

Abstract
Tumor angiogenic process is regulated by multiple proangiogenic pathways, such as vascular endothelial growth factor receptor 2 (VEGFR2) and Axl receptor tyrosine kinase (Axl). Axl is one of many important factors involved in anti-VEGF resistance. Inhibition of VEGF/VEGFR2 signaling pathway alone fails to block tumor neovascularization. Therefore, discovery of novel agents targeting multiple angiogenesis pathways is in demand. Desacetylvinblastine monohydrazide (DAVLBH), a derivative of vinblastine (VLB), has been reported exhibit an anticancer activity via its cytotoxic effect. However, little attention has been paid to the antiangiogenic properties of DAVLBH. Here, we firstly reported that DAVLBH exerted a more potent antiangiogenic effect than VLB in vitro and in vivo, which was associated with inactivation of VEGF/VEGFR2 and Gas6/Axl signaling pathways. We found that DAVLBH inhibited VEGF- and Gas6-induced HUVECs proliferation, migration, tube formation and vessel sprouts formation in vitro and ex vivo. It significantly inhibited in vivo tumor angiogenesis and tumor growth in HeLa xenografts. It also inhibited Gas6-induced pericytes recruitment to endothelial tubes accompanied with a decrease in expression and activation of Axl. Besides, it could block the compensatory up-regulating expression and activation of Axl in response to bevacizumab treatment in HUVECs. Taken together, our results suggest that DAVLBH potently inhibits angiogenesis-mediated tumor growth through blockage of the activation of VEGF/VEGFR2 and Gas6/Axl pathways and it might serve as a promising antiangiogenic agent for the cancer therapy.
Keywords: Desacetylvinblastine monohydrazide (DAVLBH), antiangiogenesis, vascular endothelial growth factor receptor 2 (VEGFR2), Axl receptor tyrosine kinase (Axl), pericytes recruitment
Introduction
Angiogenesis is essential for solid tumor growth and metastasis [1]. Without blood vessels, tumors cannot grow beyond a threshold size or metastasize to other organs [2]. Therefore, antiangiogenesis is considered an attractive approach for antitumor treatment.
The tumor angiogenic process is regulated by multiple proangiogenic factors that destabilize the integrated blood vessel, promote endothelial cell motility (proliferation, migration, invasion and tubulogenesis) and ultimately induce neovascularization [3,4]. Among these factors, VEGF plays a critical role in the angiogenesis process [5]. VEGFR2 is responsible for the major angiogenic functions of VEGF [6,7]. The phosphorylation of VEGFR2 induces the activation of multiple downstream signaling pathways, including extracellular signal-regulated kinase (ERK) and Akt, and then stimulates endothelial cell proliferation, migration and tube formation [8-10]. Therefore, blocking the VEGF/VEGFR2 signaling pathway by inhibiting endogenous VEGF release and interfering with VEGF binding to VEGFR2 has become an important antiangiogenic therapeutic strategy [11]. A number of antiangiogenic agents have been developed in recent years, including bevacizumab, a recombinant humanized anti-VEGF neutralizing antibody [12], and small-molecule inhibitors of VEGFRs such as sunitinib [13] and sorafenib [14]. These agents have been approved for the treatment of certain metastatic cancers. However, inhibition of the VEGF/VEGFR2 signaling pathway alone fails to block tumor growth and neovascularization [7], which is partly due to the abundant and complementary proangiogenic pathways that give rise to anti-VEGF resistance [15,16].
Growth arrest-specific protein 6 (Gas6) is another important proangiogenic factor that mediates the activation of Axl, Mer and Tyr3 receptor tyrosine kinase (RTK) pathways in tumor angiogenesis [17]. However, as a ligand of RTKs, Gas6 shows the highest affinity for Axl, which is overexpressed in endothelial cells and pericytes [18-20]. Gas6-mediated Axl activation promotes endothelial cell proliferation, migration, and invasion during tumor angiogenesis [21] through Akt [22,23] and ERK [24] pathways. Axl is also involved in the complicated regulation of VEGF/VEGFR2 signaling pathway [23,25]. Activation of the Gas6/Axl pathway is associated with anti-VEGF resistance, and the combination of an Axl antibody or an Axl inhibitor with anti-VEGF agents improves the antiangiogenic effect [26] or overcomes anti-VEGF resistance [27].
Desacetylvinblastine monohydrazide (DAVLBH) is a derivative of the natural product vinblastine (VLB) and exhibits powerful microtubule-destabilizing effects [28] and potent antitumor activity both in vitro and in vivo [29,30]. However, little attention has been paid to the antiangiogenic properties of DAVLBH. In the present study, we report the first evidence that DAVLBH inhibits various endothelial cellular motilities required for neovascularization in vitro and suppresses HeLa xenograft tumor growth and angiogenesis in vivo through the inhibition of both VEGF/VEGFR2 and Gas6/Axl pathways. We also demonstrate that DAVLBH inhibits bevacizumab-induced Axl expression in human umbilical vein endothelial cells (HUVECs) and affects pericyte recruitment during vessel maturation.
Materials and methods
Materials
DAVLBH (purity > 98%) was synthesized, and its structure was identified according to the methods described in the literature [29]. DAVLBH was then stored at -20°C and protected from light. Pentobarbital sodium was purchased from Merck (Darmstadt, Germany). VEGF165 is a product of Life Technology (Invitrogen, Carlsbad, CA). Matrigel was purchased from BD Biosciences (Franklin Lakes, NJ). Bevacizumab was purchased from Roche (Genentech Inc., San Francisco, CA). Antibodies against VEGFR2, p-VEGFR2Tyr1175, Axl, Akt, p-AktThr308, ERK, p-ERKThr202/Tyr204, PDGFR-β and Ki67 and a secondary antibody were purchased from Cell Signaling Technology (Danvers, MA). CD31 antibody, p-AxlY779 antibody and recombinant human Gas6 growth arrest-specific protein 6 (Gas6) were obtained from R&D Systems (Minneapolis, MN). Alexa Fluor 594 Donkey anti-Goat IgG and Alexa Fluor 488 Donkey anti-Rabbit IgG were purchased from Life Technologies (Invitrogen, Eugene, OR). The Human RTK Phosphorylation Antibody Array was purchased from RayBiotech (Atlanta, GA). The Rac1/Cdc42 Activation Assay Kit is a product of Merck Millipore (Darmstadt, Germany). The DAB substrate kit was obtained from Vector Laboratories (Burlingame, CA). Endothelial cell medium (ECM) and pericyte medium (PM) were obtained from ScienCell Research Laboratories (San Diego, CA). Other reagents were purchased from Sigma-Aldrich (St. Louis, MO).
Cell lines and cell culture
HUVECs were isolated from fresh umbilical cords using type II collagenase by adapting a previously described protocol [31] and were maintained in ECM. The cells were then characterized using CD31 immunohistochemical staining. Human brain vascular pericytes (HBVPs) were obtained from ScienCell Research Laboratories and cultured in PM. HeLa cells were purchased from Chinese Academy of Sciences Cell Bank (Shanghai, China) and cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (Life Technology, Carlsbad, CA). All cells were maintained in a humidified atmosphere containing 5% CO2 at 37°C.
Animals
Female BABL/c (nu/nu) mice were obtained from Vital River Laboratory Animal Technology Co, Ltd. (Beijing, China). Adult male Sprague-Dawley rats (weighing 220-240 g) were obtained from Guangdong Medical Experimental Animal Center (Guangzhou, China). All animals were maintained in specific pathogen free room and with free access to water and standard laboratory chow. All animal experimental procedures were approved by Laboratory Animal Ethics Committee of Jinan University (Guangzhou, China). The results of all studies involving animals were reported in accordance with the ARRIVE guidelines.
Cell proliferation assay
VEGF- and Gas6-induced HUVEC proliferation was measured by 3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as previously described [32]. Briefly, 1×104 cells per well were seeded in 96-well plates and cultured for 24 h. Adherent cells were starved with serum-free ECM for 6 h to prompt the cells to enter the resting state. Then, the cells were treated with various concentrations of DAVLBH, VLB (1.0 nM), VEGF (20 ng/mL) or Gas6 (100 ng/mL) at the same time. After incubation for 24 h, cell viability was determined using MTT assay.
Endothelial cell migration and invasion assays
The effects of DAVLBH on endothelial cell migration and invasion were evaluated with wound-healing and modified Boyden chamber assays, respectively. In the wound-healing assay, HUVECs were seeded in 6-well plates and grown to confluence. Then, they were starved with serum-free ECM for 6 h and scratched with pipette tips. After washing with phosphate-buffered saline (PBS), fresh ECM containing VLB (1.0 nM) or various concentrations of DAVLBH (0.5, 1.0 or 2.0 nM) in the presence or absence of VEGF (20 ng/mL) or Gas6 (100 ng/mL) was added. Images of cells were obtained with an Olympus IX70 inverted microscope (Shinjuku, Tokyo, Japan) after treatment for 8 h. The migrated cells were quantified by Image-Pro Plus 6.0 software (Rockville, MD). The Boyden chamber assay was performed as previously described [33] with some modifications. Briefly, the insert transwell plate was pre-coated with Matrigel for 1 h at 37°C. Then, 1×104 cells suspended in 100 μL serum-free ECM containing VLB or various concentrations of DAVLBH were added to the upper chamber and 600 μL fresh ECM, supplemented with VEGF (20 ng/mL) or Gas6 (100 ng/mL), was placed in the lower chamber. After a 24-h incubation, the upper chamber was fixed with 4% paraformaldehyde for 30 min and the cells were stained with crystal violet. The inner side of the upper chamber was scraped with a cotton swab to remove the non-invasive cells. Invasive cells on the lower surface were photographed using an Olympus IX70 inverted microscope and quantified using Image-Pro Plus 6.0.
Tube formation assay
The tube formation assay was performed as previously described with some modifications [34]. Cells (2×104) suspended in 100 μL ECM were seeded in Matrigel-coated 96-well plates and then incubated in the presence of VLB (1.0 nM) or various concentrations of DAVLBH (0.5, 1.0 or 2.0 nM) with or without VEGF (20 ng/mL) or Gas6 (100 ng/mL). After an 8-h incubation, endothelial tubes were photographed with an Olympus IX70 inverted microscope and the number of tubes, loops and branching points were calculated by Image-Pro Plus 6.0.
Aortic ring assay
Dorsal aortas were isolated from Sprague-Dawley rats after anaesthetisation with an intraperitoneal injection of 50 mg/kg pentobarbital sodium. Then, the periadventitial fat and connective tissue around the aorta were removed and the aorta was cut into 1- to 1.5-mm-long rings. The rinsed aortic rings were placed in Matrigel-coated 96-well plates and sealed with an overlay of 80 μL Matrigel. After a 2-h incubation, ECM containing VEGF (100 ng/mL) or Gas6 (300 ng/mL) was added and the rings were incubated for 48 h. Then, the aortic rings were treated with VLB (1.0 nM) or various concentrations of DAVLBH (0.5, 1 or 2 nM) for 7 days. The microvessels were photographed using an Olympus IX70 inverted microscope, and microvessel sprouts were quantified using Image-Pro Plus 6.0.
Human RTK phosphorylation antibody array
The expression of phosphorylated RTK growth factors in DAVLBH-treated HUVECs was detected using the Human RTK Phosphorylation Antibody Array. The procedures were performed following the manufacturer’s protocol. First, the membranes were blocked with blocking buffer for 1 h and then incubated overnight with 1 mg protein dissolved in 1.2 mL blocking buffer at 4°C. Then, the membranes were incubated with a biotin-conjugated secondary antibody for 2 h at room temperature, followed by incubation with FITC-conjugated streptavidin for another 2 h. The signals were directly detected and analyzed following the manufacturer’s instructions using an Array Worx fluorescence slide scanner (Applied Precision, Issaquah, WA).
Western blot analysis
Whole-cell extracts were prepared using RIPA lysis buffer. Proteins (50 μg) were resolved by electrophoresis on 8-12% SDS-polyacrylamide gels and then detected by western blotting as previously described [35]. Quantitative data were measured with ImageJ software (NIH, NY).
GTPase activation assay of Rac1 and cdc42
The GTP activities of rRac1 and rCdc42 were determined with a GST-PBD pull-down assay using the Rac1/Cdc42 Activation Assay Kit. According to the manufacturer’s protocol, HUVECs were starved with serum-free ECM and treated with different concentrations of DAVLBH for 4 h. The cells were subsequently stimulated with VEGF (20 ng/mL) or Gas6 (100 ng/mL) for 1 h. Then, the cells were lysed in Mg2+ lysis buffer. Cell extracts (500 μg) were mixed with GST-PBD (the Pak Rac1/Cdc42-binding domain) attached to beads. After incubation at 4°C overnight, the beads were washed five times with lysis buffer and boiled at 100°C for 5 min with 2× sodium dodecyl sulphate sample buffer. Immunoblotting for Rac1 and Cdc42 was performed with antibodies specifically against these proteins.
Three-dimensional co-cultures
In vitro pericyte migration to endothelial tubes was detected using a three-dimensional co-culture assay as previously described [36] with some modifications. Briefly, HUVECs were labelled with PKH 26 following the manufacturer’s instructions. Next, 3×104 HUVECs suspended in serum-free ECM were seeded in 96-well plates that had been pre-coated with 150 μL Matrigel; the cells were then incubated for 2 h to allow the tube formation. At the same time, 2×104 HBVPs that had been pre-treated with or without DAVLBH (1.0 nM) for 4 h were labeled with PKH 67, suspended in serum-free PM in the presence or absence of Gas6 (100 ng/mL) and added to the endothelial tubes for an additional 12-h incubation to allow for HBVP adhesion to the HUVEC tubes. Images were obtained with an Olympus IX70 inverted microscope at 2 and 12 h after HBVP addition. The respective excitation and emission wavelengths were 551 and 567 nm for PKH 26 and 490 and 502 nm for PKH 67.
Tumor xenograft model
HeLa cells (1×107) mixed with 200 μL 50% Matrigel in PBS (v/v) were inoculated subcutaneously into the backs of 5- to 6-week old BABL/c (nu/nu) female mice. When the tumor size grew to approximately 200 mm3, tumor-bearing mice were randomized into three groups with five mice per group. The mice were intravenously (i.v.) injected every two days with saline, VLB (0.75 μmol/kg) or DAVLBH (0.75 μmol/kg) for a total of 9 administrations. Tumor volumes were measured using a slide calliper and calculated using the following formula: a×b 2×0.5, where a refers to the longest diameter and b is the shortest diameter. At the end of the experiment, mice bearing tumors were anaesthetised with an intraperitoneal injection of 50 mg/kg pentobarbital sodium and the tumors were removed, weighed and photographed. Then, the tumors were fixed in 4% paraformaldehyde for histological and immunohistochemical examination.
Histology and immunohistochemistry
Tumors (five samples per group) were fixed in 4% paraformaldehyde for 24 h and then embedded in paraffin. Tissue sections (5 μm thick) were mounted on glass slides. For histological examination, sections were stained with haematoxylin-eosin (H&E). For immunohistochemistry, sections were incubated with anti-CD31, anti-Ki67, anti-Axl or anti-p-VEGFR2 (1:400) overnight at 4°C. The sections were rinsed with PBS and incubated with secondary antibodies, and the proteins were detected using the DAB kit following the manufacturer’s instructions. Images were collected with an Olympus IX70 inverted microscope. The Ki67 proliferation index is presented as the percentage of Ki67-positive cells/total cells per field with a 100× objective. The microvessel density (MVD) was evaluated by counting the number of vessels in three fields per slide using a 100× objective. The integrated optical density (IOD) values of Axl and p-VEGFR2 in each visual field (200× magnification) were calculated with Image-Pro Plus 6.0. The analysis was performed blind by a trained pathologist.
Immunofluorescence assay
Tumor sections were blocked for 1 h in blocking buffer (5% BSA) and then incubated with anti-CD31 and anti-PDGFR-β (1:400) antibodies overnight at 4°C. After being washed with PBS, the sections were incubated with secondary antibodies, including Alexa Fluor 488 Donkey anti-Rabbit IgG (1:2000) and Alexa Fluor 594 Donkey anti-Goat IgG (1:2000), for 1 h at room temperature. Next, the sections were mounted with Crystal Mount (GeneTex) and observed under a confocal microscope (LSM700, Zeiss). The pericyte coverage of tumor vessels was quantified by analysing the proportion of total CD31-positive endothelial lengths also positive for the pericyte marker PDGFR-β using Image-Pro Plus 6.0 software.
Statistics
The data are expressed as the mean ± SEM. GraphPad Prism 5.0 (GraphPad Software, La Jolla, CA) was used for statistical analysis. Significant differences were evaluated using one-way ANOVA followed by Tukey’s test. A difference was considered significant when P < 0.05.
Results
DAVLBH inhibits VEGF-induced HUVEC proliferation, migration, invasion, and tube formation in vitro and vessel sprout formation ex vivo
Before assessing the antiangiogenic properties of DAVLBH in vitro, the cytotoxic effect of DAVLBH on HUVECs in normal growth medium was evaluated using the MTT assay. Our results demonstrated that DAVLBH effectively reduced cell viability in a dose-dependent manner (data not shown) and induced cell cycle arrest and apoptosis at high concentrations (12.5 and 25 nM) (data not shown), whereas DAVLBH lower than 3.125 nM were non-toxic to HUVECs (data not shown). In this regard, the concentrations of 0.5, 1 and 2 nM were used in the in vitro and ex vivo antiangiogenic experiments. Next, the inhibitory effect of DAVLBH on VEGF-stimulated HUVEC proliferation was examined. As shown in Figure 1A, VEGF stimulation increased the viability of HUVECs compared with the non-VEGF treatment groups, whereas DAVLBH significantly suppressed VEGF-induced HUVEC proliferation in a dose-dependent manner.
Figure 1.
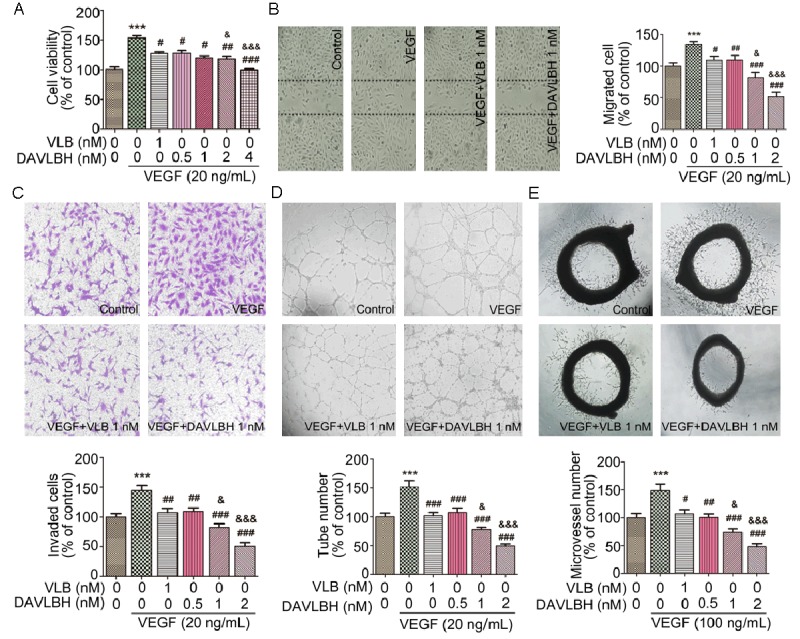
DAVLBH inhibits VEGF-induced angiogenesis in vitro and ex vivo. A. DAVLBH inhibited VEGF-induced HUVEC proliferation in a dose-dependent manner. Adherent HUVECs (1 × 104 cells per well) were starved with serum-free ECM and then treated with different concentrations of DAVLBH or VLB (1 nM) for 24 h in the presence or absence of VEGF. B. DAVLBH inhibited VEGF-induced HUVEC migration in a wound-healing assay. After starvation, monolayer cells were scratched with pipette tips and then treated with VLB or different concentrations of DAVLBH in the presence or absence of VEGF. After incubation for 8 h, the migrated cells were quantified using Image-Pro Plus 6.0 software. Representative fields were photographed (100× magnification). C. DAVLBH inhibited the VEGF-induced invasion of HUVECs in a Boyden chamber assay. HUVECs (2 × 104) were seeded in the upper chamber of the transwell and treated with VLB or different concentrations of DAVLBH. The bottom chamber was filled with medium that was supplemented with or without VEGF. After a 24-h incubation, cell invasion was quantified by counting the cells that had migrated through the membrane. Representative figures were photographed (100× magnification). D. DAVLBH inhibited the tube formation of HUVECs. HUVECs (2 × 104) treated with VLB or various concentrations of DAVLBH in the presence or absence of VEGF were seeded in Matrigel-coated 96-well plates. Tubular structures were quantified by manual counting after an 8-h incubation. Representative fields were photographed (100× magnification). E. DAVLBH inhibited microvessel sprouting in a rat aortic ring assay. Approximately 1- to 1.5-mm-thick cleaned mouse aortic rings were placed in Matrigel-coated 96-well plates and then treated with VLB or various concentrations of DAVLBH in the presence or absence of VEGF. Representative pictures were taken (40× magnification). Graphs show the quantitative effect. Quantitative data were analyzed using GraphPad Prism 5.0. The data are presented as mean ± SEM, n = 5. ***P < 0.001 compared with the control group; #P < 0.05, ##P < 0.01, and ###P < 0.001 compared with the VEGF-treated group; and &P < 0.05 and &&&P < 0.001 compared with the VLB-treated group.
The effects of DAVLBH on the VEGF-mediated migration and invasion of HUVECs were measured using a wound-healing migration assay and a transwell cell invasion assay, respectively. Our results showed that VEGF (20 ng/mL) increased the number of migrated cells and invaded cells, and the stimulatory effect of VEGF was inhibited by DAVLBH in a dose-dependent manner. The inhibitory effects of DAVLBH were more potent than those of VLB at the same concentration (1 nM) (Figure 1B, 1C). The HUVEC tube formation results, shown in Figure 1D, demonstrated that the number of tube-like structures significantly increased after stimulation with 20 ng/mL VEGF. However, the ability of endothelial cells to form tubular structures was dramatically attenuated in the presence of various concentrations of DAVLBH, and the inhibitory effect of DAVLBH was more pronounced than that of VLB at a concentration of 1 nM. Next, we explored whether DAVLBH inhibited VEGF-induced microvessel sprouting from aortic rings ex vivo. VEGF (100 ng/mL) markedly triggered microvessel sprouting and led to the formation of a complex network off microvessels around the aortic rings (Figure 1E). By contrast, DAVLBH treatment reduced the number of microvessel sprouts in a dose-dependent manner. DAVLBH also more effectively inhibited VEGF-induced vessel sprout formation than VLB at a concentration of 1 nM (Figure 1E). Taken together, these data suggest that DAVLBH significantly suppresses VEGF-induced angiogenesis both in vitro and ex vivo and that DAVLBH is more effective than VLB.
DAVLBH inhibits the expression of Axl in HUVECs
Accumulative evidence has shown that multiple growth factors and their related signaling pathways take part in angiogenesis [3,4]. To discover novel pathways that are involved in the antiangiogenic effect of DAVLBH, we screened for changes in the RTKs of HUVECs with or without DAVLBH treatment for 4 h using an RTK array. DAVLBH down-regulated the phosphorylation of several kinases, including Axl, Csk, EphB3, Fgr, Fyn, Insulin R and VEGFR2 (Figure 2A). Among these kinases, Axl, which is similar to VEGFR2, is also a transmembrane receptor and plays an important role in angiogenesis [37]. Because Axl is closely associated with the VEGF/VEGFR2 signaling pathway in tumor angiogenesis [23,25], we selected it for further investigation. Using a western blot assay, we confirmed the diminished expression of Axl in HUVECs treated with DAVLBH for 4 h at a concentration of 1 nM (Figure 2B). Axl may confer resistance to VEGF/VEGFR pathway inhibitors such as bevacizumab [27]. As shown in Figure 2C, the expression of Axl was up-regulated in HUVECs treated with bevacizumab (5 μM), but DAVLBH antagonized the bevacizumab-induced expression of Axl. These results indicate that Axl may take part in the regulation of DAVLBH-induced antiangiogenesis.
Figure 2.
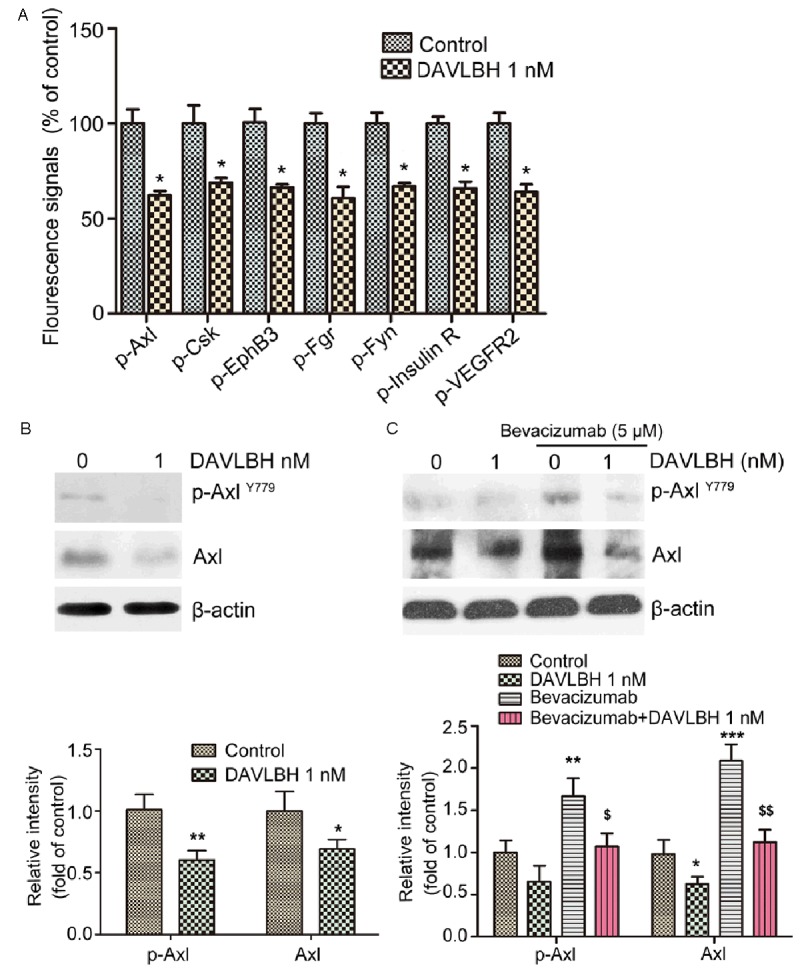
DAVLBH suppressed the expression of Axl in HUVECs. A. Quantitative analysis of the changes in human phosphorylated RTKs associated with angiogenesis in DAVLBH-treated HUVECs. B. DAVLBH inhibited the expression of Axl in normal HUVECs. HUVECs were treated with or without DAVLBH (1 nM) for 4 h. Cells were collected, lysed and subjected to western blot analysis. C. DAVLBH antagonized the increased expression of Axl in bevacizumab-treated HUVECs. Cells were continuously stimulated with bevacizumab for 48 h and then treated with or without DAVLBH for 4 h. The levels of Axl and p-Axl in the collected cell supernatants were analyzed by western blotting. The quantitative analysis of Western blot was shown. Data are ratios of p-Axl/β-actin and Axl/β-actin, and are presented as mean ± SEM, n = 5. *P < 0.05, **P < 0.01 and ***P < 0.001 compared with the control group; $P < 0.05 and $$P < 0.01, compared with the bevacizumab-treated group.
DAVLBH inhibits Gas6-induced HUVEC proliferation, migration, invasion, tube formation and vessel sprout formation in vitro and ex vivo
Given that Gas6, as a ligand of Axl, activates Axl to trigger endothelial cell proliferation, migration, and invasion during tumor angiogenesis, we assessed the inhibitory effect of DAVLBH on Gas6-induced angiogenesis in vitro. Our results revealed that DAVLBH significantly inhibited Gas6-induced HUVEC proliferation (Figure 3A), migration (Figure 3B), invasion (Figure 3C) and tube formation (Figure 3D) in a concentration-dependent manner, with effects even more potent than those of VLB at 1 nM. The DAVLBH inhibition of Gas6-induced neovascularization was further examined using a rat aortic ring assay. The results shown in Figure 3E demonstrate that DAVLBH blocked microvessel sprouting in a dose-dependent manner. At 1 nM, DAVLBH more effectively inhibited Gas6-induced neovessel sprouting than did VLB. These data suggest that DAVLBH inhibits Gas6-induced angiogenesis in vitro and ex vivo and that its efficacy is much higher than that of VLB.
Figure 3.

DAVLBH inhibited Gas6-induced angiogenesis in vitro and ex vivo. DAVLBH inhibited Gas6-stimulated HUVEC (A) proliferation, (B) migration, (C) invasion, (D) capillary-structure formation and (E) aortic ring microvessel sprouting. HUVECs or rat aortic rings were treated with VLB or various concentrations of DAVLBH in the presence or absence of Gas6. Representative figures (100× magnification in B, C, and D and 40× magnification in E) are shown, and the quantitative data were analyzed with GraphPad Prism 5.0. The data are presented as mean ± SEM, n = 5. ***P < 0.001 versus the control group; #P < 0.05, ##P < 0.01, and ###P < 0.001 versus the Gas6-treated group; and &P < 0.05, &&P < 0.01, and &&&P < 0.001 compared with the VLB-treated group.
DAVLBH inhibits the activation of VEGF/VEGFR2 and Gas6/Axl pathways in HUVECs
The above results showed that DAVLBH effectively inhibited VEGF- and Gas6-induced angiogenesis both in vitro and ex vivo. Thus, we investigated whether DAVLBH suppressed the activation of VEGF/VEGFR2, Gas6/Axl and their downstream pathways, such as Akt and ERK. As shown in Figure 4A, DAVLBH significantly blocked the VEGF-mediated phosphorylation of VEGFR2 and down-regulated the VEGFR2-mediated phosphorylation of Akt and ERK in a dose-dependent manner. In addition, DAVLBH dramatically reduced Axl expression and subsequently inhibited the Gas6-stimulated phosphorylation of Akt and ERK (Figure 4B). The VEGFR2 [38] and Axl pathways [39] both regulate Rho GTPase activity. Two prominent Rho GTPase family members, Rac1 and Cdc42, regulate cell actin cytoskeleton and motility [40]. Our results showed that VEGF and Gas6 obviously activated Rac1 and Cdc42, but this activation could be down-regulated after DAVLBH treatment (Figure 4C). Western blot analysis revealed that the in vitro antiangiogenic effect of DAVLBH was associated with the simultaneous inhibition of VEGF/VEGFR2 and Gas6/Axl signaling pathways.
Figure 4.
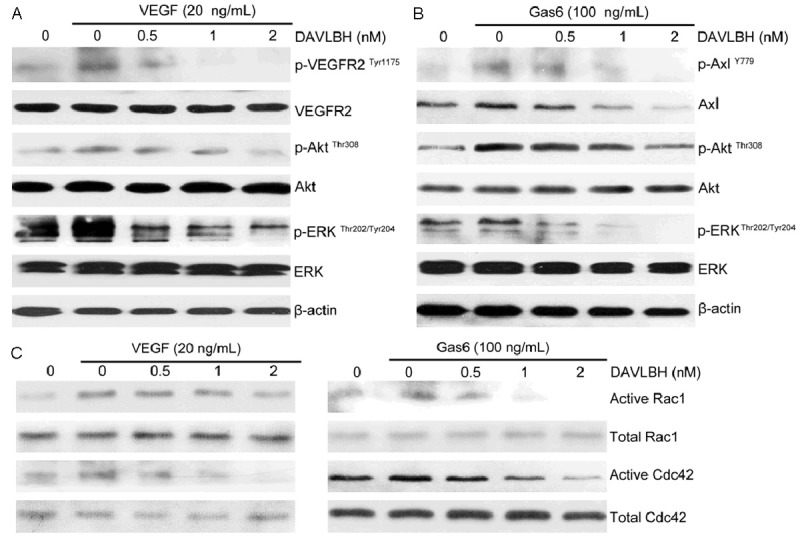
DAVLBH suppressed VEGF/VEGFR2 and Gas6/Axl signaling pathways. DAVLBH inhibited VEGFR2, Axl, Akt and ERK in (A) VEGF-treated HUVECs and (B) Gas6-treated HUVECs. HUVECs were pre-treated with various concentrations of DAVLBH for 4 h and then stimulated with VEGF or Gas6 for 1 h. Protein was collected and subjected to western blot analysis. (C) DAVLBH inhibited the VEGF- and Gas6-induced activation of Rac1 and Cdc42. HUVECs were starved with serum-free ECM and treated with different concentrations of DAVLBH for 4 h and then stimulated with VEGF or Gas6 for 1 h. The cells were then lysed, and active Rac1 and Cdc42 were pulled down and applied to western blotting.
DAVLBH inhibits Gas6-induced pericyte recruitment to endothelial tubes
Because pericytes, in which Axl is overexpressed, play an important role in the maturation of neovessels during late-stage angiogenesis, we tested the effect of DAVLBH on Gas6-induced pericyte recruitment to endothelial tubes. After HUVECs on Matrigel formed the original vessel network, HBVPs were added. In the control group, most of the seeded HBVPs migrated to the endothelial tubes within 2 h. Gas6 triggered even more HBVP adhesion to the endothelial tubes. However, in the DAVLBH group, fewer HBVPs were recruited to the endothelial tubes (Figure 5A). After a 12-h incubation, HBVP-supported endothelial tubes were well formed in the control and Gas6-treated groups. By contrast, DAVLBH inhibited Gas6-stimulated HBVP migration to the endothelial tubes and almost no HBVP-supported endothelial tubes were observed (Figure 5A). In addition, DAVLBH inhibited the Gas6-induced the expression of Axl in HBVP (Figure 5B).
Figure 5.
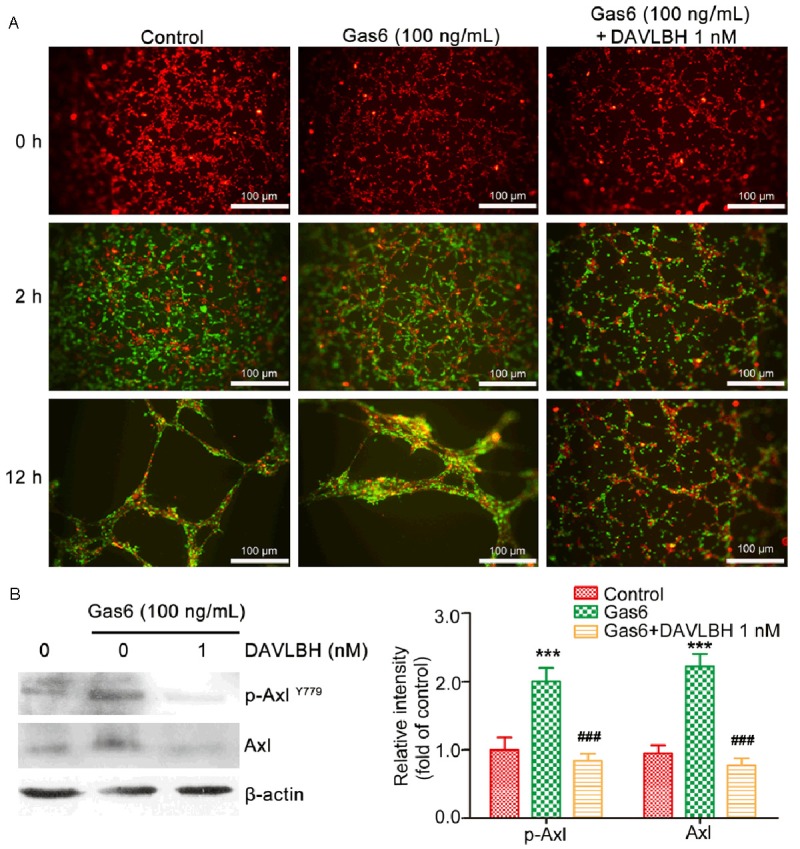
DAVLBH inhibited Gas6-induced migration of pericytes to endothelial tubes. A. Representative images of HUVECs (red) and HBVPs (green) in a three-dimensional co-culture system in vitro (scale bar = 100 μm). HBVPs that were pre-treated with or without DAVLBH for 4 h were seeded after endothelial tubes formed. The recruitment of HBVPs to HUVEC tubes was observed after the addition of HBVPs for 2 and 12 h in the presence or absence of Gas6. B. DAVLBH suppressed the expression of Axl in HBVPs. HBVPs were pre-treated with or without DAVLBH for 4 h and cultured for another 12 h in the presence or absence of Gas6. The cells were then lysed, and the proteins were subjected to western blot analysis. Data are ratios of p-Axl/β-actin and Axl/β-actin, and are presented as mean ± SEM, n = 5. ***P < 0.001 versus the control group; ###P < 0.001 versus the Gas6-treated group.
DAVLBH inhibits tumor angiogenesis and tumor growth in HeLa xenografts
To test whether DAVLBH inhibits angiogenesis-mediated tumor growth, a mouse xenograft model bearing HeLa cells was used. As shown in Figure 6A, tumor volume in the control group increased from 190.9 ± 52.6 mm3 at the beginning to 693.9 ± 198.9 mm3 at the end of treatment. VLB treatment had no noticeable effect on tumor volume compared with the control group: tumor volume increased to 631.8 ± 105.1 mm3 from 191.2 ± 28.9 mm3. However, in the DAVLBH-treated group, tumor volume increased to 371.7 ± 88.8 mm3 from 201.2 ± 37.5 mm3. The tumor weight of the DAVLBH-treated group was 309.2 ± 59.46 mg, which was much lower than that of the control and the VLB-treated groups (850.0 ± 163.5 mg and 743.4 ± 75.4 mg, respectively) (Figure 6B). In addition, as measured using Ki67 staining, the proliferation index in the DAVLBH-treated group was reduced to 31.94% compared with the control group, whereas the Ki67 proliferation index of the VLB-treated group was 37.70%, which was higher than that of the DAVLBH-treated group (Figure 6D, 6E). This result suggests that DAVLBH dramatically suppressed HeLa xenograft tumor growth and was more potent than VLB. It is important to note that DAVLBH did not lead to body weight loss at the end of treatment compared with the control group (Figure 6C), implying that DAVLBH may be low in toxicity towards tumor-bearing mice. To further evaluate the antiangiogenic effect of DAVLBH in vivo, we stained solid tumor sections with anti-CD31 antibody. Our results showed that the microvessel density (MVD) in DAVLBH-treated tumors was 11.83, which was much lower than that in the VLB-treated group (18.67) (Figure 6D, 6E). In addition, we demonstrated that the levels of Axl and p-VEGFR2 were much lower in DAVLBH-treated tumors compared with the control and VLB groups (Figure 6D, 6E). Moreover, the endothelial cells and pericytes in the tumor sections were immunostained with anti-CD31 and anti-PDGFR-β antibodies, respectively, and DAVLBH reduced the pericyte coverage compared with the control or VLB-treated groups (Figure 6D, 6E). These data suggest that DAVLBH inhibits tumor growth, which might be partly associated with the inhibition of VEGFR2- and Axl-mediated angiogenesis.
Figure 6.
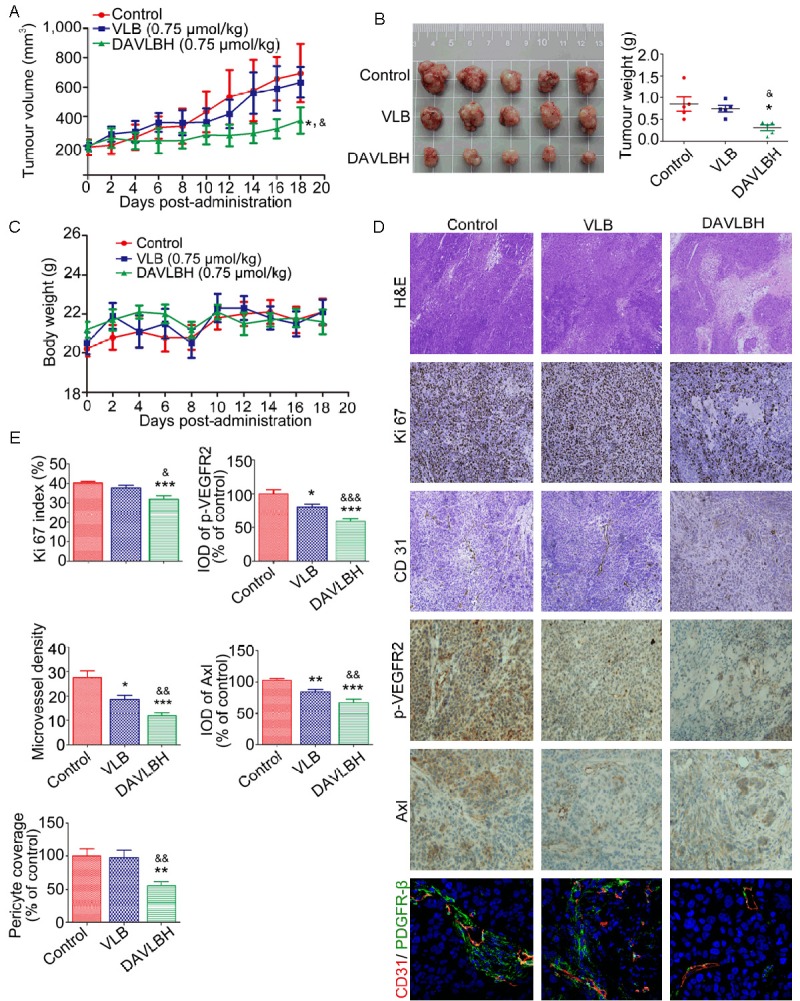
DAVLBH inhibited tumor growth and tumor angiogenesis in a HeLa xenograft model. A. DAVLBH inhibited HeLa xenograft tumor growth, as measured by tumor volume. HeLa cells (1 × 107 cells per mouse) were injected subcutaneously into 5- to 6-week-old BABL/c (nu/nu) female mice. After the tumors were established (approximately 200 mm3), mice were injected intravenously (i.v.) with saline, VLB or DAVLBH every two days for a total of 9 injections. Differences in tumor volumes on the 18th day were statistically analyzed using a one-way ANOVA, n = 5. *P < 0.05 compared with the control group; &P < 0.05 compared with the VLB-treated group. B. Tumors were removed from the mice and imaged at the end of treatment (left panel, n = 5), and the tumor weights were calculated (right panel, n = 5). C. Mouse body weight curves are shown (n = 5). No significant differences in body weights were observed between the control and the VLB- or DAVLBH-treated groups. D. Representative figures of the H&E staining, immunohistochemical and immunofluorescence analyses of tumors from mice that were sacrificed at the end of the experiment. The H&E staining is at 40× magnification; the immunohistochemical analyses with anti-Ki67 and anti-CD31 are at 100× magnification; the immunohistochemical analyses with anti-Axl and anti-p-VEGFR2 are at 200× magnification; and the immunofluorescence analyses with anti-CD31 (red), anti-PDGFR-β (green) and the nuclei (blue) are at 40× magnification. E. Graphs show the quantitative effect. The immunohistochemical and immunofluorescence results were calculated using Image-Pro Plus 6.0 software. The data are presented as mean ± SEM, n = 5. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with the control group. &P < 0.05, &&P < 0.01 and &&&P < 0.001 compared with the VLB-treated group.
Discussion
The antiangiogenic effect of VLB has been validated both in vitro and in vivo [41,42] and is credited with inhibiting VEGFR2 expression [43] and the activities of Rac1 and Cdc42 [44]. DAVLBH is a derivative of VLB [29] and possesses a much more potent microtubule depolymerization effect than VLB [28]. DAVLBH also exhibits a remarkable antitumor effect in vivo [30]. However, its antiangiogenic effect has never been tested. In the present study, we show for the first time that DAVLBH can inhibit angiogenesis more effectively than VLB in vitro and in vivo. We also demonstrate that DAVLBH inhibits not only the VEGF/VEGFR2 pathway but also the Gas6/Axl signaling pathway. Importantly, DAVLBH not only inhibited the various Gas6-induced endothelial cellular motilities required for neovascularization but also suppressed the Gas6-induced recruitment of pericytes to well-established endothelial tubes ex vivo and reduced the pericyte coverage of blood vessels in HeLa xenografts. DAVLBH also blocked the compensatory up-regulation of Axl in response to bevacizumab treatment in HUVECs. Our results indicate that DAVLBH may possesses promising applications in (a) enhancing anti-VEGF therapy, (b) overcoming anti-VEGF resistance, and (c) targeting pericyte recruitment during late-stage angiogenesis. In addition, DAVLBH, considered a cytotoxic agent [45], is the parent drug of the folate-conjugate prodrug EC145, which exerts a powerful antitumor effect on folic acid receptor (FR)-positive tumors in preclinical mouse xenografts [30]. As FR is also expressed in HUVECs, EC145 may be transported into HUVECs to exert an antiangiogenic effect. In this regard, our study provides new clues for further exploring the potent antiangiogenic and antitumor effects of EC145 and contributes to an understanding of its mechanism of action.
Previous research has shown that VEGF-induced VEGFR2 phosphorylation can activate the Akt and ERK downstream pathways, as well as Rho GTPase (Rac1 and Cdc42) activity, to further promote endothelial cell motility and angiogenesis [46-48]. In addition to the VEGF/VEGFR2 signaling pathway, the Gas6/Axl [21] pathway is a key regulator of angiogenesis. Axl is overexpressed in several human cancers [49] but is also prominent in endothelial cells [19] and vascular smooth muscle cells [50]. Gas6-activated Axl promotes tumor angiogenesis by stimulating the migration, proliferation, and survival of endothelial cells and vascular smooth muscle cells [21,51]. Inhibiting Gas6/Axl activation can dramatically suppress tumor angiogenesis [51,52]. The Akt, ERK and Rho GTPase pathways are also involved in Gas6/Axl-regulated endothelial cell survival and motility [21,53]. Because tumor angiogenesis is regulated by complex pathways in addition to VEGF/VEGFR2, suppressing VEGF/VEGFR2 signaling alone exerts only a modest antiangiogenic effect [7]. Combining therapies that target different pathways can lead to synergetic antiangiogenic effects. For example, the anti-Axl monoclonal antibody YW327.6S2 enhances the antiangiogenic effect of an anti-VEGF antibody on MDA-MB-231 and A549 xenografts [26]. The combination of bevacizumab with S49076, which is an inhibitor of MET, Axl and FGFR, exerts more potent antitumor effects on HT-29 xenografts compared with bevacizumab alone [27]. DAVLBH both inhibited the VEGF- and Gas6-induced activation of the Akt and ERK pathways and suppressed Rac1 and Cdc42 activities, which subsequently significantly altered the endothelial cell behaviours required for tumor angiogenesis. These results show that DAVLBH, a single chemical agent, can achieve overlapping antiangiogenic efficacy similar to that of combinational treatments with Gas6/Axl- and VEGF/VEGFR2-signaling-pathway inhibitors. However, the mechanism underlying the simultaneous suppression of VEGFR2 and Axl by DAVLBH remains unclear, and further study is needed.
Several recent studies have demonstrated that the Gas6/Axl and VEGF/VEGFR2 signaling pathways have complicated interactions. On the one hand, the VEGF-A-mediated activation of endothelial cell motility is antagonized by Gas6-induced Axl activation. This modulation of VEGF-A-mediated angiogenesis by Gas6/Axl may normalize angiogenesis [25]. On the other hand, Axl is essential for the VEGF-A-mediated activation of the PI3K/Akt pathway [23]. The Axl-specific inhibitor R428 significantly and consistently blocks tumor angiogenesis and VEGF-induced neovascularization [52]. In addition, recent reports have shown that Axl is in-volved in acquired resistance to VEGF/VEGFR-signaling-pathway inhibitors [27,54]. Thus, inhibiting the Axl pathway may be useful in overcoming resistance to anti-VEGF therapy. The combination of the Axl inhibitor S49076 with bevacizumab enhances the inhibitory effect of bevacizumab on the growth of bevacizumab-resistant LS-174T tumor xenografts [27]. Axl knockdown also has an additive effect with anti-VEGF to impair tube formation [21]. Because DAVLBH inhibited both the Gas6/Axl and VEGF/VEGFR2 signaling pathways, we hypothesized that DAVLBH might improve the therapeutic effect of VEGF/VEGFR2-signaling-pathway inhibitors. Our results demonstrated that DAVLBH inhibited the bevacizumab-stimulated expression of Axl. These results warrant further investigation into the potential of DAVLBH for the treatment of anti-VEGF-resistant tumors.
Pericytes play a critical role in the stabilization and maturation of neovessels in late-stage tumor angiogenesis by attaching to newly formed sprouts [55]. Both Gas6 and Axl are overexpressed in pericytes [20]. However, the influence of the Axl pathway on pericytes during angiogenesis remains unclear. Our results indicated that DAVLBH also inhibited Gas6-induced Axl expression and activation in pericytes and suppressed the Gas6-stimulated migration of pericytes towards the endothelial tubes. We also demonstrated that DAVLBH could reduce pericyte coverage in HeLa xenograft solid tumors. These results suggest that DAVLBH might inhibit not only the proliferation, migration, invasion and tube formation of HUVECs in the early stage of tumor angiogenesis but also the stabilization and maturation of the neovessels that pericytes regulate in late-stage tumor angiogenesis.
In this study, the antiangiogenic effect of DAVLBH was found to be associated with the inhibition of the activation of two transmembrane receptors, VEGFR2 and Axl, and their downstream signaling pathways. The human RTK phosphorylation antibody array indicated that, in addition to Axl and VEGFR2, the phosphorylation of Csk, Fgr, Fyn, Insulin R and EphB3 was down-regulated following DAVLBH treatment. Fyn, Fgr and Csk are cytoplasmic RTKs and affect angiogenesis through interactions with particular transmembrane receptors [56,57]. EphB3, which is a ligand for the EphB receptor, exhibits a complicated effect during angiogenesis and is not fully understood [58]. The role of Insulin R in angiogenesis is pleiotropic and has not been elucidated clearly [49]. We will also further investigate the roles of Csk, Fgr, Fyn, EphB3 and Insulin R in DAVLBH-induced angiogenesis inhibition in the future.
In conclusion, our results demonstrate that DAVLBH significantly inhibits angiogenesis-mediated HeLa xenograft tumor growth without causing body weight loss, and its antiangiogenic activity may be associated with the suppression of both the VEGF/VEGFR2 and Gas6/Axl pathways. This study provides persuasive evidence for the clinical development of DAVLBH into an antiangiogenic agent for the treatment of cervical cancer and other solid tumors.
Acknowledgements
We thank Mr. Ming-Han Xia (The First Affiliated Hospital of Jinan University) for his guidance with the immunohistochemical assay. This work was supported by the Science and Technology Program of China (2012ZX09103101-053), Guangzhou City (2011Y1-00017-11), the National Science Foundation of China (30901847 and 81573455), Guangdong Province (S2013050014183 and 2013CXZDA006), and the Program for New Century Excellent Talents in University (D. M. Zhang).
Disclosure of conflict of interest
None.
Abbreviations
- HUVECs
human umbilical vein endothelial cells
- VEGF
vascular endothelial growth factor
- VEGFR2
vascular endothelial growth factor receptor 2
- DAVLBH
desacetylvinblastine monohydrazide
- VLB
vinblastine
- ERK
extracellular signal-regulated kinase
- p-Axl
phospho-Axl
- p-VEGFR2
phospho-VEGFR2
- p-Akt
phospho-Akt
- p-ERK
phospho-ERK
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5- diphenyltetrazolium bromide
- Gas6
growth arrest-specific protein 6
- BSA
bovine serum albumin
- PDGFR-β
platelet-derived growth factor receptor-β
- MVD
microvessel density
- IOD
integrated optical density
References
- 1.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 2.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 3.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gavalas NG, Liontos M, Trachana SP, Bagratuni T, Arapinis C, Liacos C, Dimopoulos MA, Bamias A. Angiogenesis-related pathways in the pathogenesis of ovarian cancer. Int J Mol Sci. 2013;14:15885–15909. doi: 10.3390/ijms140815885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cao R, Eriksson A, Kubo H, Alitalo K, Cao Y, Thyberg J. Comparative evaluation of FGF-2-, VEGF-A-, and VEGF-C-induced angiogenesis, lymphangiogenesis, vascular fenestrations, and permeability. Circ Res. 2004;94:664–670. doi: 10.1161/01.RES.0000118600.91698.BB. [DOI] [PubMed] [Google Scholar]
- 6.Zachary I, Gliki G. Signaling transduction mechanisms mediating biological actions of the vascular endothelial growth factor family. Cardiovasc Res. 2001;49:568–581. doi: 10.1016/s0008-6363(00)00268-6. [DOI] [PubMed] [Google Scholar]
- 7.Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev. 2004;25:581–611. doi: 10.1210/er.2003-0027. [DOI] [PubMed] [Google Scholar]
- 8.Rousseau S, Houle F, Kotanides H, Witte L, Waltenberger J, Landry J, Huot J. Vascular endothelial growth factor (VEGF)-driven actinbased motility is mediated by VEGFR2 and requires concerted activation of stress-activated protein kinase 2 (SAPK2/p38) and geldanamycin-sensitive phosphorylation of focal adhesion kinase. J Biol Chem. 2000;275:10661–10672. doi: 10.1074/jbc.275.14.10661. [DOI] [PubMed] [Google Scholar]
- 9.Matsumoto T, Claesson-Welsh L. VEGF receptor signal transduction. Sci STKE. 2001;2001:re21. doi: 10.1126/stke.2001.112.re21. [DOI] [PubMed] [Google Scholar]
- 10.Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3:391–400. doi: 10.1038/nrd1381. [DOI] [PubMed] [Google Scholar]
- 11.Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007;6:273–286. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- 12.Keating GM. Bevacizumab: a review of its use in advanced cancer. Drugs. 2014;74:1891–1925. doi: 10.1007/s40265-014-0302-9. [DOI] [PubMed] [Google Scholar]
- 13.Imbulgoda A, Heng DY, Kollmannsberger C. Sunitinib in the treatment of advanced solid tumors. Recent Results Cancer Res. 2014;201:165–184. doi: 10.1007/978-3-642-54490-3_9. [DOI] [PubMed] [Google Scholar]
- 14.Hasskarl J. Sorafenib: targeting multiple tyrosine kinases in cancer. Recent Results Cancer Res. 2014;201:145–164. doi: 10.1007/978-3-642-54490-3_8. [DOI] [PubMed] [Google Scholar]
- 15.Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005;438:967–974. doi: 10.1038/nature04483. [DOI] [PubMed] [Google Scholar]
- 16.Thanapprapasr D, Hu W, Sood AK, Coleman RL. Moving beyond VEGF for anti-angiogenesis strategies in gynecologic cancer. Curr Pharm Des. 2012;18:2713–2719. doi: 10.2174/138161212800626201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gustafsson A, Bostrom AK, Ljungberg B, Axelson H, Dahlback B. Gas6 and the receptor tyrosine kinase Axl in clear cell renal cell carcinoma. PLoS One. 2009;4:e7575. doi: 10.1371/journal.pone.0007575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nagata K, Ohashi K, Nakano T, Arita H, Zong C, Hanafusa H, Mizuno K. Identification of the product of growth arrest-specific gene 6 as a common ligand for Axl, Sky, and Mer receptor tyrosine kinases. J Biol Chem. 1996;271:30022–30027. doi: 10.1074/jbc.271.47.30022. [DOI] [PubMed] [Google Scholar]
- 19.Avanzi GC, Gallicchio M, Bottarel F, Gammaitoni L, Cavalloni G, Buonfiglio D, Bragardo M, Bellomo G, Albano E, Fantozzi R, Garbarino G, Varnum B, Aglietta M, Saglio G, Dianzani U, Dianzani C. GAS6 inhibits granulocyte adhesion to endothelial cells. Blood. 1998;91:2334–2340. [PubMed] [Google Scholar]
- 20.Collett G, Wood A, Alexander MY, Varnum BC, Boot-Handford RP, Ohanian V, Ohanian J, Fridell YW, Canfield AE. Receptor tyrosine kinase Axl modulates the osteogenic differentiation of pericytes. Circ Res. 2003;92:1123–1129. doi: 10.1161/01.RES.0000074881.56564.46. [DOI] [PubMed] [Google Scholar]
- 21.Li Y, Ye X, Tan C, Hongo JA, Zha J, Liu J, Kallop D, Ludlam MJ, Pei L. Axl as a potential therapeutic target in cancer: role of Axl in tumor growth, metastasis and angiogenesis. Oncogene. 2009;28:3442–3455. doi: 10.1038/onc.2009.212. [DOI] [PubMed] [Google Scholar]
- 22.Hasanbasic I, Rajotte I, Blostein M. The role of gamma-carboxylation in the anti-apoptotic function of gas6. J Thromb Haemost. 2005;3:2790–2797. doi: 10.1111/j.1538-7836.2005.01662.x. [DOI] [PubMed] [Google Scholar]
- 23.Ruan GX, Kazlauskas A. Axl is essential for VEGF-A-dependent activation of PI3K/Akt. EMBO J. 2012;31:1692–1703. doi: 10.1038/emboj.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stenhoff J, Dahlback B, Hafizi S. Vitamin K-dependent Gas6 activates ERK kinase and stimulates growth of cardiac fibroblasts. Biochem Biophys Res Commun. 2004;319:871–878. doi: 10.1016/j.bbrc.2004.05.070. [DOI] [PubMed] [Google Scholar]
- 25.Gallicchio M, Mitola S, Valdembri D, Fantozzi R, Varnum B, Avanzi GC, Bussolino F. Inhibition of vascular endothelial growth factor receptor 2-mediated endothelial cell activation by Axl tyrosine kinase receptor. Blood. 2005;105:1970–1976. doi: 10.1182/blood-2004-04-1469. [DOI] [PubMed] [Google Scholar]
- 26.Ye X, Li Y, Stawicki S, Couto S, Eastham-Anderson J, Kallop D, Weimer R, Wu Y, Pei L. An anti-Axl monoclonal antibody attenuates xenograft tumor growth and enhances the effect of multiple anticancer therapies. Oncogene. 2010;29:5254–5264. doi: 10.1038/onc.2010.268. [DOI] [PubMed] [Google Scholar]
- 27.Burbridge MF, Bossard CJ, Saunier C, Fejes I, Bruno A, Leonce S, Ferry G, Da Violante G, Bouzom F, Cattan V, Jacquet-Bescond A, Comoglio PM, Lockhart BP, Boutin JA, Cordi A, Ortuno JC, Pierre A, Hickman JA, Cruzalegui FH, Depil S. S49076 is a novel kinase inhibitor of MET, AXL, and FGFR with strong preclinical activity alone and in association with bevacizumab. Mol Cancer Ther. 2013;12:1749–1762. doi: 10.1158/1535-7163.MCT-13-0075. [DOI] [PubMed] [Google Scholar]
- 28.Owellen RJ, Hartke CA, Dickerson RM, Hains FO. Inhibition of tubulin-microtubule polymerization by drugs of the Vinca alkaloid class. Cancer Res. 1976;36:1499–1502. [PubMed] [Google Scholar]
- 29.Barnett CJ, Cullinan GJ, Gerzon K, Hoying RC, Jones WE, Newlon WM, Poore GA, Robison RL, Sweeney MJ, Todd GC, Dyke RW, Nelson RL. Structure-activity relationships of dimeric Catharanthus alkaloids. 1. Deacetylvinblastine amide (vindesine) sulfate. J Med Chem. 1978;21:88–96. doi: 10.1021/jm00199a016. [DOI] [PubMed] [Google Scholar]
- 30.Reddy JA, Dorton R, Westrick E, Dawson A, Smith T, Xu LC, Vetzel M, Kleindl P, Vlahov IR, Leamon CP. Preclinical evaluation of EC145, a folate-vinca alkaloid conjugate. Cancer Res. 2007;67:4434–4442. doi: 10.1158/0008-5472.CAN-07-0033. [DOI] [PubMed] [Google Scholar]
- 31.Baudin B, Bruneel A, Bosselut N, Vaubourdolle M. A protocol for isolation and culture of human umbilical vein endothelial cells. Nat Protoc. 2007;2:481–485. doi: 10.1038/nprot.2007.54. [DOI] [PubMed] [Google Scholar]
- 32.Zhang DM, Liu JS, Tang MK, Yiu A, Cao HH, Jiang L, Chan JY, Tian HY, Fung KP, Ye WC. Bufotalin from Venenum Bufonis inhibits growth of multidrug resistant HepG2 cells through G2/M cell cycle arrest and apoptosis. Eur J Pharmacol. 2012;692:19–28. doi: 10.1016/j.ejphar.2012.06.045. [DOI] [PubMed] [Google Scholar]
- 33.Yi T, Yi Z, Cho SG, Luo J, Pandey MK, Aggarwal BB, Liu M. Gambogic acid inhibits angiogenesis and prostate tumor growth by suppressing vascular endothelial growth factor receptor 2 signaling. Cancer Res. 2008;68:1843–1850. doi: 10.1158/0008-5472.CAN-07-5944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pang X, Yi Z, Zhang X, Sung B, Qu W, Lian X, Aggarwal BB, Liu M. Acetyl-11-keto-betaboswellic acid inhibits prostate tumor growth by suppressing vascular endothelial growth factor receptor 2-mediated angiogenesis. Cancer Res. 2009;69:5893–5900. doi: 10.1158/0008-5472.CAN-09-0755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi JM, Bai LL, Zhang DM, Yiu A, Yin ZQ, Han WL, Liu JS, Li Y, Fu DY, Ye WC. Saxifragifolin D induces the interplay between apoptosis and autophagy in breast cancer cells through ROSdependent endoplasmic reticulum stress. Biochem Pharmacol. 2013;85:913–926. doi: 10.1016/j.bcp.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 36.Darland DC, D’Amore PA. TGF beta is required for the formation of capillary-like structures in three-dimensional cocultures of 10T1/2 and endothelial cells. Angiogenesis. 2001;4:11–20. doi: 10.1023/a:1016611824696. [DOI] [PubMed] [Google Scholar]
- 37.Korshunov VA. Axl-dependent signalling: a clinical update. Clin Sci (Lond) 2012;122:361–368. doi: 10.1042/CS20110411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Claesson-Welsh L, Welsh M. VEGFA and tumour angiogenesis. J Intern Med. 2013;273:114–127. doi: 10.1111/joim.12019. [DOI] [PubMed] [Google Scholar]
- 39.Goruppi S, Ruaro E, Varnum B, Schneider C. Gas6-mediated survival in NIH3T3 cells activates stress signalling cascade and is independent of Ras. Oncogene. 1999;18:4224–4236. doi: 10.1038/sj.onc.1202788. [DOI] [PubMed] [Google Scholar]
- 40.Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 41.Vacca A, Iurlaro M, Ribatti D, Minischetti M, Nico B, Ria R, Pellegrino A, Dammacco F. Antiangiogenesis is produced by nontoxic doses of vinblastine. Blood. 1999;94:4143–4155. [PubMed] [Google Scholar]
- 42.Schwartz EL. Antivascular actions of microtubule-binding drugs. Clin Cancer Res. 2009;15:2594–2601. doi: 10.1158/1078-0432.CCR-08-2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meissner M, Pinter A, Michailidou D, Hrgovic I, Kaprolat N, Stein M, Holtmeier W, Kaufmann R, Gille J. Microtubule-targeted drugs inhibit VEGF receptor-2 expression by both transcriptional and post-transcriptional mechanisms. J Invest Dermatol. 2008;128:2084–2091. doi: 10.1038/jid.2008.37. [DOI] [PubMed] [Google Scholar]
- 44.Bijman MN, van Nieuw Amerongen GP, Laurens N, van Hinsbergh VW, Boven E. Microtubule-targeting agents inhibit angiogenesis at subtoxic concentrations, a process associated with inhibition of Rac1 and Cdc42 activity and changes in the endothelial cytoskeleton. Mol Cancer Ther. 2006;5:2348–2357. doi: 10.1158/1535-7163.MCT-06-0242. [DOI] [PubMed] [Google Scholar]
- 45.Leamon CP, Vlahov IR, Reddy JA, Vetzel M, Santhapuram HK, You F, Bloomfield A, Dorton R, Nelson M, Kleindl P, Vaughn JF, Westrick E. Folate-vinca alkaloid conjugates for cancer therapy: a structure-activity relationship. Bioconjug Chem. 2014;25:560–568. doi: 10.1021/bc400441s. [DOI] [PubMed] [Google Scholar]
- 46.Jamora C, Fuchs E. Intercellular adhesion, signalling and the cytoskeleton. Nat Cell Biol. 2002;4:E101–108. doi: 10.1038/ncb0402-e101. [DOI] [PubMed] [Google Scholar]
- 47.Merajver SD, Usmani SZ. Multifaceted role of Rho proteins in angiogenesis. J Mammary Gland Biol Neoplasia. 2005;10:291–298. doi: 10.1007/s10911-006-9002-8. [DOI] [PubMed] [Google Scholar]
- 48.Laramee M, Chabot C, Cloutier M, Stenne R, Holgado-Madruga M, Wong AJ, Royal I. The scaffolding adapter Gab1 mediates vascular endothelial growth factor signaling and is required for endothelial cell migration and capillary formation. J Biol Chem. 2007;282:7758–7769. doi: 10.1074/jbc.M611327200. [DOI] [PubMed] [Google Scholar]
- 49.Verma A, Warner SL, Vankayalapati H, Bearss DJ, Sharma S. Targeting Axl and Mer kinases in cancer. Mol Cancer Ther. 2011;10:1763–1773. doi: 10.1158/1535-7163.MCT-11-0116. [DOI] [PubMed] [Google Scholar]
- 50.Fridell YW, Villa J Jr, Attar EC, Liu ET. GAS6 induces Axl-mediated chemotaxis of vascular smooth muscle cells. J Biol Chem. 1998;273:7123–7126. doi: 10.1074/jbc.273.12.7123. [DOI] [PubMed] [Google Scholar]
- 51.Holland SJ, Powell MJ, Franci C, Chan EW, Friera AM, Atchison RE, McLaughlin J, Swift SE, Pali ES, Yam G, Wong S, Lasaga J, Shen MR, Yu S, Xu W, Hitoshi Y, Bogenberger J, Nor JE, Payan DG, Lorens JB. Multiple roles for the receptor tyrosine kinase axl in tumor formation. Cancer Res. 2005;65:9294–9303. doi: 10.1158/0008-5472.CAN-05-0993. [DOI] [PubMed] [Google Scholar]
- 52.Holland SJ, Pan A, Franci C, Hu Y, Chang B, Li W, Duan M, Torneros A, Yu J, Heckrodt TJ, Zhang J, Ding P, Apatira A, Chua J, Brandt R, Pine P, Goff D, Singh R, Payan DG, Hitoshi Y. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010;70:1544–1554. doi: 10.1158/0008-5472.CAN-09-2997. [DOI] [PubMed] [Google Scholar]
- 53.Laurance S, Aghourian MN, Jiva Lila Z, Lemarie CA, Blostein MD. Gas6-induced tissue factor expression in endothelial cells is mediated through caveolin-1-enriched microdomains. J Thromb Haemost. 2014;12:395–408. doi: 10.1111/jth.12481. [DOI] [PubMed] [Google Scholar]
- 54.Sennino B, McDonald DM. Controlling escape from angiogenesis inhibitors. Nat Rev Cancer. 2012;12:699–709. doi: 10.1038/nrc3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stratman AN, Malotte KM, Mahan RD, Davis MJ, Davis GE. Pericyte recruitment during vasculogenic tube assembly stimulates endothelial basement membrane matrix formation. Blood. 2009;114:5091–5101. doi: 10.1182/blood-2009-05-222364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Okada M. Regulation of the SRC family kinases by Csk. Int J Biol Sci. 2012;8:1385–1397. doi: 10.7150/ijbs.5141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang X, Simerly C, Hartnett C, Schatten G, Smithgall TE. Src-family tyrosine kinase activities are essential for differentiation of human embryonic stem cells. Stem Cell Res. 2014;13:379–389. doi: 10.1016/j.scr.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Adams RH, Wilkinson GA, Weiss C, Diella F, Gale NW, Deutsch U, Risau W, Klein R. Roles of ephrinB ligands and EphB receptors in cardiovascular development: demarcation of arterial/venous domains, vascular morphogenesis, and sprouting angiogenesis. Genes Dev. 1999;13:295–306. doi: 10.1101/gad.13.3.295. [DOI] [PMC free article] [PubMed] [Google Scholar]