

Abstract
Dysregulated PI3K/Akt/mTOR (PAM) pathway signaling occurs in ~30% of human cancers, making it a rational target for new therapies; however, the effectiveness of some PAM pathway inhibitors, such as mTORC rapalogs, may be compromised by a compensatory feedback loop leading to Akt activation. In this study, the p70S6K/Akt dual inhibitor, M2698 (previously MSC2363318A), was characterized as a potential anti-cancer agent through examination of its pharmacokinetic, pharmacodynamic and metabolic properties, and anti-tumor activity. M2698 was highly potent in vitro (IC50 1 nM for p70S6K, Akt1 and Akt3 inhibition; IC50 17 nM for pGSK3β indirect inhibition) and in vivo (IC50 15 nM for pS6 indirect inhibition), and relatively selective (only 6/264 kinases had an IC50 within 10-fold of p70S6K). Orally administered M2698 crossed the blood-brain barrier in rats and mice, with brain tumor exposure 4-fold higher than non-disease brain. Dose-dependent inhibition of target substrate phosphorylation was observed in vitro and in vivo, indicating that M2698 blocked p70S6K to provide potent PAM pathway inhibition while simultaneously targeting Akt to overcome the compensatory feedback loop. M2698 demonstrated dose-dependent tumor growth inhibition in mouse xenograft models derived from PAM pathway-dysregulated human triple-negative (MDA-MB-468) and Her2-expressing breast cancer cell lines (MDA-MB-453 and JIMT-1), and reduced brain tumor burden and prolonged survival in mice with orthotopically implanted U251 glioblastoma. These findings highlight M2698 as a promising PAM pathway inhibitor whose unique mechanism of action and capacity to pass the blood-brain barrier warrant clinical investigation in cancers with PAM pathway dysregulation, and those with central nervous system involvement.
Keywords: p70S6K, Akt, PAM pathway, blood-brain barrier, MSC2363318A, M2698
Introduction
In healthy cells, the PI3K/Akt/mTOR (PAM) pathway is an important regulator of growth, proliferation and metabolism. However, hyperactivation of the PAM pathway correlates with dysregulated cell growth and survival and has been observed in ~30% of human cancers [1]. Aberrant PI3K signaling has a particularly high prevalence in glioblastoma and breast cancer [2], where genetic alterations, such as mutation or deletion of PTEN or mutation or amplification of PIK3CA, are associated with resistance to a number of anti-cancer therapies, including the epidermal growth factor receptor (EGFR) kinase inhibitors, gefitinib and erlotinib, and trastuzumab [3-6]. Inhibition of PAM pathway signaling is therefore a highly attractive prospect for new anti-cancer therapies, evidenced by the approved agents targeting the PAM pathway that have already demonstrated clinical efficacy in various solid tumor types. The mammalian target of rapamycin complex-1 (mTORC1) rapalog inhibitor, everolimus, has been approved for the treatment of hormone receptor-positive, Her2-negative breast cancer, pancreatic neuroendocrine tumors and renal cell carcinoma [7], and another rapalog, temsirolimus, is approved for renal cell carcinoma [8]. Although these agents provide clinical efficacy through mTORC inhibition, their effectiveness may be limited due to activation of a compensatory Akt feedback loop which leads to increased PAM pathway signaling [9]. Consequently, there is a need for agents that both provide effective PAM pathway inhibition and overcome the activation of the Akt feedback loop.
Signaling via the PAM pathway ultimately leads to activation of p70S6K, which in turn directly impacts cell growth and survival. Dysregulation of p70S6K can be caused by increased activity of a number of upstream kinases such as PI3K, Akt, or mTORC, but can also be produced independently of the PAM pathway via amplification of the chromosomal locus 17q23, which contains the p70S6K gene [10]. Aberrant p70S6K activity has been observed in a number of cancers, including breast cancer [11] and meningioma [12], making p70S6K a key target for PAM pathway inhibition. To that end, M2698 (previously MSC2363318A), a selective, adenosine triphosphate (ATP) competitive, dual inhibitor of p70S6K and Akt1/3, was developed. Through dually targeting both PAM pathway components, M2698 has the potential to inhibit tumor growth by blocking p70S6K activity while simultaneously blocking Akt activation emanating through the feedback loop that occurs with mTORC rapalogs. The aim of the current study was to evaluate the pharmacological profile of M2698 in vitro and in vivo, including its capacity to cross the blood-brain barrier (BBB), and to assess anti-tumor activity in animal models of human breast cancer and glioblastoma.
Materials and methods
Animals and housing
The following animals were used for determination of pharmacokinetics (PK): male and female NMRI mice (Crl: NMRI (Han); 33-to-69 days old; Charles River Laboratories, Sulzfeld, Germany), male and female Wistar Han rats (Crl: WI (Han); 10-to-13 weeks old; Charles River Laboratories, Sulzfeld, Germany); female mongrel dogs (2.5-to-4 years old; 21-23 kg; Marshall Farms, North Rose, NY, USA); female cynomolgus monkeys (Macaca fascicularis; 9-to-10 years old; 3.7-6.1 kg; female cynomolgus monkeys were imported from Mauritius and quarantined for at least 30 days before shipment to the study site).
The following mouse strains were used for subcutaneous and orthotopic xenograft models: female SCID Beige (CB17.Cg-PrkdcscidLystbg-J/Crl; 6-to-8 weeks old) and female nude (Crl: NU-Foxn1nu; 6-to-8 weeks old; Charles River Laboratories, Wilmington, MA, USA); female nude (Hsd: Athymic Nude-Fox1nu; 6-to-7 weeks old; Harlan Laboratories, Indianapolis, IN, USA).
All animals were housed under specific pathogen-free conditions, had access to standard diet and water ad libitum, and were used according to the guidelines approved by the Animal Care and Use Committee of EMD Serono and/or Molecular Imaging, Inc. (Ann Arbor, MI, USA).
Cell culture
MDA-MB-468 (triple-negative breast cancer [TNBC], PTEN loss-of-function), MDA-MB-453 (Her2-expressing), HCC1419 (Her2 amplification), and JIMT-1 (Her2+) cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). The U251 (glioblastoma) cell line was obtained from the National Cancer Institute (NCI). Cell lines used in the proliferation assay were obtained directly from ATCC, NCI, Cell Lines Service (CLS) GmbH, and the German Resource Centre for Biological Material (DSMZ) cell line collections.
Cell lines were tested by Whole Genome Array (Agilent, USA) and short tandem repeats analysis [13] to ensure absence of contamination and wrong assignment. The majority of cell lines were cultured in standard medium (RPMI 1640, DMEM or MEM) supplemented with 10% fetal bovine serum. Gene cluster and estrogen receptor (ER)/progesterone receptor (PR)/Her2 status of breast cancer cell lines have been reported previously [14,15].
Pharmacology
A radiometric protein kinase assay was used to determine the concentration of M2698 causing 50% inhibition (IC50) of target proteins. To assess kinase selectivity, M2698 was tested against a panel of 264 human kinases (including mutant kinases) at 1 µM and the percentage remaining activity (compared to a dimethyl sulfoxide [DMSO] control) was determined by a radiometric kinase activity assay that directly measures the catalytic incorporation of phosphate onto kinase substrates. For the 17 most sensitive kinases an IC50 value was determined. The assay was performed according to Merck Millipore guidelines [16].
Cell-based potency of M2698 was evaluated by determination of phosphorylation levels of the p70S6K1 and Akt substrate proteins, ribosomal protein S6 and GSK3β, respectively, in the MDA-MB-468 cell line. Cells were incubated with M2698 at concentrations of 100 pM to 25 µM or DMSO control for 2 hours. Levels of phosphorylation were determined using immunocytochemistry with primary antibodies against phosphorylated (p) S6 240/242 and pGSK3β (S9) (Cell Signaling Technologies, Danvers, MA, USA). Proteins were detected using anti-rabbit IgG (H+L), F(ab’) 2 Fragment (Alexa Fluor® 488 conjugate; Cell Signaling Technology), quantified using the Acumen Explorer (TTP Labtech, Cambridge, MA, USA), and analyzed using Genedata version 6 (Genedata, Lexington, MA, USA).
Modulation of the PAM pathway and induction of the Akt feedback loop were monitored in the cell lines HCC1419 and MDA-MB-453. Cells were incubated with 0.1% DMSO or M2698 (0.3 or 1 µM) for 24 hours, then lysed in Complete CST RIPA Buffer. Protein concentrations in cell lysates were measured using a BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA), and protein phosphorylation was analyzed by western blot.
Western blot analysis
Between 30 and 50 µg of cell or tissue lysate was used for western blot analysis. Primary antibodies against pS6 S240/244, pAkt T308, pPRAS40 T246, pGSK3 α/β (S21/9), total (t)S6, tAkt, tPRAS40, and tGSK3α/β were obtained from Cell Signaling Technologies (Danvers, MA, USA; dilutions per manufacturer recommendations). For in vitro cell lysates, proteins were detected with 1:2000 goat antirabbit IgG-HRP secondary Ab (BioRad Cat#170-6515) and BM Chemiluminescence Blotting Substrate (Roche, Indianapolis, IN, USA). For in vivo lysates, proteins were detected with AlexaFluor 680 goat α-mouse (Life Technologies, Carlsbad, CA, USA) and Rockland IRDye800 goat α-rabbit (VWR, West Chester, PA, USA) secondary antibodies (1:2000 dilution) and imaged on Li-COR Odyssey Scanner (Odyssey, Lincoln, ME, USA).
Drug metabolism and pharmacokinetics
The in vitro plasma protein binding of M2698 in human plasma and rat plasma and brain at concentrations of 0.1 to 10 µM was determined using equilibrium dialysis. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) was used to measure concentrations of M2698.
To assess brain penetration at steady state, M2698 was infused to female Wistar rats (n=2) via a catheter into the vena jugularis over 16 hours (approximately 4 times the half-life of M2698 in rats) at a total dose of 17.7 mg/kg. Following euthanasia, total concentrations of M2698 in plasma and brain were determined by LC-MS/MS. The unbound brain:plasma partition ratio (Kp, uu) was calculated from total concentrations taking into account the free fraction in rat brain and the free fraction in rat plasma, by the formula Kp uu=fu, brain * total brain concentration/fu, plasma * total plasma concentration, as previously described [17].
Human clearance was predicted using cross-species in vivo clearance (mouse, rat, dog and monkey) adjusted for relative rates of metabolism in vitro as described previously [18]. The volume of distribution at steady state (Vss) was predicted by using the average value of three prediction methods: allometric scaling using unbound volume of distribution; unbound human/dog-proportionality; and the Oie-Tozer method [19]. Plasma half-life was calculated using the equation t1/2=0.693 × Vss/CL.
In vitro cell proliferation assays
A panel of 81 cell lines derived from various human tumors with and without PAM pathway mutations were exposed to M2698 at concentrations ranging from 50 M to 0.3 nM. Cells were seeded into 96-well-plates at optimum seeding densities to ensure exponential growth for the duration of the experiment. Cells were cultured in medium containing M2698 for 72 hours before protein was precipitated by addition of 10% TCA. Cells were then stained with 0.08% wt/v sulforhodamine B (SRB) solution and washed with 1% acetic acid to remove unbound stain. Bound SRB was solubilized with 10 mM Tris base. Optical density was measured at 560 nm on a Victor 2 plate reader (Perkin Elmer, Waltham, MA). The concentration of M2698 needed to inhibit proliferation by 50% (IC50) was calculated based on non-linear curve fitting calculations performed using algorithms similar to those previously described [20] and complemented with the mean square error model.
Efficacy studies in breast cancer xenograft models
Tumor establishment and treatment
Cultured tumor cells were harvested during the log phase of growth and implanted into mice within 1 hour of collection. Tumor cells were injected subcutaneously above the right foreleg of each animal as follows: 10E6 MDA-MB-468 cells implanted into female nude (Crl: NU-Foxn1nu) mice; 10E6 MDA-MB-453 cells implanted into 100 female SCID Beige (CB17.Cg-PrkdcscidLystbg-J/Crl) mice; and 5E6 JIMT-1 cells implanted into 90 female SCID Beige mice. Tumors were allowed to grow until they had reached a predetermined size range to begin the study (18, 14 and 10 days for MDA-MB-468, MDA-MB-453 and JIMT-1 xenografts, respectively). Tumor-bearing mice were sorted into groups such that the average tumor volume for each treatment group was approximately 180 mm3, 190 mm3 and 150 mm3 for MDA-MB-468 (n=8), MDA-MB-453 (n=10) and JIMT-1 (n=10) xenografts, respectively. Animals were treated once-per-day (qd), orally (po) with vehicle (0.5% HPMC-0.25% Tween20 -pH3 in 100 mM citrate buffer) or M2698 formulated in vehicle at the following doses: 10, 20, and 30 mg/kg (MDA-MB-468 xenografts), 10 and 20 mg/kg (MDA-MB-453 xenografts), and 10 and 20 mg/kg (JIMT-1 xenografts) at a volume of 10 mL/kg for administration. Initiation of treatment was designated as Day 0. Tumor volumes and body weights were measured and recorded twice weekly and animals were euthanized on Days 28, 25 and 17 in the MDA-MB-468, MDA-MB-453 and JIMT-1 xenograft models, respectively.
Tumor volume calculations and statistical analyses
Tumor volume was calculated using tumor length (l) and width (w) measurements with the equation l*w2/2. Using electronic calipers, length was measured along the longest axis of the tumor and width was measured perpendicular to the length. Tumor volume data were log-transformed, and analyzed by repeated measures analysis of variance (RM-ANOVA) followed by Tukey’s post-hoc multiple pairwise comparisons (α=0.05) using GraphPad Prism 5. Day 0 measurements were not included in the statistical analyses.
Efficacy studies in orthotopic U251 glioblastoma models
Tumor establishment and treatment
Orthotopic U251 glioblastoma models were established and treated at Molecular Imaging, Inc. (Ann Harbor, Michigan). U251 cells were harvested during the log phase of growth and stored on ice until implantation. A total of 10E6 cells in 10 µl PBS were injected intracranially into each mouse (Hsd: Athymic Nude-Fox1nu) held in a stereotaxic frame. Tumors were allowed to grow for 18 days, then animals were divided into two groups of ~10 such that the average tumor volume was approximately 6 mg per group, as determined by magnetic resonance imaging (MRI) as described below (designated as Day 0). Animals were treated with vehicle (n=9) or M2698 in vehicle at 25 mg/kg (n=10) qd po until euthanasia at end of study (Day 50) or prior to Day 50 if weight loss exceeded 20%.
Tumor growth calculation
Tumor volume was measured on Days 17 and 31 by MRI, as described below. Day 31% T/C was defined as the MRI-determined median tumor volume of the treated group (T) divided by the MRI-determined median tumor volume of the control group (C) × 100. As tumor mass and evaluation size data did not satisfy standard testing requirements for normality and equal variance, a Kruskall-Wallace one-way ANOVA by ranks was performed. Post hoc testing for multiple comparisons was performed by using Dunn’s method.
Magnetic resonance imaging (MRI)
Tumor volume was determined via a T2-weighted fast spin-echo multi-slice sequence. Acquisition parameters were: repetition time 4 seconds, 8 echoes, echo spacing 14.5 ms, k-space centered on the fourth echo, and 2 averages. A 17 mm2 field of view was used with an image matrix of 128 × 128 and 17 contiguous, 0.5 mm thick transaxial slices covering the entire tumor volume. The images were reconstructed using custom-written scripts in MATLAB, and then segmented using the Amira image processing package. The entire tumor was manually segmented in the Amira Segmentation Editor by drawing tumor regions of interest (ROIs) for each slice of the MR images in which tumor tissue was visualized. Tumor volume was then calculated from the defined ROIs by the software.
Pharmacokinetics and pharmacodynamics
Animal treatment
To examine the PK and pharmacodynamic (PD) relationships of M2698 in breast cancer mouse models, MDA-MB-453 xenografts were established as described above. Tumors were allowed to grow for 28 days and animals were assigned to treatment groups such that the average tumor volume was approximately 462 mm3 per group. For the analysis of M2698 concentrations in plasma and tumor and inhibition of pS6 in tumor, animals were treated with vehicle or M2698 in vehicle at 20 mg/kg qd po for 4 days and terminal plasma and tumor samples were collected upon euthanasia at 4, 8 and 24 hours after the final treatment (n=3 per time point). Samples from vehicle-treated mice were collected at 4 hours after the final administration. For the characterization of the dose-response relationship among pS6 in tumor and concentrations of M2698 in plasma and tumor, animals were treated with vehicle or M2698 in vehicle at 1, 5 or 20 mg/kg po in a single administration ‘acute’ study or 5, 10 or 20 mg/kg qd po for 7 days in a ‘chronic’ study. Blood and tumor samples were collected upon euthanasia at 1, 4, 8, 24, 48 and 72 hours after the final administration for compound-treated animals and at 4 and 8 hours for the vehicle-treated animals (n=4 per time point). Tumors from animals in both studies were removed upon euthanasia, snap frozen in liquid nitrogen and stored at -80C. Plasma was harvested from whole blood and stored at -80C.
To examine the PK/PD properties of M2698 in the plasma, tumor and brain of orthotopic U251 glioblastoma models, U251 cells were implanted intracranially into mice as described above. Tumors were allowed to grow for 29 days and then animals were assigned to treatment groups such that the average tumor volume was approximately 60 mg per group. Animals were treated with a single administration of vehicle or M2698 in vehicle at 25 mg/kg qd po, and terminal plasma, brain and tumor samples were collected at 1, 4, 8 and 24 hours post-administration for compound-treated animals and at 24 hours for vehicle-treated mice (n=6 per time point).
Determination of M2698 concentrations
M2698 in plasma, tumor and extra-tumor brain tissue was measured by HPLC-MS/MS analysis. Concentrations of M2698 were determined using a standard curve (standard curve fitting was accomplished with Analyst 1.5 software from AB Sciex).
Determination of target modulation
The proportion of phosphorylated (p) and total (t) S6, PRAS40 and Akt in the subcutaneous tumors of MDA-MB-453 xenografts and in the tumor and brain of orthotopic U251 models was determined by western blot analysis, as described above.
Determination of pS6 inhibition
Inhibition of S6 by M2698 was described by the equation below, where IC50 is the plasma concentration of M2698 producing 50% inhibition of S6, C is the plasma concentration of M2698, kout (h-1) is the rate constant defining the loss of the pS6, t is time, and pS60 represents the level of pS6 at baseline (pre-treatment vehicle control). Plasma concentrations of M2698 in mice were simulated using a 1-compartment model with first order absorption determined from the tumor PK/PD experiments.
Equation 1:
dpS6/dt = kout • pS6 0 • (1-(C/(IC 50 + C)))- kout • pS6
Pharmacodynamic analysis of pS6 tumor expression was analyzed by one-way ANOVA followed by Bonferroni’s post-hoc multiple comparisons (α=0.05) using GraphPad Prism software (Version 5.02).
Results
Pharmacology
M2698 was potent towards p70S6K, Akt1 and Akt3, with an IC50 of 1 nM required to equally inhibit each of the three proteins. An acceptable level of selectivity was observed against a panel of 264 kinases, with only 6 kinases (MSK1, MSK2, PKG1a, PKG1b, PKA, and PrKX) inhibited by M2698 at an IC50 within 10 times that of p70S6K. In the human breast cancer-derived cell line, MDA-MB-468, M2698 inhibited phosphorylation of p70S6K1 and Akt substrates, S6 (IC50=11 nM) and GSK3β (IC50=17 nM), respectively, in a dose-dependent manner. Additional in vitro studies showed that following 24-hour incubation with M2698, levels of pS6 were reduced in the HCC1419 and MDA-MB-453 cell lines compared with DMSO control, confirming that p70S6K activity was inhibited by M2698. Although in both cells lines the presence of pAkt following M2698 incubation suggests that Akt was activated, phosphorylation of the pAkt substrate, PRAS40 was not increased compared with control, indicating that pAkt activity and the compensatory feedback loop were subsequently inhibited by M2698. This observation is supported by the reduction in p-GSK3α/β with M2698 versus control in MDA-MB-453 cells (Figure 1).
Figure 1.
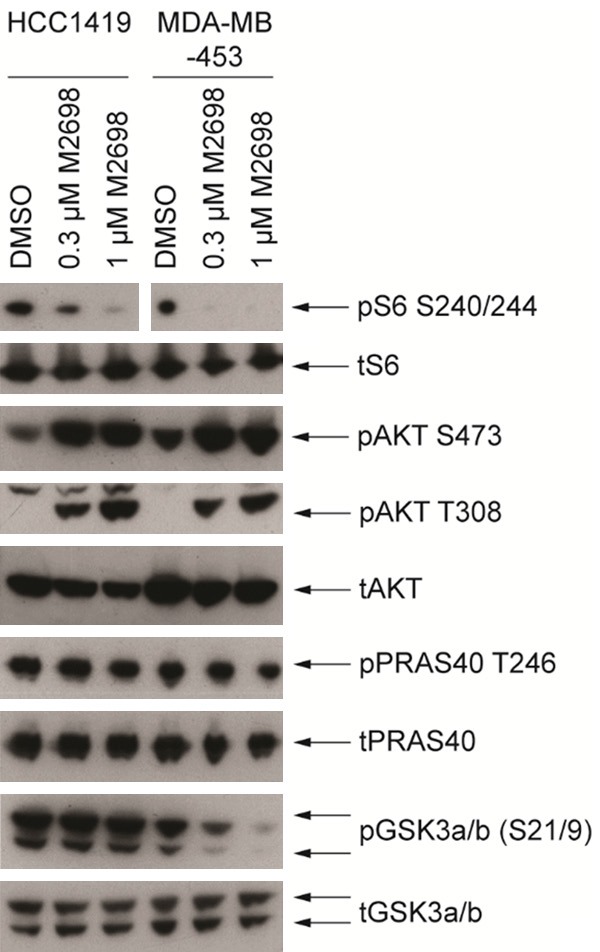
M2698 inhibited p70S6K activity, induced feedback loop phosphorylation on Akt and suppressed Akt activity in breast cancer cell lines. HCC1419 and MDA-MB-453 cells were treated with 0.3 µM or 1 µM M2698 for 24 h. PI3K pathway modulation and feedback loop induction were monitored by western blot analysis of cell lysates. DMSO, dimethysulfoxide; WCE, whole cell extract.
Protein binding, exposure in brain and predicted human pharmacokinetics
The free fraction of M2698 was 2.2% and 4.9% in human and rat plasma, respectively, with no indication of concentration dependency. This compared with 0.5% of M2698 that remained unbound in rat brain. The mean (n=2) total concentration of M2698 after 16-hour infusion in rats was 1750 ng/g and 175 ng/mL in brain and plasma, respectively. The Kp uu (ratio of free M2698 concentration in brain and plasma) was calculated to be 1.0 in rat, showing an equal distribution of M2698 between plasma and brain.
An allometric scaling approach adjusting for protein binding and in vitro clearance (using data from mouse, rat, dog and monkey; data not shown) predicted human clearance to be 0.17 L/hour/kg. Based on this predicted clearance and a predicted Vss (average of three prediction methods) of 5 L/kg, a plasma half-life for M2698 in humans of 20 hours can be expected. Bioavailability in humans was estimated to be >70%.
Pharmacokinetics and pharmacodynamics
In the MDA-MB-453 xenografted animals treated daily with M2698 at 20 mg/kg for 4 days, a tumor:plasma exposure ratio of 12:1 over 24 hours was observed for M2698 (Figure 2A). Compared with that of vehicle-treated mice, S6 phosphorylation in the tumors of mice treated with M2698 was inhibited by more than 90%, indicating a significant reduction in p70S6k activity (P<0.05). Treatment with M2698 led to increased levels of pAkt in tumor tissue compared with tumors from vehicle-treated animals, suggesting activation of Akt following p70S6k inhibition; however, no changes in phosphorylation of the Akt substrate, PRAS40, were observed, suggesting that Akt activity was subsequently blocked by M2698 (Figure 2B). The dose-response relationships between S6 phosphorylation in tumors and M2698 concentrations in plasma of treated mice bearing MDA-MB-453 tumors are shown in Figure 2C. The PK/PD relationships were characterized using an indirect response model. M2698 inhibited the phosphorylation of S6 in a dose-proportional manner over time after a single administration or daily treatments over 7 days. No PK-independent accumulation or tolerance was observed with multiple administrations. In vivo inhibition of S6 phosphorylation had an estimated IC50 value of 137 ng/mL. Correcting for mouse plasma protein binding, the unbound in vivo IC50 value was estimated to be 15 nM.
Figure 2.

Distribution of M2698 in plasma and tumors, and targeted inhibition of p70S6K. A. Mice bearing subcutaneous MDA-MB-453 human xenograft tumors (n=3 per time point) were treated with vehicle or 20 mg/kg M2698 qd po for 4 days and plasma and tumors were harvested at 4, 8 and 24 h after the final administration. Plasma and tumor concentrations of M2698 were assessed by LC-MS/MS at each time point. B. Mice treatment and sample collection was the same as described for A. Phosphorylated and total S6 (S240/244), PRAS40, and Akt (T308) were assessed from tumor protein lysates by western blot analysis at each time point. C. Mice bearing MDA-MB-453 human breast cancer xenograft tumors (n=4 per time point) were treated with vehicle or M2698 1, 5, 10, or 20 mg/kg qd po for 7 days then euthanized at 1, 4, 8, 24, 48 and 72 h after the final administration (4 h post-treatment for vehicle-treated animals). M2698 concentrations were assessed by LC-MS/MS and the proportion of phosphorylated:total S6 (pS6/tS6) in tumor and normal brain (expressed as a percentage of control) was determined by western blot analysis at each time point. The PK/PD relationships between S6 phosphorylation in tumors and concentrations of unbound M2698 in plasma were characterized using an indirect response model (Equation 1), which generated an unbound IC50 of 15 nM. h, hours; qd, once daily.
In the mouse orthotopic U251 glioblastoma model, concentrations of M2698 were 5-fold higher in the brain compared with plasma, confirming that M2698 passed through the BBB, and concentrations of M2698 were 4-fold higher in brain tumor than in the normal brain (Figure 3). Reduced levels of pS6/tS6 were observed in the brain and brain tumors of mice treated with M2698 from 4 hours post-treatment, confirming the inhibition of the p70S6k by the compound. This inhibition was more pronounced in tumors than in brain, especially 24 hours after treatment.
Figure 3.
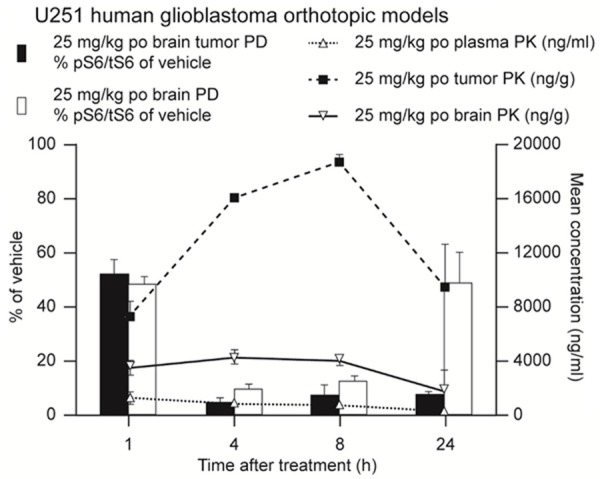
M2698 concentrations were higher in the brain than in the plasma of orthotopic U251 glioblastoma models. Mice bearing U251 human glioblastoma orthotopic tumors (n=6 per time point) were treated with a single oral administration of either vehicle or M2698 in vehicle at 25 mg/kg. Plasma, tumor and normal brain samples were obtained 1, 4, 8 and 24-h post-treatment. M2698 concentrations were assessed by LC-MS/MS and the proportion of phosphorylated: total S6 (pS6/tS6) in tumor and normal brain (expressed as a percentage of vehicle) was determined by western blot analysis at each time point. h, hours; PD, pharmacodynamics; PK, pharmacokinetics; po, oral administration.
Anti-tumor activity of M2698
In vitro M2698 inhibited proliferation in a dose-dependent manner in all 81 cell lines tested, which corresponded to multiple different tumor types, including nine that were derived from breast tumors (Figure 4). Proliferation of all nine breast cancer cell lines was inhibited by M2698, with IC50s <2 µM (range: 0.3-1.1 µM) in 6 out of 9 lines, including 2 which harbored PI3K mutations and 2 which harbored PTEN mutations. In an additional panel of breast cancer cell lines, M2698 was able to inhibit proliferation of the cells regardless of ER, PR or Her2 status (Table 1).
Figure 4.
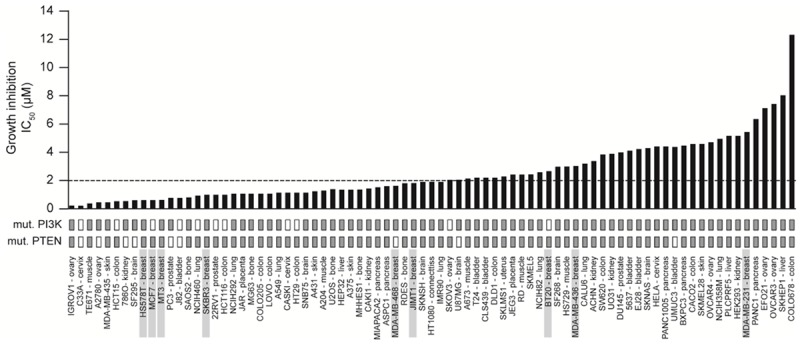
M2698 inhibited in vitro cell growth in 81 distinct human cancer cell lines. A panel of human cancer cell lines were treated with a range of concentrations of M2698 for 72 h. The sulforhodamine B (SRB) assay was used to determine cell growth inhibition. IC50 values shown are the mean of triplicate samples.
Table 1.
M2698 inhibited proliferation of breast cancer cell lines of different sub-types in vitro
| Cell line | Gene cluster | ER | PR | Her2 | IC50 (µM) |
|---|---|---|---|---|---|
| BT-474 | Lu | + | + | + | 0.02 |
| T74 D | Lu | + | + | - | 0.03 |
| ZR-75-1 | Lu | + | - | - | 0.03 |
| MDA-MB-453 | Lu | - | - | - | 0.04 |
| BT-549 | BaB | - | - | - | 0.05 |
| HCC1419 | Lu | - | - | + | 0.20 |
| MCF-7 | Lu | + | + | - | 0.52 |
| Hs578T | BaB | - | - | - | 0.62 |
| HCC1187 | BaA | - | - | - | 0.70 |
| JIMT-1 | n/a | - | - | + | 0.80 |
| SK-BR-3 | Lu | - | - | + | 1.06 |
| MDA-MB-157 | BaB | - | - | - | 1.20 |
| MDA-MB-468 | BaA | - | - | - | 2.01 |
| BT-20 | BaB | - | - | + | 2.10 |
| MDA-MB-436 | BaB | - | - | - | 2.56 |
| HCC1937 | BaA | - | - | - | 3.80 |
| MDA-MB-231 | BaB | - | - | - | 5.25 |
| HCC1143 | BaA | - | - | - | 8.50 |
BaA, Basal A; BaB, Basal B; Ca, carcinoma; ER, estrogen receptor; Lu, luminal; PR, progesterone receptor.
In vivo Treatment of mice bearing MDA-MB-468 tumors (TNBC, PTEN loss of function) resulted in dose-dependent inhibition of tumor growth (P<0.05; Figure 5A) with the highest dose of 30 mg/kg resulting in tumor regression. Similarly, treatment with M2698 at both 10 and 20 mg/kg significantly inhibited tumor growth compared with vehicle (P<0.05) in two breast cancer xenograft models, MDA-MB-453 (Her2-expressing) and JIMT-1 (Her2+), both of which harbor PI3K-activating mutations and demonstrate increased signaling through the PAM pathway [21] (Figure 5B, 5C).
Figure 5.
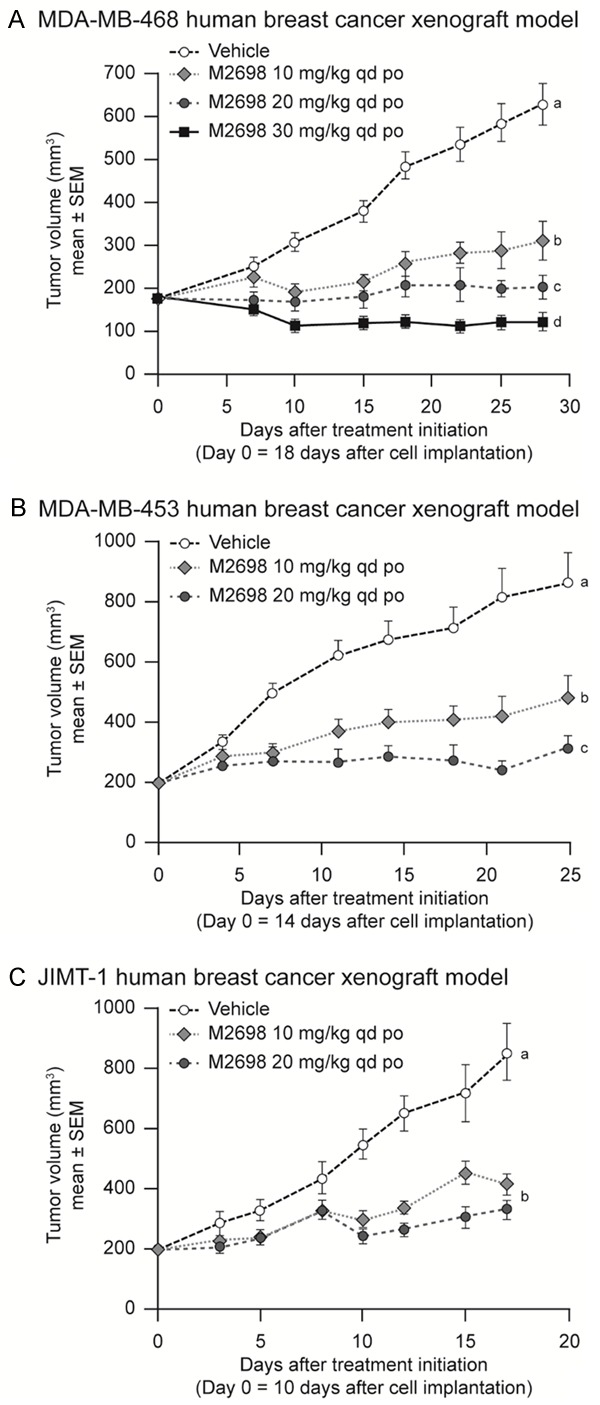
M2698 inhibited growth of human breast cancer xenografted tumors. A. Mice bearing MDA-MB-468 human TNBC xenograft tumors (n=8) were treated with vehicle or M2698 10, 20 and 30 mg/kg qd po. B. Mice bearing MDA-MB-453 Her2-expressing human breast xenograft tumors (n=10) were treated with vehicle or M2698 10 or 20 mg/kg qd po. C. Mice bearing JIMT-1 Her2+ human breast xenograft tumors (n=10) were treated with vehicle or M2698 10 or 20 mg/kg qd po. a,b,cTreatment means with the same superscript are not significantly different (P>0.05). po, oral administration; qd, once daily; SEM, standard error of the mean. IC50, concentration causing 50% inhibition; mut, mutation.
The efficacy of M2698 in the treatment of brain tumors was assessed in the orthotopic U251 model of human glioblastoma. Daily treatment of animals with M2698 at 25 mg/kg resulted in significant inhibition of tumor growth at Day 31 as measured by MRI (P<0.05; Figure 6A). Median survival was significantly increased in mice treated with M2698 compared to those treated with vehicle, from Day 18 to Day 50 (P<0.05; Figure 6B).
Figure 6.
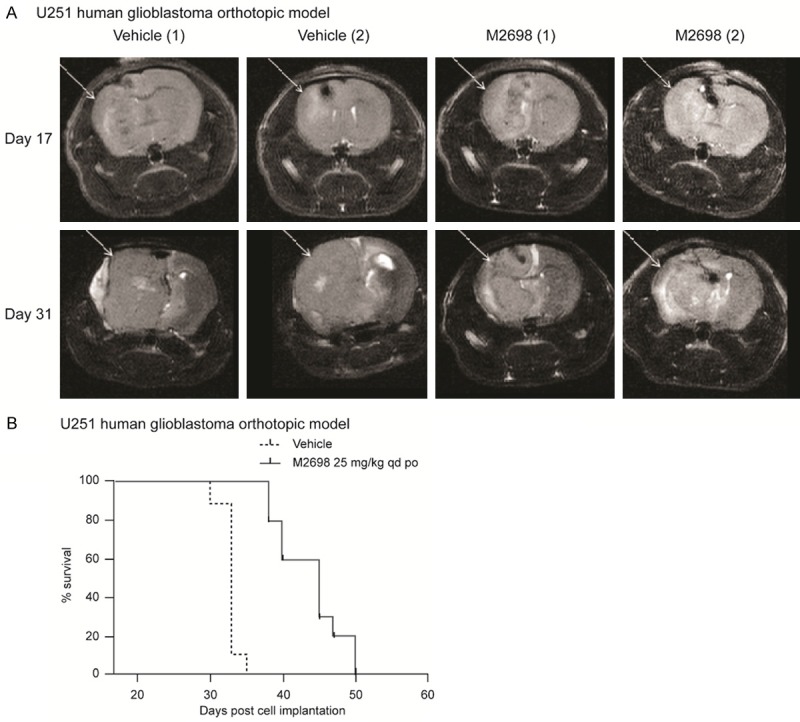
M2698 reduced tumor burden and prolonged survival in the U251 human glioblastoma xenograft mouse model. Mice bearing U251 orthotopic tumors were treated with vehicle (n=9) or M2698 25 mg/kg qd po (n=10) from Day 18 (when mean tumor mass was 6 mg) until Day 50 (end of study). A. MRI was performed on Days 17 and 31. Representative images are shown. B. Group survival curves of tumor-bearing animals treated with vehicle or M2698 25 mg/kg. qd po. po, oral administration; qd, once daily.
Discussion
With these findings we present M2698 as a potent and selective, orally bioavailable dual inhibitor of p70S6K and Akt. M2698 effectively and dose-dependently inhibited the activity of these target proteins both in vitro and in vivo in exposure-relevant PK/PD relationships and with significant distribution in vivo from plasma to tumor and brain.
Importantly, the Akt compensatory feedback loop that is induced through one mechanism of M2698, p70S6K inhibition, is effectively blocked by the other, i.e. inhibition of Akt, providing an advantage to M2698 compared with other PAM pathway inhibitors. Indeed, this blockade of the compensatory Akt feedback loop is essential for the achievement of sustained PAM pathway inhibition because, if left unchecked, activation of the feedback loop is likely to result in increased pathway signaling, as is seen with mTORC rapalogs [9]. By overcoming the negative consequences of the Akt feedback loop, treatment with M2698 has the potential to provide more durable therapeutic responses compared with agents that inhibit single PAM pathway components such as mTORC. Although its PD properties need to be confirmed in humans, this unique dual inhibitory mechanism of action therefore makes M2698 an attractive prospect for the treatment of malignancies with dysregulated PAM signaling.
Examination of the PK properties of M2698 revealed that it is orally bioavailable, with no PK-dependent accumulation observed with multiple administrations. The bioavailability of M2698 in humans is predicted to be high, based on findings in animals. The half-life of M2698 was predicted to be 20 hours in humans, which is comparable to approved PAM inhibitors everolimus (30 hours) [22] and temsirolimus (18 hours) [23]. Based on xenograft models, M2698 appears to distribute well from plasma to tumor, even in the brain, resulting in significant inhibition of the target as measured by PD analysis and significant efficacy in the brain.
In vitro, M2698 reduced proliferation of all tumor cell lines tested in a dose-dependent manner, several of which carried PI3K mutations and/or PTEN mutations. These included nine breast cancer-derived cell lines, two of which harbored PI3K mutations and two of which harbored PTEN mutations. It is worth noting that a response to M2698 was also observed in cell lines that did not carry PI3K3CA or PTEN mutations. Since the mechanism of action of M2698 suggests that upregulation of PAM pathway signaling is a prerequisite for the compound to be active, it is possible that these cells harbored other PAM pathway alterations that were not measured, such as AKT amplification. Additional characterization of the cell lines would be required to confirm this. However, in both preclinical and clinical studies, anti-tumor activity by compounds targeting various components of the PAM pathway in tumors with dysregulated PAM signaling has been documented [24-26] and the activity of M2698 in cell lines and animal models with PI3K3CA and PTEN mutations is consistent with these findings.
M2698 inhibited cellular proliferation in an additional panel of breast cancer cell lines of varying subtypes, regardless of ER, PR or Her2 expression. Although breast cancer is the most commonly occurring malignancy in women, there is still an urgent need for new treatments, particularly in TNBC and Her2+ subtypes. No targeted therapies are currently available for patients with TNBC, with chemotherapy remaining the standard of care treatment. As such, the prognosis for these patients remains poor [27,28]. Similarly, although clinical outcomes for patients with Her2+ breast cancer have improved with the availability of the biological agent, trastuzumab, many patients with metastatic disease who respond initially to this agent ultimately progress while still receiving treatment [29]. Such trastuzumab resistance in Her2+ breast cancer is often associated with dysregulated PAM signaling due to loss of PTEN, or PIK3CA mutation [4-6]. In this study, M2698 demonstrated significant anti-tumor activity in xenograft mouse models derived from both a TNBC cell line (MDA-MB-468) and two trastuzumab-resistant [21], Her2+ breast cancer cell lines (MDA-MB-453 and JIMT-1). In the TNBC model, daily oral M2698 treatment at the highest dose tested (30 mg/kg) resulted in tumor regression, and treatment at lower doses (10 and 20 mg/kg) resulted in tumor stasis and growth inhibition. M2698 inhibited tumor growth in the Her2+ models (MDA-MB-453 and JIMT-1) at both doses tested (10 and 20 mg/kg).
The capacity of M2698 to cross the BBB is of particular relevance to human breast cancer because of its high rates of secondary central nervous system (CNS) metastases [30]. Her2 expression in particular is considered to be a risk factor for CNS metastasis in breast cancer, and the prevalence of this manifestation is further exacerbated by the widespread use of trastuzumab, which cannot penetrate the BBB [31,32]. There is, therefore, a need for new therapies that are able to cross the BBB. Indeed, this requirement is not exclusive to Her2+ breast cancer; the ability to pass the BBB is a prerequisite for compounds targeting any type of CNS malignancy, the majority of which are notoriously difficult to treat due to the lack of agents which have this property [30]. Glioblastoma is one such CNS tumor for which treatment options are limited, and prognosis is extremely poor (5-year survival rate is <10%); yet glioblastoma is the most common malignant primary brain cancer in adults [30,33,34]. As with trastuzumab-resistant Her2+ breast cancer, there is also a strong association between resistance to targeted therapies and PAM pathway dysregulation in glioblastoma [3-6]. A PAM pathway inhibitor that crosses the BBB, such as M2698, may therefore represent a rational approach to treating these types of cancer.
To investigate the anti-tumor effects of M2698 on CNS tumors, the compound was administered to athymic mice bearing orthotopically implanted U251 glioblastoma brain tumors, resulting in significant inhibition of tumor growth and prolonged median survival. These findings provide preclinical evidence that may translate into significant anti-tumor activity against CNS tumors in the clinic.
In conclusion, M2698 is an orally bioavailable, potent, selective dual inhibitor of p70S6K and Akt that effectively blocks PAM pathway signaling while at the same time overcoming the negative consequences of the Akt feedback loop. M2698 demonstrated anti-tumor activity in xenograft models of breast cancer and crossed the BBB in an orthotopically implanted model of human glioblastoma, where it simultaneously reduced tumor growth and prolonged survival. These studies provide a strong rationale for the further exploration of M2698 in cancers with frequent PAM pathway genomic alterations, including breast cancer, and malignancies associated with CNS tumors. As such, a phase I dose escalation trial in patients with advanced malignancies (ClinicalTrials.gov identifier: NCT01971515) is currently ongoing, the findings of which will inform further phase Ib studies.
Acknowledgements
Studies were funded by EMD Serono and Merck KGaA. U251 orthotopic studies were performed at Molecular Imaging, Inc., Ann Arbor, MI. Medical writing assistance was provided by Emily Heath, Bioscript Science, Macclesfield, UK, and funded by Merck KGaA, Darmstadt, Germany.
Disclosure of conflict of interest
AM, EW and PS are former employees of EMD Serono. HT, XL, AC and BH are employees of EMD Serono.
References
- 1.Pal I, Mandal M. PI3K and Akt as molecular targets for cancer therapy: current clinical outcomes. Acta pharmacologica Sinica. 2012;33:1441–58. doi: 10.1038/aps.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, Lu KV, Yoshimoto K, Huang JH, Chute DJ, Riggs BL, Horvath S, Liau LM, Cavenee WK, Rao PN, Beroukhim R, Peck TC, Lee JC, Sellers WR, Stokoe D, Prados M, Cloughesy TF, Sawyers CL, Mischel PS. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353:2012–24. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
- 4.Esteva FJ, Guo H, Zhang S, Santa-Maria C, Stone S, Lanchbury JS, Sahin AA, Hortobagyi GN, Yu D. PTEN, PIK3CA, p-AKT, and p-p70S6K status: association with trastuzumab response and survival in patients with HER2-positive metastatic breast cancer. Am J Pathol. 2010;177:1647–56. doi: 10.2353/ajpath.2010.090885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, Klos KS, Li P, Monia BP, Nguyen NT, Hortobagyi GN, Hung MC, Yu D. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6:117–27. doi: 10.1016/j.ccr.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 6.Chandarlapaty S, Sakr RA, Giri D, Patil S, Heguy A, Morrow M, Modi S, Norton L, Rosen N, Hudis C, King TA. Frequent mutational activation of the PI3K-AKT pathway in trastuzumab-resistant breast cancer. Clin Cancer Res. 2012;18:6784–91. doi: 10.1158/1078-0432.CCR-12-1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Administration UFaD. Everolimus 2012 [03 February 2016]. Available from: http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm313008.htm.
- 8.US Food and Drug Administration. Temsirolimus 2007 [03 February 2016]. Available from: http://www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDER/ucm129247.htm.
- 9.O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, Baselga J, Rosen N. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–8. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang S, Bjornsti MA, Houghton PJ. Rapamycins: mechanism of action and cellular resistance. Cancer Biol Ther. 2003;2:222–32. doi: 10.4161/cbt.2.3.360. [DOI] [PubMed] [Google Scholar]
- 11.Barlund M, Monni O, Kononen J, Cornelison R, Torhorst J, Sauter G, Kallioniemi OP, Kallioniemi A. Multiple genes at 17q23 undergo amplification and overexpression in breast cancer. Cancer Res. 2000;60:5340–4. [PubMed] [Google Scholar]
- 12.Surace EI, Lusis E, Haipek CA, Gutmann DH. Functional significance of S6K overexpression in meningioma progression. Ann Neurol. 2004;56:295–8. doi: 10.1002/ana.20201. [DOI] [PubMed] [Google Scholar]
- 13.Masters JR, Thomson JA, Daly-Burns B, Reid YA, Dirks WG, Packer P, Toji LH, Ohno T, Tanabe H, Arlett CF, Kelland LR, Harrison M, Virmani A, Ward TH, Ayres KL, Debenham PG. Short tandem repeat profiling provides an international reference standard for human cell lines. Proc Natl Acad Sci U S A. 2001;98:8012–7. doi: 10.1073/pnas.121616198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, Speed T, Spellman PT, DeVries S, Lapuk A, Wang NJ, Kuo WL, Stilwell JL, Pinkel D, Albertson DG, Waldman FM, McCormick F, Dickson RB, Johnson MD, Lippman M, Ethier S, Gazdar A, Gray JW. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–27. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hurvitz SA, Kalous O, Conklin D, Desai AJ, Dering J, Anderson L, O’Brien NA, Kolarova T, Finn RS, Linnartz R, Chen D, Slamon DJ. In vitro activity of the mTOR inhibitor everolimus, in a large panel of breast cancer cell lines and analysis for predictors of response. Breast Cancer Res Treat. 2015;149:669–80. doi: 10.1007/s10549-015-3282-x. [DOI] [PubMed] [Google Scholar]
- 16.Gao Y, Davies SP, Augustin M, Woodward A, Patel UA, Kovelman R, Harvey KJ. A broad activity screen in support of a chemogenomic map for kinase signalling research and drug discovery. Biochem J. 2013;451:313–28. doi: 10.1042/BJ20121418. [DOI] [PubMed] [Google Scholar]
- 17.Kalvass JC, Maurer TS. Influence of nonspecific brain and plasma binding on CNS exposure: implications for rational drug discovery. Biopharm Drug Dispos. 2002;23:327–38. doi: 10.1002/bdd.325. [DOI] [PubMed] [Google Scholar]
- 18.Lave T, Coassolo P, Reigner B. Prediction of hepatic metabolic clearance based on interspecies allometric scaling techniques and in vitro-in vivo correlations. Clin Pharmacokinet. 1999;36:211–31. doi: 10.2165/00003088-199936030-00003. [DOI] [PubMed] [Google Scholar]
- 19.Oie S, Tozer TN. Effect of altered plasma protein binding on apparent volume of distribution. J Pharm Sci. 1979;68:1203–5. doi: 10.1002/jps.2600680948. [DOI] [PubMed] [Google Scholar]
- 20.DeLean A, Munson PJ, Rodbard D. Simultaneous analysis of families of sigmoidal curves: application to bioassay, radioligand assay, and physiological dose-response curves. Am J Physiol. 1978;235:E97–102. doi: 10.1152/ajpendo.1978.235.2.E97. [DOI] [PubMed] [Google Scholar]
- 21.O’Brien NA, Browne BC, Chow L, Wang Y, Ginther C, Arboleda J, Duffy MJ, Crown J, O’Donovan N, Slamon DJ. Activated phosphoinositide 3-kinase/AKT signaling confers resistance to trastuzumab but not lapatinib. Mol Cancer Ther. 2010;9:1489–502. doi: 10.1158/1535-7163.MCT-09-1171. [DOI] [PubMed] [Google Scholar]
- 22.O’Donnell A, Faivre S, Burris HA 3rd, Rea D, Papadimitrakopoulou V, Shand N, Lane HA, Hazell K, Zoellner U, Kovarik JM, Brock C, Jones S, Raymond E, Judson I. Phase I pharmacokinetic and pharmacodynamic study of the oral mammalian target of rapamycin inhibitor everolimus in patients with advanced solid tumors. J. Clin. Oncol. 2008;26:1588–95. doi: 10.1200/JCO.2007.14.0988. [DOI] [PubMed] [Google Scholar]
- 23. Torisel: Summary of product characteristics 2012. Available from: http://www.google.co.uk/url?sa=t&rct=j&q=&esrc=s&source=web&cd=1&ved=0ahUKEwiTqZSkq9bKAhXE0hoKHYbSBIQQFggiMAA&url=http%3A%2F%2Fwww.ema.europa.eu%2Fdocs%2Fen_GB%2Fdocument_library%2FEPAR_-_Product_Information%2Fhuman%2F000799%2FWC500039912.pdf&usg=AFQjCNEv-rUoT-FpGJBJ4oSiObDWXu7Etw&sig2=c2hz_88HdbS2zwdg0ht_fQ&bvm=bv.113034660,d.d2s.
- 24.Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR signaling in cancer. Front Oncol. 2014;4:64. doi: 10.3389/fonc.2014.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hudes G, Carducci M, Tomczak P, Dutcher J, Figlin R, Kapoor A, Staroslawska E, Sosman J, McDermott D, Bodrogi I, Kovacevic Z, Lesovoy V, Schmidt-Wolf IG, Barbarash O, Gokmen E, O’Toole T, Lustgarten S, Moore L, Motzer RJ, Global AT. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356:2271–81. doi: 10.1056/NEJMoa066838. [DOI] [PubMed] [Google Scholar]
- 26.Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, Grünwald V, Thompson JA, Figlin RA, Hollaender N, Urbanowitz G, Berg WJ, Kay A, Lebwohl D, Ravaud A RECORD-1 Study Group. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372:449–56. doi: 10.1016/S0140-6736(08)61039-9. [DOI] [PubMed] [Google Scholar]
- 27.Crown J, O’Shaughnessy J, Gullo G. Emerging targeted therapies in triple-negative breast cancer. Ann Oncol. 2012;23(Suppl 6):vi56–65. doi: 10.1093/annonc/mds196. [DOI] [PubMed] [Google Scholar]
- 28.Wahba HA, El-Hadaad HA. Current approaches in treatment of triple-negative breast cancer. Cancer Biol Med. 2015;12:106–16. doi: 10.7497/j.issn.2095-3941.2015.0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nahta R, Esteva FJ. HER2 therapy: molecular mechanisms of trastuzumab resistance. Breast Cancer Res. 2006;8:215. doi: 10.1186/bcr1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.El-Habashy SE, Nazief AM, Adkins CE, Wen MM, El-Kamel AH, Hamdan AM, Hanafy AS, Terrell TO, Mohammad AS, Lockman PR, Nounou MI. Novel treatment strategies for brain tumors and metastases. Pharm Pat Anal. 2014;3:279–96. doi: 10.4155/ppa.14.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stemmler J SM, Willems A, Bernhard H, Harbeck N, Heinemann V. Brain metastases in HER2-overexpressing metastatic breast cancer: comparative analysis of trastuzumab levels in serum and cerebrospinal fluid [abstract 1525] . J. Clin. Oncol. 2006;24:64s. [Google Scholar]
- 32.Lin NU, Winer EP. Brain metastases: the HER2 paradigm. Clin Cancer Res. 2007;13:1648–55. doi: 10.1158/1078-0432.CCR-06-2478. [DOI] [PubMed] [Google Scholar]
- 33.Ostrom QT, Gittleman H, Liao P, Rouse C, Chen Y, Dowling J, Wolinsky Y, Kruchko C, Barnholtz-Sloan J. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007-2011. Neuro Oncol. 2014;16(Suppl 4):iv1–63. doi: 10.1093/neuonc/nou223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]