

Abstract
Cardiac hypertrophy is a common pathological alteration in heart disease, which has been reported to be connected with serine/threonine protein phosphatases that control the dephosphorylation of a variety of cardiac proteins. Herein, we generated protein phosphatase type 2A knockout expressing a tamoxifen-inducible Cre recombinase protein fused to two mutant estrogen-receptor ligand-binding domains (MerCreMer) under the control of the a-myosin heavy chain promoter. Cardiac function of mice was determined by echocardiography. Decrease in PP2A activity leads to increased cardiomyocyte hypertrophy and fibrosis. Loss of PP2ACα leads to the heart failure, including the changes of EF, FS, LV, ANP and BNP. On the molecular level, knockout mice shows increased expression of B55a and B56e at 60 days after tamoxifen injection. Additionally, the regulation of the Akt/GSK3β/β-catenin pathway is severely disturbed in knockout mice. In conclusion, cardiomyocyte specific deletion of PP2A gene causes the cardiac hypertrophy. We will use the knockout mice to generate a type of cardiomyocyte hypertrophy mouse model with myocardial fibrosis.
Keywords: PP2A, hypertrophy, knockout mouse model
Introduction
In the pathology of Cardiac hypertrophy, most cardiac proteins have a dynamic balance between their phosphorylation state and dephosphorylation state through the action of multiple kinases and phosphatases. These kinases and phosphatases control multiple physiological processes and play important roles in cardiac pathophysiology. Protein phosphatase type 2A (PP2A), which constitutes a large family of serine/threonine protein phosphatases, controls the dephosphorylation of a variety of cardiac proteins in cardiac pathophysiology, thereby fine-tuning cardiac electrophysiology and function [1]. PP2A Heterotrimeric PP2A holoenzymes are constituted with a catalytic C subunit, a structural A subunit, and a regulatory B-type subunit [2,3], which accounts for greater than 90% of protein dephosphorylation activity in the heart together with PP1 [4]. The A structure subunit recruits the C catalytic subunit to form the core dimer, which acts as a scaffold for B subunits of the enzyme [5]. PP2A activity are due to the tyrosine or serine/threonine phosphorylation of not only the catalytic C subunit, but also the regulatory B subunit, especially those of the B’ family [6,7]. The regulatory B subunits play key roles in controlling PP2A substrate specificity, cellular localization, and enzymatic activity [8]. Four B subunit families have been identified (PR55 or B, PR61 or B’, PR72/130 or B” and PR93/PR110 or B”’). Each family of B subunits contains several isoforms that can bind the AC dimer in a mutually exclusive manner [9].
Total myocardial serine/threonine protein phosphatase activity is mainly the result of both PP1 and PP2A activity [10]. PP2A is a Ser/Thr phosphatase that exists across many eukaryotic cell types. Transgenic mouse model overexpressing PP2Acα under control of the α-myosin heavy chain promotor show necrotic isles within the myocardium, cardiac hypertrophy, and left ventricular fibrosis [11]. PP2A overexpressing mice develop dilated cardiomyopathy which is associated with the disruption of the signaling cascade of Akt/GSK3β/β-catenin [12]. However, PP2Aca deficiency in mice resulted in embryonic lethality by embryonic day 6.5 [13].
In this study, the Cre/loxP system is used to achieve cardiac-specific PP2ACα exon 2 knockouts in adult mice. In the Myh6-MerCreMer (MCM) model, the Cre fusion protein is expressed and controlled by the human α-myosin heavy chain gene promoter [14]. As determined by the palindromic core sequence of the loxP site, an important coding exon 2 is flanked (floxed) by loxP sites ppp2caflox/flox mice that are in the same orientation and thus inactivate PP2ACα. While Cre recombinase is expressed, the region between the two loxP sites is deleted and the PP2ACα exon2 gene is temporally inactivated.
Cardiac-specific induction of Cre recombinase in Myh6-MerCreMer mice, employing a widely used protocol (5 days of injection of TAM at a dose of 40 mg/kg), results in severe, transient systolic dysfunction associated with depletion of cardiac mitochondrial ATP content [15]. The PP2ACα knockout mice were treated with tamoxifen by intraperitoneal injection once a day for 3 days at a dosage of 20 mg/kg per day. Ppp2caflox/+; Myh6-MerCreMer mice were used as control group to avoid any potential nonspecific effects of Cre activation in the heart. In knockout mice heart, B55a and B56e express increased with significant difference compared with the control group. The regulation of the Akt/GSK3β/β-catenin pathway is severely disturbed in knockout mice hearts.
These types of knockout mice have experience cardiac hypertrophy and myocardial fibrosis as adults; therefore, we generated a unique mouse model to study the role of PP2ACα in cardiac hypertrophy. Moreover, this mouse model can be used to further research of PP2A as a potential therapeutic target for cardiac hypertrophy.
Materials and methods
Animals
All animal protocols were approved by the Animal Care and Use Committee of the Model Animal Research Center of Nanjing Medical University. This study was approved by the Institutional Animal Care and Use Committee of Nanjing Medical University (Number: NAJMU- IACRCUC-20100601001). All mice were fed a standard diet and housed in specific pathogen-free barrier facilities. To induce Cre recombination, cardiac specific knockout mice as well as wild control mice at 3 month, were treated with tamoxifen (Sigma, USA) by intraperitoneal injection once a day for 3 days at a dosage of 20 mg/kg per day. Mice were killed two months after the end of tamoxifen administration.
Generation of knockout and control group mice
Genomic DNA was extracted from a 2 mm tail clip using the PBND extraction method (http://www.jax.org/imr/tail_nonorg.html). In brief, mouse tails were digested with 40 mg Proteinase K (Invitrogen) in 200 ml PBND buffer at 55°C for 16 h. After heating at 96°C for 10 min to inactivate Proteinase K, and the tail digest was used as template DNA for PCR analysis. To detect expression of the CD11b-cre transgene, the following primers were used: CreF (5’-CGTACTGACGGTGGGAGAAT-3’) and CreR (5’-TGCATGATCTCCGGTAT TGA-3’). Flox was detected by ppp2ca-loxp1-F1 (5’-TACTTCAGTGTTACT GACACAGACTC-3’) And ppp2ca-loxp1-R1 (5’-GTGAAAACAGGTCATTCC CAT-3’). PCR reactions were performed in PCR buffer (25 ml) containing 0.2 mM dNTP (Promega), forward and reverse primer (0.2 mM each), template DNA (1 ml), DMSO (5%; v/v, Merck), MgCl2 (4 mM), and 0.25 ml Ampli Taq Gold TM (Roche). Amplification was performed as follows: initial denaturation, 94°C for 5 min; 35 PCR cycles, 94°C for 30 sec, 55°C for 30 sec, and 72°C for 30 sec; and final extension, 72°C for 10 min. Amplification of the Myh6-MerCreMer transgene generated a 374 bp DNA fragment.
RT-PCR and quantitative real-time PCR analyses
Total RNA were isolated from mice heart and cardiomyocytes were dissolving in Trizol reagent (Invitrogen, USA). RT-PCR was performed using PrimeScriptTM (TAKATA, Japan). The cDNA of PP2ACα was detected by primers Mpp2ca-P5F and Mpp2ca-P5R. The PCR products were 623-bp (control) and 423-bp (knockout). Expression levels of mRNA of ANP (atrial natriuretic peptide), BNP (Brain natriuretic peptide) α-MHC (α-myosin heavy chain) and β-MHC (β-myosin heavy chain) were determined using quantitative real-time PCR and β-actin was used for normalization. The sequences of the specific primers used for PCR are listed in Table 1.
Table 1.
Sequences of the specific primers used for PCR
| Genes | Primers | Sequences |
|---|---|---|
| Mpp2ca-P5 | Forwards | 5’-GGTCAAGAGCCTCTGCGAGAA-3’ |
| Reverse | 5’-CCGGTCATGGCACCAGTTAT-3’ | |
| β-actin | Forwards | 5’-GAGAAGATCTGGCACCACACC-3’ |
| Reverse | 5’-GCATACAGGGACAGCACAGC-3’ | |
| PP2A-Cα | Forwards | 5’-ATGGACGAGAAGTTGTTCACC-3’ |
| Reverse | 5’-CAGTGACTGGACATCGAACCT-3’ | |
| PP2A-Cβ | Forwards | 5’-GAGGGTACTACTCTGTGGAGAC-3’ |
| Reverse | 5’-CCGGCTTTCGTGATTTCCT-3’ | |
| PP2A-B55α | Forwards | 5’-AGGAGGGAATGATATTCAGTGGT-3’ |
| Reverse | 5’-CCTGTGGCTAGTAATTCTCCAGA-3’ | |
| PP2A-B55β | Forwards | 5’-TGCCTTATATCTTCAGACCTCCA-3’ |
| Reverse | 5’-AATGTCAGCTTCAGTATGGCAG-3’ | |
| PP2A-B55δ | Forwards | 5’-GGAAGCCGACATCATCTCCAC-3’ |
| Reverse | 5’-GTGAGCGCGGCCTTTATTCT-3’ | |
| PP2A-B56α | Forwards | 5’-ATTGAAGAGCCGCTTTTTAAGCA-3’ |
| Reverse | 5’-TGAGGGTTTTCAGCACATTGT-3’ | |
| PP2A-B56β | Forwards | 5’-GGGCCTACATCCGCAAACA-3’ |
| Reverse | 5’-GGATCAGGACTCGAACCAGG-3’ | |
| PP2A-B56γ | Forwards | 5’-CCCGTGGTCCTTCTCCATATT-3’ |
| Reverse | 5’-GCAACACTGACGTAACTTCTGG-3’ | |
| PP2A-B56γ2 | Forwards | 5’-TTCTCCACATTCGAGATGTTCCT-3’ |
| Reverse | 5’-GCGCTTTACTTCCTTCCACTT-3’ | |
| PP2A-B56δ | Forwards | 5’-CCCAGTCTCAGTCACCATCAT-3’ |
| Reverse | 5’-GCGTCGTTCTTTCTTGACAATC-3’ | |
| PP2A-B56ε | Forwards | 5’-GACGGATTTTCTCGGAAGTCC-3’ |
| Reverse | 5’-GAGGTTGGAACGTCTTTCAGC-3’ | |
| ANF | Forwards | 5’-AGGAGAAGATGCCGGTAGAAGA-3’ |
| Reverse | 5’-GCTTCCTCAGTCTGCTCACTCA-3’ | |
| BNP | Forwards | 5’-AAGCTGCTGGAGCTGATAAGA-3’ |
| Reverse | 5’-GTTACAGCCCAAACGACTGAC-3’ | |
| α-MHC | Forwards | 5’-CCACTTCTCCTTGGTCCACTATG-3’ |
| Reverse | 5’-ACAAACCCACCACCGTCTCA-3’ | |
| β-MHC | Forwards | 5’-CCTCCTCACATCTTCTCCATCTCT-3’ |
| Reverse | 5’-CTCCGGATTCTCCGGTGAT-3’ |
Protein phosphatase assay
Protein phosphatase 2A activity was measured in whole heart homogenate with Serine/Threonine Phosphatase Assay System (Product number: V2460). The non-radioactive Serine/Threonine Phosphatase Assay System provides a fast, convenient and flexible method for measuring protein serine/threonine phosphatase activity. This system determines the amount of free phosphate generated in a reaction by measuring the absorbance of a molybdate: malachite green: phosphate complex. The system allows the use of a variety of buffer conditions and substrates, including naturally phosphorylated proteins orsynthetic phosphopeptides.
Western blot
Protein extraction was quantified according to standard procedures, and proteins underwent electrophoretic separation (20 μg protein/lane) and electroblotting onto PVDF membranes. Hearts were homogenized in hypotonic solution. A mix of protease inhibitors (Roche) were also added to the buffer. For the present studies, the following primary antibodies were used: anti-PP2AC, GAPDH, AKT, P-AKT(S473), P-AKT (T308), phospho-GSK3β (ser9), β-catenin, P-β-catenin (Ser552) (Cell signaling).
Echocardiography
Adult animals were lightly anesthetized before undergoing echocardiography or hemodynamic analysis. Echocardiography of neonates was performed while conscious. Mice were anesthetized with tribromoethanol (0.18 mL/10 g of body weight) and echocardiographic images were obtained using a VisualSonics Vevo 2100 system (Canada). M-mode echocardiography of the left ventricle was recorded at the tip of the mitral valve apparatus using a 30-MHz transducer (Vevo 2100, Canada).
Tissue preparation and histological analyses
Hearts were excised and washed in cold PBS carefully, then freed of connective tissue. Each heart was weighted and heart weight was normalized by body weight (HW/BW ratio). For collagen quantification, paraffin sections (6 µm) of myocardial sections were also stained with Masson’s trichrome. Normal cardiac myocytes were stained dark red, and fibrotic areas were stained blue.
Statistical analysis
Significance was determined using an unpaired two-way Student’s t-test (GraphPad Prism 5). Statistical comparisons between two different groups were performed with unpaired student’s t-test, ANOVA followed by post-hoc testing (LSD) was used for all comparisons between multiple groups. *P < 0.05, **P<0.01 was considered significant.
Result
Induced PP2ACα knockout in the working myocardium of adult hearts
A tamoxifen (Tam)-inducible, myocardial cell-specific PP2ACα (exon 2,200 bp) knockout mouse model was used to investigate the role of PP2A in myocardial fibrosis introduced heart failure. PP2ACα knockout mice were obtained by crossing homozygous PP2ACαlox/lox mice carrying the loxP sites across exon 2 with heterozygous Myh6-MerCreMer (MCM) mice carrying the Cre recombinase sequence downstream of the cardiac-specific α-myosin heavy chain promoter (Figure 1A). In our study, knockout and control groups consisted of PP2ACαlox/+, Myh6-MerCreMer transgenic mice treated with tamoxifen at 3 month (Figure 1B). Total RNA were isolated from mice’s hearts and the mutant form of PP2ACα knockout mRNA in these mice was verified by RT-PCR, which was 200 bp shorter than control wild type, PP2ACα gene level was decreased in knockout mice by quantitative real-time PCR examine (Figure 1C). To examine PP2ACα at the protein level, western blot was performed to detect the catalytic subunits of PP2A. Figure 1D shows that the protein levels of PP2AC decrease significantly in knockout mice. Phosphatase activity was found elevated in knockout in comparison to control controls (Figure 1E), PP2A activity was reduced significantly. These results indicate that PP2A activity impaired knockout mice were constructed by breeding the PP2ACαlox/lox mice with Myh6-MerCreMer mice.
Figure 1.
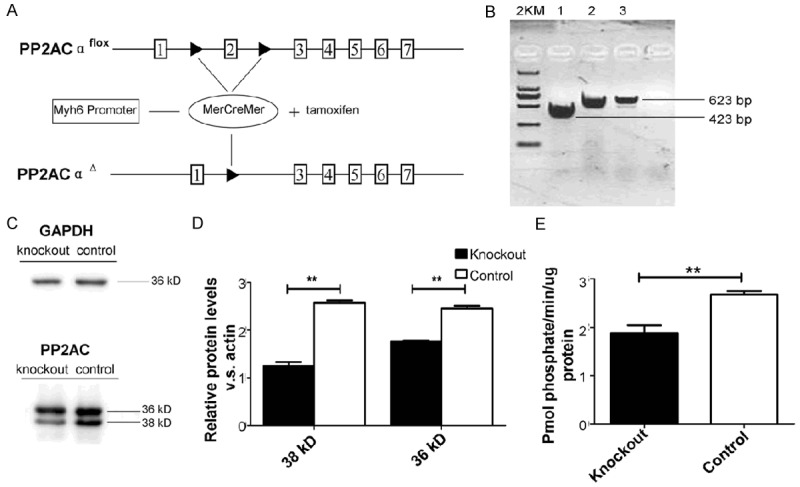
Construction and identification of PP2ACα knockout mice. A. The recombination process and the loxP sites used for PP2ACα knockout. B. Prim PP2A-P5F and PP2A-P5R were used to detect PP2ACα cDNA. The solid line represents detecting PP2ACα cDNA was 200 bp shorter than the control. Quantitative real-time PCR assay was carried out to examine the expression levels of PP2ACα gene. C. Western blot demonstrates PP2AC protein levels (36 and 38 kDa) of mice heart protein extracts from the knockout group and the control group 10 days after tamoxifen injection. D. Statistical analysis for the western blot. E. Phosphatase activity was elevated both in knockout and control groups. n ≥ 6 in each group, *P < 0.05, **P < 0.01.
Loss of PP2ACα leads to heart failure
We next investigated the cardiac phenotype of both knockout and control group mice at 3 month. Echocardiography was used to evaluate cardiac chamber size and function in vivo among knockout and control mice before tamoxifen injection, 3, 10, 17 and 60 days after injection. The wall motion in knockout hearts was drastically attenuated and the blood flow in the left ventricular chamber was not properly maintained start at about 10 days after injection (Figure 2A). Calculated ejection fraction (EF%), fractional shortening (FS%) and a measure of systolic function indicated that the cardiac function in PP2ACα knockout mice was impaired gradually after tamoxifen injection and eventually went into a decompensation stage also start at about 10 days after injection. We found that left ventricular mass (LV Mass) and left ventricular end-diastolic volumes (LV Vol), important descriptors of cardiac status, in knockout mice were larger than in control mice since 10 days after injection (Figure 2B). Cardiac hypertrophy is frequently associated with gene re-expression during fetal and perinatal development, and with the upregulation of some cardiac proteins, such as ANP (atrial natriuretic peptide), BNP (brain natriuretic peptide), α-MHC (α-myosin heavy chain) and β-MHC (β-myosin heavy chain). Quantitative real-time PCR of myocardial cell RNA showed that these genes were significantly upregulated in the knockout mice hearts compared with controls at 60 days after injection. Hypertrophy markers were highly expressed in knockout heart (Figure 2C).
Figure 2.
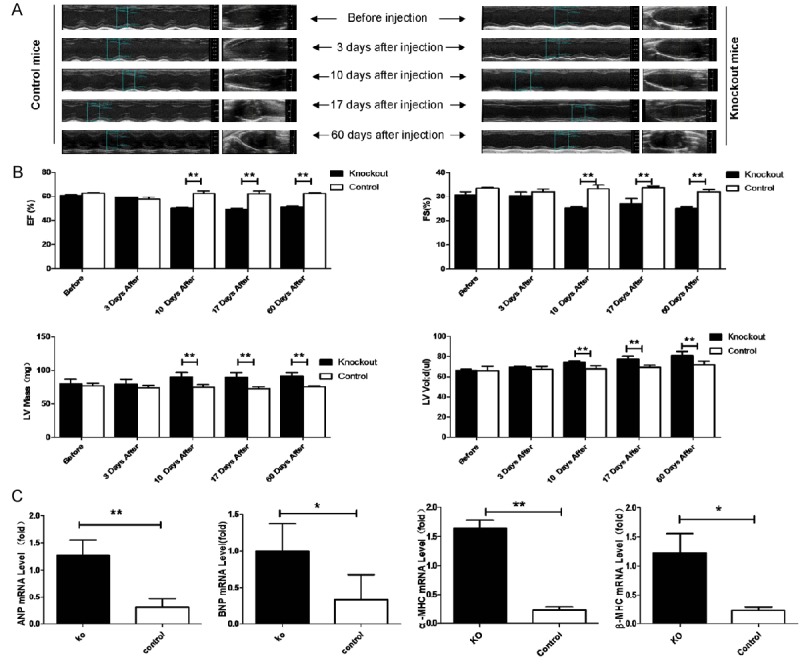
Echocardiographic assessment of knockout and control wild type mice. A. Echocardiography was performed before tamoxifen injection, 3 days after injection, 10 days after injection, 17 days after injection, 60 days after injection. B. Ejection fraction (EF%), fraction shortening (FS%), left ventricular mass (LV Mass [mg]), left ventricular end-diastolic volumes (LV vol [μl]) were calculated. C. Quantitative real-time PCR assay was carried out to examine the expression levels of fetal genes including A. ANF, B. BNP, and C. βMHC. n ≥ 6 in each group, *P < 0.05, **P < 0.01.
Morphological features of PP2ACα knockout mice
Mice were killed at 60 days after tamoxifen injection. Knockout mouse heart volume increased significantly compared with control mice (Figure 3A). Compared with knockout and control mice, heart to body ratio (H/B) of both mice (Figure 3B) was in accordance with LV Mass and LV vol (Figure 2B). Figure 3C shows Masson’s trichrome staining of a representative heart tissue section from a mouse. The amount of collagen fibrils in knockout mice was significantly higher compared to control mice (Figure 3C). Pathologic cardiac remodeling of myocardial fibers in knockout mice is caused by compensatory response to long-term cardiac function defecting.
Figure 3.
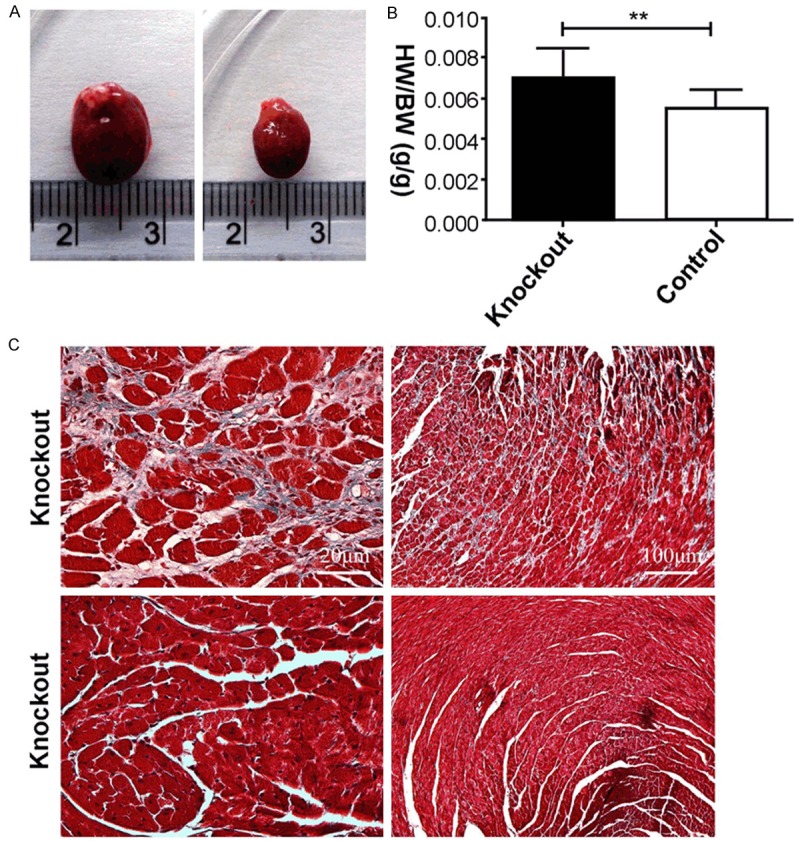
Morphological features of PP2ACα knockout mice at 60 days after injection. A. Overall appearance of knockout and control mice. B. Heart to body ratio (H/B) of knockout and control mice. C. Myocardial fibrosis induced by PP2ACa knockout and evaluated by Masson trichrome staining; indicative heart section from knockout mice and the control group; n ≥ 6 in each group,*P < 0.05, **P < 0.01.
PP2A activity decreased leads to B subunits changed and Akt/GSK3β/β-catenin pathway disrupted
PP2A protein B subunits expression were detected from adult mice hearts. RNA extracted from mice heart, was reverse transcribed into cDNA. PP2A-B subunits expression were tested by qPCR with β-actin as a reference. The results showed in knockout mice heart, B55a and B56e express increased with significant difference compared with the control group. B55b, B56a, B56b, B56c1, B56c2, B56d and B56e, also express higher, while the expression of B55b decreased (Figure 4A).
Figure 4.
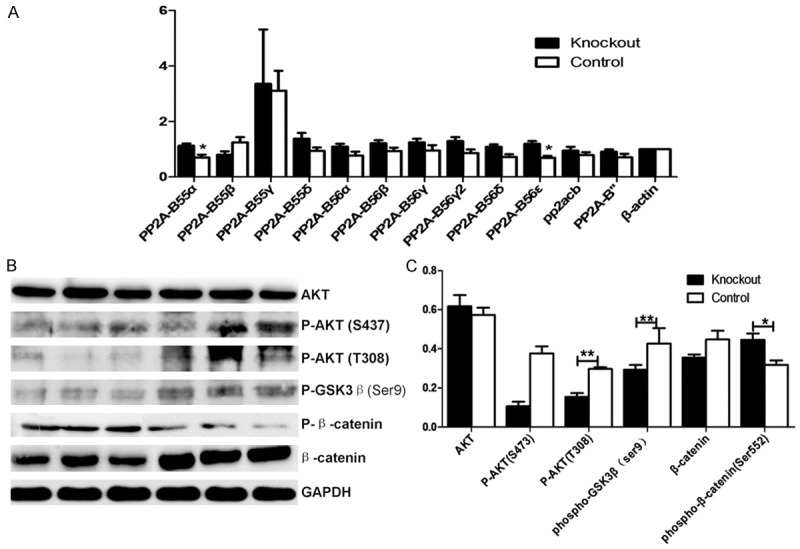
PP2A B subunits changed and Akt/GSK3β/β-catenin pathway disrupted. A. Quantitative real-time PCR assay was carried out to examine the expression levels of PP2ACb genes. B. Western blot of AKT, P-AKT (S473), P-AKT (T308), phospho-GSK3β (ser9), β-catenin, P-β-catenin (Ser552) from mice heart protein extracts from the knockout group and the control group 10 days after tamoxifen injection. C. Statistical analysis for the western blot. n ≥ 6 in each group,*P < 0.05, **P < 0.01.
The B55a subunit directly binds to the β-catenin destruction complex, where PP2A dephosphorylates β-catenin and induces its degradation [16,17]. Hence, we tried to explore the involvement of members of the Wnt signaling pathway in our infarctionmodel (Figure 4B). While we found no differences regarding the total amount of Akt between the different groups, there was a significant decrease of Akt-phosphorylation in knockout mice (Figure 4C). GSK-3β, the β-catenin negative regulator, is activated both directly and through AKT inhibition by PP2A. In knockout mice, no significant difference of phosphorylated GSK3β was detected. Controversially, we found significantly lower concentrations of β-catenin in PP2A knockout mice with an increase of β-catenin phosphorylated on Ser552. These findings indicate that the otherwise close regulation of β-catenin by Akt and GSK3β is lost under lower activity of PP2A.
Discussion
PP2A is ubiquitously expressed in almost every tissue, is globular in structure. The present study highlights the importance of PP2A for proper cardiac function in adult mice. Protein phosphatases contribute to cardiac structural remodeling, most notably to hypertrophy. Former research of heart-specific PP2A overexpression using a mutant PP2A scaffolding subunit mouse model showed that it was unable to bind regulatory subunits, and the mice then experienced cardiac hypertrophy, dilatation, and contractile dysfunction [18-20]. L-type Ca2+ currents decreased in this model, but arrhythmogenesis was not assessed. Other effects of PP2A modulation on cardiac disease have been less extensively characterized [21,22]. For instance, expression of casein kinase-2 interacting protein-1 (CKIP-1), a modulator of PP2A and PP1, can reduce PP2A-mediated dephosphorylation of histone deacetylase type-4, and then promote cardiac hypertrophy and fibrosis in mice [23]. Reduced PP2A activity in vivo may contribute to heart failure-related structural remodeling, but the precise roles of PP1/PP2A in atrial hypertrophy and fibrosis remain largely unknown [24].
We chose to induce Cre recombinase by tamoxifen at 3 month in knockout mice to avoid mice dying during the postnatal period [25]. The generation of mice with cardiac PP2A activity decreased was initially driven by the intention to further understand the causal relationship between increased phosphatase expression and the development of heart failure. PP2A transgenic mice are characterized by an impaired cardiac pump function indicating that the development of dilated cardiomyopathy. Next, we measured calculated EF%, FS%, left ventricular mass (LV Mass) and left ventricular end-diastolic volumes (LV Vol) in mice using M-mode echocardiography before tamoxifen injection, 3 days after injection, 10 days after injection, 17 days after injection, 60 days after injection. The cardiomyocyte-specific PP2ACα knockout induced cardiac hypertrophy since 10 days after injection, as indicated by the significant increase in absolute H/B weight ratio at 60 days after injection. Myocardial hypertrophy in this model was characterized by increased size of cardiomyocytes and expression of ANP, BNP, α-MHC and β-MHC genes [26].
Quantitative real-time PCR of myocardial cell RNA showed that these genes were significantly upregulated in the knockout mice hearts compared with controls at 60 days after injection. Hypertrophy markers were highly expressed in knockout heart. In the heart, PP1, PP2A and calcineurin make up almost 90% of protein phosphatase activity [24,27]. PP2ACα knockout mice experienced cardiac hypertrophy and myocardial fibrosis as adults, thus heart dysfunction observed in our mouse model was caused by decreased PP2A activity. We used Masson’s trichrome staining to test the presence of collagen fibrils inside the myocardium and particularly between the capillaries and the myocardium cell. Induced hypertrophy, may be the cause of the perivascular myocardial fibrosis that we noted.
Different B subunits interact via the same or overlapping sites within the A subunit of the core dimer. The association of these B subunits with the core AC dimer is mutually exclusive [28-30]. The PP2A holoenzyme’s substrate specificity, enzymatic activity, and/or cellular localization can be modulated by the B regulatory subunit [31].
PP2A-B subunits expression were tested by qPCR with β-actin as a reference. The results showed in knockout mice heart, B55a and B56e express increased with significant difference compared with the control group. B55b, B56a, B56b, B56c1, B56c2, B56d, B56e, B’’also express higher without significant, while the expression of B55b decreased. Each family of B subunits contains several isoforms that can bind the AC dimer in a mutually exclusive manner [32,33]. Overexpression of B56 family members inhibits secondary axis induced by Wnt and impairs endogenous axis specification during Xenopus embryonic development. Based on epistasis analysis, B56s were placed downstream of Casein Kinase I, GSK3, and Axin, but upstream of ß-catenin in the Wnt pathway. Axin interacts with B56-containing PP2As [34,35]. The B55a subunit directly binds to the β-catenin destruction complex, where PP2A dephosphorylates β-catenin and induces its degradation [36].
Total amount of Akt were at the same level of the PP2A knockout mice and the control. But phosphorylated Akt and phosphorylated GSK3β decreased, and β-catenin was hyperphosphorylated and the total amount of β-catenin was reduced in PP2A knockout mice. Wnt signaling, by inhibiting GSK3β mediated phosphorylation and cytoplasmic β-catenin degradation to introduce EMT (Epithelial-Mesenchymal Transition), has a fundamental role in cardiac remodeling and is unquestionable dependent on β-catenin balance [37]. Former research suggested a link between Wnt signaling and PP2A activity. β-catenin stability and activity are controlled at various phosphorylation sites: Phosphorylation at Ser45 through casein kinase I promotes β-catenin degradation [38]. Phosphorylation at Ser33/Ser37/Thr41 through GSK3β also promotes degradation of β-catenin [39]. Phosphorylation of β-catenin at Ser552 through protein kinase A, Akt or cAMP dependent protein kinase does not alter β-catenin degradation but promotes transcriptional activity [40]. GSK3β phosphorylation in knockout mice decreased while constitutive-active cardiac GSK3β is protected from adverse remodeling [41]. These findings may be related to the disruption of β-catenin/GSK3β balance and the downstream effectors of Wnt signaling.
During the postnatal stage, PP2ACα ablation in mouse hearts causes premature death via heart failure; mice died on postnatal day 13 [42]. One major advantage of our PP2ACα knockout mouse model is that the activity of the Cre recombinase can be controlled in a temporal fashion by tamoxifen administration. The adult knockout mice were observed in vivo echocardiography with significantly reduced cardiac function. Moreover, loss of PP2ACα results in cardiac hypertrophy. We found a B55a and B56e express increased in knockout mice. While there were no differences regarding the total amount of Akt between the two different groups, and phosphorylated Akt and phosphorylated GSK3β decreased. It turned out that β-catenin was hyperphosphorylated and the total amount of β-catenin was reduced in PP2A knockout mice. Our PP2ACα knockout mouse model can be used for further research investigating clinical targeting of phosphatases to treat cardiovascular diseases and related arrhythmias.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 31171443).
Disclosure of conflict of interest
None.
References
- 1.Ling S, Sun Q, Li Y, Zhang L, Zhang P, Wang X, Tian C, Li Q, Song J, Liu H, Kan G, Cao H, Huang Z, Nie J, Bai Y, Chen S, Li Y, He F, Zhang L, Li Y. CKIP-1 inhibits cardiac hypertrophy by regulating class II histone deacetylase phosphorylation through recruiting PP2A. Circulation. 2012;126:3028–3040. doi: 10.1161/CIRCULATIONAHA.112.102780. [DOI] [PubMed] [Google Scholar]
- 2.Gu P, Qi X, Zhou Y, Wang Y, Gao X. Generation of Ppp2Ca and Ppp2Cb conditional null alleles in mouse. Genesis. 2012;50:429–436. doi: 10.1002/dvg.20815. [DOI] [PubMed] [Google Scholar]
- 3.Pan X, Chen X, Tong X, Tang C, Li J. Ppp2ca knockout in mice spermatogenesis. Reproduction. 2015;149:385–391. doi: 10.1530/REP-14-0231. [DOI] [PubMed] [Google Scholar]
- 4.Luss H, Klein-Wiele O, Boknik P, Herzig S, Knapp J, Linck B, Muller FU, Scheld HH, Schmid C, Schmitz W, Neumann J. Regional expression of protein phosphatase type 1 and 2A catalytic subunit isoforms in the human heart. J Mol Cell Cardiol. 2000;32:2349–2359. doi: 10.1006/jmcc.2000.1265. [DOI] [PubMed] [Google Scholar]
- 5.Janssens V, Longin S, Goris J. PP2A holoenzyme assembly: in cauda venenum (the sting is in the tail) Trends Biochem Sci. 2008;33:1131121. doi: 10.1016/j.tibs.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 6.Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J. 2001;353:417–439. doi: 10.1042/0264-6021:3530417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Janssens V, Derua R, Zwaenepoel K, Waelkens E, Goris J. Specific regulation of protein phosphatase 2A PR72/B” subunits by calpain. Biochem Biophys Res Commun. 2009;386:676–681. doi: 10.1016/j.bbrc.2009.06.096. [DOI] [PubMed] [Google Scholar]
- 8.Cho US, Morrone S, Sablina AA, Arroyo JD, Hahn WC, Xu W. Structural basis of PP2A inhibition by small t antigen. PLoS Biol. 2007;5:e202. doi: 10.1371/journal.pbio.0050202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lechward K, Awotunde OS, Swiatek W, Muszynska G. Protein phosphatase 2A: variety of forms and diversity of functions. Acta Biochim Pol. 2001;48:921–933. [PubMed] [Google Scholar]
- 10.Herzig S, Neumann J. Effects of serine/threonine protein phosphatases on ion channels in excitable membranes. Physiol Rev. 2000;80:173–210. doi: 10.1152/physrev.2000.80.1.173. [DOI] [PubMed] [Google Scholar]
- 11.Carr AN, Schmidt AG, Suzuki Y, del Monte F, Sato Y, Lanner C, Breeden K, Jing SL, Allen PB, Greengard P, Yatani A, Hoit BD, Grupp IL, Hajjar RJ, DePaoli-Roach AA, Kranias EG. Type 1 phosphatase, a negative regulator of cardiac function. Mol Cell Biol. 2002;22:4124–4135. doi: 10.1128/MCB.22.12.4124-4135.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoehn M, Zhang Y, Xu J, Gergs U, Boknik P, Werdan K, Neumann J, Ebelt H. Overexpression of protein phosphatase 2A in a murine model of chronic myocardial infarction leads to increased adverse remodeling but restores the regulation of β-catenin by glycogen synthase kinase 3β. Int J Cardiol. 2015;183:39–46. doi: 10.1016/j.ijcard.2015.01.087. [DOI] [PubMed] [Google Scholar]
- 13.Dong D, Li L, Gu P, Jin T, Wen M, Yuan C, Gao X, Liu C, Zhang Z. Profiling metabolic remodeling in PP2Aca deficiency and chronic pressure overload mouse hearts. FEBS Lett. 2015;589:3631–3639. doi: 10.1016/j.febslet.2015.10.016. [DOI] [PubMed] [Google Scholar]
- 14.Hall ME, Smith G, Hall JE, Stec DE. Systolic dysfunction in cardiac-specific ligand-inducible MerCreMer transgenic mice. Am J Physiol Heart Circ Physiol. 2011;301:H253–260. doi: 10.1152/ajpheart.00786.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J. 2011;353:417–439. doi: 10.1042/0264-6021:3530417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin X, Xu X. Distinct functions of Wnt/betacatenin signaling in KV development and cardiac asymmetry. Development. 2009;136:207–217. doi: 10.1242/dev.029561. [DOI] [PubMed] [Google Scholar]
- 17.Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- 18.Haq S, Michael A, Andreucci M, Bhattacharya K, Dotto P, Walters B, Woodgett J, Kilter H, Force T. Stabilization of beta-catenin by a Wntindependent mechanism regulates cardiomyocyte growth. Proc Natl Acad Sci U S A. 2003;100:4610–4615. doi: 10.1073/pnas.0835895100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baruscotti M, Bucchi A, Viscomi C, Mandelli G, Consalez G, Gnecchi-Rusconi T, Montano N, Casali KR, Micheloni S, Barbuti A, DiFrancesco D. Deep bradycardia and heart block caused by inducible cardiac-specific knockout of the pacemaker channel gene Hcn4. Proc Natl Acad Sci U S A. 2011;108:1705–1710. doi: 10.1073/pnas.1010122108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nicolaou P, Hajjar RJ, Kranias EG. Role of protein phosphatase-1 inhibitor-1 in cardiac physiology and pathophysiology. J Mol Cell Cardiol. 2009;47:365–371. doi: 10.1016/j.yjmcc.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hall DD, Feekes JA, Arachchige Don AS, Shi M, Hamid J, Chen L, Strack S, Zamponi GW, Horne MC, Hell JW. Binding of protein phosphatase 2A to the L-type calcium channel Cav1.2 next to Ser1928, its main PKA site, is critical for Ser1928 dephosphorylation. Biochemistry. 2006;45:3448–3459. doi: 10.1021/bi051593z. [DOI] [PubMed] [Google Scholar]
- 22.Xu H, Ginsburg KS, Hall DD, Zimmermann M, Stein IS, Zhang M, Tandan S, Hill JA, Horne MC, Bers D, Hell JW. Targeting of protein phosphatases PP2A and PP2B to the C-terminus of the L-type calcium channel Ca v1.2. Biochemistry. 2010;49:10298–10307. doi: 10.1021/bi101018c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ling S, Sun Q, Li Y, Zhang L, Zhang P, Wang X, Tian C, Li Q, Song J, Liu H, Kan G, Cao H, Huang Z, Nie J, Bai Y, Chen S, Li Y, He F, Zhang L, Li Y. CKIP-1 inhibits cardiac hypertrophy by regulating class II histone deacetylase phosphorylation through recruiting PP2A. Circulation. 2012;126:3028–3040. doi: 10.1161/CIRCULATIONAHA.112.102780. [DOI] [PubMed] [Google Scholar]
- 24.Heijman J, Dewenter M, El-Armouche A, Dobrev D. Function and regulation of serine/threonine phosphatases in the healthy and diseased heart. J Mol Cell Cardiol. 2013;64:90–98. doi: 10.1016/j.yjmcc.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 25.Takeda M, Briggs LE, Wakimoto H, Marks MH, Warren SA, Lu JT, Weinberg EO, Robertson KD, Chien KR, Kasahara H. Slow progressive conduction and contraction defects in loss in Nkx2-5 mice after cardiomyocyte terminal differentiation. Lab Invest. 2009;89:983–993. doi: 10.1038/labinvest.2009.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, Penninger JM, Molkentin JD. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res. 2001;89:20–25. doi: 10.1161/hh1301.092687. [DOI] [PubMed] [Google Scholar]
- 27.Carr AN, Schmidt AG, Suzuki Y, del Monte F, Sato Y, Lanner C, Breeden K, Jing SL, Allen PB, Greengard P, Yatani A, Hoit BD, Grupp IL, Hajjar RJ, DePaoli-Roach AA, Kranias EG. Type 1 phosphatase, a negative regulator of cardiac function. Mol Cell Biol. 2002;22:4124–4135. doi: 10.1128/MCB.22.12.4124-4135.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Janssens V, Derua R, Zwaenepoel K, Waelkens E, Goris J. Specific regulation of protein phosphatase 2A PR72/B’’ subunits by calpain. Biochem Biophys Res Commun. 2009;386:676–681. doi: 10.1016/j.bbrc.2009.06.096. [DOI] [PubMed] [Google Scholar]
- 29.Park JH, Sung HY, Lee JY, Kim HJ, Ahn JH, Jo I. B56alpha subunit of protein phosphatase 2A mediates retinoic acid-induced decreases in phosphorylation of endothelial nitric oxide synthase at serine 1179 and nitric oxide production in bovine aortic endothelial cells. Biochem Biophys Res Commun. 2013;430:476–481. doi: 10.1016/j.bbrc.2012.12.011. [DOI] [PubMed] [Google Scholar]
- 30.Zwaenepoel K, Louis JV, Goris J, Janssens V. Diversity in genomic organisation, developmental regulation and distribution of the murine PR72/B” subunits of protein phosphatase 2A. BMC Genomics. 2008;9:393. doi: 10.1186/1471-2164-9-393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahn JH, Sung JY, McAvoy T, Nishi A, Janssens V, Goris J, Greengard P, Nairn AC. The B’’/PR72 subunit mediates Ca2+-dependent dephosphorylation of DARPP-32 by protein phosphatase 2A. Proc Natl Acad Sci U S A. 2007;104:9876–9881. doi: 10.1073/pnas.0703589104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lechward K, Awotunde OS, Swiatek W, Muszynska G. Protein phosphatase 2A: variety of forms and diversity of functions. Acta Biochim Pol. 2001;48:921–933. [PubMed] [Google Scholar]
- 33.Perrotti D, Neviani P. Protein phosphatase 2A: a target for anticancer therapy. Lancet Oncol. 2013;14:e229–238. doi: 10.1016/S1470-2045(12)70558-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao ZH, Seeling JM, Hill V, Yochum A, Virshup DM. Casein kinase I phosphorylates and destabilizes the beta-catenin degradation complex. Proc Natl Acad Sci U S A. 2002;99:1182–1187. doi: 10.1073/pnas.032468199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li X, Yost HJ, Virshup DM, Seeling JM. Protein phosphatase 2A and its B56 regulatory subunit inhibit Wnt signaling in Xenopus. EMBO J. 2001;20:4122–4131. doi: 10.1093/emboj/20.15.4122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 37.Gao ZH, Seeling JM, Hill V, Yochum A, Virshup DM. Casein kinase I phosphorylates and destabilizes the beta-catenin degradation complex. Proc Natl Acad Sci U S A. 2002;99:1182–1187. doi: 10.1073/pnas.032468199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao J, Yue W, Zhu MJ, Sreeiavan N, Du M. AMP-activated protein kinase (AMPK) crosstalks with canonical Wnt signaling via phosphorylation of beta-catenin at Ser 552. Biochem Biophys Res Commun. 2010;395:146–151. doi: 10.1016/j.bbrc.2010.03.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taurin S, Sandbo N, Qin Y, Browning D, Dulin NO. Phosphorylation of betacatenin by cyclic AMP-dependent protein kinase. J Biol Chem. 2006;281:9971–9976. doi: 10.1074/jbc.M508778200. [DOI] [PubMed] [Google Scholar]
- 40.Fang D, Hawke D, Zheng Y, Xia Y, Meisenhelder J, Nika H, Mills GB, Kobayashi R, Hunter T, Lu Z. Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J Biol Chem. 2007;282:11221–11229. doi: 10.1074/jbc.M611871200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Antos CL, McKinsey TA, Frey N, Kutschke W, McAnally J, Shelton JM, Richardson JA, Hill JA, Olson EN. Activated glycogen synthase-3 beta suppresses cardiac hypertrophy in vivo. Proc Natl Acad Sci U S A. 2002;99:907–912. doi: 10.1073/pnas.231619298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen W, Gu P, Jiang X, Ruan HB, Li C, Gao X. Protein phosphatase 2A catalytic subunit alpha (PP2Acalpha) maintains survival of committed erythroid cells in fetal liver erythropoiesis through the STAT5 pathway. Am J Pathol. 2011;178:2333–2343. doi: 10.1016/j.ajpath.2011.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]