishii H. Alcohol-related cancers and aldehyde dehydrogenase-2 in Japanese alcoholics. Carcinogenesis. 1998;19:1383–1387. doi: 10.1093/carcin/19.8.1383. [DOI] [PubMed] [Google Scholar]
- 23.Amanuma Y, Ohashi S, Itatani Y, Tsurumaki M, Matsuda S, Kikuchi O, Nakai Y, Miyamoto S, Oyama T, Kawamoto T, Whelan KA, Nakagawa H, Chiba T, Matsuda T, Muto M. Protective role of ALDH2 against acetaldehyde-derived DNA damage in oesophageal squamous epithelium. Sci Rep. 2015;5:14142. doi: 10.1038/srep14142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Osei-Sarfo K, Urvalek AM, Tang XH, Scognamiglio T, Gudas LJ. Initiation of esophageal squamous cell carcinoma (ESCC) in a murine 4-nitroquinoline-1-oxide and alcohol carcinogenesis model. Oncotarget. 2015;6:6040–6052. doi: 10.18632/oncotarget.3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 26.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, Kominami E, Tanaka K, Chiba T. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diakopoulos KN, Lesina M, Wormann S, Song L, Aichler M, Schild L, Artati A, Romisch-Margl W, Wartmann T, Fischer R, Kabiri Y, Zischka H, Halangk W, Demir IE, Pilsak C, Walch A, Mantzoros CS, Steiner JM, Erkan M, Schmid RM, Witt H, Adamski J, Algul H. Impaired autophagy induces chronic atrophic pancreatitis in mice via sex- and nutrition-dependent processes. Gastroenterology. 2015;148:626–638. e617. doi: 10.1053/j.gastro.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 29.Dolganiuc A, Thomes PG, Ding WX, Lemasters JJ, Donohue TM Jr. Autophagy in alcoholinduced liver diseases. Alcoholism, clinical and experimental research. 2012;36:1301–1308. doi: 10.1111/j.1530-0277.2012.01742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Piano MR, Phillips SA. Alcoholic cardiomyopathy: pathophysiologic insights. Cardiovasc Toxicol. 2014;14:291–308. doi: 10.1007/s12012-014-9252-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kitagawa K, Kawamoto T, Kunugita N, Tsukiyama T, Okamoto K, Yoshida A, Nakayama K, Nakayama K. Aldehyde dehydrogenase (ALDH) 2 associates with oxidation of methoxyacetaldehyde; in vitro analysis with liver subcellular fraction derived from human and Aldh2 gene targeting mouse. FEBS Lett. 2000;476:306–311. doi: 10.1016/s0014-5793(00)01710-5. [DOI] [PubMed] [Google Scholar]
- 32.N’Diaye EN, Kajihara KK, Hsieh I, Morisaki H, Debnath J, Brown EJ. PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep. 2009;10:173–9. doi: 10.1038/embor.2008.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takaoka M, Harada H, Deramaudt TB, Oyama K, Andl CD, Johnstone CN, Rhoades B, Enders GH, Opitz OG, Nakagawa H. Ha-Ras(G12V) induces senescence in primary and immortalized human esophageal keratinocytes with p53 dysfunction. Oncogene. 2004;23:6760–6768. doi: 10.1038/sj.onc.1207923. [DOI] [PubMed] [Google Scholar]
- 34.Kinugasa H, Whelan KA, Tanaka K, Natsuizaka M, Long A, Guo A, Chang S, Kagawa S, Srinivasan S, Guha M, Yamamoto K, St Clair DK, Avadhani NG, Diehl JA, Nakagawa H. Mitochondrial SOD2 regulates epithelial-mesenchymal transition and cell populations defined by differential CD44 expression. Oncogene. 2015;34:5229–5239. doi: 10.1038/onc.2014.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Natsuizaka M, Kinugasa H, Kagawa S, Whelan KA, Naganuma S, Subramanian H, Chang S, Nakagawa KJ, Rustgi NL, Kita Y, Natsugoe S, Basu D, Gimotty PA, Klein-Szanto AJ, Diehl JA, Nakagawa H. IGFBP3 promotes esophageal cancer growth by suppressing oxidative stress in hypoxic tumor microenvironment. Am J Cancer Res. 2014;4:29–41. [PMC free article] [PubMed] [Google Scholar]
- 36.Ohashi S, Natsuizaka M, Yashiro-Ohtani Y, Kalman RA, Nakagawa M, Wu L, Klein-Szanto AJ, Herlyn M, Diehl JA, Katz JP, Pear WS, Seykora JT, Nakagawa H. NOTCH1 and NOTCH3 coordinate esophageal squamous differentiation through a CSL-dependent transcriptional network. Gastroenterology. 2010;139:2113–2123. doi: 10.1053/j.gastro.2010.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shin HW, Umber BJ, Meinardi S, Leu SY, Zaldivar F, Blake DR, Cooper DM. Acetaldehyde and hexanaldehyde from cultured white cells. J Transl Med. 2009;7:31. doi: 10.1186/1479-5876-7-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thapaliya S, Runkana A, McMullen MR, Nagy LE, McDonald C, Naga Prasad SV, Dasarathy S. Alcohol-induced autophagy contributes to loss in skeletal muscle mass. Autophagy. 2014;10:677–690. doi: 10.4161/auto.27918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ding WX, Li M, Chen X, Ni HM, Lin CW, Gao W, Lu B, Stolz DB, Clemens DL, Yin XM. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology. 2010;139:1740–1752. doi: 10.1053/j.gastro.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rautou PE, Mansouri A, Lebrec D, Durand F, Valla D, Moreau R. Autophagy in liver diseases. J Hepatol. 2010;53:1123–1134. doi: 10.1016/j.jhep.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 41.Guo R, Hu N, Kandadi MR, Ren J. Facilitated ethanol metabolism promotes cardiomyocyte contractile dysfunction through autophagy in murine hearts. Autophagy. 2012;8:593–608. doi: 10.4161/auto.18997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, Dipaola RS, Karantza-Wadsworth V, White E. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–1075. doi: 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hori K, Miyamoto S, Yukawa Y, Muto M, Chiba T, Matsuda T. Stability of acetaldehyde-derived DNA adduct in vitro. Biochem Biophys Res Commun. 2012;423:642–646. doi: 10.1016/j.bbrc.2012.05.158. [DOI] [PubMed] [Google Scholar]
- 44.Seitz HK, Stickel F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat Rev Cancer. 2007;7:599–612. doi: 10.1038/nrc2191. [DOI] [PubMed] [Google Scholar]
- 45.Yu HS, Oyama T, Matsuda T, Isse T, Yamaguchi T, Tanaka M, Tsuji M, Kawamoto T. The effect of ethanol on the formation of N2-ethylidene-dG adducts in mice: implications for alcohol-related carcinogenicity of the oral cavity and esophagus. Biomarkers. 2012;17:269–274. doi: 10.3109/1354750X.2012.666675. [DOI] [PubMed] [Google Scholar]
- 46.Aita VM, Liang XH, Murty VV, Pincus DL, Yu W, Cayanis E, Kalachikov S, Gilliam TC, Levine B. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics. 1999;59:59–65. doi: 10.1006/geno.1999.5851. [DOI] [PubMed] [Google Scholar]
- 47.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100:15077–15082. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Langevin F, Crossan GP, Rosado IV, Arends MJ, Patel KJ. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature. 2011;475:53–58. doi: 10.1038/nature10192. [DOI] [PubMed] [Google Scholar]
- 49.Akbari MR, Malekzadeh R, Lepage P, Roquis D, Sadjadi AR, Aghcheli K, Yazdanbod A, Shakeri R, Bashiri J, Sotoudeh M, Pourshams A, Ghadirian P, Narod SA. Mutations in Fanconi anemia genes and the risk of esophageal cancer. Hum Genet. 2011;129:573–582. doi: 10.1007/s00439-011-0951-7. [DOI] [PubMed] [Google Scholar]
- 50.van Zeeburg HJ, Snijders PJ, Wu T, Gluckman E, Soulier J, Surralles J, Castella M, van der Wal JE, Wennerberg J, Califano J, Velleuer E, Dietrich R, Ebell W, Bloemena E, Joenje H, Leemans CR, Brakenhoff RH. Clinical and molecular characteristics of squamous cell carcinomas from Fanconi anemia patients. J Natl Cancer Inst. 2008;100:1649–1653. doi: 10.1093/jnci/djn366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rosenberg PS, Greene MH, Alter BP. Cancer incidence in persons with Fanconi anemia. Blood. 2003;101:822–826. doi: 10.1182/blood-2002-05-1498. [DOI] [PubMed] [Google Scholar]
- 52.Wu JJ, Quijano C, Chen E, Liu H, Cao L, Fergusson MM, Rovira II, Gutkind S, Daniels MP, Komatsu M, Finkel T. Mitochondrial dysfunction and oxidative stress mediate the physiological impairment induced by the disruption of autophagy. Aging. 2009;1:425–437. doi: 10.18632/aging.100038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kahai S, Lee SC, Lee DY, Yang J, Li M, Wang CH, Jiang Z, Zhang Y, Peng C, Yang BB. MicroRNA miR-378 regulates nephronectin expression modulating osteoblast differentiation by targeting GalNT-7. PLoS One. 2009;4:e7535. doi: 10.1371/journal.pone.0007535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jiang M, Wei Q, Dong G, Komatsu M, Su Y, Dong Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012;82:1271–83. doi: 10.1038/ki.2012.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Duran A, Amanchy R, Linares JF, Joshi J, Abu-Baker S, Porollo A, Hansen M, Moscat J, Diaz-Meco MT. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell. 2011;44:134–46. doi: 10.1016/j.molcel.2011.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, Moscat J. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer cell. 2008;13:343–54. doi: 10.1016/j.ccr.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 57.Lee HM, Shin DM, Yuk JM, Shi G, Choi DK, Lee SH, Huang SM, Kim JM, Kim CD, Lee JH, Jo EK. Autophagy negatively regulates keratinocyte inflammatory responses via scaffolding protein p62/SQSTM1. J Immunol. 2011;186:1248–1258. doi: 10.4049/jimmunol.1001954. [DOI] [PubMed] [Google Scholar]
- 58.Sanz L, Diaz-Meco MT, Nakano H, Moscat J. The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. EMBO J. 2000;19:1576–86. doi: 10.1093/emboj/19.7.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Samuels IS, Seibenhener ML, Neidigh KB, Wooten MW. Nerve growth factor stimulates the interaction of ZIP/p62 with atypical protein kinase C and targets endosomal localization: evidence for regulation of nerve growth factorinduced differentiation. J Cell Biochem. 2001;82:452–466. doi: 10.1002/jcb.1177. [DOI] [PubMed] [Google Scholar]
- 60.Wooten MW, Seibenhener ML, Mamidipudi V, Diaz-Meco MT, Barker PA, Moscat J. The atypical protein kinase C-interacting protein p62 is a scaffold for NF-kappaB activation by nerve growth factor. J Biol Chem. 2001;276:7709–7712. doi: 10.1074/jbc.C000869200. [DOI] [PubMed] [Google Scholar]
- 61.Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, Kim M, Nishito Y, Iemura S, Natsume T, Ueno T, Kominami E, Motohashi H, Tanaka K, Yamamoto M. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 62.Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T, Sun Z, White E, Zhang DD. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol. 2010;30:3275–85. doi: 10.1128/MCB.00248-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A, McMahon M, Hayes JD, Johansen T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010;285:22576–91. doi: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jiang M, Ku WY, Zhou Z, Dellon ES, Falk GW, Nakagawa H, Wang ML, Liu K, Wang J, Katzka DA, Peters JH, Lan X, Que J. BMP-driven NRF2 activation in esophageal basal cell differentiation and eosinophilic esophagitis. J Clin Invest. 2015;125:1557–68. doi: 10.1172/JCI78850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schafer M, Farwanah H, Willrodt AH, Huebner AJ, Sandhoff K, Roop D, Hohl D, Bloch W, Werner S. Nrf2 links epidermal barrier function with antioxidant defense. EMBO Mol Med. 2012;4:364–379. doi: 10.1002/emmm.201200219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Millonig G, Wang Y, Homann N, Bernhardt F, Qin H, Mueller S, Bartsch H, Seitz HK. Ethanol-mediated carcinogenesis in the human esophagus implicates CYP2E1 induction and the generation of carcinogenic DNA-lesions. Int J Cancer. 2011;128:533–540. doi: 10.1002/ijc.25604. [DOI] [PubMed] [Google Scholar]
- 67.Bor S, Caymaz-Bor C, Tobey NA, Abdulnour-Nakhoul S, Marten E, Orlando RC. Effect of ethanol on the structure and function of rabbit esophageal epithelium. Am J Physiol. 1998;274:G819–826. doi: 10.1152/ajpgi.1998.274.5.G819. [DOI] [PubMed] [Google Scholar]
- 68.Ma H, Guo R, Yu L, Zhang Y, Ren J. Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: role of autophagy paradox and toxic aldehyde. Eur Heart J. 2011;32:1025–1038. doi: 10.1093/eurheartj/ehq253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ge W, Yuan M, Ceylan AF, Wang X, Ren J. Mitochondrial aldehyde dehydrogenase protects against doxorubicin cardiotoxicity through a transient receptor potential channel vanilloid 1-mediated mechanism. Biochim Biophys Acta. 2016;1862:622–34. doi: 10.1016/j.bbadis.2015.12.014. [DOI] [PubMed] [Google Scholar]
- 70.Ge W, Ren J. mTOR-STAT3-notch signalling contributes to ALDH2-induced protection against cardiac contractile dysfunction and autophagy under alcoholism. J Cell Mol Med. 2012;16:616–626. doi: 10.1111/j.1582-4934.2011.01347.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Guo Y, Yu W, Sun D, Wang J, Li C, Zhang R, Babcock SA, Li Y, Liu M, Ma M, Shen M, Zeng C, Li N, He W, Zou Q, Zhang Y, Wang H. A novel protective mechanism for mitochondrial aldehyde dehydrogenase (ALDH2) in type i diabetes-induced cardiac dysfunction: role of AMPKregulated autophagy. Biochim Biophys Acta. 2015;1852:319–331. doi: 10.1016/j.bbadis.2014.05.017. [DOI] [PubMed] [Google Scholar]
- 72.Kode A, Yang SR, Rahman I. Differential effects of cigarette smoke on oxidative stress and proinflammatory cytokine release in primary human airway epithelial cells and in a variety of transformed alveolar epithelial cells. Respir Res. 2006;7:132. doi: 10.1186/1465-9921-7-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Budas GR, Disatnik MH, Chen CH, Mochly-Rosen D. Activation of aldehyde dehydrogenase 2 (ALDH2) confers cardioprotection in protein kinase C epsilon (PKCvarepsilon) knockout mice. J Mol Cell Cardiol. 2010;48:757–764. doi: 10.1016/j.yjmcc.2009.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sun L, Ferreira JC, Mochly-Rosen D. ALDH2 activator inhibits increased myocardial infarction injury by nitroglycerin tolerance. Sci Transl Med. 2011;3:107ra111. doi: 10.1126/scitranslmed.3002067. [DOI] [PMC free article] [PubMed] [Google Scholar]