

Nanoparticles are promising structures for targeted drug delivery and molecular imaging because the size, shape, composition, and surface chemistry of the nanocarrier can be designed to control nanoparticle behavior in vivo.[1,2] The nanocarrier aspect ratio (AR, defined as length divided by width) is a critical design parameter, but its impact on cell targeting specificity is not well understood due to conflicting data.[3–9] We have therefore developed a novel, bottom-up approach for the synthesis of nanoparticles (AR1), dimers (AR2), and assemblies (AR3–AR8). We focused on different iterations of dimers and individual nanoparticles that target cancer cells and show that the uptake of these structures is AR-dependent. They were formed by generating multifunctional assemblies containing targeting ligands (specifically cyclic RGD peptides that target integrins overexpressed on cancer cells) and fluorescent dyes (for optical imaging) that were either displayed homogeneously on the dimer or restricted to a single nanoparticle component. These formulations were compared with untargeted assemblies and individual particles. We found that symmetrical rather than topo-selective presentation of RGD peptides and the dimerization of nanoparticles (AR2) increased the efficiency of cancer cell targeting fourfold compared with individual particles (AR1).
The development pipeline for nanoparticle platform technologies is expanding rapidly but little is known about the fundamental principles of nanomaterial–cell interactions. The size, shape, composition, and surface chemistry of the nanocarrier are known to affect its biodistribution, interaction with cells, and intracellular trafficking. For example, mammalian cell membranes are negatively charged, thus positively charged nanomaterials interact with them more strongly than their negatively charged counterparts.[3,9,10] PEGylation can be used to camouflage nanoparticles and inhibit cellular uptake, and receptor targeting is an effective strategy to enhance cell interactions and to confer cell-type specificity.[11] However, it is unclear how nonspherical nanomaterials interact with cells, i.e., some reports indicate that higher-AR materials interact with cells more efficiently,[3,9] whereas other reports disagree.[5–8] As well as potential differences in the experimental setup, the comparison of shape-based effects is challenging because the surface properties of the nanoparticles also differ. For example, the zeta potential of gold rods (+17 to +24 mV) differs significantly from that of gold spheres (−38 to −18 mV).[9] A method is needed that avoids confounding variables such as surface charge, allowing parameters such as size, shape, and flexibility to be evaluated in isolation.
We therefore synthesized plant virus-based nanoparticle assemblies with diverse but defined ARs by linking multiple copies of identical particles to maintain the charge and surface properties while varying the shape. The application of viruses in the medical sector is gaining recognition and many novel types of viruses, including plant viruses, are undergoing preclinical development.[12–14] Plant viruses are natural, biodegradable carrier systems that can be produced on a large scale in plants, but they do not pose a risk of infection in mammals. They can easily be modified by genetic engineering or chemical conjugation, enabling the well-ordered, multivalent display of functional groups on their external and internal surfaces.[15] We used Cowpea mosaic virus (CPMV) as a model system. The icosahedral capsid comprises 60 copies of an asymmetric unit composed of large (L) and small (S) coat protein subunits and has pseudo T = 3 symmetry.
Particle cluster formation was tightly controlled to prevent aggregation. This was achieved by initially producing symmetry-broken (Janus-type) particles containing functional groups on one side only.[16] Two methods were used, as shown in Scheme 1 (detailed experimental procedures are provided in the Supporting Information). Briefly, in the first method, surface lysine residues were converted into thiols[17] and the particles were bound to a thiol-activated solid-phase support through the formation of disulfide bonds, allowing the remaining unbound thiols to be pacified with iodoacetic acid (IAA)[18] before the symmetry-broken particles were released under reducing conditions to yield asymmetric CPMV nanoparticles with topo-selective thiols. In the second method, surface lysine residues were bound to homobifunctional polyethylene glycol–N-hydroxysuccinimide ester (PEG–NHS) linkers containing disulfide bonds that had previously been reacted with an amine-functionalized solid support. Reduction of the disulfide bonds with tris(2-carboxyethyl)phosphine (TCEP) produced asymmetric particles with thiol groups on one side.
Scheme 1.
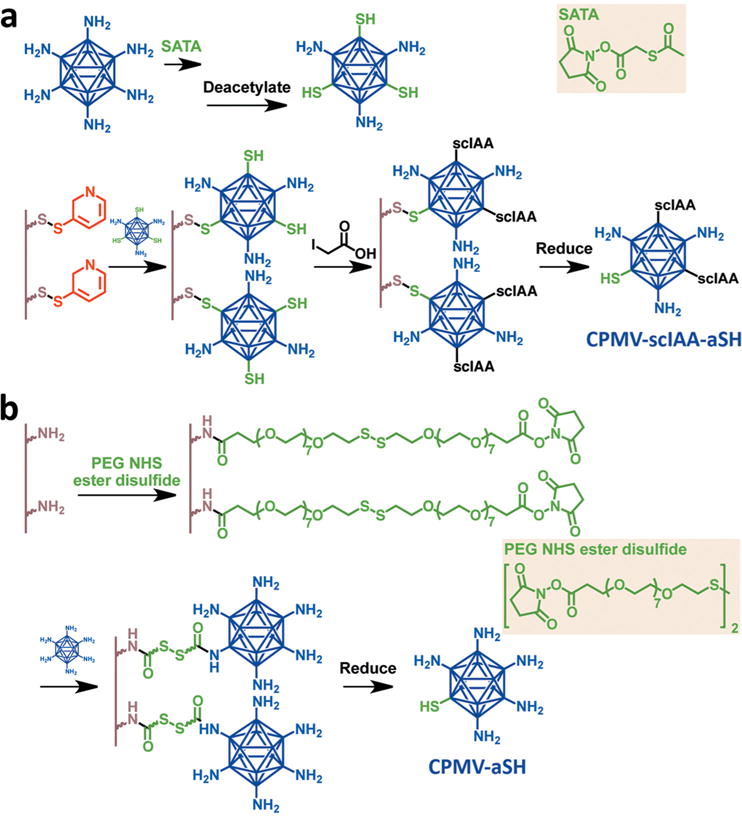
Symmetry-breaking of CPMV particles using solid-state supports. a) Method 1: thiol groups are introduced, CPMV is bound to the support, and free thiols are pacified. b) Method 2: CPMV is initially bound to the support using a linker with a disulfide bond and then released by reduction to expose thiol groups.
The lysine residues are close to the surface of the CPMV particle (see Figure S1, Supporting Information), so the efficiency of conjugation to the solid support depends on the linker size. Although efficient binding was achieved using the homobifunctional PEG–NHS ester linker, which contains seven PEG spacers on either side of the disulfide bond and therefore measures 10 nm in length, negligible binding was achieved using the shorter 3,3′-dithiobis(sulfosuccinimidylpropionate) (DTSSP) linker lacking a PEG spacer (1.2 nm). Both methods yielded symmetry-broken nanoparticles (Figure 2), but we favored the second method because more lysines were available for downstream functionalization and the PEG spacer enhanced the coupling efficiency when the nanoparticles were joined together into mesoscale assemblies.
Figure 2.
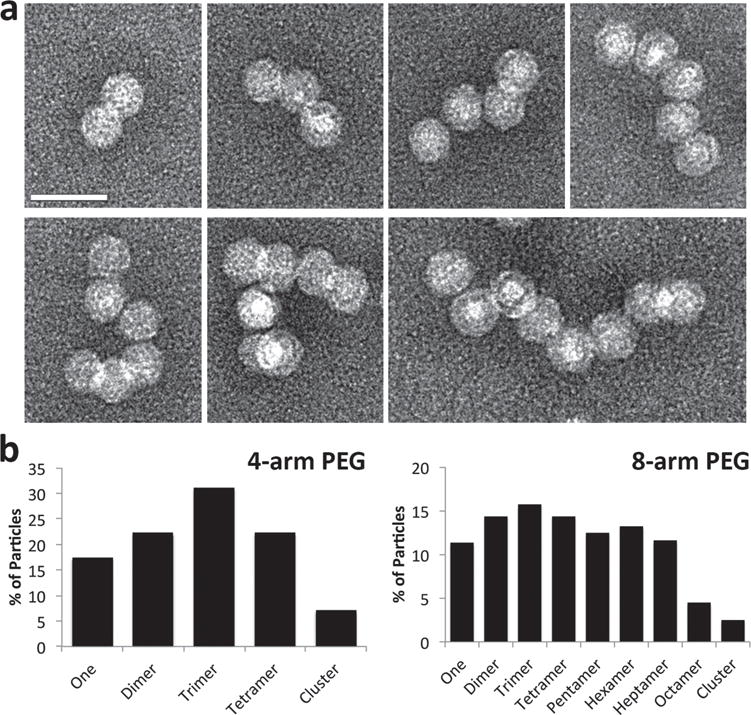
Formation of networks and chains using the four-arm and eight-arm PEG linkers. a) TEM images showing CPMV nanoparticle arrangements in the range AR2–AR8 assembled using eight-arm PEG maleimide. Scale bar = 50 nm. b) Statistics show percentage of particles found in clusters of various aspect ratios, based on the observation of at least 250 particles and with relaxed requirements allowing particles to be within 30 nm of each other.
The number of thiol groups on each particle was determined by conjugating maleimide-functionalized fluorophores followed by UV/visible spectroscopy. This revealed an average of five thiol groups per particle after binding and release from the resin (Figures S2 and S3, Supporting Information). We propose that these thiols are displayed around a single fivefold axis of the CPMV particle near the interface of the small and large coat proteins, where the most reactive lysine residues 38 and 99 are located (Figure S1, Supporting Information). Symmetry-breaking and the topo-selective display of thiols was confirmed by conjugating biotin labels to solvent-exposed thiol groups on symmetrical and asymmetrical CPMV nanoparticles followed by the addition of avidin or immunogold staining (Figure 1). Visual inspection and transmission electron microscopy (TEM) showed that the symmetrical particles formed large aggregates when presented with avidin, reflecting continuous chains of avidin–biotin interactions, whereas the symmetry-broken CPMV nanoparticles remained well dispersed. Due to the directionality conferred by symmetry-breaking, no other interactions can occur after avidin has bound up to four asymmetric CPMV, thus preventing aggregation. Based on steric hindrance between avidin and symmetry-broken CPMV, it is unlikely that oligomerization occurs either. Formation of dimers or oligomers was not observed. Immunogold staining using 5-nm gold-labeled anti-biotin antibodies demonstrated universal binding to the symmetrical CPMV particles but unilateral binding to the symmetry-broken particles (Figure 1 and Figure S4, Supporting Information). The degree of labeling was quantified over multiple images, with an average of five gold particles surrounding the symmetric particles and 0.5 surrounding the asymmetric particles. The orientation of the particles upon adsorption to the TEM grid determined whether or not the biotin was exposed and available for immunogold labeling, which explains why the average for the asymmetric particles is <1.
Figure 1.
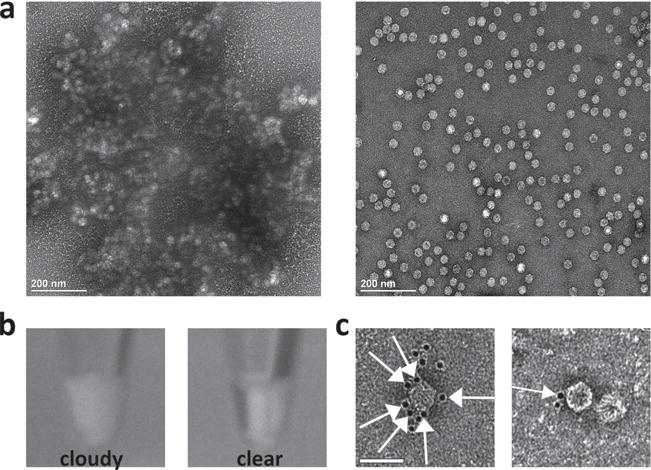
Characterization of symmetry-broken particles. a) TEM images show that avidin causes the aggregation of symmetric CPMV particles modified with biotin (left) but not the asymmetric CPMV particles displaying topo-selective biotin (right). b) Visual inspection of the avidin-containing samples confirmed the aggregation of symmetric CPMV particles (left), but not asymmetric CPMV particles (right). c) Immunogold staining using gold-labeled anti-biotin antibodies showed universal labeling of symmetric CPMV particles modified with biotin (left), but unilateral labeling of asymmetric CPMV particles displaying topo-selective biotin (right). Orange arrows show the localization of the gold particles. Scale bar = 50 nm.
It is possible that the asymmetric particles can associate over time through formation of disulfide bonds between thiol groups, but no significant association was observed by TEM even after several months of storage. This potentially reflects the position of the thiol groups in the cavity formed where the small and large coat proteins assemble, thus reducing their exposure and availability for interaction (Figure S1, Supporting Information).
The asymmetric CPMV nanoparticles were used as building blocks for the self-assembly of structures ranging from AR2 to AR8. Dimers (AR2) were produced using different excess amounts of homobifunctional maleimide PEG linkers with a molecular weight of 2000 Da. Subsequent TEM analysis showed that dimer formation occurred in the presence of a tenfold molar excess of linker. The reaction, imaging, and statistical analysis were replicated several times, in each case counting at least 250 particles. There was little variation in the results, with each reaction producing an ≈80% yield of CPMV dimers (Figure S5, Supporting Information).
To form larger structures, we used four-arm and eight-arm PEG maleimide linkers with molecular weights of 20 and 40 kDa, respectively (Figure S6, Supporting Information). Each arm was 5000 Da (≈50 nm in length with a Flory dimension RF = 6 nm), so that there was enough spacing between the arms for multiple CPMV particles to assemble side-by-side. This was particularly important for the eight-arm PEG linker because there is only 5.6 Å along the backbone between each arm. An exact stoichiometric ratio of four symmetry-broken CPMV particles per four-arm linker and eight per eight-arm linker was used in the coupling reactions. Although lower excesses of the linkers resulted in many free particles, greater excesses resulted in less efficient chain and network formation because the particles bound to separate linkers. The reactions were repeated several times as above and monitored by TEM (Figure 2 and Figure S7, Supporting Information).
The multiple-arm PEG linkers produced a distribution of assemblies ranging from AR2 to AR8 with consistent results among different batches (a minimum of 250 particles was counted for each batch). The histograms appear to resemble a Poisson distribution, with the greatest proportion of the particles coordinated into dimers or trimers for the four-arm PEG and mainly trimers for the eight-arm PEG. There is a good representation of each arrangement of particles, with at least 20% of each type of cluster for the four-arm PEG and at least 10% of each for the eight-arm PEG (Figure 2). We attempted to purify the assemblies by density gradient centrifugation, size exclusion chromatography, and electrophoresis but have not yet optimized the methods to produce acceptable yields of purified nanoparticle assemblies with distinct aspect ratios. Nevertheless, CPMV dimers could be produced with a yield of 80%, allowing us to compare AR1 versus AR2 particles in terms of the efficiency of cellular uptake.
AR1 and AR2 nanoparticles were synthesized and modified with the near infrared dye AlexaFluor 647 (A647) for imaging and the cyclic RGD peptide for cell targeting. RGD is a well-known peptide motif that effectively targets cells displaying integrins αvβ3 and αvβ5, which are overexpressed on many cancer cell types and promote malignancy by stimulating angiogenesis and metastasis.[19] We have previously shown that CPMV nanoparticles displaying RGD peptides are effectively targeted to cancer cells.[20]
Dimers displaying A647 with and without RGD were produced using the three-step process shown in Figure S8 (detailed experimental procedures are provided in the Supporting Information). Briefly, CPMV was first labeled with A647 and/or RGD using a combination of NHS ester chemistry and copper(I)-catalyzed azide–alkyne cycloaddition. The particles were functionalized with dyes before labeling with RGD to ensure the extent of dye labeling was consistent for CPMV-A647 and CPMV-A647-RGD particles. UV/visible spectroscopy revealed that 20 dye molecules were attached per particle, agarose gel electrophoresis confirmed successful labeling of the particles with dyes and RGD, and SDS-PAGE followed by densitometric analysis indicated the presence of 20–30 RGD ligands per particle (Figure S9, Supporting Information). Particles displaying dyes and RGD at a low density allowed the formation of symmetry-broken particles in the next step using method 2 as described above, resulting in similar yields as achieved using unmodified particles. Finally, the particles were linked together to form homodimers using PEG linkers as described above. Heterodimer formation was achieved by first reacting CPMV-A647-aSH with a large, 1000 molar, excess of the bifunctional linker to saturate binding sites and prevent dimer formation, followed by purifying the particles after 1 h to remove excess linker, then reacting with CPMV-RGD-aSH (see Figures S8 and S11, Supporting Information). The resulting library of dimers included homodimers of CPMV-A647 and CPMV-A647-RGD as well as CPMV-A647/CPMV-A647-RGD heterodimers. TEM imaging confirmed the formation of dimers with yields of ≈80% (Figures S10 and S11, Supporting Information).
Flow cytometry was carried out using HeLa cells, a cervical cancer cell line expressing αvβ3 and αvβ5,[21] to compare the efficiency of cellular uptake for the different formulations of single particles and dimers (Figure 3). Single particles were added to the cells at a concentration of 100 000 particles per cell. The dimers were added at two different concentrations: one to ensure that the CPMV concentration was the same across all formulations (i.e., one dimer corresponding to two single particles, denoted as 1:2) and the other in which the nanoparticle concentration was kept constant (i.e., one dimer corresponding to one single particle, denoted as 1:1). In other words, 1:2 signifies that 50 000 dimers were added per cell (equivalent to 100 000 CPMV particles per cell for each formulation), while 1:1 indicates that 100 000 CPMV dimers were added per cell. This was particularly important for comparisons involving the CPMV-A647/CPMV-A647-RGD heterodimer because the dye concentration would be half that of all the other formulations if the CPMV concentration is kept constant.
Figure 3.
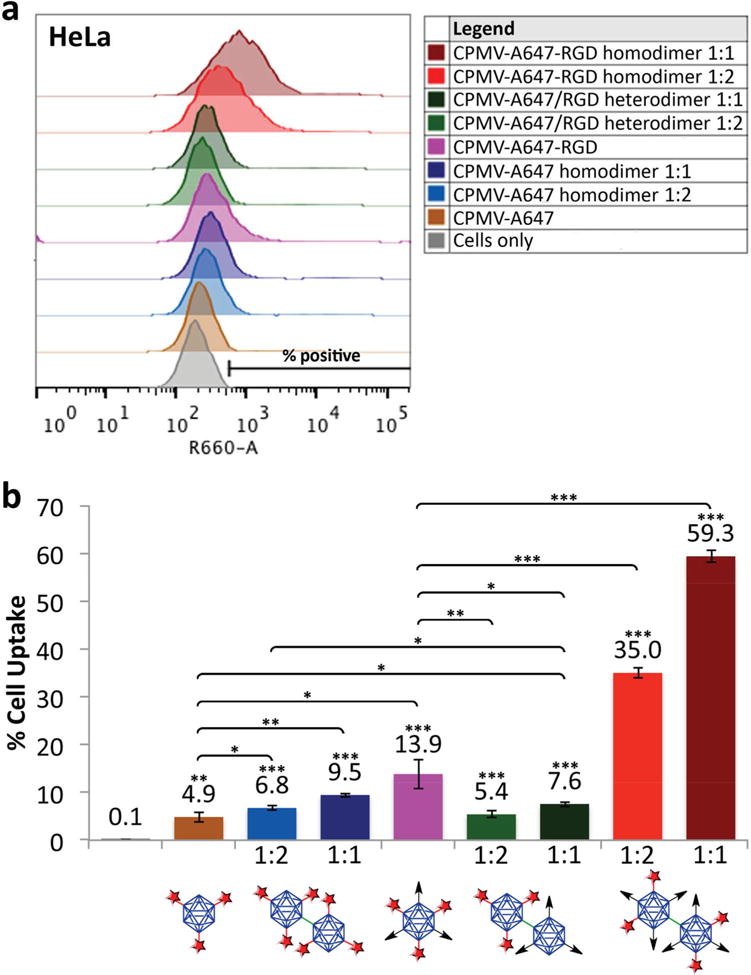
Flow cytometry histograms showing the efficiency of cellular uptake. The ratios 1:2 and 1:1 indicate the ratio of dimers:single CPMV nanoparticles. a) Cells to the right of the cells-only control region were considered positive for particle uptake. b) Proportion of positive cells shown as percentages (*p < 0.05, **p < 0.01, ***p < 0.001). Unpaired asterisks denote statistical significance compared with the cells-only control. CPMV with red star denotes CPMV-A647, CPMV with black arrow denotes CPMV-RGD.
We found that the addition of RGD even at low densities increased the efficiency of cell targeting threefold when focusing on single-particle formulations, reflecting the presence of integrins αvβ3 and αvβ5 on the surface of HeLa cells. We then compared homodimers (AR2) to their equivalent single-particle formulations (AR1). The CPMV-A647 and CPMV-A647-RGD dimers were both taken up more efficiently than their single-particle counterparts, for example, the uptake of homodimeric CPMV-A647-RGD was >2.5-fold more efficient than the corresponding single particles. These observations probably reflect the combined influence of size, AR, and ligand density. Competition between hydrodynamic driving forces and receptor diffusion kinetics ensures that the optimal particle radius for cellular uptake is 30 nm (viruses were used as model systems in several of these studies).[22,23] CPMV has a radius of 15 nm, so dimers (60 nm × 30 nm) lie closer to the optimum and are likely to be internalized preferentially.
We next investigated the behavior of the CPMV-A647/CPMV-A647-RGD heterodimer and found that it was internalized more efficiently than the CPMV-A647 single particles/homodimers due to the presence of RGD, but less efficiently than CPMV-A647-RGD single particles. The other half of the dimer may actually be in the way of some of the RGD displayed by the heterodimer, thus resulting in less ligand–receptor interactions compared with the CPMV-A647-RGD single particles. It is also likely that the symmetric distribution of ligands increases the probability of interaction with integrin, whereas the unilateral presentation of the ligand makes such interactions orientation-dependent. The CPMV-A647-RGD homodimer outperformed the heterodimer to an even greater extent, with 6.5–7.8 times more efficient uptake. This probably reflected a combination of symmetrical presentation, the larger overall number of ligands, and the optimal size of the particle. A larger number of ligands increases the avidity of the particle and thus promotes internalization.[24] Cells are typically 10–100 times larger than nanoparticles, so the cell surface tends to be relatively flat in comparison. The homodimer structure may therefore interact with a larger number of binding sites on the cell surface, thus increasing targeting sensitivity and specificity. In addition, as membrane wrapping occurs, the CPMV-A647-RGD homodimer presents more sites for attachment to induce internalization along with receptor clustering and diffusion. We performed competition assays using free RGD ligand to confirm that CPMV-RGD monomers and dimers are indeed targeted to the cells, and that cell uptake is a result of integrin receptor binding (Figure S13, Supporting Information). Furthermore, colloidal stability of monomers versus dimers suspended in cell culture media was confirmed (Figure S12, Supporting Information), therefore, ruling out that increased cell uptake of dimers is caused by aggregation or sedimentation. From these observations, we conclude that enhanced uptake was achieved as a synergistic contribution of receptor targeting, spatial orientation of targeting ligands, and aspect ratio (AR2 > AR1).
In summary, we have successfully engineered symmetry-broken CPMV particles and used a collection of PEG-based polymers to link the particles together to form novel assemblies, allowing the behavior of multifunctional dimers to be investigated in vitro. We are currently focusing on the separation of different particle formulations because purification is necessary to achieve large-scale synthesis for downstream applications. State-of-the-art methods for nanoparticle assembly use solid-state, layer-by-layer approaches that are inefficient and produce low yields of longer chains. The ability to synthesize libraries of networks in one reaction followed by the separation of individual components would therefore be ideal for future nanotechnology applications.
Multifunctional assemblies were formed displaying targeting ligands (specifically cyclic RGD peptides that bind to integrins overexpressed on cancer cells) and fluorescent dyes (for optical imaging) either homogeneously or concentrated on a single nanoparticle component. We compared functionally equivalent dimers and monomers with either symmetric or topo-selective RGD display and found that dimers symmetrically displaying the peptide-targeted cancer cells more efficiently than corresponding monomers and topo-selective dimers. This indicated that cancer cell targeting is promoted by synergistic enhancement based on a combination of optimized receptor targeting and nanoparticle AR. The rules for effective nanocarrier design are complex because the nanocarrier interacts with multiple binding partners, cells, and biological barriers on its way to the target site. Understanding the impact of AR on cell targeting is only one piece of this puzzle.
Supplementary Material
Acknowledgments
This work was funded by a grant from the National Science Foundation, CMMI NM 333651 (to N.F.S.).
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Amy M. Wen, Department of Biomedical Engineering, Schools of Medicine and Engineering, Case Western Reserve University, 10900 Euclid Avenue, Cleveland, Ohio 44106, USA
Prof. Nicole F. Steinmetz, Email: nicole.steinmetz@case.edu, Departments of Biomedical Engineering, Radiology, Materials Science and Engineering & Macromolecular Science and Engineering, Schools of Medicine and Engineering, Case Western Reserve University, 10900 Euclid Avenue, Cleveland, Ohio 44106, USA.
References
- 1.Decuzzi P, Godin B, Tanaka T, Lee SY, Chiappini C, Liu X, Ferrari M. J Controlled Release. 2010;141:320. doi: 10.1016/j.jconrel.2009.10.014. [DOI] [PubMed] [Google Scholar]
- 2.Wen AM, Rambhia PH, French RH, Steinmetz NF. J Biol Phys. 2013;39:301. doi: 10.1007/s10867-013-9314-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gratton SEA, Ropp PA, Pohlhaus PD, Luft JC, Madden VJ, Napier ME, DeSimone JM. Proc Natl Acad Sci USA. 2008;105:11613. doi: 10.1073/pnas.0801763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barua S, Yoo JW, Kolhar P, Wakankar A, Gokarn YR, Mitragotri S. Proc Natl Acad Sci USA. 2013;110:3270. doi: 10.1073/pnas.1216893110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nan A, Bai X, Son SJ, Lee SB, Ghandehari H. Nano Lett. 2008;8:2150. doi: 10.1021/nl0802741. [DOI] [PubMed] [Google Scholar]
- 6.Chithrani BD, Chan WCW. Nano Lett. 2007;7:1542. doi: 10.1021/nl070363y. [DOI] [PubMed] [Google Scholar]
- 7.Chithrani BD, Ghazani AA, Chan WCW. Nano Lett. 2006;6:662. doi: 10.1021/nl052396o. [DOI] [PubMed] [Google Scholar]
- 8.Schaeublin NM, Braydich-Stolle LK, Maurer EI, Park K, MacCuspie RI, Afrooz AR, Vaia RA, Saleh NB, Hussain SM. Langmuir. 2012;28:3248. doi: 10.1021/la204081m. [DOI] [PubMed] [Google Scholar]
- 9.Arnida, Malugin A, Ghandehari H. J Appl Toxicol. 2010;30:212. doi: 10.1002/jat.1486. [DOI] [PubMed] [Google Scholar]
- 10.Arnida, Janát-Amsbury MM, Ray A, Peterson CM, Ghandehari H. Eur J Pharm Biopharm. 2011;77:417. doi: 10.1016/j.ejpb.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hillaireau H, Couvreur P. Cell Mol Life Sci. 2009;66:2873. doi: 10.1007/s00018-009-0053-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chitale R. J Natl Cancer Inst. 2009;101:222. doi: 10.1093/jnci/djp014. [DOI] [PubMed] [Google Scholar]
- 13.Liu TC, Galanis E, Kirn D. Nat Clin Pract Oncol. 2007;4:101. doi: 10.1038/ncponc0736. [DOI] [PubMed] [Google Scholar]
- 14.Shirakawa T. Drug News Perspect. 2009;22:140. doi: 10.1358/dnp.2009.22.3.1354090. [DOI] [PubMed] [Google Scholar]
- 15.Pokorski JK, Steinmetz NF. Mol Pharm. 2011;8:29. doi: 10.1021/mp100225y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kang S, Suci PA, Broomell CC, Iwahori K, Kobayashi M, Yamashita I, Young M, Douglas T. Nano Lett. 2009;9:2360. doi: 10.1021/nl9009028. [DOI] [PubMed] [Google Scholar]
- 17.Steinmetz NF, Evans DJ, Lomonossoff GP. ChemBioChem. 2007;8:1131. doi: 10.1002/cbic.200700126. [DOI] [PubMed] [Google Scholar]
- 18.Klem MT, Willits D, Young M, Douglas T. J Am Chem Soc. 2003;125:10806. doi: 10.1021/ja0363718. [DOI] [PubMed] [Google Scholar]
- 19.Desgrosellier JS, Cheresh DA. Nat Rev Cancer. 2010;10:9. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hovlid ML, Steinmetz NF, Laufer B, Lau JL, Kuzelka J, Wang Q, Hyypia T, Nemerow GR, Kessler H, Manchester M, Finn MG. Nanoscale. 2012;4:3698. doi: 10.1039/c2nr30571b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Werner J, Decarlo CA, Escott N, Zehbe I, Ulanova M. Innate Immun. 2012;18:55. doi: 10.1177/1753425910392934. [DOI] [PubMed] [Google Scholar]
- 22.Gao H, Shi W, Freund LB. Proc Natl Acad Sci USA. 2005;102:9469. doi: 10.1073/pnas.0503879102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang W, Kim BYS, Rutka JT, Chan WCW. Nat Nanotechnol. 2008;3:145. doi: 10.1038/nnano.2008.30. [DOI] [PubMed] [Google Scholar]
- 24.Boturyn D, Coll JL, Garanger E, Favrot MC, Dumy P. J Am Chem Soc. 2004;126:5730. doi: 10.1021/ja049926n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.