

Abstract
CDDO-Me has exhibited a potent anticancer effect in human esophageal squamous cell carcinoma (ESCC) cells in our previous study, but the molecular interactome remains elusive. We applied the approach of stable-isotope labeling by amino acids in cell culture (SILAC) to assess the proteomic responses of CDDO-Me treatment in human ESCC Ec109 cells. The data were subsequently validated using Western blot assay. The results of our study revealed that CDDO-Me increased the expression level of 543 protein molecules, but decreased the expression level of 709 protein molecules in Ec109 cells. Among these modulated protein molecules, calcium/calmodulin-dependent protein kinase type II subunit α (CaMKIIα) was highly expressed in all tested ESCC cell lines, whereas its expression levels were substantially lower in normal control cell line. Its silencing by small interfering RNA inhibited CDDO-Me induced apoptosis and autophagy in ESCC cells. Collectively, these data demonstrate that the therapeutic response of CDDO-Me in the human ESCC cells is mediated by CaMKIIα.
Keywords: CDDO-Me, CaMKIIα, esophageal squamous cell carcinoma, SILAC
Introduction
Esophageal cancer ranks ninth for cancer incidence and sixth for cancer death in the worldwide [1,2]. More than 90% of esophageal cancers are either squamous cell carcinomas, which is more common in the developing countries, or adenocarcinomas [3]. Surgical treatment is the mainstay of therapy for patients with early stage esophageal squamous cell carcinoma (ESCC). For locally advanced or metastatic ESCC, chemotherapy is the most commonly used treatment modality [4-6]. However, most chemotherapeutic agents have limited effects on prolong overall survival of ESCC patients due to drug resistance and severe side effects. Thus, the development of efficacious and safe agents for ESCC therapyis an urgent need [7,8].
CDDO-Me [2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oic acid methyl ester], a semisynthetic oleanane triterpenoid, is an orally available, first-in-class antioxidant inflammation modulator [9]. Interestingly, CDDO-Me has also shown promising activities against several types of cancers in both laboratory test and clinical trials [10,11]. In our previous study, the results showed that CDDO-Me suppressed the proliferation and arrested cells in G2/M phase, and induced apoptosis and autophagy in human ESCC Ec109 and KYSE70 cells. Furthermore, CDDO-Me also inhibited cell invasion, epithelial-mesenchymal transition (EMT), and stemness in Ec109 and KYSE70 cells. These results indicate that CDDO-Me is a promising anticancer agent against ESCC [12]. However, to further improve CDDO-Me efficacy in the treatment of ESCC cells, it is of great importance to globally understand and uncover the molecular targets and related signaling pathways involved in the anticancer effect of CDDO-Me.
Stable-isotope labeling by amino acids in cell culture (SILAC) is a practical and powerful approach to uncover the global proteomic response to drug treatment and other interventions [13-15]. To fully understand the CDDO-Me-induced protein alterations and identify novel targets of CDDO-Me, we here employed SILAC-based proteomics technology to screen new targets whose knockdown could impact the CDDO-Me mediated growth inhibition in human ESCC cells. The results demonstrate that CaMKIIα is one of the secondary targets to enhance the efficacy of CDDO-Me in human ESCC cells.
Materials and methods
Cell culture and treatment
Human ESCC Ec109 cells was obtained from AddexBio Inc. (San Diego, CA, USA), KYSE70 and KYSE30 cells were obtained from Sigma-Aldrich Co (St Louis, MO, USA). The normal human esophageal epithelial cell line Het-1A was obtained from American Type Culture Collection (Manassas, VA, USA). The Ec109, KYSE70 and KYSE30 cells were cultured in RPMI-1640 medium with 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% heat-inactivated FBS, and Het-1A was cultured in BEGM™ BulletKit™ (Lonza Group Ltd. Co., Walkersville, MD, USA). Cells were maintained in a humidified atmosphere with 5% CO2 at 37°C, with medium renewal at every 2-3 days. CDDO-Me was dissolved in Dimethyl sulfoxide (DMSO) with a stock concentration of 10 mM, and was freshly diluted to the desired concentrations with culture medium with 0.05% (v/v) final concentration of DMSO. All cells were seeded into the plates for 24 hours to achieve a confluence of ~80% before CDDO-Me treatment.
SILAC approach
SILAC approach was performed as described previously [16]. Briefly, Ec109 cells was cultured in the medium with (heavy) or without (light) stable isotope-labeled amino acids (13C6 L-lysine and 13C6 15N4 L-arginine) and 10% dialyzed FBS. Cells were propagated in SILAC medium for more than six generations to ensure nearly 100% incorporation of labeled amino acids. After that, cells were treated with 0.5 μM CDDO-Me for 24 hours together with stable isotope-labeled amino acids. Then, Ec109 cell samples were harvested and lysed with hot lysis buffer (100 mM Tris base, 4% sodium dodecyl sulfate [SDS], and 100 mM dithiothreitol). The samples were centrifuged and collected after sonication for 3 seconds with six pulses. After the measurement of protein concentration, equal amounts of heavy and light protein samples were combined. The peptide mixtures were then analyzed using the hybrid linear ion trap-Orbitrap (LTQ Orbitrap XLTM; Thermo Fisher Scientific Inc.) following protein digestion and desalt. Liquid chromatography-tandem mass spectrometry was performed using a 10 cm long, 75 μm (inner diameter) reversed-phase column packed with 5 μm diameter C18 material having a pore size of 300 Å (New Objective Inc., Woburn, MA, USA) with a gradient mobile phase of 2%-40% acetonitrile in 0.1% formic acid at 200 μL per minute for 125 minutes. The peptide SILAC ratio was calculated using MaxQuant version 1.2.0.13. Scaffold 4.3.2 from Proteome Software Inc. was used for protein IDs identification, and Ingenuity Pathway Analysis (IPA) from QIAGEN was used for pathway analysis.
Western blot assay
Protein extracts (30 μg) prepared with radioimmunoprecipitation (RIPA) lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 0.1% SDS, 1% NP-40, 0.5% sodium deoxycholate, 1 mM PMSF, 100 mM leupeptin, and 2 mg/mL aprotinin, pH 8.0) were resolved on SDS-polyacrylamide gel after boiling at 95°C for 5 minutes, and transferred to an immobilon polyvinylidene difluoride membrane by electroblotting. The membrane was then incubated in a blocking buffer containing 5% non-fat dry milk in Tris-buffered saline and Tween 20 (TBST) buffer for 1 hour at room temperature followed by overnight incubation at 4°C with the indicated primary antibody and then blotted with appropriate horseradish peroxidase-conjugated secondary anti-mouse or anti-rabbit antibody. Visualization was performed using an enhanced chemiluminescence kit (BioRad Inc., Hercules, CA, USA) with electro-chemiluminescence substrate. Blot densitometry analysis was performed using Image J (National Institute of Health, Bethesda, MD, USA).
RNA interference and transfection
Ec109 and KYSE30 cells were plated at a density of 2×104 cells/cm2 in six-well plates. CaMKIIα and control non-functional small interfering RNA (siRNA) (Santa Cruz Biotechnology, Dallas, TX, USA) were transfected into the cells using Lipofectamine 2000 as recommended by the manufacturer (Invitrogen, Waltham, MA, USA). The medium was changed 24 hours post-transfection. After that, cells were treated with the indicated drugs. The knockdown efficiency was determined by Western blot analysis.
Flow cytometry
Cell apoptosis and autophagy were determined with flow cytometer as previously described [12]. Apoptosis was detected using Annexin V/7-AAD (7-amino-actinomycin D) staining. Cells were washed once in phosphate-buffered saline (PBS) and then mixed with 5 μL of annexin V:PE reagent and 5 μL of 7-AAD and incubated for 15 minutes at room temperature in the dark. Samples were analyzed by flow cytometer (Becton Dickinson Immunocytometry Systems, San Jose, CA, USA) within 1 hour. In autophagy analysis, cells were stained with Cyto-ID® Green (Enzo Life Sciences Inc., Farmingdale, NY, USA) for 30 minutes at 37°C and the stained cells were immediately analyzed via flow cytometry using Beckman-Coulter Kaluza software. The flow cytometer collected 10,000 events. The percentage of cells with Cyto-ID® staining was used to represent the relative formation or accumulation of autophagosomes. Results are expressed as the percentage of Cyto-ID®-positive cells of total live cells.
Confocal fluorescence microscopy
Confocal fluorescence microscopy was performed to further examine the cellular autophagy using a Cyto-ID® autophagy detection kit. In brief, cells were washed twice with 1× assay buffer and stained with Cyto-ID® detection agent according to the manufacturer’s instructions. The confocal images were recorded with a Leica TCS SP2 laser scanning confocal microscope (Leica Microsystems, Wetzlar, Germany) at wavelengths of 405/488 nm. The intensity of cell fluorescence was calculated for the corrected total cell fluorescence using the program Image J.
Statistical analysis
Statistical calculations were performed using Prism 5.0 (GraphPad Software Inc., San Diego, CA, USA). All values were presented as mean ± standard deviation from at least three separate experiments. Multiple comparisons were evaluated by one-way analysis of variance followed by Tukey’s multiple comparison. Data with p-values <0.05 were determined to be statistically significant.
Results
Proteomic responses of CDDO-Me treatment in Ec109 cells
In order to reveal the protein molecules that modulate the response of ESCC cells to CDDO-Me treatment, we performed the SILAC approach to identify the molecular targets of CDDO-Me. Ec109 cells was treated with 0.5 μM CDDO-Me for 24 hours and then subjected samples of the cells to quantitative proteomic analysis. The results showed that CDDO-Me increased the expression level of 543 protein molecules, but decreased the expression level of 709 protein molecules in Ec109 cells. These proteins were involved in a number of important cellular processes, including cell proliferation, redox homeostasis, cell metabolism, cell migration and invasion, cell survival, and cell death. The top 20 upregulated protein molecules were list in Table 1. Subsequently, we used IPA software to analyze the biological functions and interaction pathways for those protein molecules identified in the quantitative proteomic analysis. The results showed that there were a number of signaling pathways were regulated by CDDO-Me in Ec109 cells. The top ten targeted signaling pathways were the eukaryotic initiation factor (eIF) 2 signaling pathway, mitochondrial dysfunction pathway, oxidative phosphorylation pathway, eIF4 and p70S6 kinase (p70S6K) signaling pathway, mammalian target of rapamycin (mTOR) signaling pathway, protein ubiquitination pathway, ras-related nuclear protein (RAN) signaling pathway, granzyme A signaling pathway, protein kinase A signaling pathway, and nuclear factor (erythroid-derived 2)-like 2 (Nrf2)-mediated oxidative stress response signaling pathway (Figure 1).
Table 1.
Top 20 upregulated protein molecules modulated by CDDO-Me in Ec109 cells
| Gene name | Description | P value | Fold change |
|---|---|---|---|
| VKORC1 | Vitamin K epoxide reductase complex subunit 1 | 0.001799 | 3.6099 |
| CAMK2 | Calcium/calmodulin-dependent protein kinase type II | 2.84E-41 | 2.8928 |
| NPLOC4 | Nuclear protein localization protein 4 homolog | 2.94E-20 | 2.4761 |
| PSME3 | Proteasome activator complex subunit 3 | 8.4E-11 | 2.2741 |
| DNM2 | Dynamin-2 | 7.98E-05 | 2.1467 |
| UBQLN2 | Ubiquilin-2 | 3.76E-05 | 2.1463 |
| HSPA1A | Heat shock 70 kDa protein 1A | 0 | 2.0698 |
| DCTPP1 | dCTP pyrophosphatase 1 | 0.000669 | 2.0631 |
| EPB41L1 | Band 4.1-like protein 1 | 6.78E-37 | 2.0533 |
| ME1 | NADP-dependent malic enzyme; Malic enzyme | 4.47E-16 | 2.0398 |
| HYPK | Huntingtin-interacting protein K | 0.000128 | 2.0314 |
| FKBP3 | Peptidyl-prolyl cis-trans isomerase FKBP3 | 3.68E-05 | 1.8642 |
| SUPV3L1 | ATP-dependent RNA helicase SUPV3L1, mitochondrial | 8.07E-16 | 1.8281 |
| PIN1 | Peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 | 1.45E-08 | 1.7548 |
| SERPINH1 | Serpin H1 | 1.19E-08 | 1.7279 |
| MRPS26 | 28S ribosomal protein S26, mitochondrial | 0.003095 | 1.7194 |
| TMEM85 | Transmembrane protein 85 | 0.00048 | 1.6919 |
| KRR1 | KRR1 small subunit processome component homolog | 0.000297 | 1.6851 |
| FTH1 | Ferritin heavy chain; Ferritin | 8.4E-08 | 1.6757 |
| PRPF40A | Pre-mRNA-processing factor 40 homolog A | 3.13E-06 | 1.6393 |
Figure 1.
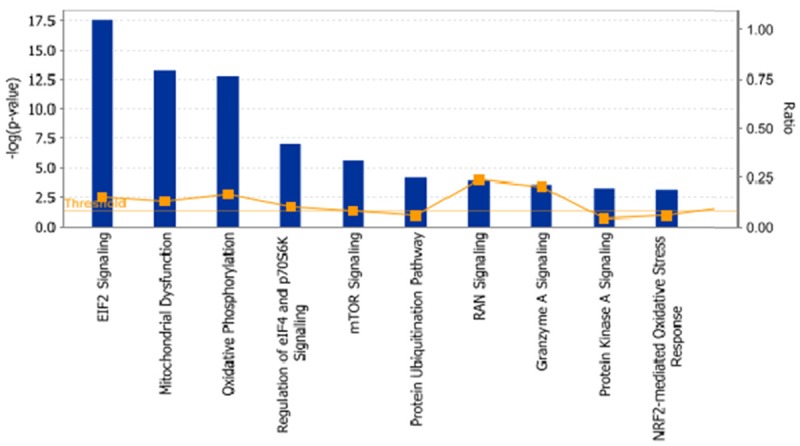
The top ten canonical signaling pathways regulated by CDDO-Me in Ec109 cells analyzed by ingenuity pathway analysis. Abbreviations: CDDO-Me, methyl ester of 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid; eIF2, eukaryotic initiation factor 2; p70S6K, p70S6 kinase; mTOR, mammalian target of rapamycin; RAN, ras-related nuclear protein; Nrf2, nuclear factor (erythroid-derived 2)-like 2.
Validation of the upregulated protein molecules which modulated by CDDO-Me in ESCC cells
In order to verify the quantitative proteomic data, we analyzed the expression level of vitamin K epoxide reductase complex subunit 1 (VKORC1), CaMKII, nuclear protein localization protein 4 homolog (NPLOC4), proteasome activator complex subunit 3 (PSME3), and Dynamin 2 in three human ESCC cell lines and one normal control by Western blot assay (Figure 2). The results showed that the expression level of VKORC1 and Dynamin 2 were similar between malignant cell lines and the normal control. CaMKII, NPLOC4, and PSME3 were highly expressed in human ESCC cells lines and lower protein levels were detected in Het-1A. However, only CaMKIIα was highly expressed in all tested ESCC cell lines, whereas its expression levels were substantially lower in normal control cell lines. Thus, CaMKIIα was selected as the potential therapeutic target and evaluated in the subsequent study.
Figure 2.
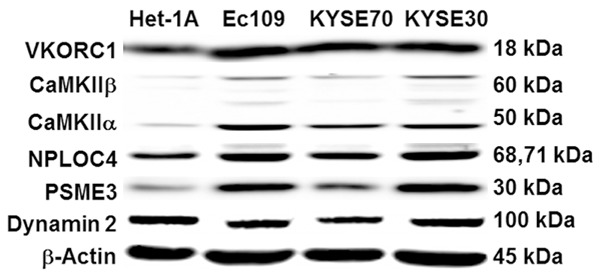
Representative blots of protein levels of VKORC1, CaMKIIα, NPLOC4, PSME3, and Dynamin 2 in various human ESCC cell lines and normal human esophageal epithelial cell line (Het-1A) were determined using Western blot analysis. β-Actin served as loading controls. Abbreviations: VKORC1, vitamin K epoxide reductase complex subunit 1; CaMKIIα, calcium/calmodulin-dependent protein kinase type II subunit alapha; NPLOC4, nuclear protein localization protein 4 homolog; PSME3, proteasome activator complex subunit 3; ESCC, esophageal squamous cell carcinoma.
Knockdown of CaMKIIα suppressed apoptosis and autophagy which induced by CDDO-Me
The biology functions and involved diseases of CaMKII were analyzed by IPA. The results showed that CaMKII played a critical role in cell death and survival, DNA replication, recombination, and repair and cell cycle regulation (Table 2). In addition, as shown in Figure 1, CDDO-Me showed an ability to modulate the mitochondrial dysfunction pathway (Figure 3) and the mTOR signaling pathway (Figure 4) in Ec109 cells. So the interaction role of CaMKIIα and CDDO-Me in the apoptosis and autophagy induction in human ESCC cell lines was validated in the subsequent study.
Table 2.
The biology function and involved disease of CaMKII analyzed by ingenuity pathway analysis
| Category | Function Annotation | p-value | Molecules |
|---|---|---|---|
| Cell Death and Survival | Cell death | 4.50E-07 | ABCD1, AKAP8, ANXA2, AP2B1, BAG3, BAX, C1QBP, CAMK2, CAPNS1, CASP14, CAT, CBFB, CDA, CDC37, CHTOP, COX5A, CSNK1A1, CSNK2A2, CSTA, CSTB, DDX17, DNMT1, DYNC1H1, EHD4, EIF3B, EIF3G, EIF3I, EIF4E, EIF5A, ELAVL1, EPHA2, FIS1, FKBP8, GJB2, GLRX, GPX1, HIST1H1C, HMGA1, HNRNPA1, HNRNPK, HPRT1, HSP90AA1, HYPK, IGF2R, ILF2, IMPDH2, ISG15, ITPR3, KPNA2, M6PR, MANF, MCTS1, MIF, MLKL, MRPL41, MRPS30, MSN, MT2A, NCKAP1, NDUFAB1, NME1, NUP62, OLFM1, PAK2, PAM16, PARK7, PCMT1, PCNA, PELP1, PNPT1, PPIB, PPP1CC, PRDX1, PRDX6, PRKAR1A, PSMB1, PSME3, PTRH2, PURA, PXN, RBM3, RBX1, RHOG, RPL10, RPS19, RRM2, S100A10, S100A11, S100P, SEC61G, SF3B6, SLC25A5, SLC3A2, SMARCC1, SPR, SRSF1, STMN1, SUPV3L1, TARDBP, TIMM50, TMED10, TMSB10/TMSB4X, TOP1, TPD52, TPM3, TUFM, UBE2M, VAPA, VASP, XRCC5, YARS |
| Cell death of tumor cell lines | 2.16E-06 | ANXA2, BAG3, BAX, CAMK2, CAPNS1, CASP14, CAT, CDA, CHTOP, COX5A, CSNK1A1, CSNK2A2, CSTA, DYNC1H1, EIF3G, EIF4E, ELAVL1, EPHA2, FKBP8, GLRX, GPX1, HMGA1, HNRNPA1, HNRNPK, HYPK, IGF2R, ILF2, ITPR3, KPNA2, MIF, MLKL, MSN, MT2A, NME1, PAK2, PARK7, PCNA, PELP1, PRDX1, PRKAR1A, PTRH2, RBX1, RPS19, RRM2, S100A11, SEC61G, SLC25A5, SLC3A2, SMARCC1, SRSF1, STMN1, TARDBP, TMED10, TMSB10/TMSB4X, TOP1, TPD52, TUFM, XRCC5, YARS | |
| Necrosis | 4.54E-07 | ANXA2, AP2B1, BAG3, BAX, CAMK2, CAPNS1, CASP14, CAT, CBFB, CDA, CDC37, CHTOP, COX5A, CSNK1A1, CSNK2A2, CSTA, DDX17, DNMT1, DYNC1H1, EHD4, EIF3B, EIF3G, EIF3I, EIF4E, ELAVL1, EPHA2, FKBP8, GJB2, GLRX, GPX1, HIST1H1C, HMGA1, HNRNPA1, HNRNPK, HPRT1, HSP90AA1, HYPK, IGF2R, ILF2, ISG15, ITPR3, KPNA2, M6PR, MANF, MCTS1, MIF, MLKL, MSN, MT2A, NDUFAB1, NME1, PAK2, PARK7, PCNA, PELP1, PNPT1, PPP1CC, PRDX1, PRDX6, PRKAR1A, PSMB1, PSME3, PTRH2, PURA, PXN, RBM3, RBX1, RHOG, RPL10, RPS19, RRM2, S100A10, S100A11, S100P, SEC61G, SF3B6, SLC25A5, SLC3A2, SMARCC1, SRSF1, STMN1, TARDBP, TMED10, TMSB10/TMSB4X, TOP1, TPD52, TUFM, UBE2M, VAPA, XRCC5, YARS | |
| Apoptosis of tumor cell lines | 1.35E-06 | ANXA2, BAG3, BAX, CAMK2, CAPNS1, CASP14, CAT, COX5A, CSNK1A1, CSNK2A2, CSTA, DYNC1H1, EIF4E, ELAVL1, EPHA2, FKBP8, GLRX, GPX1, HMGA1, HNRNPA1, HNRNPK, HYPK, IGF2R, ITPR3, KPNA2, MIF, MSN, MT2A, NME1, PAK2, PARK7, PCNA, PELP1, PRDX1, PRKAR1A, PTRH2, RBX1, RPS19, RRM2, S100A11, SEC61G, SLC25A5, SMARCC1, SRSF1, STMN1, TMED10, TMSB10/TMSB4X, TOP1, TPD52, XRCC5 | |
| Apoptosis | 1.19E-05 | AKAP8, ANXA2, BAG3, BAX, C1QBP, CAMK2, CAPNS1, CASP14, CAT, CBFB, CDC37, COX5A, CSNK1A1, CSNK2A2, CSTA, CSTB, DDX17, DNMT1, DYNC1H1, EHD4, EIF3B, EIF3I, EIF4E, EIF5A, ELAVL1, EPHA2, FIS1, FKBP8, GLRX, GPX1, HIST1H1C, HMGA1, HNRNPA1, HNRNPK, HSP90AA1, HYPK, IGF2R, ISG15, ITPR3, KPNA2, MCTS1, MIF, MRPL41, MRPS30, MSN, MT2A, NCKAP1, NME1, NUP62, PAK2, PAM16, PARK7, PCMT1, PCNA, PELP1, PNPT1, PPP1CC, PRDX1, PRDX6, PRKAR1A, PSMB1, PTRH2, PXN, RBM3, RBX1, RHOG, RPL10, RPS19, RRM2, S100A10, S100A11, SEC61G, SF3B6, SLC25A5, SMARCC1, SRSF1, STMN1, SUPV3L1, TARDBP, TMED10, TMSB10/TMSB4X, TOP1, TPD52, UBE2M, VASP, XRCC5, YARS | |
| Cell viability | 5.92E-03 | AP2B1, BAG3, BAX, CAMK2, CAT, CBFB, DNMT1, EHD4, EIF4A3, EIF4E, ELAVL1, GPX1, HMGA1, HNRNPUL2, IGF2BP3, IGF2R, MIF, NME1, NOP58, PAK2, PARK7, PCNA, PPP1CC, PRDX6, PRKACB, PRKAR1A, PRPF8, PSMA1, PSME3, RPL27, RPL38, RRM2, S100P, SMC3, SNRPA1, SNRPD1, STMN1, TARDBP, TOP1, XRCC5 | |
| Cell survival | 3.32E-03 | AP2B1, BAG3, BANF1, BAX, CAMK2, CAT, CBFB, DNMT1, EHD4, EIF4A3, EIF4E, ELAVL1, EPHA2, GLRX, GPX1, HMGA1, HNRNPUL2, IGF2BP3, IGF2R, MIF, NME1, NOP58, PAK2, PARK7, PCNA, PPIB, PPP1CC, PRDX6, PRKACB, PRKAR1A, PRPF8, PSMA1, PSME3, RPL27, RPL38, RRM2, S100P, SMC3, SNRPA1, SNRPD1, STMN1, TARDBP, TOP1, XRCC5 | |
| DNA Replication, Recombination, and Repair | Metabolism of DNA | 3.27E-04 | BAX, CAMK2, GPX1, HMGA1, HNRNPA1, KPNA2, NME1, PAM16, PCNA, POLE3, PPIB, PPP1CC, PRDX6, PURA, PXN, S100A11, SMC3, TOP1, XRCC5 |
| Cell Cycle | Cell cycle progression | 1.20E-02 | AKAP8, BAX, BCCIP, C1QBP, CAMK2, CDC37, CSNK1A1, CSNK2A2, DDX17, DNMT1, DYNC1H1, EIF4E, GPX1, HYPK, MARCKS, MIF, NME1, PAK2, PCNA, PELP1, PRDX1, PRKAR1A, PURA, RBM3, RBX1, RCC1, RPS15A, SMC3, STMN1, TOP1, TRIM25, TXLNG |
Figure 3.

Mitochondrial dysfunction signaling pathway regulated by CDDO-Me in Ec109 cells. Notes: Ec109 cells were treated with 0.5 μM CDDO-Me for 24 hours and the protein samples were subject to quantitative proteomic analysis. Red indicates an upregulation; green indicates a downregulation. The intensity of green and red molecule colors indicates the degree of down- or upregulation, respectively. Solid arrows indicate direct interaction and dashed arrows indicate indirect interaction. Abbreviations: CDDO-Me, methyl ester of 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid.
Figure 4.

mTOR signaling pathway regulated by CDDO-Me in Ec109 cells. Notes: Ec109 cells were treated with 0.5 μM CDDO-Me for 24 hours and the protein samples were subject to quantitative proteomic analysis. Red indicates an upregulation; green indicates a downregulation. The intensity of green and red molecule colors indicates the degree of down- or upregulation, respectively. Solid arrows indicate direct interaction and dashed arrows indicate indirect interaction. Abbreviation: CDDO-Me, methyl ester of 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid; mTOR, mammalian target of rapamycin.
In order to test the synthetic lethal effect between CaMKIIα and CDDO-Me in human ESCC cells, we transfected two representative human ESCC cells, Ec109 and KYSE30, with siRNAs to selectively knockdown the CaMKIIα expression in the presence or absence of 0.5 μM CDDO-Me. The expression level of CaMKIIα was downregulated successfully in Ec109 and KYSE30 cells by specific siRNAs after 24 hours transfection.
The combination of CaMKIIα siRNA and CDDO-Me decreased about 80%-90% of cellular apoptosis compared with CDDO-Me alone in KYSE30 cells (Figure 5A). However, the CaMKIIα siRNA did not show any significant effect on cellular apoptosis in Ec109 cells by the intervention of CaMKIIα siRNA alone or combination with CDDO-Me. B-cell lymphoma-2 (Bcl-2), Bcl-2-associated X protein (bax), and cleaved caspase-3 are major proteins involved in the apoptosis which induced via mitochondrial dysfunction pathway [17]. In order to validate the flow cytometry results, we examined the expression of these key proteins by Western blot assay. The results demonstrated that the combination of CaMKIIα siRNA and CDDO-Me consistently decreased the level of bax and cleaved caspase-3 and increased the level of bcl-2 compared with CDDO-Me alone in KYSE30 cells (Figure 5B). These results confirmed that CaMKIIα was a negative modulator of CDDO-Me induced apoptosis in some human ESCC cells.
Figure 5.

The effect of knockdown of CaMKIIα on the CDDO-Me induced apoptosis in human ESCC cells. A. Percentages of specific cell populations showed in dot plots and apoptotic cells showed in bar graphs for the silencing of CaMKIIα in Ec109 and KYSE30 cells with or without 0.5 μM CDDO-Me treatment for 24 hours. B. Representative blots of bcl-2, bax, and cleaved caspase-3 for the proteinlysate samples, which were prepared from Ec109 and KYSE30 cells treated with CaMKIIα siRNAs or 0.5 μM CDDO-Me for 24 hours. Abbreviation: CaMKIIα, calcium/calmodulin-dependent protein kinase type II subunit alapha; CDDO-Me, methyl ester of 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid; ESCC, esophageal squamous cell carcinoma; bcl-2, B-cell lymphoma-2; bax, bcl-2 associated X protein; siRNA , small interfering RNA.
To further illustrate the synthetic lethal effect between CaMKIIα and CDDO-Me in human ESCC cells, we evaluated the effect of silencing CaMKIIα on the induction of autophagy by flow cytometry in Ec109 and KYSE30 cells firstly. As the shown in Figure 6A, the combination of CDDO-Me and CaMKIIα siRNA substantially decreased the percentage of autophagic cells to 18.72% and 35.01% in Ec109 and KYSE30 cells, respectively, compared with CDDO-Me treatment alone. And these results was confirmed by the confocal microscopy analysis subsequently (Figure 6B). Microtubule-associated protein 1A/1B-light chain 3 (LC3) and beclin 1 are two essential modifiers of the autophagic process and are commonly used to monitor autophagy activity combined with other biochemical factors [18,19]. In current study, the expression level of LC3 and beclin 1 were also determined using western blotting assay. We found the combination of CDDO-Me and CaMKIIα siRNA suppressed the conversation of LC3I/II and also the expression level of beclin 1 (Figure 6C). This indicated that the downregulation of CaMKIIα might partially contribute to CDDO-Me induced autophagy in human ESCC cells.
Figure 6.
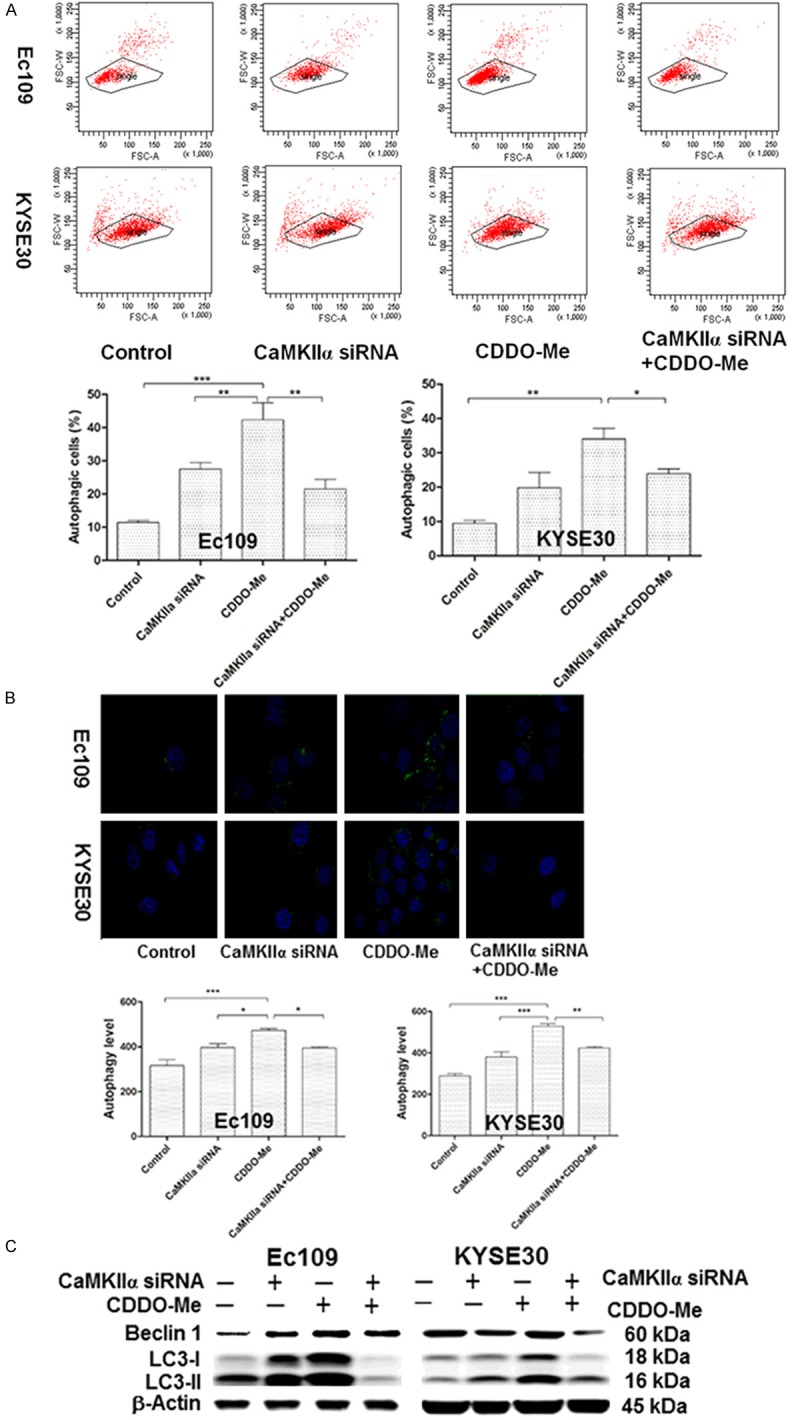
The effect of knockdown of CaMKIIα on the CDDO-Me induced autophagy in human ESCC cells. A. Percentages of specific cell populations showed in dot plots and autophagic cells showed in bar graphs for the silencing of CaMKIIα in Ec109 and KYSE30 cells with or without 0.5 μM CDDO-Me treatment for 24 hours. B. CDDO-Me-induced autophagic death in the silencing of CaMKIIα in Ec109 and KYSE30 cellsdetermined by confocal microscopy. The level of autophagy was evaluated using a lysosome-specific fluorescence dye. The confocal microscopic images of autophagic Ec109 and KYSE 30 cells (stained in green) are also shown. C. Representative blots of beclin-1 and LC3I/II for the proteinlysate samples, which were prepared from Ec109 and KYSE30 cells treated with CaMKIIα siRNAs or 0.5 μM CDDO-Me for 24 hours. Abbreviation: CaMKIIα, calcium/calmodulin-dependent protein kinase type II subunit alapha; CDDO-Me, methyl ester of 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid; ESCC, esophageal squamous cell carcinoma; siRNA, small interfering RNA; LC 3, microtubule-associated protein 1A/1B-light chain 3.
Collectively, our data showed that downregulating the CaMKIIα could decrease CDDO-Me mediated apoptosis and autophagy induction in human ESCC cells. The therapeutic response of CDDO-Me in the ESCC cells is mediated by CaMKIIα.
Discussion
Esophageal cancer is one of the most common types of cancer worldwide and is generally associated with a poor prognosis [1,5]. There is an increasing interest in new agents and therapies for the treatment of esophageal cancer. CDDO-Me, a synthetic triterpenoid derivatives, has been found to inhibit the growth of various cancers in preclinical and clinical studies [9,10]. In our previous study, we demonstrated that CDDO-Me induced cell cycle arrest, apoptosis, and autophagy, and inhibited reactive oxygen species (ROS) level, cell invasion, EMT, and stemness in ESCC cells. CDDO-Me might be a potent and promising agent to combat esophageal cancer [12]. However, the global potential molecular targets and the possible mechanisms involved are not fully identified as yet.
The SILAC-based proteomic approach can quantitatively and comprehensively evaluate the effect of a given compound and identify its potential molecular targets. In the present study, we used a quantitative SILAC-based proteomic approach to evaluate the responses of human ESCC Ec109 cells to treatment with CDDO-Me. The results showed that 543 protein molecules were upregulated and 709 protein molecules were downregulated by the treatment of CDDO-Me in Ec109 cells. In the subsequent IPA analysis, the results revealed that CDDO-Me modulated lots of important signaling pathways, which in turn resulted in an alteration in cell proliferation, redox homeostasis, cell metabolism, cell migration and invasion, cell survival, and cell death with the involvement of a number of function proteins. In order to validate the quantitative proteomic study results, we selected VKORC1, CaMKII, NPLOC4, PSME3, and Dynamin 2 as the secondary targets of CDDO-Me for further analysis. The validation study results showed that only CaMKIIα was highly expressed in all tested ESCC cell lines, whereas its expression levels were substantially lower in normal control cell lines. Thus, CaMKIIα was selected as the potential therapeutic target and evaluated in the subsequent study.
CaMKII is a multifunctional serine/threonine kinases best known for its critical role in learning and memory [20]. There are four different CaMKII genes, and each gene encodes a distinct CaMKII isoform (α, β, γ, and δ). All CaMKII isoforms appear to share common regulatory mechanisms and protein targets but differ in tissue distribution [21]. CaMKII phosphorylates nearly 40 different proteins, including enzymes, ion channels, kinases, and transcription factors and plays a critical role in the regulation of proliferation, cell cycle, invasion and metastasis, and apoptosis of various cancer cells.Recent studies suggested that high levels of CaMKII also expressed in variety of malignant diseases [22]. In previous study, we demonstrate that apoptosis and autophagy are two predominant cell death routes regulated by CDDO-Me in human ESCC cells. In addition, our proteomic analysis in current study showed that mitochondrial dysfunction pathway and mTOR signaling pathway were two of the top five signaling pathways regulated by CDDO-Me in Ec109 cells. So the role of CaMKIIα in the CDDO-Me induced apoptosis and autophagy have been verified by subsequent experiments. The validation study further confirmed that the silencing of CaMKIIα could substantially impact the sensitivity of ESCC cells to CDDO-Me via apoptosis or autophagy signaling pathway. Our findings therefore suggest that CaMKIIα is a potential target of CDDO-Me in the treatment of ESCC, although further in vivo validation studies are needed.
In conclusion, using this SILAC-based proteomic analysis, a number of protein molecules and signaling pathway were newly found to be altered in response to CDDO-Me treatment in human ESCC cells. Among these identified proteins, VKORC1, CaMKII, NPLOC4, PSME3, and Dynamin 2 were further validated. The validation study further confirmed that CaMKIIα is a modulator of CDDO-Me therapeutic response in human ESCC cells. Our findings provide new insights into the understanding of mechanisms of CDDO-Me induced anticancer effect and new molecule targets in the modulation of CDDO-Me therapy efficacy in human ESCC cells.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81502094) and Scientific Program for Overseas Chinese Scholar of MOHRSS of China (2015). They also appreciate the guidance and technical assistance for this study from Shu-Feng Zhou, PhD, Zhi-Wei Zhou, PhD, and Kent Seeley, PhD, University of South Florida, Tampa, FL 33612, USA.
Disclosure of conflict of interest
None.
References
- 1.Global Burden of Disease Cancer Collaboration. Fitzmaurice C, Dicker D, Pain A, Hamavid H, Moradi-Lakeh M, MacIntyre MF, Allen C, Hansen G, Woodbrook R, Wolfe C, Hamadeh RR, Moore A, Werdecker A, Gessner BD, Te Ao B, McMahon B, Karimkhani C, Yu C, Cooke GS, Schwebel DC, Carpenter DO, Pereira DM, Nash D, Kazi DS, De Leo D, Plass D, Ukwaja KN, Thurston GD, Yun Jin K, Simard EP, Mills E, Park EK, Catalá-López F, deVeber G, Gotay C, Khan G, Hosgood HD 3rd, Santos IS, Leasher JL, Singh J, Leigh J, Jonas JB, Sanabria J, Beardsley J, Jacobsen KH, Takahashi K, Franklin RC, Ronfani L, Montico M, Naldi L, Tonelli M, Geleijnse J, Petzold M, Shrime MG, Younis M, Yonemoto N, Breitborde N, Yip P, Pourmalek F, Lotufo PA, Esteghamati A, Hankey GJ, Ali R, Lunevicius R, Malekzadeh R, Dellavalle R, Weintraub R, Lucas R, Hay R, Rojas-Rueda D, Westerman R, Sepanlou SG, Nolte S, Patten S, Weichenthal S, Abera SF, Fereshtehnejad SM, Shiue I, Driscoll T, Vasankari T, Alsharif U, Rahimi-Movaghar V, Vlassov VV, Marcenes WS, Mekonnen W, Melaku YA, Yano Y, Artaman A, Campos I, MacLachlan J, Mueller U, Kim D, Trillini M, Eshrati B, Williams HC, Shibuya K, Dandona R, Murthy K, Cowie B, Amare AT, Antonio CA, Castañeda-Orjuela C, van Gool CH, Violante F, Oh IH, Deribe K, Soreide K, Knibbs L, Kereselidze M, Green M, Cardenas R, Roy N, Tillmann T, Li Y, Krueger H, Monasta L, Dey S, Sheikhbahaei S, Hafezi-Nejad N, Kumar GA, Sreeramareddy CT, Dandona L, Wang H, Vollset SE, Mokdad A, Salomon JA, Lozano R, Vos T, Forouzanfar M, Lopez A, Murray C, Naghavi M. The Global Burden of Cancer 2013. JAMA Oncol. 2015;1:505–27. doi: 10.1001/jamaoncol.2015.0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 3.Daly JM, Fry WA, Little AG, Winchester DP, McKee RF, Stewart AK, Fremgen AM. Esophageal cancer: results of an American College of Surgeons Patient Care Evaluation Study. J Am Coll Surg. 2000;190:562–72. doi: 10.1016/s1072-7515(00)00238-6. discussion 572-3. [DOI] [PubMed] [Google Scholar]
- 4.Enzinger PC, Mayer RJ. Esophageal cancer. N Engl J Med. 2003;349:2241–52. doi: 10.1056/NEJMra035010. [DOI] [PubMed] [Google Scholar]
- 5.Lagergren J, Lagergren P. Recent developments in esophageal adenocarcinoma. CA Cancer J Clin. 2013;63:232–48. doi: 10.3322/caac.21185. [DOI] [PubMed] [Google Scholar]
- 6.D’Journo XB, Thomas PA. Current management of esophageal cancer. J Thorac Dis. 2014;6(Suppl 2):S253–64. doi: 10.3978/j.issn.2072-1439.2014.04.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mariette C, Robb WB, Piessen G, Adenis A. Neoadjuvant chemoradiation in oesophageal cancer. Lancet Oncol. 2015;16:1008–9. doi: 10.1016/S1470-2045(15)00127-8. [DOI] [PubMed] [Google Scholar]
- 8.Belkhiri A, El-Rifai W. Advances in targeted therapies and new promising targets in esophageal cancer. Oncotarget. 2015;6:1348–58. doi: 10.18632/oncotarget.2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liby KT, Sporn MB. Synthetic oleanane triterpenoids: multifunctional drugs with a broad range of applications for prevention and treatment of chronic disease. Pharmacol Rev. 2012;64:972–1003. doi: 10.1124/pr.111.004846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang YY, Zhe H, Zhao R. Preclinical evidences toward the use of triterpenoid CDDO-Me for solid cancer prevention and treatment. Mol Cancer. 2014;13:30. doi: 10.1186/1476-4598-13-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hong DS, Kurzrock R, Supko JG, He X, Naing A, Wheler J, Lawrence D, Eder JP, Meyer CJ, Ferguson DA, Mier J, Konopleva M, Konoplev S, Andreeff M, Kufe D, Lazarus H, Shapiro GI, Dezube BJ. A phase I first-in-human trial of bardoxolone methyl in patients with advanced solid tumors and lymphomas. Clin Cancer Res. 2012;18:3396–406. doi: 10.1158/1078-0432.CCR-11-2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang YY, Yang YX, Zhao R, Pan ST, Zhe H, He ZX, Duan W, Zhang X, Yang T, Qiu JX, Zhou SF. Bardoxolone methyl induces apoptosis and autophagy and inhibits epithelial-to-mesenchymal transition and stemness in esophageal squamous cancer cells. Drug Des Devel Ther. 2015;9:993–1026. doi: 10.2147/DDDT.S73493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ong SE, Mann M. Stable isotope labeling by amino acids in cell culture for quantitative proteomics. Methods Mol Biol. 2007;359:37–52. doi: 10.1007/978-1-59745-255-7_3. [DOI] [PubMed] [Google Scholar]
- 14.Chen X, Wei S, Ji Y, Guo X, Yang F. Quantitative proteomics using SILAC: Principles, applications, and developments. Proteomics. 2015;15:3175–92. doi: 10.1002/pmic.201500108. [DOI] [PubMed] [Google Scholar]
- 15.Hoedt E, Zhang G, Neubert TA. Stable isotope labeling by amino acids in cell culture (SILAC) for quantitative proteomics. Adv Exp Med Biol. 2014;806:93–106. doi: 10.1007/978-3-319-06068-2_5. [DOI] [PubMed] [Google Scholar]
- 16.Pan ST, Zhou ZW, He ZX, Zhang X, Yang T, Yang YX, Wang D, Qiu JX, Zhou SF. Proteomic response to 5, 6-dimethyl xanthenone 4-acetic acid (DMXAA, vadimezan) in human non-small cell lung cancer A549 cells determined by the stable-isotope labeling by amino acids in cell culture (SILAC) approach. Drug Des Devel Ther. 2015;9:937–68. doi: 10.2147/DDDT.S76021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Llambi F, Green DR. Apoptosis and oncogenesis: give and take in the BCL-2 family. Curr Opin Genet Dev. 2011;21:12–20. doi: 10.1016/j.gde.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell. 2007;130:165–78. doi: 10.1016/j.cell.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 19.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–6. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 20.Hoelz A, Nairn AC, Kuriyan J. Crystal structure of a tetradecameric assembly of the association domain of Ca2+/calmodulin-dependent kinase II. Mol Cell. 2003;11:1241–51. doi: 10.1016/s1097-2765(03)00171-0. [DOI] [PubMed] [Google Scholar]
- 21.Gaertner TR, Kolodziej SJ, Wang D, Kobayashi R, Koomen JM, Stoops JK, Waxham MN. Comparative analyses of the three-dimensional structures and enzymatic properties of alpha, beta, gamma and delta isoforms of Ca2+-calmodulin-dependent protein kinase II. J Biol Chem. 2004;279:12484–94. doi: 10.1074/jbc.M313597200. [DOI] [PubMed] [Google Scholar]
- 22.Wang YY, Zhao R, Zhe H. The emerging role of CaMKII in cancer. Oncotarget. 2015;6:11725–34. doi: 10.18632/oncotarget.3955. [DOI] [PMC free article] [PubMed] [Google Scholar]