

Abstract
In response to DNA damage lesions due to cellular stress, DNA damage response (DDR) pathways are activated to promote cell survival and genetic stability or unrepaired lesion-induced cell death. Current cancer treatments predominantly utilize DNA damaging agents, such as irradiation and chemotherapy drugs, to inhibit cancer cell proliferation and induce cell death through the activation of DDR. However, a portion of cancer patients is reported to develop therapeutic resistance to these DDR-inducing agents. One significant resistance mechanism in cancer cells is oncogenic kinase overexpression, which promotes cell survival by enhancing DNA damage repair pathways and evading cell cycle arrest. Among the oncogenic kinases, overexpression of receptor tyrosine kinases (RTKs) is reported in many of solid tumors, and numerous clinical trials targeting RTKs are currently in progress. As the emerging trend in cancer treatment combines DNA damaging agents and RTK inhibitors, it is important to understand the substrates of RTKs relative to the DDR pathways. In addition, alteration of RTK expression and their phosphorylated substrates can serve as biomarkers to stratify patients for combination therapies. In this review, we summarize the deleterious effects of RTKs on the DDR pathways and the emerging biomarkers for personalized therapy.
Keywords: Receptor tyrosine kinase, DNA damage, DNA repair, epidermal growth factor receptor, signaling
Introduction
DNA damage stimuli can be divided into two classes, endogenous and environmental, based on the site of the stimulus’ origin [1]. Endogenous DNA damage generates chemical changes in DNA structure leading to mutagenic events such as deamination of bases resulting from hydrolytic and oxidative events inside the cell. Environmental DNA damage can result from either physical or chemical agents outside the cells [1]. The incidence of DNA damage occurs frequently in normal cells. It is estimated that the error rate of the DNA replication machinery is at least 10-8 in Escherichia coli and human [2,3]. In addition to replication errors, DNA breaks mainly caused by reactive oxygen species (ROS) are estimated to be 105 events per day [4,5]. Thus, DNA damage response (DDR) is required to correct mistakes in DNA and is also responsible for eliminating cells with irreparable deleterious damage.
DNA damage and DDR are highly related to the formation and treatment of cancer. During carcinogenesis, the inefficiency and infidelity of the DDR pathway are the main causes of oncogenic events, such as DNA mutations, translocations, and epigenetic modifications, which correlate DDR to cancer risks [1,6-9]. In cancer treatment, both radiotherapy and chemotherapy utilize DNA damaging agents that eliminate cancer cells by inducing DDR. Capitalizing on the deficiency of DDR in cancerous cells, the treatment of cancer with DNA damaging agents is an effective means of inducing massive DNA lesions and programmed cell death in the cells unable to resolve the damage. However, resistance to these types of treatment is reported in patients, and the crosstalk between DDR and altered receptor tyrosine kinase (RTK) signaling pathways in solid tumors is thought to be an important contributor to the development of chemotherapy resistance [10-13]. Overexpression of RTKs also contributes to tumor progression through promotion of cell survival, metastasis and stimulation of angiogenesis [14]. While many inhibitors targeting RTKs are already in clinical trials or clinical use [14], it is important to understand how RTKs promote cell survival upon DNA damage to develop combination therapies to enhance treatment efficacy.
DNA damage response
Once the DNA damage sensor protein machinery detects DNA damage lesions, it recruits mediators and numerous transducer and effector proteins to ensure that the transcription and translation processes are paused by cell cycle arrest and to initiate DNA damage repair or apoptosis (Figure 1 and Table 1) [1]. The main mediators in DDR pathways are members of the phosphatidylinositol 3-kinase-like protein kinases family, including ataxia-telangiectasia mutated (ATM), ATM and Rad 3-related (ATR), and DNA-dependent protein kinase (DNA-PK). When DNA damage lesions are recognized by a sensor protein, these mediators are recruited to the damage site and phosphorylate downstream proteins that are involved in all aspects of DDR. In addition to these mediators, poly (ADP-ribose) polymerases (PARPs), a large enzyme family with multiple functions, also play important roles in DDR [15,16]. PARP1 and PARP2 are activated by DNA single-strand break (SSB) and DNA double-strand breaks (DSB) and can poly(ADP-ribos)ylate (PARylate) different substrates under different genotoxic stress to further elevate DDR response [17].
Figure 1.
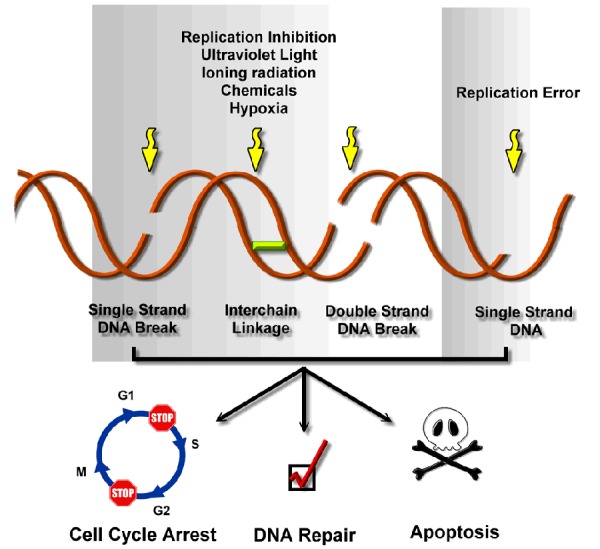
DNA damage reagents and DNA damage response. DNA is vulnerable to both exogenous and endogenous DNA damage reagents, including replication error, replication inhibition, ultraviolet (UV) light and cancer treatments such us irradiation therapy and chemotherapy. Exposure to these DNA damage reagents leads to DNA damage including DNA single-strand break (SSB), DNA interstrand cross-linking (ICL), DNA double-strand break (DSB) as well as single-strand DNA lesion (ssDNA). The DNA damage lesions then trigger the signal cascade which results in DDR primarily through delayed cell cycle from G1 to S phase (G1/S arrest) or from G2 to M phase (G2/M arrest) and as well as triggering DNA damage repair pathways. After successfully repaired, the cell cycle arrest is released and the cells will survive. However, the severe DNA damage adducts or DNA damage repair failure will eventually leads to apoptotic cell death.
Table 1.
Sources of DNA damage and major repair pathways
| Sources of damage | Spontaneous reactions | X-ray | UV | Replication inhibitor | Replication errors | ||||
| AAs | ROS | Aromatic groups | UV | ||||||
| AAs | Hydrocarbons | IR | |||||||
| 5-FU | Platin | ||||||||
| ROS | |||||||||
| Hypoxia | |||||||||
| DNA damage types | O6mG | 8-oxoG | CPD | DNA crosslink | Base mismatch | ||||
| Pyrimidine dimer | AP site | 6,4-P.P. | dsDNA break | Insertion | |||||
| Uracil | DNA crosslink | Deletion | |||||||
|
| |||||||||
| Repair pathway | Direct reverse | BER | NER | HR | NHEJ | MMR | |||
|
|
|
|
|||||||
| GG-NER | TC-NER | ATR | ATM | C-NHEJ | A-NHEJ | ||||
|
| |||||||||
| Sensor/Initiator | MGMT | OGG1 | XPE | RNA Pol I/II | ATRIP | MRN | Ku70 | PARP | MSH2 |
| AGT | PARP | XPC | CSA | PARP | Ku80 | MSH3 | |||
| HR23B | CSB | MLH1 | |||||||
| PARP | |||||||||
| Transducer/Effector | XRCC1 | RPA | ATR | ATM | DNA-PK | Fan1 | |||
| APE1/2 | XPA | Chk1 | Chk2 | WRN | PNKP | ||||
| XPC | Artemis | XRCC1 | |||||||
| TFIIH | XRCC4 | ||||||||
| XPB | XLF | ||||||||
| XPD | |||||||||
| XPG | |||||||||
| XPF | |||||||||
| ERCC1 | |||||||||
| MRN | |||||||||
| Rad51 | |||||||||
| BRCA 1/2 | |||||||||
| XRCC 2/3 | |||||||||
| P53 | |||||||||
| Rad52 | |||||||||
| Rad54 | |||||||||
| Elongation/Ligase | PCNA | PCNA | PCNA | DNA Polβ | Lig III | RFC | |||
| DNA Polβ | RFC | Lig I | Lig IV | PCNA | |||||
| Lig III | FEN1 | DNA Pol δ/ε | EXO1 | ||||||
| Lig I | DNA Pol δ | ||||||||
| DNA Pol δ/ε | Lig I/IV | ||||||||
Abbreviations: AAs, alkylating agents; ROS, reactive oxygen species; 5-FU, 5-Fluorouracil; IR, ioninzing radiation; O6mG, O6-methylguanine; 8-oxoG, 8-oxoguanine; CPD, cyclo-butane pyrimidine dimer; 6,4-P.P, 6-4 photoproduct.
Cell cycle arrest
The cell cycle is subdivided into G1, S, G2, and M phase. In brief, cells increase in size and prepare for DNA synthesis during G1 phase and undergo DNA replication during S phase. Then, cells continue to grow and prepare for mitosis in G2 phase before dividing in M phase. To ensure genomic stability, eukaryotic cells develop cell cycle checkpoints that pause cell division in response to environmental stress, DNA damage, and improper DNA replication [18]; this process is referred as cell cycle arrest. In mammalian cells, there are two major signaling pathways that control cell cycle arrest in response to DNA damaging stress: the ATM pathway, which is responsible for DSB throughout the cell division cycle and the ATR pathway, which is responsible for both DSB as well as replication forks [18,19].
DNA damage repair and therapeutic DNA damaging agents
In 1974, researchers had already realized that the integrity of DNA is vulnerable and that the repair mechanisms are crucial to maintain genomic stability. Dr. Francis Crick stated in The double helix: a personal view that “… one could hardly discuss mutation without considering repair at the same time” [20]. The DNA damage repair pathways are composed of base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), single-strand annealing (SSA), homologous recombination repair (HR) and non-homologous end joining repair (NHEJ) [1].
Repair of base alternation and small DNA damage adducts
Base alternation and small DNA damage adducts, covalent DNA-chemical binding structures, can be caused by low concentration of reactive oxygen species (ROS) as well as alkylation agents and DNA crosslinking agents. Small DNA damage adducts can be easily repaired in normal cells compared with DSBs, but the failure to repair these adducts’ fidelity may lead to oncogenic mutations. In cancer treatment, low doses of ionizing radiation (IR) and low linear energy transfer γ-radiation [21] can generate low concentrations of ROS whereas a large number of chemotherapeutic drugs are alkylating agents, including nitrogen mustards (mechlorethamine, cyclophosphamide, and ifosfamide), nitrosourease (streptozocin, carmustine and lomustine), alkyl sulfonates (busulfan), triazine (dacarbazine and temozolomide) and ehylenimines (thiotepa and altretamine) [22].
Base excision repair
BER is mainly responsible for small lesions caused by endogenous DNA damage, such as oxidation, hydroxylation, deamination, or methylation, and is considered to be the most frequently used DNA damage repair pathway [1,23]. Abnormal DNA bases are detected and excised by lesion-specific DNA glycosylases, such as OGG1 and MYH, creating apurinic, apyrimidinic, or abasic sites (AP sites) [1]. For AP sites limited to a single base, the short patch BER endonuclease APE1 generates a single nucleotide gap at the AP site and recruits DNA polymerase β as well as XRCC1-DNA ligase to fill the gap. For extensive AP sites (2-10 bases), long patch BER with FEN1 endonuclease and proliferating cell nuclear antigen (PCNA)-DNA polymerase δ/ε complex are used to repair the lesions [1]. Genetic variants of ADPART, XRCC1, APE1 proteins in BER are reported to increase the risk of squamous cell carcinoma [24,25] and bladder cancer [26]. APE1 and XRCC1 polymorphisms have been reported to correlate with gastric cancer [27] and with risk of lung adenocarcinoma [28,29], respectively. The nitrogen (N-) and oxygen (O-) alkylated DNA bases caused by alkylating agents as well as oxidative DNA bases induced by ROS are repaired by BER [30].
PARP participates in many DNA repair pathways including BER, NER, HR and NHEJ [31,32] but predominantly functions in the BER pathway. Although PARP is not essential in the BER pathway, the treatment of PARP inhibitor has successfully converted the base lesion into a SSB [33], which is a more severe type of DNA damage that can be developed into lethal DSB lesions during DNA replication [15,34]. PARP inhibitors, for example, olaparib, can induce synthetic lethality in DSB repair-deficient cancer cells, such as BRCA-mutated cells, and benefit patients with BRCA1/2-mutated breast or ovarian cancer [15,35-37].
Nucleotide excision repair
NER mainly tackles a variety of helix-distorting lesions that impede transcription and replication by interfering with base pairing [1,23]. Global genome NER (GG-NER) repairs helix-distorting lesions and prevents mutagenesis. Transcription-coupled NER (TC-NER) repairs transcription-blocking lesions to prevent perturbed gene transcription. The damage recognition steps are different between these NER mechanisms: in GG-NER, the lesions are detected by XPC/hHR23B and XPE protein complex, whereas the RNA polymerase/CSA/CSB/HMGN1 protein complex is responsible for lesion detection in TC-NER [1]. After lesion recognition, XPA proteins are recruited and bind to DNA around 20 base pair upstream of the DNA damage adduct. The DNA double helix around the DNA damage adduct is then unwound by a multi-protein complex, TFIIH. Single-stranded DNA (ssDNA) resulting from the unwinding process is stabilized by the RPA protein, and the DNA adduct excision steps are completed by the XPF-ERCC1 and XPG proteins. NER repairs DNA lesions caused by various endogenous and environmental DNA damaging agents, including UV irradiation [38], platin-based chemotherapy drugs, e.g., cisplatin and carboplatin [39], and carcinogens, such as benzopyrene [40]. Defects in NER result in diseases, such as xeroderma pigmentosum (XP), Cockayne syndrome, and trichothiodystrophy [1]. Among the NER-related diseases, XP patients, but not Cockayne syndrome or trichothiodystrophy patients, exhibit a higher incidence of skin cancer. For example, XP group A patients, a subpopulation of XP patients, are more prone to basal cell and squamous cell carcinoma, and melanoma [41,42].
Mismatch repair
MMR is designed to resolve mispaired or modified bases as well as insertion or deletion loops. Heterodimers of the MSH2/MSH6 complex recognize mismatched pairs and single-base loops whereas the MSH2/MSH3 complex recognizes insertion/deletion loops. This damage recognition complex then recruits and interacts with MLH1/PMS2 and EXO1 endonuclease to excise the newly synthesized strand after mismatch/loop. DNA is then resynthesized by PCNA, RPA, and DNA polymerase δ/ε complex [1]. Germline mutations in MLH1 and MSH2 have been shown to contribute to hereditary non-polyposis colorectal cancer [43], and defects in MSH6 are known to cause atypical hereditary non-polyposis colorectal cancer. Germline variants in DNA polymerase ε are also associated with MMR-deficient colorectal cancer [44]. MMR deficiency testing can predict the prognosis of colorectal cancer and stratify patients for adjuvant chemotherapy [45].
Repair of DNA double-strand breaks
DSBs are considered to be lethal DNA damage lesions that must be repaired before cell continues to grow and proliferate. Currently, radiotherapy and most chemotherapies aim at creating irreparable DSBs in cancerous cells. In cancer treatments, radiotherapies, such as ionizing radiation, induce high concentrations of ROS [21]. Topoisomerase poisons, such as doxorubicin and daunorubicin, can cause DSBs [46].
Non-homologous end joining
NHEJ is important for DSB repair in DNA damage repair as well as for V(D)J recombination in T and B cells [47]. NHEJ functions in DSB repair throughout the cell cycle, especially in G0/G1 phase, and is highly conserved from prokaryotes to eukaryotes, demonstrating its mechanistic flexibility and tolerance for various structures of DNA ends [48-50]. NHEJ is a highly mutagenic repair pathway in that it ligates two ends at the DSB site together regardless of the homology of the DNA sequence [50]. NHEJ can be divided into two pathways, canonical NHEJ (C-NHEJ) and alternative NHEJ (A-NHEJ), according to the resection of DNA ends at the breakage site and the proteins involved [47]. For C-NHEJ, Ku proteins bind to the broken ends of DSBs and recruit DAN-PK as well as 53BP1 and the Mre11 complex to the damage site. The breakage sites are then processed by Artemis and are simply ligated in cis by the XRCC4/Ligase IV/XLF complex [47]. The direct ligation process in C-NHEJ alters the DNA sequence at the damage site, resulting in more mutations as extra nucleotides are excised before ligation. For A-NHEJ, the DNA breakage ends are recognized by PARP1, which recruits the Mre11 complex to the damage site before a few nucleotides are excised by CtIP-mediated end resection. The gap can then be filled and ligated by the XRCC1/Ligase III/Ligase I complex [47].
Homologous recombination repair
HR repair is the predominant type of DSB repair that occurs in late S and G2 phases of the cell cycle [51]. The HR pathway utilizes a DNA template strand with significant sequence homology to the damaged strand; therefore, this repair pathway is considered to be error-free and non-mutagenic [1]. The regulation and flexibility of the Mre11 nuclease activities are important in controlling the repair pathway choice during DSB repair [52]. The HR pathway initiates binding of the Mre11-Rad50-Nbs1 protein complex (MRN) to the DSB site and the cyclin-dependent kinase (CDK)-dependent activation of the CtIP protein, which regulates Mre11-mediated end resection of DNA [52,53]. After initial resection, the Exo1-DNA2-Sgs1 complex is responsible for further DNA resection, and the ssDNA is protected by the RPA proteins [51]. The RPA proteins are then replaced by Rad51 in a BRCA1/BRCA2 dependent strand invasion process, and the pairing of homologous sequence is completed and extended with the help of Rad52, Rad54 and WRN complex proteins [54]. The junctions at homologous pairing site are then resolved by the BLM/TOPIII/Mus81 complex [54].
Cancer cells are highly proliferative and divide more frequently than cells in normal tissue. Genomic integrity in S and G2 phase is required before cells divide; therefore, inhibiting HR and initiating DSB in HR-deficient cells are both efficient ways to inhibit cancer cell proliferation by trapping cells in the G2/M cell cycle checkpoint. Chemicals that serve as HR inhibitors are often involved in regulating protein expression, nuclear localization, and recruitment of HR proteins. For example, inhibitors of histone deacetylation and HSP90 can block HR by diminishing the expression of BRCA2 [55] and Rad51 [56]. There are also cancer cells that have HR-deficiency. For example, BRCA1/2 germline mutations are reported in many patients with solid tumors, especially in hereditary breast and ovarian cancer patients [57,58]. The deficiency of HR leads to sensitization of patients to DNA damaging agents such as cisplatin and PARP1 inhibitors [59-63].
Single-strand annealing
SSA is an error prone repair mechanism that is initiated when DSBs occur between two repeated intra-strand DNA sequences. The ERCC1/XPF complex is responsible for the DNA excision step in SSA [64]. After excision of the 5’-ends and exposing regions of homology, the homologous strands of DNA must be paired through SSA, as in HR. Unlike HR, the RAD52 and RAD59 proteins play a predominant role in the SSA DNA binding step instead of RAD51 [65]. SSA is reduced in G1-arrested cells, but it is not clear whether the SSA pathway is cell cycle dependent because it is not under control of ATM, ATR, or DNA-PK [64]. Although SSA utilizes homologous pairing of repeated DNA sequence, it excises the repeated sequence closer to the site of the DNA break and could be mutagenic. However, the detailed mechanism and regulation of SSA is still unclear. Therefore, the importance of SSA in cancer formation and progression cannot be clearly addressed at this point in time.
RTK signaling
RTKs are highly conserved in protein structure, activation mechanisms, and downstream regulations from C. elegans to humans [66]. In human, there are 20 RTK subfamilies composed of 58 known RTKs [67], which activate both the canonical and non-canonical signaling pathways [67]. In the canonical signaling cascade, RTKs are localized at the cell surface membrane where they are activated by ligands from outside of the cell. Subsequently, they recruit downstream substrate proteins to the membrane, and the substrate proteins are phosphorylated to recruit more proteins to transduce the signaling cascade in the cell. In non-canonical RTK signaling pathways, the RTKs are internalized into the cells after activation and translocate from cell surface into the cytosol and nucleus where they phosphorylate their substrates [67].
Canonical RTK signaling cascade
For the canonical signaling cascade, receptors are initially activated through conformational changes induced by ligand binding, and their kinase activity is further elevated through receptor oligomerization [68]. These trans auto-phosphorylated tyrosine sites then serve as docking sites for downstream cytosolic adaptor proteins containing phosphotyrosine-binding and Src homology-2 (SH2) domains [69]. Different adaptor proteins trigger different signaling pathways, including the RAS and PI3K protein-mediated pathways. In the RAS-mediated pathway, Grb-2 protein serves as an adaptor protein that binds to and is activated by phospho-RTK before recruiting and activating the RAS protein. The activated RAS protein then triggers the signaling cascade comprised of RAF, MEK and ERK proteins to enhance cell proliferation and transformation through regulating transcription factors and cell cycle regulatory proteins, such as AP1 and cyclin D1 [70,71]. In the PI3K-mediated pathway, the p85 regulatory subunit of PI3K serves as an adaptor protein to the activated RTK, and the p110 catalytic subunit of PI3K further phosphorylates downstream proteins, such as protein kinase C (PKC) and AKT [72,73]. The activated PKC signaling pathways promote proliferation, survival and metastasis of potential of cancer cells [74,75]; activated AKT signaling pathways also promote cell proliferation and survival [76]. Canonical RTK signaling is featured by its signaling redundancy among RTKs and the signaling crosstalk among the downstream pathways. Although RTKs can regulate common downstream proteins, these signaling pathways also feature signaling crosstalk among different parallel pathways. One RTK can transactivate other RTKs to regulate signaling cascade. For example, insulin-like growth factor (IGF-1)-induced IGF-1 receptor (IGF-1R) activation can also transactivate EGFR by promoting protease dependent-release of EGFR ligand. Transactivated EGFR accounts for the majority of RAS signaling induced by IGF [77]. RAS stimulates PI3K activity whereas AKT inhibits RAF activity. Both RAS-mediated and AKT-mediated signaling pathways can regulate common downstream proteins, such as mTOR [78]. Although the RTK signal redundancy and crosstalk suggest that downstream signaling proteins may be better targets than RTK itself, inhibitors blocking these downstream signaling proteins can affect both normal and cancer cells. To minimize the cytotoxicity effect on normal cells, targeting cancer cell-specific mutated protein and cancer cell-addicted RTK may be more practical.
Non-canonical RTK signaling pathway
In canonical RTK signaling, the activated receptors are internalized and are either recycled to the cell surface or subjected to lysosomal degradation. Although the majority of internalized RTKs are recycled [79], not all of the other internalized RTKs are degraded. A small portion of internalized RTKs, for example, about two percent of internalized EGFR, can phosphorylate non-canonical cytosolic and nuclear substrates and can trigger non-canonical RTK signaling pathways [67,80]. The first nuclear localized RTK was discovered in 1984 [81], and the non-canonical functions of nuclear localized RTKs were characterized by 1994 [82]. To date, more than 14 RTK subfamilies have been reported to play an important role in promoting cell proliferation, survival, DNA repair and drug resistance through non-canonical nuclear signaling pathways [67]. For instance, activated EGFR and HER2 are internalized into the cell and translocates into nucleus through retrograde trafficking mechanisms to endoplasmic reticulum (ER) before translocate from ER to nucleus through membrane-bound trafficking mechanism known as integral trafficking from the (ER) to the nuclear envelope transport (INTERNET) pathway [67,83-85]. Nuclear EGFR then serves as a transcription co-activator to promote proliferation, inflammation, survival, and drug resistance by interacting with transcription factors, e.g., E2F1, STAT3/5, and RHA, enhancing the expression of proteins such as cyclin D1, iNOS, c-Myc, B-Myb and BCRP [86-91]. Nuclear EGFR also functions as signaling transduction kinase to promote proliferation, DNA repair, and cell survival by phosphorylating PCNA, Histone H4, ATM, and DNA-PK [10,80,92-95]. On the other hand, fibroblast growth factor receptor (FGFR) is reported to transport to ER via a retrograde pathway and then translocate from ER into the nucleus in a membrane-vesicle independent mechanism which is a part of integrative nuclear FGFR-1 signaling (INFS) [67]. Nuclear FGFR then serves as transcription regulator by interacting with CREB-binding protein and STAT5 [96].
Regulation of RTK signaling on DDR and therapeutic resistance
Mutations in the RTKs as well as dysregulation of its downstream signaling proteins can impair normal DDR. Some RTKs are reported to translocate into the nucleus and their nuclear substrates includes DDR related, RTKs have been implicated in DDR regulation as the canonical RTK downstream proteins have been shown to correlate with DDR regulation (Figure 2). RAS constitutive activation and/or mutation are observed frequently in human cancers, and the K-RAS encoding gene is particularly vulnerable to chemical carcinogens [97,98]. Oncogenic activation of K-RAS leads to an accumulation of replication stress by orchestrating wild-type H- and N-RAS signaling, and triggers the ATR/Chk1 pathways to evade G2 cell-cycle arrest [99]. Oncogenic K-RAS also promotes A-NHEJ by upregulating the expression of DNA ligase III, PARP1, and XRCC1 in leukemia cancer model [100]. The AKT-mediated signaling pathway also regulates DDR. It has been reported that when cells are pretreated with Chk1 inhibitor, inactivation of AKT/PKB pathway can restore radiation-induced Chk1 activation at late G2 cell cycle arrest [101]. Other than Chk1, AKT is known to inhibit TopBP1 and BRCA1 even though it also positively regulates ATM, ATR, and DNA-PK (reviewed in [102]). In addition to the RAS and AKT pathways, some RTKs can also regulate DDR through other pathways as discussed in the following sections.
Figure 2.
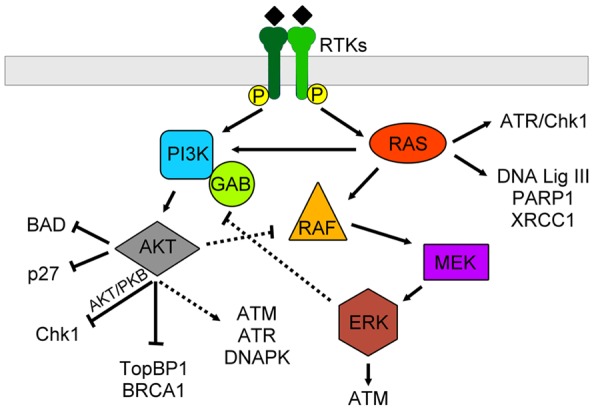
RTKs mediate DDR through canonical AKT and RAS pathways. In general, RTKs can activate both AKT and RAS pathways. Crosstalk between these two pathways can occur through AKT-RAF and ERK-GAB interactions. The downstream effects of the AKT pathway include inhibition of apoptosis through BAD and p27, inhibition of cell cycle progression through Chk1, downregulation of DNA damage repair through BRCA1, and indirect upregulation of DNA damage repair through ATM, ATR and DNAPK. Meanwhile, RAS itself can activate cell cycle arrest through ATR and Chk1 while promote DNA damage repair through expression of DNA ligase III, PARP1 and XRCC1. Also, the downstream of RAS pathway can activate ATM to promote DDR.
Regulation of DDR by the ErbB family
The ErbB family is composed of four receptors, ErbB1 (epidermal growth factor receptor, EGFR), ErbB2 (HER2), ErbB3 (HER3), and ErbB4 (HER4). Among them, EGFR and HER2 have been shown to regulate DDR and contribute to therapeutic resistance through both canonical and non-canonical signaling pathways. Using EGFR siRNAs and EGFR small molecule inhibitors, Wei et al. demonstrated that EGFR-mediated AKT/ERK pathway upregulates cell cycle regulatory proteins, including cyclin A, B, E, and CDK 1/2, in carcinogenic metal-induced proliferation of triple-negative breast cancer cells (Figure 3) [103]. The RAS/MEK/ERK pathway promotes EGFR-mediated radioprotection [104] by affecting gene transcription of the DNA repair proteins. The expression levels of the base repair DNA ligase XRCC1 and the DNA adduct excision protein ERCC1 upregulated under radiation treatment can be attenuated by EGFR inhibitor [105,106]. By utilizing small molecule inhibitors, radiation-induced and EGFR-mediated XRCC1 upregulation was shown to depend on the RAS/MEK/ERK pathway whereas normal XRCC1 expression is affected by EGFR-mediated PI3K/AKT pathway (Figure 3) [107].
Figure 3.
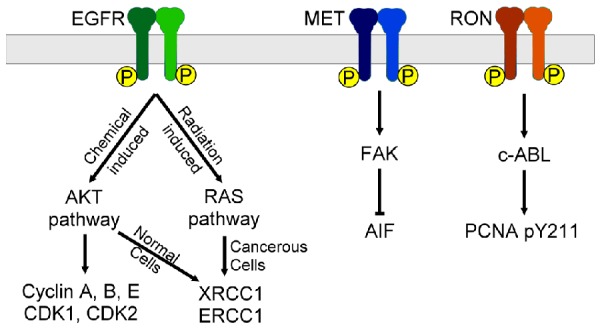
Some RTKs regulate DDR through specific canonical pathways. The chemical-induced EGFR activation will activate AKT pathway to upregulate the expression of cyclin A, B, E and CDK 1, 2 to promote cell cycle. On the other hand, the radiation-induced EGFR activation will activate RAS pathways to increase expression of XRCC1 and ERCC1 in cancer cells. MET can downregulate the expression of AIF specifically through the FAK pathway. Ron promotes PCNA Y211 phosphorylation through c-ABL mediated pathway.
Nuclear EGFR also plays an important role in DNA damage repair, including MMR, NHEJ and HR (Figure 4). For instance, nuclear EGFR can phosphorylate histone H2B and histone H4. Specifically, EGFR phosphorylates histone H4 at Y-72 to regulate histone H4 methylation [93]. EGF, as well as arsenic, can stimulate nuclear EGFR-mediated phosphorylation and stabilization PCNA via Y211. Phosphorylated PCNA Y211, which has been shown to correlate with poor patient survival, promotes cell proliferation as well as inhibits the endonuclease activity of MutLα, which leads to inhibition of MMR [10,80,108]. Yu et al. demonstrated that PCNA-derived peptide blocks the EGFR-PCNA complex and suppresses the growth of breast cancer cells [109]. Nuclear EGFR plays a role in HR in many aspects. EGFR phosphorylates ATM at Y370; depletion of EGFR abolishes ATM-mediated foci formation and HR; the ATM-EGFR interaction can be blocked by gefitinib, an EGFR inhibitor [92]. EGFR also interacts with BRCA1 to facilitate HR; the EGFR-BRCA1 interaction as well as BRCA1 nuclear translocation can be blocked by the EGFR inhibitor, lapatinib [110]. These interactions provide molecular basis for the combination therapy of EGFR inhibitor with PARP inhibitor, which induces synthetic lethality in tumor cells, as demonstrated in breast and ovarian cancers [110-112]. Radiation also enhances EGFR nuclear translocation [95,113]. Nuclear accumulation of EGFR contributes to radio-protection and interferes with DNA repair through interacting and regulating activity of DNAPK [114-117]. Treatment of EGFR monoclonal antibody, cetuximab (C225), promotes the interaction between EGFR, DNAPK, and Ku proteins, which results in a redistribution of DNAPK from the nucleus to cytosol, a critical step in the radiosensitizing role of EGFR blockade [118-120]. EGFR blockade also inhibits cell growth via p27 and maintains cells in G1 phase, which has been shown to also contribute to the radiosensitizing effect of EGFR [121,122]. In addition to EGFR, HER2 also regulates cell cycle regulation by binding to and colocalizing with cyclin B-bound CDC2 protein. Phosphorylation of CDC2 by HER2 at Y15 then delays entry of cells into M phase and contributes taxol resistance in HER2-overexpressing cancer cells [123]. Inhibition of HER3 also sensitizes cancer cells to radiation therapy by blocking AKT phosphorylation [124], and dual inhibition of EGFR and HER3 can overcome cross-resistance to EGFR inhibition and radiation [125,126].
Figure 4.
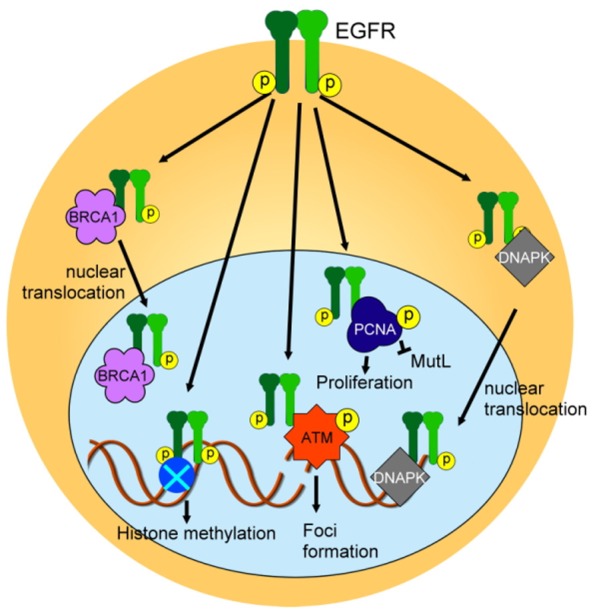
EGFR also mediates DDR through non-canonical signaling pathways. EGFR interacts with BRCA1 and DNAPK to promote their translocation into the nucleus. EGFR can also phosphorylate histone, ATM and PCNA to promote histone methylation, foci formation, and proliferation whereas EGFR-mediated PCNA phosphorylation inhibits MutL activity.
MET family regulated DDR
Two RTKs in the MET family, MET (also known as hepatocyte growth factor (HGF) receptor) and Ron (also known as macrophage stimulating 1 (MST1) receptor) also regulate DDR. In lung adenocarcinoma, HGF-induced MET activation inhibits apoptosis through the canonical pathway. Chen et al. demonstrated that the FAK-/- mouse embryonic fibroblast cells express higher levels of apoptosis-inducing factor (AIF), which correlates with better therapeutic response to cisplatin treatment. Moreover, AIF expression and cisplatin sensitivity were increased in cells when binding of MET to FAK was impeded or when MET inactivated [127], suggesting that this FAK-regulated AIF expression is downstream of MET signaling in lung adenocarcinoma cells. MET is also reported to directly phosphorylate PARP1 at Y907 site [128]. Phosphorylated PARP1 is more resistant to the PARP inhibitor, veliparib, and the combination of MET inhibitor and veliparib increased breast cancer cell killing effect. Contrary to MET, Ron phosphorylates PCNA at Y211 through the canonical signaling pathway by activating Ron downstream kinase, c-Abl, an adaptor protein containing SH2 domain [129]. These findings suggested a functional redundancy between Ron receptor and nuclear EGFR on PCNA Y211 regulation.
Future prospect
Although both radio- and chemotherapies have demonstrated significant efficacy in cancer treatment, there are still some patients who have poor response to these treatment methods. Therefore, it is important to address the biological basis of resistance to cancer treatment and to improve the efficacy of radio- or chemotherapies, such as through combination with targeted therapies. In this aspect, there is an urgent and unmet need to develop biomarkers for personalized medicine [130]. As DNA damage adducts and repair capabilities have proven success in predicting cancer risk and therapeutic response, stratifying patients according to the DDR status has emerged as a treatment modality for cancer [45,131]. While mutations in DDR proteins can serve as predictive markers in cancer treatment [132], DDR proteins are also epigenetically regulated. Thus, the regulatory modifications of DDR proteins as well as mutations and malfunctions of other molecular players involved in DDR are also important factors to consider when stratifying patients for treatment.
Among the potential biomarkers, RTK-related DDR protein phosphorylation is an ideal marker in cancer treatment because RTKs are involved in DDR regulation whereas most of cancer cells develop oncogenic addiction to upregulated RTK signaling pathways. Targeting RTKs, thus, may overcome RTK-mediated therapeutic resistance discussed above. For example, patients with pY211-PCNA and pY370-ATM may benefit from treatments combining EGFR inhibitor with current chemo- or radio- therapies; patients with pY907-PARP1 may benefit from treatment combining MET inhibitor with PARP1 inhibitor. Since oncogenic addiction is important to distinguish the killing effect of RTK inhibitors between normal cells and cancer cells [133], in theory, combining DNA damaging agents with RTK inhibitors can selectively increase genotoxic effects in cancer cells. As the molecular mechanisms underlying RTK-mediated DDR are not yet fully understood, more investigations are needed to further characterize the therapeutic potential of personalized combination therapy targeting this regulatory pathway.
Acknowledgements
We thank Drs. Richard Behringer, Rachel Miller, and Eric Swindell for helpful comments on the manuscript. The work was supported by the following grants: National Institutes of Health (CA109311, CA099031, and CCSG CA016672); National Breast Cancer Foundation, Inc.; Patel Memorial Breast Cancer Endowment Fund; The University of Texas MD Anderson-China Medical University and Hospital Sister Institution Fund; Ministry of Science and Technology, International Research-intensive Centers of Excellence in Taiwan (I-RiCE; MOST 105-2911-I-002-302); Ministry of Health and Welfare, China Medical University Hospital Cancer Research Center of Excellence (MOHW105-TDU-B-212-134003); and Center for Biological Pathways. We apologize to the authors whose original work could not be cited due to space limitation.
Disclosure of conflict of interest
None.
References
- 1.Friedberg EC. DNA repair and mutagenesis. Washington, D.C: ASM Press; 2006. [Google Scholar]
- 2.Johnson RE, Washington MT, Prakash S, Prakash L. Fidelity of human DNA polymerase eta. J Biol Chem. 2000;275:7447–7450. doi: 10.1074/jbc.275.11.7447. [DOI] [PubMed] [Google Scholar]
- 3.Schaaper RM. Base selection, proofreading, and mismatch repair during DNA replication in Escherichia coli. J Biol Chem. 1993;268:23762–23765. [PubMed] [Google Scholar]
- 4.Kuzminov A. Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc Natl Acad Sci U S A. 2001;98:8241–8246. doi: 10.1073/pnas.131009198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jackson AL, Loeb LA. The contribution of endogenous sources of DNA damage to the multiple mutations in cancer. Mutat Res. 2001;477:7–21. doi: 10.1016/s0027-5107(01)00091-4. [DOI] [PubMed] [Google Scholar]
- 6.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoeijmakers JH. Genome maintenance mechanisms are critical for preventing cancer as well as other aging-associated diseases. Mech Ageing Dev. 2007;128:460–462. doi: 10.1016/j.mad.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 8.Goode EL, Ulrich CM, Potter JD. Polymorphisms in DNA repair genes and associations with cancer risk. Cancer Epidemiol Biomarkers Prev. 2002;11:1513–1530. [PubMed] [Google Scholar]
- 9.Peltomaki P. Role of DNA mismatch repair defects in the pathogenesis of human cancer. J. Clin. Oncol. 2003;21:1174–1179. doi: 10.1200/JCO.2003.04.060. [DOI] [PubMed] [Google Scholar]
- 10.Ortega J, Li JY, Lee S, Tong D, Gu L, Li GM. Phosphorylation of PCNA by EGFR inhibits mismatch repair and promotes misincorporation during DNA synthesis. Proc Natl Acad Sci U S A. 2015;112:5667–5672. doi: 10.1073/pnas.1417711112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen DJ, Nirodi CS. The epidermal growth factor receptor: a role in repair of radiation-induced DNA damage. Clin Cancer Res. 2007;13:6555–6560. doi: 10.1158/1078-0432.CCR-07-1610. [DOI] [PubMed] [Google Scholar]
- 12.Medova M, Aebersold DM, Zimmer Y. The Molecular Crosstalk between the MET Receptor Tyrosine Kinase and the DNA Damage Response-Biological and Clinical Aspects. Cancers (Basel) 2013;6:1–27. doi: 10.3390/cancers6010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Skorski T. Oncogenic tyrosine kinases and the DNA-damage response. Nat Rev Cancer. 2002;2:351–360. doi: 10.1038/nrc799. [DOI] [PubMed] [Google Scholar]
- 14.Eckstein N, Roper L, Haas B, Potthast H, Hermes U, Unkrig C, Naumann-Winter F, Enzmann H. Clinical pharmacology of tyrosine kinase inhibitors becoming generic drugs: the regulatory perspective. J Exp Clin Cancer Res. 2014;33:15. doi: 10.1186/1756-9966-33-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scott CL, Swisher EM, Kaufmann SH. Poly (ADP-ribose) polymerase inhibitors: recent advances and future development. J. Clin. Oncol. 2015;33:1397–1406. doi: 10.1200/JCO.2014.58.8848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ame JC, Spenlehauer C, de Murcia G. The PARP superfamily. Bioessays. 2004;26:882–893. doi: 10.1002/bies.20085. [DOI] [PubMed] [Google Scholar]
- 17.Jungmichel S, Rosenthal F, Altmeyer M, Lukas J, Hottiger MO, Nielsen ML. Proteome-wide identification of poly(ADP-Ribosyl)ation targets in different genotoxic stress responses. Mol Cell. 2013;52:272–285. doi: 10.1016/j.molcel.2013.08.026. [DOI] [PubMed] [Google Scholar]
- 18.Nojima H. G1 and S-phase checkpoints, chromosome instability, and cancer. Methods Mol Biol. 2004;280:3–49. doi: 10.1385/1-59259-788-2:003. [DOI] [PubMed] [Google Scholar]
- 19.Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 20.Crick F. The double helix: a personal view. Nature. 1974;248:766–769. doi: 10.1038/248766a0. [DOI] [PubMed] [Google Scholar]
- 21.Lomax ME, Folkes LK, O’Neill P. Biological consequences of radiation-induced DNA damage: relevance to radiotherapy. Clin Oncol (R Coll Radiol) 2013;25:578–585. doi: 10.1016/j.clon.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 22.Huitema AD, Smits KD, Mathot RA, Schellens JH, Rodenhuis S, Beijnen JH. The clinical pharmacology of alkylating agents in highdose chemotherapy. Anticancer Drugs. 2000;11:515–533. doi: 10.1097/00001813-200008000-00002. [DOI] [PubMed] [Google Scholar]
- 23.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 24.Yu H, Liu Z, Huang YJ, Yin M, Wang LE, Wei Q. Association between single nucleotide polymorphisms in ERCC4 and risk of squamous cell carcinoma of the head and neck. PLoS One. 2012;7:e41853. doi: 10.1371/journal.pone.0041853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li C, Hu Z, Lu J, Liu Z, Wang LE, El-Naggar AK, Sturgis EM, Spitz MR, Wei Q. Genetic polymorphisms in DNA base-excision repair genes ADPRT, XRCC1, and APE1 and the risk of squamous cell carcinoma of the head and neck. Cancer. 2007;110:867–875. doi: 10.1002/cncr.22861. [DOI] [PubMed] [Google Scholar]
- 26.Wang M, Qin C, Zhu J, Yuan L, Fu G, Zhang Z, Yin C. Genetic variants of XRCC1, APE1, and ADPRT genes and risk of bladder cancer. DNA Cell Biol. 2010;29:303–311. doi: 10.1089/dna.2009.0969. [DOI] [PubMed] [Google Scholar]
- 27.Hu D, Lin X, Zhang H, Zheng X, Niu W. APEX nuclease (multifunctional DNA repair enzyme) 1 gene Asp148Glu polymorphism and cancer risk: a meta-analysis involving 58 articles and 48903 participants. PLoS One. 2013;8:e83527. doi: 10.1371/journal.pone.0083527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Divine KK, Gilliland FD, Crowell RE, Stidley CA, Bocklage TJ, Cook DL, Belinsky SA. The XRCC1 399 glutamine allele is a risk factor for adenocarcinoma of the lung. Mutat Res. 2001;461:273–278. doi: 10.1016/s0921-8777(00)00059-8. [DOI] [PubMed] [Google Scholar]
- 29.Schneider J, Classen V, Helmig S. XRCC1 polymorphism and lung cancer risk. Expert Rev Mol Diagn. 2008;8:761–780. doi: 10.1586/14737159.8.6.761. [DOI] [PubMed] [Google Scholar]
- 30.Krokan HE, Bjoras M. Base excision repair. Cold Spring Harb Perspect Biol. 2013;5:a012583. doi: 10.1101/cshperspect.a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.King BS, Cooper KL, Liu KJ, Hudson LG. Poly(ADP-ribose) contributes to an association between poly(ADP-ribose) polymerase-1 and xeroderma pigmentosum complementation group A in nucleotide excision repair. J Biol Chem. 2012;287:39824–39833. doi: 10.1074/jbc.M112.393504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Herceg Z, Wang ZQ. Functions of poly(ADPribose) polymerase (PARP) in DNA repair, genomic integrity and cell death. Mutat Res. 2001;477:97–110. doi: 10.1016/s0027-5107(01)00111-7. [DOI] [PubMed] [Google Scholar]
- 33.Strom CE, Johansson F, Uhlen M, Szigyarto CA, Erixon K, Helleday T. Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res. 2011;39:3166–3175. doi: 10.1093/nar/gkq1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Montoni A, Robu M, Pouliot E, Shah GM. Resistance to PARP-Inhibitors in Cancer Therapy. Front Pharmacol. 2013;4:18. doi: 10.3389/fphar.2013.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee JM, Ledermann JA, Kohn EC. PARP Inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies. Ann Oncol. 2014;25:32–40. doi: 10.1093/annonc/mdt384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, Scott C, Weitzel JN, Oaknin A, Loman N, Lu K, Schmutzler RK, Matulonis U, Wickens M, Tutt A. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376:245–251. doi: 10.1016/S0140-6736(10)60893-8. [DOI] [PubMed] [Google Scholar]
- 37.Bixel K, Hays JL. Olaparib in the management of ovarian cancer. Pharmgenomics Pers Med. 2015;8:127–135. doi: 10.2147/PGPM.S62809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scott AD, Waters R. Inducible nucleotide excision repair (NER) of UV-induced cyclobutane pyrimidine dimers in the cell cycle of the budding yeast Saccharomyces cerevisiae: evidence that inducible NER is confined to the G1 phase of the mitotic cell cycle. Mol Gen Genet. 1997;254:43–53. doi: 10.1007/s004380050389. [DOI] [PubMed] [Google Scholar]
- 39.Reed E. Platinum-DNA adduct, nucleotide excision repair and platinum based anti-cancer chemotherapy. Cancer Treat Rev. 1998;24:331–344. doi: 10.1016/s0305-7372(98)90056-1. [DOI] [PubMed] [Google Scholar]
- 40.Hess MT, Gunz D, Luneva N, Geacintov NE, Naegeli H. Base pair conformation-dependent excision of benzo[a] pyrene diol epoxide-guanine adducts by human nucleotide excision repair enzymes. Mol Cell Biol. 1997;17:7069–7076. doi: 10.1128/mcb.17.12.7069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kraemer KH, Lee MM, Andrews AD, Lambert WC. The role of sunlight and DNA repair in melanoma and nonmelanoma skin cancer. The xeroderma pigmentosum paradigm. Arch Dermatol. 1994;130:1018–1021. [PubMed] [Google Scholar]
- 42.Lai JP, Liu YC, Alimchandani M, Liu Q, Aung PP, Matsuda K, Lee CC, Tsokos M, Hewitt S, Rushing EJ, Tamura D, Levens DL, Digiovanna JJ, Fine HA, Patronas N, Khan SG, Kleiner DE, Oberholtzer JC, Quezado MM, Kraemer KH. The influence of DNA repair on neurological degeneration, cachexia, skin cancer and internal neoplasms: autopsy report of four xeroderma pigmentosum patients (XP-A, XP-C and XP-D) Acta Neuropathol Commun. 2013;1:4. doi: 10.1186/2051-5960-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Poulogiannis G, Frayling IM, Arends MJ. DNA mismatch repair deficiency in sporadic colorectal cancer and Lynch syndrome. Histopathology. 2010;56:167–179. doi: 10.1111/j.1365-2559.2009.03392.x. [DOI] [PubMed] [Google Scholar]
- 44.Elsayed FA, Kets CM, Ruano D, van den Akker B, Mensenkamp AR, Schrumpf M, Nielsen M, Wijnen JT, Tops CM, Ligtenberg MJ, Vasen HF, Hes FJ, Morreau H, van Wezel T. Germline variants in POLE are associated with early onset mismatch repair deficient colorectal cancer. Eur J Hum Genet. 2015;23:1080–1084. doi: 10.1038/ejhg.2014.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Assasi N, Blackhouse G, Campbell K, Weeks L, Levine M. Mismatch Repair Deficiency Testing for Patients with Colorectal Cancer: A Clinical and Cost-Effectiveness Evaluation. Ottawa (ON): 2015. [PubMed] [Google Scholar]
- 46.Cheung-Ong K, Giaever G, Nislow C. DNAdamaging agents in cancer chemotherapy: serendipity and chemical biology. Chem Biol. 2013;20:648–659. doi: 10.1016/j.chembiol.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 47.Deriano L, Roth DB. Modernizing the nonhomologous end-joining repertoire: alternative and classical NHEJ share the stage. Annu Rev Genet. 2013;47:433–455. doi: 10.1146/annurev-genet-110711-155540. [DOI] [PubMed] [Google Scholar]
- 48.Aravind L, Koonin EV. Prokaryotic homologs of the eukaryotic DNA-end-binding protein Ku, novel domains in the Ku protein and prediction of a prokaryotic double-strand break repair system. Genome Res. 2001;11:1365–1374. doi: 10.1101/gr.181001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gu J, Lieber MR. Mechanistic flexibility as a conserved theme across 3 billion years of nonhomologous DNA end-joining. Genes Dev. 2008;22:411–415. doi: 10.1101/gad.1646608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krajewska M, Fehrmann RS, de Vries EG, van Vugt MA. Regulators of homologous recombination repair as novel targets for cancer treatment. Front Genet. 2015;6:96. doi: 10.3389/fgene.2015.00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shibata A, Moiani D, Arvai AS, Perry J, Harding SM, Genois MM, Maity R, van Rossum-Fikkert S, Kertokalio A, Romoli F, Ismail A, Ismalaj E, Petricci E, Neale MJ, Bristow RG, Masson JY, Wyman C, Jeggo PA, Tainer JA. DNA doublestrand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol Cell. 2014;53:7–18. doi: 10.1016/j.molcel.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lafrance-Vanasse J, Williams GJ, Tainer JA. Envisioning the dynamics and flexibility of Mre11-Rad50-Nbs1 complex to decipher its roles in DNA replication and repair. Prog Biophys Mol Biol. 2015;117:182–193. doi: 10.1016/j.pbiomolbio.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li X, Heyer WD. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008;18:99–113. doi: 10.1038/cr.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Noguchi M, Yu D, Hirayama R, Ninomiya Y, Sekine E, Kubota N, Ando K, Okayasu R. Inhibition of homologous recombination repair in irradiated tumor cells pretreated with Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Biochem Biophys Res Commun. 2006;351:658–663. doi: 10.1016/j.bbrc.2006.10.094. [DOI] [PubMed] [Google Scholar]
- 56.Adimoolam S, Sirisawad M, Chen J, Thiemann P, Ford JM, Buggy JJ. HDAC inhibitor PCI-24781 decreases RAD51 expression and inhibits homologous recombination. Proc Natl Acad Sci U S A. 2007;104:19482–19487. doi: 10.1073/pnas.0707828104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Toss A, Tomasello C, Razzaboni E, Contu G, Grandi G, Cagnacci A, Schilder RJ, Cortesi L. Hereditary ovarian cancer: not only BRCA 1 and 2 genes. Biomed Res Int. 2015;2015:341723. doi: 10.1155/2015/341723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wittersheim M, Buttner R, Markiefka B. Genotype/Phenotype correlations in patients with hereditary breast cancer. Breast Care (Basel) 2015;10:22–26. doi: 10.1159/000380900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Taniguchi T, Tischkowitz M, Ameziane N, Hodgson SV, Mathew CG, Joenje H, Mok SC, D’Andrea AD. Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat Med. 2003;9:568–574. doi: 10.1038/nm852. [DOI] [PubMed] [Google Scholar]
- 60.Byrski T, Gronwald J, Huzarski T, Grzybowska E, Budryk M, Stawicka M, Mierzwa T, Szwiec M, Wisniowski R, Siolek M, Narod SA, Lubinski J Polish Hereditary Breast Cancer Consortium. Response to neo-adjuvant chemotherapy in women with BRCA1-positive breast cancers. Breast Cancer Res Treat. 2008;108:289–296. doi: 10.1007/s10549-007-9600-1. [DOI] [PubMed] [Google Scholar]
- 61.Byrski T, Gronwald J, Huzarski T, Grzybowska E, Budryk M, Stawicka M, Mierzwa T, Szwiec M, Wisniowski R, Siolek M, Dent R, Lubinski J, Narod S. Pathologic complete response rates in young women with BRCA1-positive breast cancers after neoadjuvant chemotherapy. J. Clin. Oncol. 2010;28:375–379. doi: 10.1200/JCO.2008.20.7019. [DOI] [PubMed] [Google Scholar]
- 62.Benafif S, Hall M. An update on PARP inhibitors for the treatment of cancer. Onco Targets Ther. 2015;8:519–528. doi: 10.2147/OTT.S30793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Muggia F, Safra T. ‘BRCAness’ and its implications for platinum action in gynecologic cancer. Anticancer Res. 2014;34:551–556. [PMC free article] [PubMed] [Google Scholar]
- 64.Al-Minawi AZ, Saleh-Gohari N, Helleday T. The ERCC1/XPF endonuclease is required for efficient single-strand annealing and gene conversion in mammalian cells. Nucleic Acids Res. 2008;36:1–9. doi: 10.1093/nar/gkm888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pastink A, Eeken JC, Lohman PH. Genomic integrity and the repair of double-strand DNA breaks. Mutat Res. 2001;480-481:37–50. doi: 10.1016/s0027-5107(01)00167-1. [DOI] [PubMed] [Google Scholar]
- 66.Sundaram MV. Canonical RTK-Ras-ERK signaling and related alternative pathways. WormBook. 2013:1–38. doi: 10.1895/wormbook.1.80.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen MK, Hung MC. Proteolytic cleavage, trafficking, and functions of nuclear receptor tyrosine kinases. FEBS J. 2015;282:3693–3721. doi: 10.1111/febs.13342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schlessinger J. Signal transduction by allosteric receptor oligomerization. Trends Biochem Sci. 1988;13:443–447. doi: 10.1016/0968-0004(88)90219-8. [DOI] [PubMed] [Google Scholar]
- 69.Sudol M. From Src Homology domains to other signaling modules: proposal of the ‘protein recognition code’. Oncogene. 1998;17:1469–1474. doi: 10.1038/sj.onc.1202182. [DOI] [PubMed] [Google Scholar]
- 70.Hollenhorst PC. RAS/ERK pathway transcriptional regulation through ETS/AP-1 binding sites. Small GTPases. 2012;3:154–158. doi: 10.4161/sgtp.19630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hitomi M, Stacey DW. Cyclin D1 production in cycling cells depends on ras in a cell-cyclespecific manner. Curr Biol. 1999;9:1075–1084. doi: 10.1016/s0960-9822(99)80476-x. [DOI] [PubMed] [Google Scholar]
- 72.Holt KH, Olson L, Moye-Rowley WS, Pessin JE. Phosphatidylinositol 3-kinase activation is mediated by high-affinity interactions between distinct domains within the p110 and p85 subunits. Mol Cell Biol. 1994;14:42–49. doi: 10.1128/mcb.14.1.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 74.Kim J, Thorne SH, Sun L, Huang B, Mochly-Rosen D. Sustained inhibition of PKCalpha reduces intravasation and lung seeding during mammary tumor metastasis in an in vivo mouse model. Oncogene. 2011;30:323–333. doi: 10.1038/onc.2010.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jain K, Basu A. The Multifunctional Protein Kinase C-epsilon in Cancer Development and Progression. Cancers (Basel) 2014;6:860–878. doi: 10.3390/cancers6020860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Alexandra Leary EA, Patricia Pautier, Catherine Lhomme. The PI3K/Akt/mTOR Pathway in Ovarian Cancer: Biological Rationale and Therapeutic Opportunities, Ovarian Cancer - A Clinical and Translational Update. InTech; 2013. [Google Scholar]
- 77.Roudabush FL, Pierce KL, Maudsley S, Khan KD, Luttrell LM. Transactivation of the EGF receptor mediates IGF-1-stimulated shc phosphorylation and ERK1/2 activation in COS-7 cells. J Biol Chem. 2000;275:22583–22589. doi: 10.1074/jbc.M002915200. [DOI] [PubMed] [Google Scholar]
- 78.Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci. 2011;36:320–328. doi: 10.1016/j.tibs.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sorkin A, Krolenko S, Kudrjavtceva N, Lazebnik J, Teslenko L, Soderquist AM, Nikolsky N. Recycling of epidermal growth factor-receptor complexes in A431 cells: identification of dual pathways. J Cell Biol. 1991;112:55–63. doi: 10.1083/jcb.112.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang SC, Nakajima Y, Yu YL, Xia W, Chen CT, Yang CC, McIntush EW, Li LY, Hawke DH, Kobayashi R, Hung MC. Tyrosine phosphorylation controls PCNA function through protein stability. Nat Cell Biol. 2006;8:1359–1368. doi: 10.1038/ncb1501. [DOI] [PubMed] [Google Scholar]
- 81.Gusterson B, Cowley G, Smith JA, Ozanne B. Cellular localisation of human epidermal growth factor receptor. Cell Biol Int Rep. 1984;8:649–658. doi: 10.1016/0309-1651(84)90045-6. [DOI] [PubMed] [Google Scholar]
- 82.Xie Y, Hung MC. Nuclear localization of p185neu tyrosine kinase and its association with transcriptional transactivation. Biochem Biophys Res Commun. 1994;203:1589–1598. doi: 10.1006/bbrc.1994.2368. [DOI] [PubMed] [Google Scholar]
- 83.Perillo EP, Liu YL, Huynh K, Liu C, Chou CK, Hung MC, Yeh HC, Dunn AK. Deep and high-resolution three-dimensional tracking of single particles using nonlinear and multiplexed illumination. Nat Commun. 2015;6:7874. doi: 10.1038/ncomms8874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lin SY, Makino K, Xia W, Matin A, Wen Y, Kwong KY, Bourguignon L, Hung MC. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol. 2001;3:802–808. doi: 10.1038/ncb0901-802. [DOI] [PubMed] [Google Scholar]
- 85.Wang YN, Lee HH, Lee HJ, Du Y, Yamaguchi H, Hung MC. Membrane-bound trafficking regulates nuclear transport of integral epidermal growth factor receptor (EGFR) and ErbB-2. J Biol Chem. 2012;287:16869–16879. doi: 10.1074/jbc.M111.314799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jaganathan S, Yue P, Paladino DC, Bogdanovic J, Huo Q, Turkson J. A functional nuclear epidermal growth factor receptor, SRC and Stat3 heteromeric complex in pancreatic cancer cells. PLoS One. 2011;6:e19605. doi: 10.1371/journal.pone.0019605. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 87.Huang WC, Chen YJ, Li LY, Wei YL, Hsu SC, Tsai SL, Chiu PC, Huang WP, Wang YN, Chen CH, Chang WC, Chang WC, Chen AJ, Tsai CH, Hung MC. Nuclear translocation of epidermal growth factor receptor by Akt-dependent phosphorylation enhances breast cancer-resistant protein expression in gefitinib-resistant cells. J Biol Chem. 2011;286:20558–20568. doi: 10.1074/jbc.M111.240796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lo HW, Hsu SC, Ali-Seyed M, Gunduz M, Xia W, Wei Y, Bartholomeusz G, Shih JY, Hung MC. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell. 2005;7:575–589. doi: 10.1016/j.ccr.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 89.Hanada N, Lo HW, Day CP, Pan Y, Nakajima Y, Hung MC. Co-regulation of B-Myb expression by E2F1 and EGF receptor. Mol Carcinog. 2006;45:10–17. doi: 10.1002/mc.20147. [DOI] [PubMed] [Google Scholar]
- 90.Hung LY, Tseng JT, Lee YC, Xia W, Wang YN, Wu ML, Chuang YH, Lai CH, Chang WC. Nuclear epidermal growth factor receptor (EGFR) interacts with signal transducer and activator of transcription 5 (STAT5) in activating Aurora-A gene expression. Nucleic Acids Res. 2008;36:4337–4351. doi: 10.1093/nar/gkn417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lo HW, Cao X, Zhu H, Ali-Osman F. Cyclooxygenase-2 is a novel transcriptional target of the nuclear EGFR-STAT3 and EGFRvIII-STAT3 signaling axes. Mol Cancer Res. 2010;8:232–245. doi: 10.1158/1541-7786.MCR-09-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lee HJ, Lan L, Peng G, Chang WC, Hsu MC, Wang YN, Cheng CC, Wei L, Nakajima S, Chang SS, Liao HW, Chen CH, Lavin M, Ang KK, Lin SY, Hung MC. Tyrosine 370 phosphorylation of ATM positively regulates DNA damage response. Cell Res. 2015;25:225–236. doi: 10.1038/cr.2015.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chou RH, Wang YN, Hsieh YH, Li LY, Xia W, Chang WC, Chang LC, Cheng CC, Lai CC, Hsu JL, Chang WJ, Chiang SY, Lee HJ, Liao HW, Chuang PH, Chen HY, Wang HL, Kuo SC, Chen CH, Yu YL, Hung MC. EGFR modulates DNA synthesis and repair through Tyr phosphorylation of histone H4. Dev Cell. 2014;30:224–237. doi: 10.1016/j.devcel.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dittmann K, Mayer C, Fehrenbacher B, Schaller M, Raju U, Milas L, Chen DJ, Kehlbach R, Rodemann HP. Radiation-induced epidermal growth factor receptor nuclear import is linked to activation of DNA-dependent protein kinase. J Biol Chem. 2005;280:31182–31189. doi: 10.1074/jbc.M506591200. [DOI] [PubMed] [Google Scholar]
- 95.Dittmann K, Mayer C, Kehlbach R, Rodemann HP. Radiation-induced caveolin-1 associated EGFR internalization is linked with nuclear EGFR transport and activation of DNA-PK. Mol Cancer. 2008;7:69. doi: 10.1186/1476-4598-7-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dunham-Ems SM, Lee YW, Stachowiak EK, Pudavar H, Claus P, Prasad PN, Stachowiak MK. Fibroblast growth factor receptor-1 (FGFR1) nuclear dynamics reveal a novel mechanism in transcription control. Mol Biol Cell. 2009;20:2401–2412. doi: 10.1091/mbc.E08-06-0600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Montagut C, Settleman J. Targeting the RAF-MEK-ERK pathway in cancer therapy. Cancer Lett. 2009;283:125–134. doi: 10.1016/j.canlet.2009.01.022. [DOI] [PubMed] [Google Scholar]
- 98.Feng Z, Hu W, Chen JX, Pao A, Li H, Rom W, Hung MC, Tang MS. Preferential DNA damage and poor repair determine ras gene mutational hotspot in human cancer. J Natl Cancer Inst. 2002;94:1527–1536. doi: 10.1093/jnci/94.20.1527. [DOI] [PubMed] [Google Scholar]
- 99.Grabocka E, Pylayeva-Gupta Y, Jones MJ, Lubkov V, Yemanaberhan E, Taylor L, Jeng HH, Bar-Sagi D. Wild-type H- and N-Ras promote mutant K-Ras-driven tumorigenesis by modulating the DNA damage response. Cancer Cell. 2014;25:243–256. doi: 10.1016/j.ccr.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hahnel PS, Enders B, Sasca D, Roos WP, Kaina B, Bullinger L, Theobald M, Kindler T. Targeting components of the alternative NHEJ pathway sensitizes KRAS mutant leukemic cells to chemotherapy. Blood. 2014;123:2355–2366. doi: 10.1182/blood-2013-01-477620. [DOI] [PubMed] [Google Scholar]
- 101.Xu N, Hegarat N, Black EJ, Scott MT, Hochegger H, Gillespie DA. Akt/PKB suppresses DNA damage processing and checkpoint activation in late G2. J Cell Biol. 2010;190:297–305. doi: 10.1083/jcb.201003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Xu N, Lao Y, Zhang Y, Gillespie DA. Akt: a double-edged sword in cell proliferation and genome stability. J Oncol. 2012;2012:951724. doi: 10.1155/2012/951724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wei Z, Song X, Shaikh ZA. Cadmium promotes the proliferation of triple-negative breast cancer cells through EGFR-mediated cell cycle regulation. Toxicol Appl Pharmacol. 2015;289:98–108. doi: 10.1016/j.taap.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Weidhaas JB, Eisenmann DM, Holub JM, Nallur SV. A conserved RAS/mitogen-activated protein kinase pathway regulates DNA damage-induced cell death postirradiation in Radelegans. Cancer Res. 2006;66:10434–10438. doi: 10.1158/0008-5472.CAN-06-2182. [DOI] [PubMed] [Google Scholar]
- 105.Yacoub A, McKinstry R, Hinman D, Chung T, Dent P, Hagan MP. Epidermal growth factor and ionizing radiation up-regulate the DNA repair genes XRCC1 and ERCC1 in DU145 and LNCaP prostate carcinoma through MAPK signaling. Radiat Res. 2003;159:439–452. doi: 10.1667/0033-7587(2003)159[0439:egfair]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 106.Yacoub A, Park JS, Qiao L, Dent P, Hagan MP. MAPK dependence of DNA damage repair: ionizing radiation and the induction of expression of the DNA repair genes XRCC1 and ERCC1 in DU145 human prostate carcinoma cells in a MEK1/2 dependent fashion. Int J Radiat Biol. 2001;77:1067–1078. doi: 10.1080/09553000110069317. [DOI] [PubMed] [Google Scholar]
- 107.Toulany M, Dittmann K, Fehrenbacher B, Schaller M, Baumann M, Rodemann HP. PI3K-Akt signaling regulates basal, but MAPkinase signaling regulates radiation-induced XRCC1 expression in human tumor cells in vitro. DNA Repair (Amst) 2008;7:1746–1756. doi: 10.1016/j.dnarep.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 108.Tong D, Ortega J, Kim C, Huang J, Gu L, Li GM. Arsenic Inhibits DNA Mismatch Repair by Promoting EGFR Expression and PCNA Phosphorylation. J Biol Chem. 2015;290:14536–14541. doi: 10.1074/jbc.M115.641399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yu YL, Chou RH, Liang JH, Chang WJ, Su KJ, Tseng YJ, Huang WC, Wang SC, Hung MC. Targeting the EGFR/PCNA signaling suppresses tumor growth of triple-negative breast cancer cells with cell-penetrating PCNA peptides. PLoS One. 2013;8:e61362. doi: 10.1371/journal.pone.0061362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nowsheen S, Cooper T, Stanley JA, Yang ES. Synthetic lethal interactions between EGFR and PARP inhibition in human triple negative breast cancer cells. PLoS One. 2012;7:e46614. doi: 10.1371/journal.pone.0046614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Al-Ejeh F, Shi W, Miranda M, Simpson PT, Vargas AC, Song S, Wiegmans AP, Swarbrick A, Welm AL, Brown MP, Chenevix-Trench G, Lakhani SR, Khanna KK. Treatment of triple-negative breast cancer using anti-EGFR-directed radioimmunotherapy combined with radiosensitizing chemotherapy and PARP inhibitor. J Nucl Med. 2013;54:913–921. doi: 10.2967/jnumed.112.111534. [DOI] [PubMed] [Google Scholar]
- 112.Sui H, Shi C, Yan Z, Li H. Combination of erlotinib and a PARP inhibitor inhibits growth of A2780 tumor xenografts due to increased autophagy. Drug Des Devel Ther. 2015;9:3183–3190. doi: 10.2147/DDDT.S82035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dittmann K, Mayer C, Kehlbach R, Rothmund MC, Peter Rodemann H. Radiation-induced lipid peroxidation activates src kinase and triggers nuclear EGFR transport. Radiother Oncol. 2009;92:379–382. doi: 10.1016/j.radonc.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 114.Wanner G, Mayer C, Kehlbach R, Rodemann HP, Dittmann K. Activation of protein kinase Cepsilon stimulates DNA-repair via epidermal growth factor receptor nuclear accumulation. Radiother Oncol. 2008;86:383–390. doi: 10.1016/j.radonc.2007.10.041. [DOI] [PubMed] [Google Scholar]
- 115.Yu YL, Chou RH, Wu CH, Wang YN, Chang WJ, Tseng YJ, Chang WC, Lai CC, Lee HJ, Huo L, Chen CH, Hung MC. Nuclear EGFR suppresses ribonuclease activity of polynucleotide phosphorylase through DNAPK-mediated phosphorylation at serine 776. J Biol Chem. 2012;287:31015–31026. doi: 10.1074/jbc.M112.358077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dittmann K, Mayer C, Kehlbach R, Rodemann HP. The radioprotector Bowman-Birk proteinase inhibitor stimulates DNA repair via epidermal growth factor receptor phosphorylation and nuclear transport. Radiother Oncol. 2008;86:375–382. doi: 10.1016/j.radonc.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 117.Dittmann K, Mayer C, Wanner G, Kehlbach R, Rodemann HP. The radioprotector O-phospho-tyrosine stimulates DNA-repair via epidermal growth factor receptor- and DNA-dependent kinase phosphorylation. Radiother Oncol. 2007;84:328–334. doi: 10.1016/j.radonc.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 118.Bandyopadhyay D, Mandal M, Adam L, Mendelsohn J, Kumar R. Physical interaction between epidermal growth factor receptor and DNA-dependent protein kinase in mammalian cells. J Biol Chem. 1998;273:1568–1573. doi: 10.1074/jbc.273.3.1568. [DOI] [PubMed] [Google Scholar]
- 119.Huang SM, Harari PM. Modulation of radiation response after epidermal growth factor receptor blockade in squamous cell carcinomas: inhibition of damage repair, cell cycle kinetics, and tumor angiogenesis. Clin Cancer Res. 2000;6:2166–2174. [PubMed] [Google Scholar]
- 120.Dittmann K, Mayer C, Rodemann HP. Inhibition of radiation-induced EGFR nuclear import by C225 (Cetuximab) suppresses DNA-PK activity. Radiother Oncol. 2005;76:157–161. doi: 10.1016/j.radonc.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 121.Chinnaiyan P, Huang S, Vallabhaneni G, Armstrong E, Varambally S, Tomlins SA, Chinnaiyan AM, Harari PM. Mechanisms of enhanced radiation response following epidermal growth factor receptor signaling inhibition by erlotinib (Tarceva) Cancer Res. 2005;65:3328–3335. doi: 10.1158/0008-5472.CAN-04-3547. [DOI] [PubMed] [Google Scholar]
- 122.Huang SM, Bock JM, Harari PM. Epidermal growth factor receptor blockade with C225 modulates proliferation, apoptosis, and radiosensitivity in squamous cell carcinomas of the head and neck. Cancer Res. 1999;59:1935–1940. [PubMed] [Google Scholar]
- 123.Tan M, Jing T, Lan KH, Neal CL, Li P, Lee S, Fang D, Nagata Y, Liu J, Arlinghaus R, Hung MC, Yu D. Phosphorylation on tyrosine-15 of p34(Cdc2) by ErbB2 inhibits p34(Cdc2) activation and is involved in resistance to taxol-induced apoptosis. Mol Cell. 2002;9:993–1004. doi: 10.1016/s1097-2765(02)00510-5. [DOI] [PubMed] [Google Scholar]
- 124.Li C, Brand TM, Iida M, Huang S, Armstrong EA, van der Kogel A, Wheeler DL. Human epidermal growth factor receptor 3 (HER3) blockade with U3-1287/AMG888 enhances the efficacy of radiation therapy in lung and head and neck carcinoma. Discov Med. 2013;16:79–92. [PMC free article] [PubMed] [Google Scholar]
- 125.Li C, Huang S, Armstrong EA, Francis DM, Werner LR, Sliwkowski MX, van der Kogel A, Harari PM. Antitumor Effects of MEHD7945A, a Dual-Specific Antibody against EGFR and HER3, in Combination with Radiation in Lung and Head and Neck Cancers. Mol Cancer Ther. 2015;14:2049–2059. doi: 10.1158/1535-7163.MCT-15-0155. [DOI] [PubMed] [Google Scholar]
- 126.Huang S, Li C, Armstrong EA, Peet CR, Saker J, Amler LC, Sliwkowski MX, Harari PM. Dual targeting of EGFR and HER3 with MEHD7945A overcomes acquired resistance to EGFR inhibitors and radiation. Cancer Res. 2013;73:824–833. doi: 10.1158/0008-5472.CAN-12-1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chen JT, Huang CY, Chiang YY, Chen WH, Chiou SH, Chen CY, Chow KC. HGF increases cisplatin resistance via down-regulation of AIF in lung cancer cells. Am J Respir Cell Mol Biol. 2008;38:559–565. doi: 10.1165/rcmb.2007-0001OC. [DOI] [PubMed] [Google Scholar]
- 128.Du Y, Yamaguchi H, Wei Y, Hsu JL, Wang HL, Hsu YH, Lin WC, Yu WH, Leonard PG, Lee GRt, Chen MK, Nakai K, Hsu MC, Chen CT, Sun Y, Wu Y, Chang WC, Huang WC, Liu CL, Chang YC, Chen CH, Park M, Jones P, Hortobagyi GN, Hung MC. Blocking c-Met-mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nat Med. 2016;22:194–201. doi: 10.1038/nm.4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhao H, Chen MS, Lo YH, Waltz SE, Wang J, Ho PC, Vasiliauskas J, Plattner R, Wang YL, Wang SC. The Ron receptor tyrosine kinase activates c-Abl to promote cell proliferation through tyrosine phosphorylation of PCNA in breast cancer. Oncogene. 2014;33:1429–1437. doi: 10.1038/onc.2013.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Frank R, Hargreaves R. Clinical biomarkers in drug discovery and development. Nat Rev Drug Discov. 2003;2:566–580. doi: 10.1038/nrd1130. [DOI] [PubMed] [Google Scholar]
- 131.Schabath MB, Spitz MR, Grossman HB, Zhang K, Dinney CP, Zheng PJ, Wu X. Genetic instability in bladder cancer assessed by the comet assay. J Natl Cancer Inst. 2003;95:540–547. doi: 10.1093/jnci/95.7.540. [DOI] [PubMed] [Google Scholar]
- 132.Kan C, Zhang J. BRCA1 Mutation: A Predictive Marker for Radiation Therapy? Int J Radiat Oncol Biol Phys. 2015;93:281–293. doi: 10.1016/j.ijrobp.2015.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]