

Abstract
Endothelial cell injury and subsequent death play an essential role in the pathogenesis of atherosclerosis. Autophagy of endothelial cells antagonizes the development of atherosclerosis, whereas the underlying molecular mechanisms are unclear. MicroRNA-129-5p (miR-129-5p) is a well-defined tumor suppressorin some types of cancer, while it is unknown whether miR-129-5p may also play a role in the development of atherosclerosis. Here, we addressed this question in the current study. We examined the levels of endothelial cell autophagy in ApoE (-/-) mice suppled with high-fat diet (HFD), a mouse model for atherosclerosis (simplified as HFD mice). We analyzed the levels of Beclin-1 and the levels of miR-129-5p in the purified CD31+ endothelial cells from mouse aorta. Prediction of the binding between miR-129-5p and 3’-UTR of Beclin-1 mRNA was performed by bioinformatics analyses and confirmed by a dual luciferase reporter assay. The effects of miR-129-5p were further analyzed in an in vitro model using oxidized low-density lipoprotein (ox-LDL)-treated human aortic endothelial cells (HAECs). We found that HFD mice developed atherosclerosisin 12 weeks, while the control ApoE (-/-) mice that had received normal diet (simplified as CTL mice) did not. Compared to CTL mice, HFD mice had significantly lower levels of endothelial cell autophagy, resulting from decreases in Beclin-1 protein, but not mRNA. The decreases in Beclin-1 in endothelial cells were due to HFD-induced increases inmiR-129-5p, which suppressed the translation of Beclin-1 mRNA via 3’-UTR binding. These in vivo findings were reproduced in vitro on ox-LDL-treated HAECs. Together, these data suggest that upregulation of miR-129-5p by HFD may impair the protective effects of endothelial cell autophagy against development of atherosclerosis through suppressing protein translation of Beclin-1.
Keywords: Atherosclerosis, endothelial cell autophagy, ApoE (-/-), high fat diet (HFD), ox-LDL, Beclin-1, miR-129-5p
Introduction
Atherosclerosis is a chronic inflammatory disease in which lipids and fibrous elements are deposited in the arterial wall large and medium-sized arteries to activate both the innate and adaptive immune systems. Atherosclerosis is the primary cause of heart disease and stroke, which accounts for more than half of all deaths in aged people [1,2]. The progression of atherosclerosis includes early accumulation of cholesterol-engorged macrophages in the sub-endothelial matrix, accumulation of lipid-rich necrotic debris, elevated numbers of smooth muscle cells (SMCs), and subsequent development of fibrosis, which lead to formation of complex plaques with calcification, ulceration and hemorrhage eventually [1,2].
Apolipoprotein E (ApoE) is a 34-kDa secreted protein, as a well-known strong suppressor for atherosclerosis [3,4]. ApoE not only regulates lipoprotein cholesterol transport and controls cellular lipid regulation, but also potently inhibits inflammation [3,4]. For example, ApoE-deficient (ApoE -/-) mice display enhanced chronic inflammation in response to spontaneous and diet-induced hypercholesterolemia, and display enhanced acute immune response when challenged with bacterial lipopolysaccharide (LPS) [3-9]. High fat diet (HFD) induces development of atherosclerosis in ApoE -/- mice in 12 weeks [10-12].
Endothelial cell injury is a major step for the pathological progression of atherosclerosis [13-17]. Upon injury, endothelial cell autophagy may occur to protect the cells from being damaged, while the failure or inhibition of autophagy results in endothelial cell apoptosis, leading to the break-down of the integrity of endothelium to facilitate the local lipid deposition into atherogenesis, plaque instability, and even acute coronary occlusion and sudden death [13,18-23]. Nevertheless, our understanding of the mechanisms that control the autophagy of endothelial cells is still limited.
Autophagy is a catabolic pathway that degrades and recycles cellular compartments for cell survival at various stresses, whereas its failure often leads to cell death [24]. Microtubule-associated protein 1A/1B-light chain 3 (LC3) is a soluble cellular protein. During autophagy, autophagosomes engulf cytoplasmic components, resulting in conjugation of a cytosolic form of LC3 (LC3-I) to phosphatidylethanolamine to form LC3-phosphatidylethanolamine conjugate (LC3-II). Thus, the ratio of LC3-II to LC3-I represents the autophagic activity [24-26]. Beclin-1, ATG7 and ATG5 are several key autophagy-associated proteins that strongly induce autophagy in an independent or coordinated manner [27]. Autophagy has been shown to play a critical role in endothelial cells to prevent development of atherosclerosis [13,18-23]. Nevertheless, the underlying mechanisms have not been completely elucidated.
MicroRNAs (miRNAs) are small non-coding RNAs that regulate protein translation through their base-pairing with the 3’-untranslated region (3’-UTR) of the target mRNAs. MiRNAs play essential roles in regulating atherosclerosis [5,28,29]. Among all miRNAs, miR-129-5p has been shown to suppress carcinogenesis of various cancers [30-38]. Nevertheless, a role of miR-129-5p in the atherosclerosis has not been studied.
Here, we found that high-fat-diet (HFD) -treated ApoE (-/-) mice developed atherosclerosis in 12 weeks, while the control ApoE (-/-) mice that had received normal diet (simplified as CTL mice) did not. Compared to CTL mice, HFD mice had significantly lower levels of endothelial cell autophagy, resulting from decreases in Beclin-1 protein, but not mRNA. The decreases in Beclin-1 in endothelial cells were due to HFD-induced increases in miR-129-5p, which suppressed the translation of Beclin-1 mRNA via 3’-UTR binding. These in vivo findings were reproduced in vitro on oxidized low-density lipoprotein (ox-LDL)-treated human aortic endothelial cells (HAECs).
Materials and methods
Animal models and quantification of atherosclerotic lesions
The study was approved by the Animal Care and Use Committee of Third Military Medical University. All experimental procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals, published by the US National Institutes of Health. All experiments were conducted under the supervision of the facility’s Institutional Animal Care and Use Committee according to an Institutional Animal Care and Use Committee-approved protocol. Ten-week-old male ApoE -/- mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA) or bred in-house and maintained under sterile conditions and standard animal room conditions (temperature, 21 ± 1°C; humidity, 55-60%). The animals were randomly divided into two groups: the normal-diet group (CTL) and the high-fatdiet (HFD) group. The animals of the HFD group were maintained for 12 weeks to induce atherosclerosis, after which the aortas were excised from the mice. The aortic roots along with the basal portion of the heart were fixed with 4% paraformaldehyde for 4 hours, cryo-protected in 30% sucrose for 12 hours, and then embedded in OCT compound. The tissue was cross-sectioned into sections of 6 μm thickness. Atherosclerotic lesions of the aortic root were examined by H&E staining. Oil red O staining was performed according to the manufacturer’s instructions to show the lipid deposition with an Oil red O staining kit (Abcam, Cambridge, MA, USA). Quantification of the images was measured using NIH ImageJ software (Bethesda, MD, USA). The data were calculated from 5 mice for each group. For each mouse, 3 slides that were 20 µm apart from each other were used for quantification.
Cell culture, treatment and transfection
Normal human aortic endothelial cells (HAECs) were purchased from American Type Culture Collection (ATCC PCS-100-011, Rockville, MD, USA) and cultured in Endothelial Cell Media supplemented with endothelial cell growth factors, 5% fetal bovine serum (FBS, Invitrogen, CA, Carlsbad, USA) and 1% penicillin/streptomycin (Invitrogen) at 37°C with 5% CO2. HAECs were transiently transfected with miR-129-5p mimics, antisense for miR-129-5p (as-miR-129-5p) or null controls (RiboBio Co., Ltd., Guangzhou, Guangdong, China), using Lipofectamine 2000 reagent (Invitrogen), according to the manufacturer’s instructions. The transfection efficiency was nearly 100%. One day after cell transfection, the cells were treated with or without 100 µg/ml oxidized low-density lipoprotein (ox-LDL, Beijing Xiesheng Bio-Technology Limited, Beijing, China). After drug treatment, the cells were used for flow cytometry or protein/RNA extraction.
Cell counting kit-8 (CCK-8) assay
The CCK-8 detection kit (Sigma-Aldrich, St. Louis, MO, USA) was used to measure cell viability according to the manufacturer’s instructions. Briefly, cells were seeded in a 96-well microplate at a density of 5×104/ml. After 24 h, cells were treated with resveratrol. Subsequently, CCK-8 solution (20 ml/well) was added and the plate was incubated at 37°C for 2 hours. The viable cells were counted by absorbance measurements with a monochromator microplate reader at a wavelength of 450 nm. The optical density value was reported as the percentage of cell viability in relation to the control group (set as 100%).
Flow cytometry
For analyses and isolation of CD31+ cells, the aorta was dissociated by 10 μg/ml trypsin (Sigma-Aldrich) and 10 μg/ml DNase (Roche, Nutley, NJ, USA) for 35 minutes. After filtration at 30 μm, the single cell digests from mouse aorta were incubated with PE-cy7-CD31 (Becton-Dickinson Biosciences, San Jose, CA, USA) and then sorted on a FACSAria (Becton-Dickinson Biosciences).
Quantitative RT-PCR
Aorta intimal RNA was isolated from mouse aortas. After cleaning with ice-cold PBS, mouse aortas were flushed with TRIzol reagent (Invitrogen) using an insulin syringe, and the eluate was collected in a 1.5 ml tube and prepared for RNA extraction. Total RNA and miRNAs were extracted from tissue or cultured cells with miRNeasy mini kit or RNeasy kit (Qiagen, Hilden, Germany), respectively. Complementary DNA (cDNA) was randomly primed from 2 μg of total RNA using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). RT-qPCR was subsequently performed in triplicate with QuantiTect SYBR Green PCR Kit (Qiagen). All primers were purchased from Qiagen. Data were collected and analyzed using 2-ΔΔCt method for quantification of the relative mRNA expression levels. Values of genes were first normalized against α-tubulin, and then compared to the experimental controls.
Western blotting
The cells were lysed with RIPA buffer containing protease and phosphatase inhibitors (cOmplete ULTRA Tablets, Roche, Nutley, NJ, USA). After centrifugation, the supernatant was collected and quantified. The proteins were then separated by SDS-PAGE and transferred to nitrocellulose membranes. After blocking with 5% non-fat milk, the membranes were probed with rat anti-CD31 (Becton-Dickinson Biosciences), rabbitanti-ATG5, rabbit anti-LC3, rabbit-anti-Beclin-1, rabbit-anti-ATG7 and rabbit-anti-α-tubulin (Cell Signaling Technology, Danvers, MA, USA). Secondary antibodiesare HRP-conjugated against rat or rabbit (Jackson ImmunoResearch Labs, West Grove, PA, USA). The protein levels were first normalized to α-tubulin, and then normalized to the experimental controls. Densitometry of Western blots was quantified with NIH ImageJ software.
MiRNA target prediction and 3’-UTR luciferase-reporter assay
MiRNAs targets were predicted as has been described before, using the algorithms TargetSan (https://www.targetscan.org) [39]. The Beclin-1 3’-UTR reporter plasmid (pRL-Beclin-1) was purchased from Creative Biogene (Shirley, NY, USA). MiR-129-5p-modified HAECs were transfected with 0.5 μg pRL-Beclin-1 by Lipofectamine 2000 (5×104 cells per well). Cells were collected 48 hours after transfection for assay using the dual-luciferase reporter assay system gene assay kit (Promega, Madison, WI, USA), according to the manufacturer’s instructions.
Statistical analyses
The data in this study are shown as the mean ± S.D. Differences among groups were analyzed using one-away ANOVA with a Bonferroni correction, followed by Fisher’ Exact Test for comparison of two groups (GraphPad Prism, GraphPad Software, Inc. La Jolla, CA, USA). p<0.05 was considered significant.
Result
HFD induces atherosclerosis in ApoE (-/-) mice
Here, we gave a high-fat diet (HFD) to treat ApoE (-/-) mice (simplified as HFD mice) to induce experimental atherosclerosis in 12 weeks, which was confirmed by increases in aortic lesion size (Figure 1A), and by increases in lipid content in aortic sinus (Figure 1B), compared to littermate ApoE (-/-) mice that had received normal diet (CTL) as a control. Thus, HFD induces atherosclerosis in ApoE (-/-) mice. In order to analyze endothelial cells, we dissociated mouse aorta and purified endothelial cells based on CD31 labeling by flow cytometry (Figure 1C).
Figure 1.
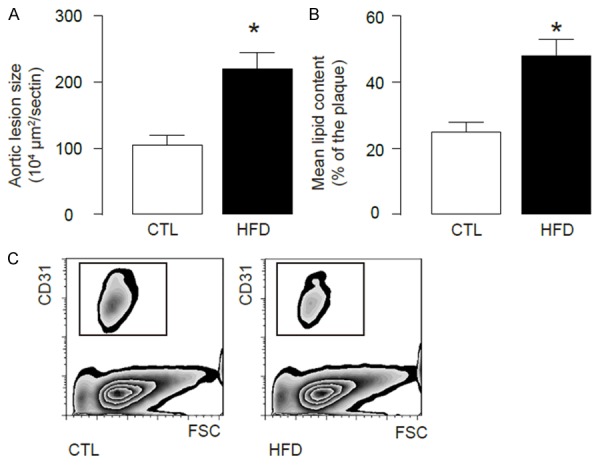
HFD induces atherosclerosis in ApoE (-/-) mice. We used ApoE (-/-) mice treated with high-fat diet (HFD; simplified as HFD mice) for analyzing endothelial cell apoptosis. ApoE (-/-) mice that had received normal diet (CTL) were used as a control. (A, B) After a 12-week HFD treatment, analysis of H&E-stained histological sections of the aortic sinus showed a significant increase in aortic lesion size (A), and analysis of Oil-red-O-stained histological sections of the aortic sinus showed a significant increase in lipid content (B). (C) The aortas were dissociated and purified for endothelial cells based on CD31 labeling by flow cytometry. *p<0.05. N=5.
HFD impairs endothelial cell autophagy in ApoE (-/-) mice
Then we analyzed the autophagy levels in endothelial cells from HFD and CTL mice. We found that the LC3-II/I ratio significantly decreased in endothelial cells from HFD mice, than those from CTL mice, shown by representative immunoblots (Figure 2A), and by quantification (Figure 2B). Then we analyzed the levels of major autophagy-activators ATG7, ATG5 and Beclin-1 in endothelial cells from these mice, and found no difference in the levels of ATG7 and ATG5 between endothelial cells from HFD and CTL mice, shown by representative immunoblots (Figure 2A), and by quantification (Figure 2C, 2D). However, we detected a significant decrease in the levels of Beclin-1 in endothelial cells from HFD mice, compared to those from CTL mice, shown by representative immunoblots (Figure 2A), and by quantification (Figure 2E). These data suggest that HFD may inhibit Beclin-1 to impair endothelial cell autophagy. Interestingly, the mRNA level of Beclin-1 was not altered in endothelial cells from HFD mice, compared to those from CTL mice (Figure 2F). These data suggest that the effects of HFD on endothelial cell Beclin-1 may be through post-transcriptional control.
Figure 2.
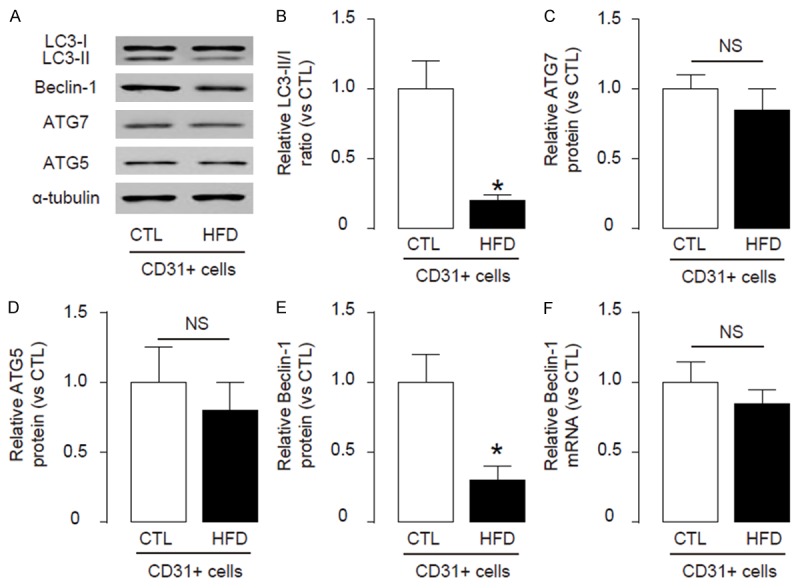
HFD impairs endothelial cell autophagy in ApoE (-/-) mice. (A-E) Immunoblots for apoptosis-associated genes LC3-II/I, ATG7, ATG5 and Beclin-1 in endothelial cells from HFD and CTL mice, shown by representative blots (A), and by quantification for LC3-II/I (B), ATG7 (C), ATG5 (D) and Beclin-1 (E). (F) RT-qPCR for mRNA of Beclin-1. *p<0.05. NS: non-significant. N=5.
HFD impairs endothelial cell autophagy in ApoE (-/-) mice through suppressing the translation of Beclin-1 mRNA by miR-129-5p
Next, we examined the underlying mechanisms. Since post-transcriptional control of gene expression is usually regulated miRNAs, we used bioinformatics analyses to screenallmiRNAs that target Beclin-1 and altered their levels in endothelial cells in HFD mice. Specifically, we found that miR-129-5p targets 3’-UTR of Beclin-1 mRNA at one binding site of base pair 97th to 106th (Figure 3A). Moreover, miR-129-5p levels in endothelial cells significantly increased after HFD treatment (Figure 3B). Hence, we modified miR-129-5p levels in human aortic endothelial cells (HAECs), by transfection of the cells with miR-129-5p mimics, antisense for miR-129-5p (as-miR-129-5p) or null controls (null). First, modification of miR-129-5p levels in HAECs was confirmed by RT-qPCR (Figure 3C). Next, the intact 3’-UTR of Beclin-1 mRNA was cloned into a luciferase reporter plasmid, and used for transfection of miR-129-5p-modified HAECs. We found that miR-129-5p significantly reduced the luciferase activity of 3’-UTR reporter for Beclin-1, while as-miR-129-5p significantly increased the luciferase activity of 3’-UTR reporter for Beclin-1, compared to null control (Figure 3D). Together, these data suggest that HFD impairs endothelial cell autophagy in ApoE (-/-) mice through suppressing the translation of Beclin-1 mRNA by miR-129-5p.
Figure 3.
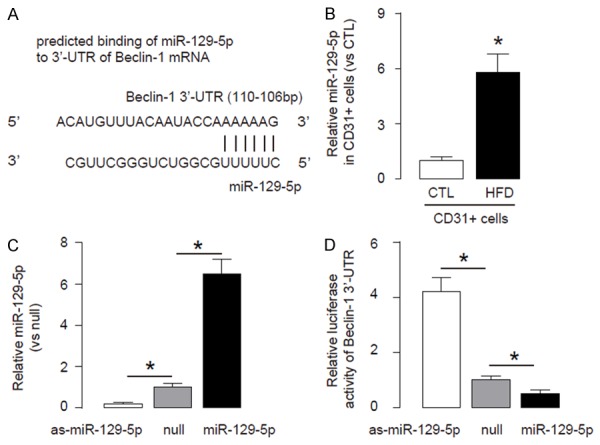
HFD impairs endothelial cell autophagy in ApoE (-/-) mice through suppressing the translation of Beclin-1 mRNA by miR-129-5p. A. Bioinformatics analyses showing that miR-129-5p targets 3’-UTR of Beclin-1 mRNA at one binding site of base pair 2820th to 2826th. B. MiR-129-5p levels in endothelial cells from HFD or CTL mice. C. RT-qPCR for miR30 in miR-129-5p-modified human aortic endothelial cells (HAECs), prepared by transfection of the cells with miR-129-5p mimics, antisense for miR-129-5p (as-miR-129-5p) or null controls (null). D. The intact 3’UTR of Beclin-1 mRNA was cloned into a luciferase reporter plasmid, and used for transfection of miR-129-5p-modified HAECs. The luciferase activities were quantified. *p<0.05. N=5.
Ox-LDL-treatmentsimilarly impairs cell autophagy in HAECs
To confirm the findings in HFDmice, we used an in vitro model of atherosclerosis, in which HAECs were treated with or without 100 µg/ml oxidized low-density lipoprotein (ox-LDL) to induce cell injury similar to in vivo model. First, we confirmed that ox-LDL treatment resulted in a significant reduction in viable cell number in an CCK-8 assay (Figure 4A). Moreover, ox-LDL significantly decreased the ratio of LC3II/I and Beclin-1levels (Figure 4B, 4D), without altering Beclin-1mRNA (Figure 4E). Finally, the levels of miR-129-5p were significantly increased by ox-LDL (Figure 4F). Together, these data suggest that ox-LDL-treated HAECs reproduceour findings in HFD on mice. Thus, our study suggest that the failure of atherosclerosis-associated endothelial cell autophagy may result from HFD-induced downregulation of Beclin-1, through increased miR-129-5p that binds and suppresses translation of Beclin-1 mRNA (Figure 5).
Figure 4.
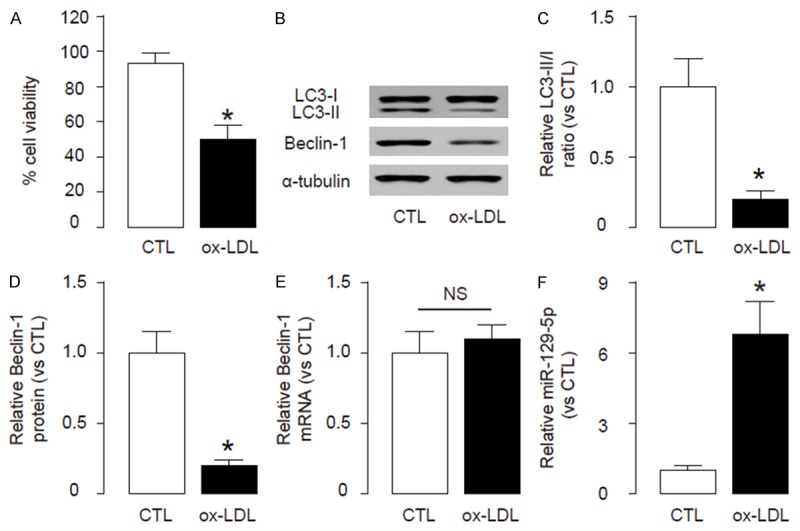
Ox-LDL-treatment similarly impairs cell autophagy in HAECs. To confirm the findings in HFD mice, we used an in vitro model of atherosclerosis, in which HAECs were treated with or without 100 µg/ml oxidized low-density lipoprotein (ox-LDL). (A) Quantification of viable cell number in an CCK-8 assay. (B-E) Immunoblots for apoptosis-associated genes LC3-II/I and Beclin-1 in endothelial cells from HFD and CTL mice, shown by representative blots (B), and by quantification for LC3-II/I (C) and Beclin-1 (D). (E, F) RT-qPCR for mRNA of Beclin-1 (E) and miR-129-5p (F). *p<0.05. NS: non-significant. N=5.
Figure 5.
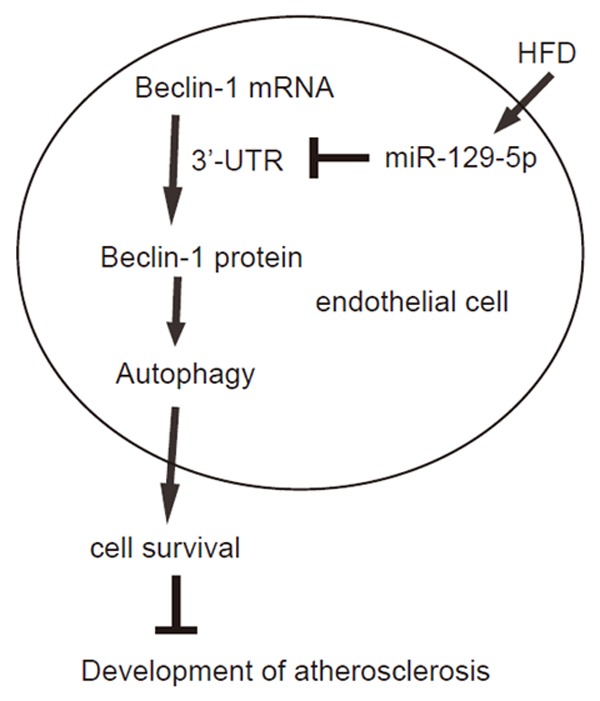
Schematic of the model. The failure of atherosclerosis-associated endothelial cell autophagy may result from HFD-induced downregulation of Beclin-1, through increased miR-129-5p that binds and suppresses translation of Beclin-1 mRNA.
Discussion
Atherosclerosis is a complex immune-inflammatory disease involving arteries of medium-to-large size and its pathogenesis is coordinated among endothelial cells, macrophages and smooth muscle cells. First of all, injury of the vascular endothelium is a proven initial step in the pathogenesis of atherosclerosis. High glucose is a potent pro-atherosclerotic factor that has been widely used to induce experimental atherosclerosis in vivo, while in vivo application of ox-LDL is able to cause endothelial cell injury that is observed during development of atherosclerosis.
Aberrantly expressed miRNAs have recently been found to be involved in the development of endothelial cell survival/death, supported by many recent reports. Specially, Pan et al. reported that cardiomyocyte autophagy was regulated by Beclin-1 and miR-129-5p in cardiac disease model. They used a dual luciferase reporter assay to confirm that Beclin-1 was a target gene of miR-129-5pa. These previous studies encouraged us to examine a possible role of miRNAs in the development of atherosclerosis through modulation of autophagy-related proteins.
Here, we found that HFD significantly impaired endothelia cell autophagy, and this failure of autophagy significantly increased endothelial cell death, through augment in cell apoptosis. Importantly, we found that Beclin-1 was the effector protein as a target of HFD, and most interestingly, its protein but not mRNA was changed by HFD. Of note, here the seemingly slight increases in mRNA (not reaching significance) may be resulting from a feedback of requirement of increasing the protein levels that continuously failed. Since it is unlikely that Beclin-1 may be regulated by phosphorylation or acetylation in our experimental setting, we hypothesized that it might be regulated by miRNAs.
All Beclin-1-targeting miRNAs were then determined by bioinformatics analyses, and we screened all these miRNAs and found that miR-129-5p levels significantly increased in CD31+ endothelial cells from HFD mice. In addition, we used an in vitro atherosclerosis model to confirm this model in endothelial cell apoptosis.
Previous studies have shown controversy on the exact role of autophagy in the heart under pathological conditions [40]. Moreover, some of these studies have demonstrated seemingly different results from our work [41-46]. However, we think that these seemingly contradictory data are actually reflections of the complexity of autophagy in the regulation of development of atherosclerosis. Since autophagy could be a double-edged sword, in which modest levels of autophagy could improve the survival of the cells under insults, while the augment of autophagy level could lead to autophagy-associated cell death. Moreover, the differences in the exact HFD protocols, mouse strains (here ApoE-knockout mice), doses and frequency of ox-LDL, and analyzed target cells (here purified endothelial cells rather than mixed cell types in some studies) may modify the relative levels of autophagy- and apoptosis- associated proteins in various studies, leading to seemingly paradoxical results. Nevertheless, these studies demonstrate a delicate control of the levels of autophagy may be crucial for the development of atherosclerosis.
Together, our study identified a new role for miR-129-5p in the regulation of endothelial cell autophagy during atherosclerosis, and shed new insight into development of innovative therapy by targeting miR-129-5p in atherosclerosis treatment.
Disclosure of conflict of interest
None.
References
- 1.Schmitz G, Grandl M. Lipid homeostasis in macrophages - implications for atherosclerosis. Rev Physiol Biochem Pharmacol. 2008;160:93–125. doi: 10.1007/112_2008_802. [DOI] [PubMed] [Google Scholar]
- 2.Liehn EA, Zernecke A, Postea O, Weber C. Chemokines: inflammatory mediators of atherosclerosis. Arch Physiol Biochem. 2006;112:229–238. doi: 10.1080/13813450601093583. [DOI] [PubMed] [Google Scholar]
- 3.Zhi K, Li M, Zhang X, Gao Z, Bai J, Wu Y, Zhou S, Li M, Qu L. α4β7 Integrin (LPAM-1) is upregulated at atherosclerotic lesions and is involved in atherosclerosis progression. Cell Physiol Biochem. 2014;33:1876–1887. doi: 10.1159/000362965. [DOI] [PubMed] [Google Scholar]
- 4.Dou L, Lu Y, Shen T, Huang X, Man Y, Wang S, Li J. Panax notogingseng saponins suppress RAGE/MAPK signaling and NF-kappaB activation in apolipoprotein-E-deficient atherosclerosis-prone mice. Cell Physiol Biochem. 2012;29:875–882. doi: 10.1159/000315061. [DOI] [PubMed] [Google Scholar]
- 5.Yang Y, Yang L, Liang X, Zhu G. MicroRNA-155 Promotes Atherosclerosis Inflammation via Targeting SOCS1. Cell Physiol Biochem. 2015;36:1371–1381. doi: 10.1159/000430303. [DOI] [PubMed] [Google Scholar]
- 6.Jia EZ, An FH, Chen ZH, Li LH, Mao HW, Li ZY, Liu Z, Gu Y, Zhu TB, Wang LS, Li CJ, Ma WZ, Yang ZJ. Hemoglobin A1c risk score for the prediction of coronary artery disease in subjects with angiographically diagnosed coronary atherosclerosis. Cell Physiol Biochem. 2014;34:672–680. doi: 10.1159/000363032. [DOI] [PubMed] [Google Scholar]
- 7.Sattler K, Lehmann I, Graler M, Brocker-Preuss M, Erbel R, Heusch G, Levkau B. HDLbound sphingosine 1-phosphate (S1P) predicts the severity of coronary artery atherosclerosis. Cell Physiol Biochem. 2014;34:172–184. doi: 10.1159/000362993. [DOI] [PubMed] [Google Scholar]
- 8.Lin J, Chang W, Dong J, Zhang F, Mohabeer N, Kushwaha KK, Wang L, Su Y, Fang H, Li D. Thymic stromal lymphopoietin over-expressed in human atherosclerosis: potential role in Th17 differentiation. Cell Physiol Biochem. 2013;31:305–318. doi: 10.1159/000343369. [DOI] [PubMed] [Google Scholar]
- 9.Gu Y, Liu Z, Li L, Guo CY, Li CJ, Wang LS, Yang ZJ, Ma WZ, Jia EZ. OLR1, PON1 and MTHFR gene polymorphisms, conventional risk factors and the severity of coronary atherosclerosis in a Chinese Han population. Cell Physiol Biochem. 2013;31:143–152. doi: 10.1159/000343356. [DOI] [PubMed] [Google Scholar]
- 10.Watson AM, Li J, Samijono D, Bierhaus A, Thomas MC, Jandeleit-Dahm KA, Cooper ME. Quinapril treatment abolishes diabetesassociated atherosclerosis in RAGE/apolipoprotein E double knockout mice. Atherosclerosis. 2014;235:444–448. doi: 10.1016/j.atherosclerosis.2014.05.945. [DOI] [PubMed] [Google Scholar]
- 11.Chatterjee S, Bedja D, Mishra S, Amuzie C, Avolio A, Kass DA, Berkowitz D, Renehan M. Inhibition of glycosphingolipid synthesis ameliorates atherosclerosis and arterial stiffness in apolipoprotein E-/- mice and rabbits fed a high-fat and -cholesterol diet. Circulation. 2014;129:2403–2413. doi: 10.1161/CIRCULATIONAHA.113.007559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cavigiolio G, Jayaraman S. Proteolysis of apolipoprotein A-I by secretory phospholipase A(2): a new link between inflammation and atherosclerosis. J Biol Chem. 2014;289:10011–10023. doi: 10.1074/jbc.M113.525717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peng N, Meng N, Wang S, Zhao F, Zhao J, Su L, Zhang S, Zhang Y, Zhao B, Miao J. An activator of mTOR inhibits oxLDL-induced autophagy and apoptosis in vascular endothelial cells and restricts atherosclerosis in apolipoprotein E(-)/(-) mice. Sci Rep. 2014;4:5519. doi: 10.1038/srep05519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun C, Wu MH, Yuan SY. Nonmuscle myosin light-chain kinase deficiency attenuates atherosclerosis in apolipoprotein E-deficient mice via reduced endothelial barrier dysfunction and monocyte migration. Circulation. 2011;124:48–57. doi: 10.1161/CIRCULATIONAHA.110.988915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leppanen P, Koota S, Kholova I, Koponen J, Fieber C, Eriksson U, Alitalo K, Yla-Herttuala S. Gene transfers of vascular endothelial growth factor-A, vascular endothelial growth factor-B, vascular endothelial growth factor-C, and vascular endothelial growth factor-D have no effects on atherosclerosis in hypercholesterolemic low-density lipoprotein-receptor/apolipoprotein B48-deficient mice. Circulation. 2005;112:1347–1352. doi: 10.1161/CIRCULATIONAHA.105.534107. [DOI] [PubMed] [Google Scholar]
- 16.Foteinos G, Afzal AR, Mandal K, Jahangiri M, Xu Q. Anti-heat shock protein 60 autoantibodies induce atherosclerosis in apolipoprotein E-deficient mice via endothelial damage. Circulation. 2005;112:1206–1213. doi: 10.1161/CIRCULATIONAHA.105.547414. [DOI] [PubMed] [Google Scholar]
- 17.Barton M, Haudenschild CC, d’Uscio LV, Shaw S, Munter K, Luscher TF. Endothelin ETA receptor blockade restores NO-mediated endothelial function and inhibits atherosclerosis in apolipoprotein E-deficient mice. Proc Natl Acad Sci U S A. 1998;95:14367–14372. doi: 10.1073/pnas.95.24.14367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xiong Y, Yepuri G, Forbiteh M, Yu Y, Montani JP, Yang Z, Ming XF. ARG2 impairs endothelial autophagy through regulation of MTOR and PRKAA/AMPK signaling in advanced atherosclerosis. Autophagy. 2014;10:2223–2238. doi: 10.4161/15548627.2014.981789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding Z, Liu S, Wang X, Dai Y, Khaidakov M, Romeo F, Mehta JL. LOX-1, oxidant stress, mtDNA damage, autophagy, and immune response in atherosclerosis. Can J Physiol Pharmacol. 2014;92:524–530. doi: 10.1139/cjpp-2013-0420. [DOI] [PubMed] [Google Scholar]
- 20.Martinet W, De Meyer GR. Autophagy in atherosclerosis. Curr Atheroscler Rep. 2008;10:216–223. doi: 10.1007/s11883-008-0034-y. [DOI] [PubMed] [Google Scholar]
- 21.Wang L, Li H, Zhang J, Lu W, Zhao J, Su L, Zhao B, Zhang Y, Zhang S, Miao J. Phosphatidylethanolamine binding protein 1 in vacular endothelial cell autophagy and atherosclerosis. J Physiol. 2013;591:5005–5015. doi: 10.1113/jphysiol.2013.262667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ding Z, Liu S, Wang X, Khaidakov M, Dai Y, Mehta JL. Oxidant stress in mitochondrial DNA damage, autophagy and inflammation in atherosclerosis. Sci Rep. 2013;3:1077. doi: 10.1038/srep01077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ding WX. Uncoupling AMPK from autophagy: a foe that hinders the beneficial effects of metformin treatment on metabolic syndrome-associated atherosclerosis? Focus on “glucose and palmitate uncouple AMPK from autophagy in human aortic endothelial cells”. Am J Physiol Cell Physiol. 2015;308:C246–248. doi: 10.1152/ajpcell.00375.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157:65–75. doi: 10.1016/j.cell.2014.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo JY, Xia B, White E. Autophagy-mediated tumor promotion. Cell. 2013;155:1216–1219. doi: 10.1016/j.cell.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12:401–410. doi: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Toba H, Cortez D, Lindsey ML, Chilton RJ. Applications of miRNA technology for atherosclerosis. Curr Atheroscler Rep. 2014;16:386. doi: 10.1007/s11883-013-0386-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu XD, Zeng K, Liu WL, Gao YG, Gong CS, Zhang CX, Chen YQ. Effect of aerobic exercise on miRNA-TLR4 signaling in atherosclerosis. Int J Sports Med. 2014;35:344–350. doi: 10.1055/s-0033-1349075. [DOI] [PubMed] [Google Scholar]
- 30.Brest P, Lassalle S, Hofman V, Bordone O, Gavric Tanga V, Bonnetaud C, Moreilhon C, Rios G, Santini J, Barbry P, Svanborg C, Mograbi B, Mari B, Hofman P. MiR-129-5p is required for histone deacetylase inhibitor-induced cell death in thyroid cancer cells. Endocr Relat Cancer. 2011;18:711–719. doi: 10.1530/ERC-10-0257. [DOI] [PubMed] [Google Scholar]
- 31.Li M, Tian L, Wang L, Yao H, Zhang J, Lu J, Sun Y, Gao X, Xiao H, Liu M. Down-regulation of miR-129-5p inhibits growth and induces apoptosis in laryngeal squamous cell carcinoma by targeting APC. PLoS One. 2013;8:e77829. doi: 10.1371/journal.pone.0077829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zongaro S, Hukema R, D’Antoni S, Davidovic L, Barbry P, Catania MV, Willemsen R, Mari B, Bardoni B. The 3’ UTR of FMR1 mRNA is a target of miR-101, miR-129-5p and miR-221: implications for the molecular pathology of FXTAS at the synapse. Hum Mol Genet. 2013;22:1971–1982. doi: 10.1093/hmg/ddt044. [DOI] [PubMed] [Google Scholar]
- 33.Dossing KB, Binderup T, Kaczkowski B, Jacobsen A, Rossing M, Winther O, Federspiel B, Knigge U, Kjaer A, Friis-Hansen L. Down-Regulation of miR-129-5p and the let-7 Family in Neuroendocrine Tumors and Metastases Leads to Up-Regulation of Their Targets Egr1, G3bp1, Hmga2 and Bach1. Genes (Basel) 2014;6:1–21. doi: 10.3390/genes6010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duan L, Hao X, Liu Z, Zhang Y, Zhang G. MiR-129-5p is down-regulated and involved in the growth, apoptosis and migration of medullary thyroid carcinoma cells through targeting RET. FEBS Lett. 2014;588:1644–1651. doi: 10.1016/j.febslet.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 35.Wu Q, Yang Z, Xia L, Nie Y, Wu K, Shi Y, Fan D. Methylation of miR-129-5p CpG island modulates multi-drug resistance in gastric cancer by targeting ABC transporters. Oncotarget. 2014;5:11552–11563. doi: 10.18632/oncotarget.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu C, Shao Y, Xia T, Yang Y, Dai J, Luo L, Zhang X, Sun W, Song H, Xiao B, Guo J. lncRNAAC130710 targeting by miR-129-5p is upregulated in gastric cancer and associates with poor prognosis. Tumour Biol. 2014;35:9701–9706. doi: 10.1007/s13277-014-2274-5. [DOI] [PubMed] [Google Scholar]
- 37.Long XH, Zhou YF, Peng AF, Zhang ZH, Chen XY, Chen WZ, Liu JM, Huang SH, Liu ZL. Demethylation-mediated miR-129-5p up-regulation inhibits malignant phenotype of osteogenic osteosarcoma by targeting Homo sapiens valosin-containing protein (VCP) Tumour Biol. 2015;36:3799–3806. doi: 10.1007/s13277-014-3021-7. [DOI] [PubMed] [Google Scholar]
- 38.Yu Y, Zhao Y, Sun XH, Ge J, Zhang B, Wang X, Cao XC. Down-regulation of miR-129-5p via the Twist1-Snail feedback loop stimulates the epithelial-mesenchymal transition and is associated with poor prognosis in breast cancer. Oncotarget. 2015;6:34423–34436. doi: 10.18632/oncotarget.5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coronnello C, Benos PV. ComiR: Combinatorial microRNA target prediction tool. Nucleic Acids Res. 2013;41:W159–164. doi: 10.1093/nar/gkt379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nemchenko A, Chiong M, Turer A, Lavandero S, Hill JA. Autophagy as a therapeutic target in cardiovascular disease. J Mol Cell Cardiol. 2011;51:584–593. doi: 10.1016/j.yjmcc.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu X, Hua Y, Nair S, Zhang Y, Ren J. Akt2 knockout preserves cardiac function in highfat diet-induced obesity by rescuing cardiac autophagosome maturation. J Mol Cell Biol. 2013;5:61–63. doi: 10.1093/jmcb/mjs055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haar L, Ren X, Liu Y, Koch SE, Goines J, Tranter M, Engevik MA, Nieman M, Rubinstein J, Jones WK. Acute consumption of a high-fat diet prior to ischemia-reperfusion results in cardioprotection through NF-kappaB-dependent regulation of autophagic pathways. Am J Physiol Heart Circ Physiol. 2014;307:H1705–1713. doi: 10.1152/ajpheart.00271.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chu KY, O’Reilly L, Ramm G, Biden TJ. Highfat diet increases autophagic flux in pancreatic beta cells in vivo and ex vivo in mice. Diabetologia. 2015;58:2074–2078. doi: 10.1007/s00125-015-3665-x. [DOI] [PubMed] [Google Scholar]
- 44.Guo R, Zhang Y, Turdi S, Ren J. Adiponectin knockout accentuates high fat diet-induced obesity and cardiac dysfunction: role of autophagy. Biochim Biophys Acta. 2013;1832:1136–1148. doi: 10.1016/j.bbadis.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang YL, Cao YJ, You SJ, Li RX, Liu HH, Liu CF. [Protective effects of autophagy against oxidized LDL-induced injury in endothelial cells] . Zhonghua Yi Xue Za Zhi. 2010;90:2792–2796. [PubMed] [Google Scholar]
- 46.Ding Z, Liu S, Sun C, Chen Z, Fan Y, Deng X, Wang X, Mehta JL. Concentration polarization of ox-LDL activates autophagy and apoptosis via regulating LOX-1 expression. Sci Rep. 2013;3:2091. doi: 10.1038/srep02091. [DOI] [PMC free article] [PubMed] [Google Scholar]