

Abstract
Traumatic brain injury (TBI) is a major cause of injury-related deaths, and the mechanism of TBI has become a research focus, but little is known about the mechanism of microRNAs in TBI. The aim of this study is the role of microRNA-22 (miR-22) in TBI-induced neuronal cell apoptosis. Rat cortical neurons were cultured and the TBI model was induced by scratch injury in vitro, before which miR-22 level was altered by transfection of agomir or antagomir. Lactate dehydrogenase (LDH) release and TUNEL assays were performed to examine neuronal cell injury and apoptosis. The activity of caspase 3 (CASP3) and level changes of several apoptosis factors including B-cell lymphoma 2 (BCL2), BCL2-associated X protein (BAX), phosphatase and tensin homolog (PTEN) and v-AKT murine thymoma viral oncogene homolog 1 (AKT1) were detected. Results showed that TBI model cells possessed a downregulated miR-22 level (P < 0.001) and more LDH release and apoptotic cells indicating the aggravated neuronal cell injury and apoptosis induced by TBI. miR-22 agomir attenuated neuronal cell injury and apoptosis of the TBI model. It also caused the corresponding changes in CASP3 activity and other apoptosis factors, with cleaved CASP3, BAX and PTEN inhibited and BCL2 and phosphorylated AKT1 promoted, while miR-22 antagomir had the opposite effects. So miR-22 has neuroprotective roles of attenuating neuronal cell injury and apoptosis induced by TBI, which may be associated with its regulation on apoptosis factors. This study reveals miR-22 as a potential approach to TBI treatment and detailed mechanism remains to be uncovered.
Keywords: Traumatic brain injury, microRNA-22, neuronal cell apoptosis, caspase 3, apoptosis factor
Introduction
Traumatic brain injury (TBI) or intracranial injury caused by external mechanical forces to the brain has become a severe public health problem worldwide. It accounts for one third of injury-related deaths in the United States [1] and is more common in young adults, particularly males [2]. Causes of TBI includes falls, traffic accidents and violence [2]. In athletes, sports-induced TBI damages axons and induces regenerative and degenerative tissue responses in the brain, and repeated concussion may eventually develop into chronic traumatic encephalopathy [3]. Neurological influences of TBI encompass both primary injury events that occur at the time of trauma and secondary injury events contributing to subsequent neuronal cell death and tissue damage [4]. These injuries and sequelae bring great inconvenience and threats to patients’ daily life [5].
The mechanism research on TBI has become a focus recently, which may help to prevent tissue damage of secondary injuries and improve outcome of TBI. For example, the role of bradykinins and substance P in modulating blood-brain barrier permeability after trauma suggests a promising approach to managing brain edema [6]. Inflammatory factors like interleukin 1α, β and tumor necrosis factor are promising targets for attenuating apoptosis or inflammation in TBI [7]. The pivotal functions of cell apoptosis factors, such as caspase 3 (CASP3), B-cell lymphoma 2 (BCL2) and BCL2-associated X protein (BAX), have also been revealed in neuronal cell death of TBI [8,9].
microRNAs (miRNAs) are small noncoding RNAs that recognize and bind to the specific sites of mRNAs to regulate gene expression post-transcriptionally. Because of their vital roles in gene modulation, the involvement of some miRNAs in TBI mechanism has been revealed in recent studies. For example, miR-16, miR-92a and miR-765 are plasma-derived miRNA biomarkers for severe TBI [10], and elevated miR-214 and miR-376a are signatures for mild TBI [11]. miR-711 upregulation promotes neuronal cell death in controlled cortical impact mouse model [12], while miR-23a and miR-27a may protect neuronal cells from TBI-induced apoptosis [13].
Several studies have uncovered the role of miR-22 in regulating cell apoptosis in diseases like myocardial ischemia-reperfusion injury [14], colorectal cancer [15] and lung cancer [16]. However, little is known about its roles in neuronal cell apoptosis. So this study aims to investigate the effect of miR-22 on TBI-induced neuronal cell apoptosis. Rat cortical neuronal cells were cultured and treated by scratch injury to study TBI in vitro. miR-22 level was altered by cell transfection with its agomir or antagomir, and then cell injury, apoptosis and the activity of CASP3 and other apoptotic factors were examined by lactate dehydrogenase (LDH) assay, TdT-mediated dUTP nick-end labeling (TUNEL), western blot and qRT-PCR. This study will provide a general understanding for miR-22 in TBI and a potential therapeutic strategy for this disease.
Materials and methods
Primary culture of rat cortical neuronal cells
Three newborn Sprague-Dawley rats (3 days old, Vital River Laboratories, Beijing, China) were anesthetized for cortex sampling according to previous studies [17,18]. The cortices were collected under sterile conditions, minced, and digested in 0.125% trypsin (Gibco, Carlsbad, CA) for 30 min at 37°C. Dulbecco’s modified Eagle medium (DMEM)-F12 supplemented with fetal bovine serum (Gibco) was added to terminate the digestion. The suspension was filtered and centrifuged at 3000 g for 10 min to collect cells which were resuspended in DMEM-F12 and adjusted to 1 × 106 cells/mL. The cells were seeded in 96-well plates pre-coated with poly-L-lysine (10 mg/L, Sigma-Aldrich, Shanghai, China) for adherence. After 72 h, arabinosylcytosine (5 μg/mL, Sigma-Aldrich) was added to prevent neurogliocyte growth. The medium was changed after 24 h and refreshed every 72 h afterwards. Cells were incubated in humidified atmosphere with 5% CO2 at 37°C.
Cell transfection
Cells were divided into 5 groups: control (untreated cells), TBI (cells treated by TBI), TBI + A (cells treated by TBI and transfected with miR-22 agomir), TBI + AN (cells treated by TBI and transfected with miR-22 antagomir), TBI + blank (cells treated by TBI and transfected with blank control). The rno-miR-22-3p-specific agomir, antagomir and the blank control for transfection were synthesized by RiboBio (Guangzhou, China). The cells cultured for over 7 days were used for transfection. The specific agomir, antagomir and blank control (50 nM) were added to cells in 96-well plates for 48-h-incubation based on the manufacturer’s instruction.
Cell model of TBI
In vitro TBI model of rat cortical neuronal cells was constructed by scratch injury according to previous studies [19,20]. At 24 h after transfection, cells of the five groups were seeded in 6-well plates and cultured for 48 h for adherence. The medium was replaced at 1 h before TBI. Cross-shaped scratches were made with a 10 μL pipette tip on the cell layer, with 5-mm space between lines. No scratch was made in the control group. After 48 h, the five cell groups were used for further analysis.
LDH release assay
Cell injury was analyzed by release of LDH using LDH-Cytotoxicity Assay Kit (BioVision, Milpitas, CA) according to the manufacturer’s instructions. At 48 h after TBI, the culture medium of each cell group was collected. The reaction catalyzed by LDH produced formazan, and the optical density (OD) at 490 nm was measured by a microplate reader SpectraMax i3x (Molecular Devices, Silicon Valley, CA) and compared to the control group.
TUNEL assay
The adherent cells of each group were washed in phosphate buffered saline (PBS) for TUNEL assay to analyze cell apoptosis using One Step TUNEL Cell Apoptosis Assay Kit (Solarbio, Beijing, China). The cells were fixed in 4% paraformaldehyde (Vetec, Shanghai, China) for 30 min, washed in PBS and incubated in ice-cold 0.1% Triton X-100 for 2 min. Fresh TUNEL detection buffer was added and the cells were incubated in dark at 37°C for 60 min, after which the cells were washed in PBS again. Fluorescence signals were observed with a fluorescence microscope (Olympus, Tokyo, Japan). The percent of fluorescein isothiocyanate (FITC)-positive cells was calculated.
CASP3 activity analysis
The relative activity of CASP3 was detected by Caspase 3 Colorimetric Assay Kit (Leagene, Beijing, China) according to the manufacturer’s instructions. Briefly, at 48 h after TBI, the cells of each group were collected, digested by trypsin and washed in PBS. Caspase Lysis buffer was added to resuspend and lyse the cells on ice. After centrifugation, the supernatant was transferred to detect the OD of p-nitroanilide at 405 nm. OD of the four TBI groups was compared to the control group.
Western blot
Cells were lysed in Radio Immunoprecipitation Assay (Beyotime, Shanghai, China) for protein extraction. Protein samples were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes. Blots were blocked in 5% skim milk for 2 h at room temperature and incubated in the specific primary antibodies for cleaved CASP3 (ab179517, Abcam, Cambridge, UK), BCL2 (ab59348), BAX (ab32503), phosphatase and tensin homolog (PTEN, ab32199), v-AKT murine thymoma viral oncogene homolog 1 (AKT1, ab25893) or phosphorylated AKT1 (p-AKT1, ab81283) overnight at 4°C. After washed in PBS, blots were incubated in horse reddish peroxidase-conjugated secondary antibodies for 1 h at room temperature. Positive signals were developed by ECL Plus Western Blotting Substrate (Piece, Carlsbad, CA) and analyzed by ImageJ 1.49 (National Institutes of Health, Bethesda, MD). GAPDH was used as an internal reference.
qRT-PCR
Total RNAs of cells were extracted by TRIzol (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. DNA contamination was removed by DNase I (Invitrogen). miRNAs were extracted using miRNeasy Mini Kit (Qiagen, Shenzhen, China). The complementary DNAs (cDNAs) were synthesized from 1 μg of total RNAs using PrimeScript Reverse Transcriptase (Takara, Dalian, China) and random primers or the specific primer for rno-miR-22-3p (5’-CTC AAC TGG TGT CGT GGA GTC GGC AAT TCA GTT GAG ACA GTT CT-3’). qRT-PCR was performed on QuantStudio 6 Flex Realtime PCR system (Applied Biosystems, Carlsbad, CA) with each reaction system containing 20 ng of cDNAs and the specific primers for rno-miR-22-3p (Fw: 5’-ACA CTC CAG CTG GGA AGC TGC CAG TTG AAG-3’ and Rv: 5’-TGG TGT CGT GGA GTC G-3’) or Pten (Fw: 5’-GGA AAG GAC GAC TGG TGT A-3’ and Rv: 5’-TGC CAC TGG TCT GTA ATC CA-3’). Gapdh (Fw: 5’-CGC ATT GCC AGA CAT ATC AGC-3’ and Rv: 5’-AGG TGA AGC AGG CTC AAT CAA-3’) and pre-U6 (Fw: 5’-CTC GCT TCG GCA GCA CA-3’ and Rv: 5’-AAC GCT TCA CGA ATT TGC GT-3’) were used as the internal reference for Pten and miR-22 expression, respectively. Data were calculated by the 2-ΔΔCt method.
Statistical analysis
In this study, 5 replicates of each cell group were analyzed. Detection on each replicate was performed three times, and results were represented as the mean ± standard deviation. Statistical analysis was carried out by SPSS 20 (IBM, New York, USA). Data were first analyzed by F test for homogeneity of variance and then t test for statistical significance. The difference with P < 0.05 was considered significant between groups.
Results
miR-22 is down-regulated in cortical neuronal cells of TBI cell model
After TBI model construction in the primary cultured rat cortical neuronal cells, miR-22 was detected by qRT-PCR, and results showed a significant down-regulated miR-22 level in the TBI group compared to the control (P < 0.001, Figure 1). Then miR-22 expression was altered by transfecting its specific agomir or antagomir to TBI model cells. The detection indicated the successful promotion and inhibition of miR-22 level with significant differences compared to the blank control (P < 0.001). Thus these five cell groups were used in further experiments.
Figure 1.
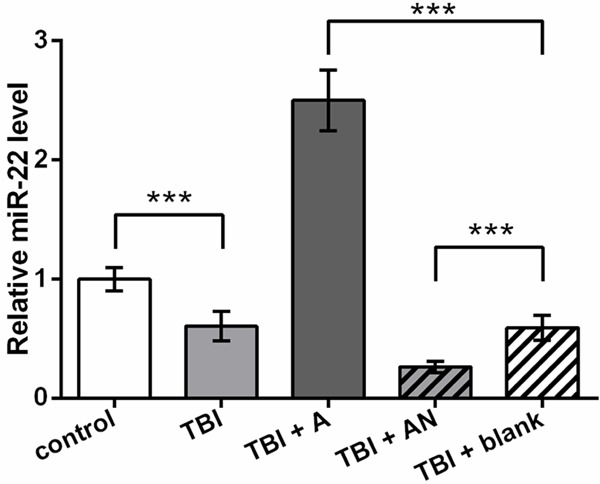
Down-regulated miR-22 level in TBI cell model. control, cells without treatment. TBI, TBI cell model. TBI + A, cells transfected with miR-22 agomir and injured by scratches. TBI + AN, cells transfected with miR-22 antagomir and injured by scratches. TBI + blank, transfection control cells injured by scratches. ***P < 0.001. TBI, traumatic brain injury.
miR-22 inhibits cortical neuronal cells injury and apoptosis of TBI cell model
Injured cells release LDH which is a relatively stable enzyme in culture medium, so the release of LDH to the culture medium of TBI model cells was detected to assess cell injury. Results showed that TBI model cells released more LDH compared to the control (P < 0.001, Figure 2A). miR-22 overexpression significantly reduced LDH release and its knockdown increased LDH level in the medium (P < 0.001). So it seemed that miR-22 could attenuate TBI-induced neuronal cell injury.
Figure 2.
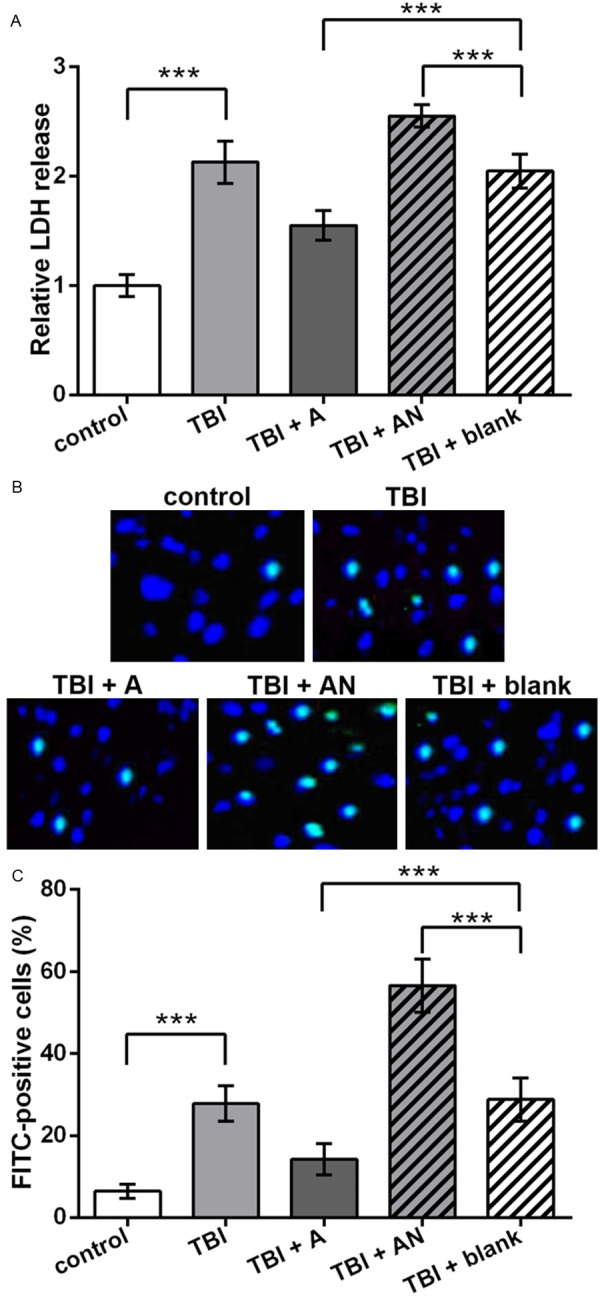
Neuronal cell injury and apoptosis promoted by TBI and inhibited by miR-22. control, cells without treatment. TBI, TBI cell model. TBI + A, cells transfected with miR-22 agomir and injured by scratches. TBI + AN, cells transfected with miR-22 antagomir and injured by scratches. TBI + blank, transfection control cells injured by scratches. A. Relative LDH release in the culture medium which indicates cell injury. B. Pictures of TUNEL assay indicating apoptotic neuronal cells (bright blue). C. Histogram of the average percent of apoptotic neuronal cells (FITC-positive) in each group. **P < 0.01. ***P < 0.001. LDH, lactate dehydrogenase. TUNEL, TdT-mediated dUTP nick-end labeling. FITC, fluorescein isothiocyanate.
Meanwhile, cell apoptosis was detected by TUNEL assay and similar changing patterns were found. TBI increased FITC-positive cell number compared to the control (Figure 2B). miR-22 overexpression decreased cell apoptosis and miR-22 knockdown increased apoptotic cell number compared to the blank control. Significant differences in the apoptotic cell percent were found between groups when considering replicated experiments (P < 0.001, Figure 2C). So miR-22 might inhibit cortical neuronal cell apoptosis in the TBI cell model.
miR-22 regulates PTEN/AKT signaling and downstream apoptosis factors
Based on the above results, miR-22 was likely to inhibit cell apoptosis in the TBI cell model, so a series of apoptosis factors were analyzed to investigate the possible regulatory mechanism of miR-22. The relative activity of CASP3, a key executor of cell apoptosis, was analyzed, and results showed that TBI elevated CASP3 activity (P < 0.001, Figure 3A). Compared to the TBI + blank group, miR-22 agomir inhibited, and miR-22 antagomir promoted CASP3 activity (P < 0.001). Western blot further verified that the cleaved CASP3 level possessed the similar changes to the activity assay (Figure 3B), which supported that TBI induced neuronal cell apoptosis and that miR-22 inhibited cell apoptosis in the TBI model.
Figure 3.
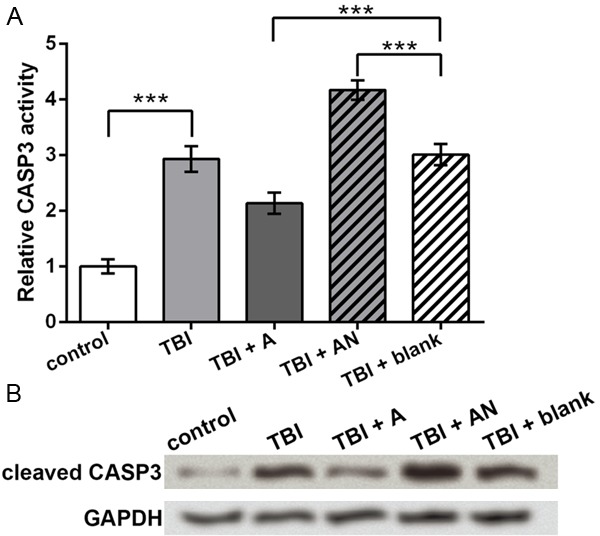
CASP3 activity regulated by miR-22. control, cells without treatment. TBI, TBI cell model. TBI + A, cells transfected with miR-22 agomir and injured by scratches. TBI + AN, cells transfected with miR-22 antagomir and injured by scratches. TBI + blank, transfection control cells injured by scratches. A. Relative CASP3 activity indicated by OD of p-nitroanilide at 405 nm. B. Western blot showing the protein level of cleaved CASP3. GAPDH is an internal reference. ***P < 0.001. CASP3, caspase 3.
Then BCL2 and BAX protein were detected for their vital position in cell apoptosis pathways. Western blot showed that BCL2 was inhibited and BAX was promoted by TBI (Figure 4A). In the transfection groups, miR-22 agomir increased and miR-22 antagomir decreased BCL2 level, while BAX showed the opposite changing pattern. Significant differences were found between groups when comparing the ratio of BCL2 and BAX, which was markedly decreased by TBI (P < 0.001, Figure 4B), elevated by miR-22 agomir and reduced by miR-22 antagomir (P <0.01). So miR-22 could regulate BCL2 and BAX in the TBI cell model.
Figure 4.
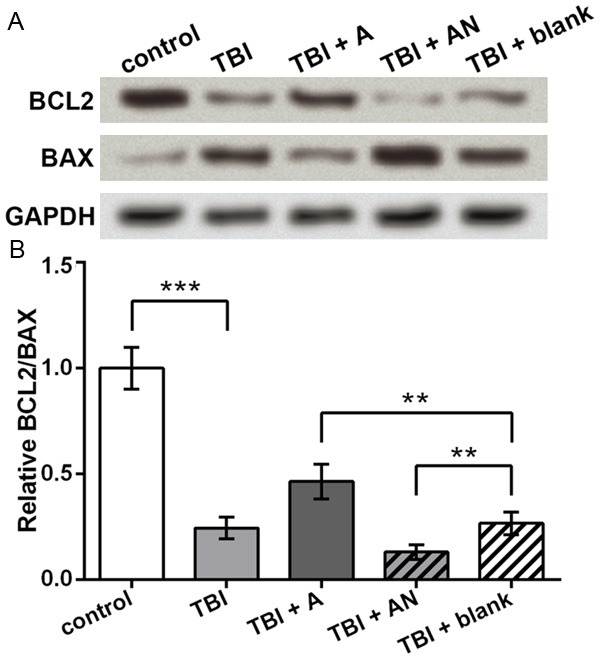
Protein levels of BCL2 and BAX regulated by miR-22. control, cells without treatment. TBI, TBI cell model. TBI + A, cells transfected with miR-22 agomir and injured by scratches. TBI + AN, cells transfected with miR-22 antagomir and injured by scratches. TBI + blank, transfection control cells injured by scratches. A. Western blot showing protein levels of BCL2 and BAX. GAPDH is an internal reference. B. Ratio of BCL2 and BAX based on the western blot results. **P < 0.01. ***P < 0.001. BCL2, B-cell lymphoma 2. BAX, BCL2-associated X protein.
Upstream of BCL2 and BAX, AKT1 was detected for its central position during cell apoptosis. As predicted, the total AKT1 level did not change obviously among groups (Figure 5A), but p-AKT1 was inhibited by TBI and promoted by miR-22 agomir, which was consistent with the cell apoptosis changes in each group. Since the phosphorylation of AKT1 can be suppressed by PTEN, so PTEN expression was also detected. Its protein level showed the opposite changing pattern to p-AKT1 (Figure 5A), which was in accordance with its influence on AKT1. Pten mRNA level in these groups was similar to its protein level, which was significantly elevated by TBI and inhibited by miR-22 agomir (P < 0.001, Figure 5B). Taken together, miR-22 could regulate apoptosis factors including PTEN, p-AKT1, BCL2, BAX and cleaved CASP3, which might be a possible regulatory mechanism of miR-22 in cortical neuronal cells during TBI.
Figure 5.
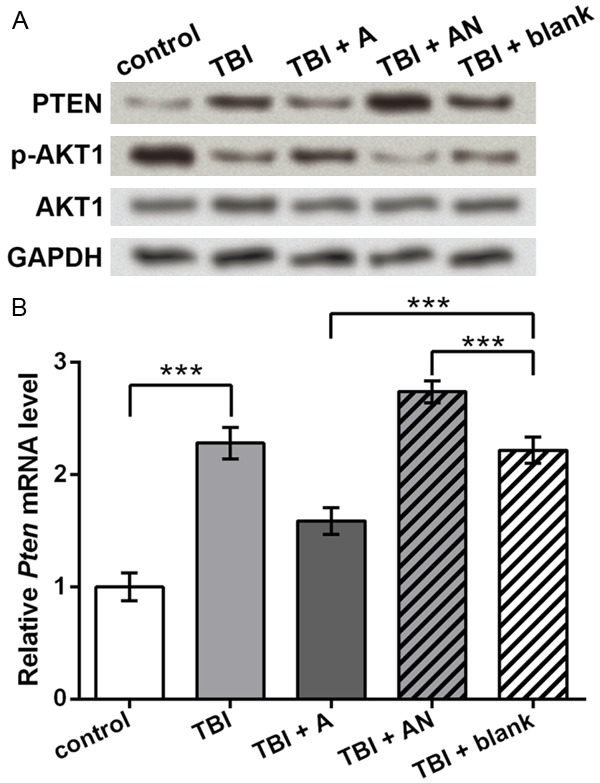
Expression of PTEN and AKT1 regulated by miR-22. control, cells without treatment. TBI, TBI cell model. TBI + A, cells transfected with miR-22 agomir and injured by scratches. TBI + AN, cells transfected with miR-22 antagomir and injured by scratches. TBI + blank, transfection control cells injured by scratches. A. Western blot showing protein levels of PTEN, p-AKT1 and AKT1. GAPDH is an internal reference. B. qRT-PCR showing Pten mRNA levels in each group. ***P < 0.001. PTEN, phosphatase and tensin homolog. AKT1, v-AKT murine thymoma viral oncogene homolog 1. p-AKT1, phosphorylated AKT1.
Discussion
miRNAs have shown great potentials in treating various diseases for their ability to regulate gene expression. In this study, miR-22 level is detected to be down-regulated in the TBI model of rat cortical neuronal cells. Up-regulation of miR-22 in TBI cell model could attenuate cell injury, apoptosis and a series of apoptosis factors including CASP3, BCL2, BAX, PTEN and AKT1.
Compared to the control group, the neuronal cells in the TBI model group released relatively more LDH to the culture medium, which indicated a more severe cell injury degree. Besides, TUNEL assay showed a higher apoptotic cell percent in the TBI model. However, the impacts induced by scratch injury were attenuated when miR-22 was up-regulated by the agomir, and aggravated by miR-22 antagomir, which implied the potential neuroprotective functions of miR-22 in attenuating neuronal cell injury and apoptosis during TBI. Similar results have been reported in the cell model of cerebral ischemia-reperfusion injury and rat myocardial ischemia-reperfusion model, where miR-22 reduces the apoptotic rate of cortical neurons, suppresses LDH release and apoptosis of cardiomyocytes; miR-22 also improves neuronal viability in an in vitro model of brain aging [14,18,21]. But disparate roles of miR-22 are revealed in hepatocellular carcinoma cells where it may inhibit cell proliferation [22]. So the function of miR-22 is not likely to be conserved in different cell types, which needs further verification.
The CASP3 activity assay, western blot and qRT-PCR results of this study showed that miR-22 inhibited CASP3 activity, BAX and PTEN expression, and promoted BCL2 and p-AKT1. As one of the three apoptosis executioners in caspase family, CASP3 can be activated by cytochrome c released from mitochondria and an important apoptosis activator, CASP9 [23]. Release of cytochrome c is monitored by BCL2 family members such as pro-survival BCL2 and pro-apoptotic protein BAX [24]. The balance between BCL2 and BAX is crucial for cell survival, and the BCL2/BAX ratio is thus becoming an indicator for cell apoptosis [25,26]. Moreover, PTEN is a further upstream regulator which inhibits the phosphorylation of AKT1 and induces cell apoptosis [27,28]. Based on the information, the regulation of miR-22 on these apoptosis factors indicates that miR-22 may regulate the PTEN/AKT1 signaling and downstream BCL2, BAX and CASP3, which is one of the possible mechanisms for its neuroprotective roles in TBI.
Up-regulation of miR-22 inhibited PTEN expression in both mRNA and protein levels as detected in this study. One possible reason may be that miR-22 hinders the translation process of PTEN production as well as the stability of Pten mRNA, as reported in some mammalian miRNAs [29,30]. Though rno-miR-22-3p was predicted to target the sequence GGCAGCUA in Pten mRNA 3’ untranslated region, the direct interaction and the regulatory mechanism between miR-22 and Pten need further investigation.
In summary, miR-22 is down-regulated in the TBI cell model of this study. miR-22 is capable of attenuating neuronal cell injury and apoptosis induced by TBI. Its regulation on the PTEN/AKT1 signaling and key apoptosis factors may be associated with its neuroprotective roles in the TBI cell model. This study facilitates the relative research on miRNAs and TBI treatment, and more detailed mechanism analyses are necessary.
Disclosure of conflict of interest
None.
References
- 1.Faul M, Xu L, Wald MM, Coronado V, Dellinger AM. Traumatic brain injury in the United States: national estimates of prevalence and incidence, 2002-2006. Injury Prevention. 2011;16:A268–A268. [Google Scholar]
- 2.Maas AI, Stocchetti N, Bullock R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008;7:728–741. doi: 10.1016/S1474-4422(08)70164-9. [DOI] [PubMed] [Google Scholar]
- 3.Blennow K, Hardy J, Zetterberg H. The neuropathology and neurobiology of traumatic brain injury. Neuron. 2012;76:886–899. doi: 10.1016/j.neuron.2012.11.021. [DOI] [PubMed] [Google Scholar]
- 4.Loane DJ, Faden AI. Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci. 2010;31:596–604. doi: 10.1016/j.tips.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Richman JE, Baglieri AM, Cho O. Tinted lenses in the treatment of visual stress in a patient with a traumatic brain injury a case report. Journal of Behavioral Optometry. 2007;18:149–153. [Google Scholar]
- 6.Donkin JJ, Vink R. Mechanisms of cerebral edema in traumatic brain injury: therapeutic developments. Curr Opin Neurol. 2010;23:293–299. doi: 10.1097/WCO.0b013e328337f451. [DOI] [PubMed] [Google Scholar]
- 7.Shojo H, Kaneko Y, Mabuchi T, Kibayashi K, Adachi N, Borlongan CV. Genetic and histologic evidence implicates role of inflammation in traumatic brain injury-induced apoptosis in the rat cerebral cortex following moderate fluid percussion injury. Neuroscience. 2010;171:1273–1282. doi: 10.1016/j.neuroscience.2010.10.018. [DOI] [PubMed] [Google Scholar]
- 8.Clark RS, Kochanek PM, Watkins SC, Chen M, Dixon CE, Seidberg NA, Melick J, Loeffert JE, Nathaniel PD, Jin KL, Graham SH. Caspase-3 Mediated Neuronal Death After Traumatic Brain Injury in Rats. J Neurochem. 2001;74:740–753. doi: 10.1046/j.1471-4159.2000.740740.x. [DOI] [PubMed] [Google Scholar]
- 9.Wennersten A, Holmin S, Mathiesen T. Characterization of Bax and Bcl-2 in apoptosis after experimental traumatic brain injury in the rat. Acta Neuropathologica. 2003;105:281–288. doi: 10.1007/s00401-002-0649-y. [DOI] [PubMed] [Google Scholar]
- 10.Redell JB, Moore AN, Ward NH 3rd, Hergenroeder GW, Dash PK. Human traumatic brain injury alters plasma microRNA levels. J Neurotrauma. 2010;27:2147–2156. doi: 10.1089/neu.2010.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma A, Chandran R, Barry ES, Bhomia M, Hutchison MA, Balakathiresan NS, Grunberg NE, Maheshwari RK. Identification of serum microRNA signatures for diagnosis of mild traumatic brain injury in a closed head injury model. PLoS One. 2014;9:e112019. doi: 10.1371/journal.pone.0112019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sabirzhanov B, Stoica BA, Zhao Z, Loane DJ, Wu J, Dorsey SG, Faden AI. miR-711 upregulation induces neuronal cell death after traumatic brain injury. Cell Death Differ. 2016;23:654–68. doi: 10.1038/cdd.2015.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sabirzhanov B, Zhao Z, Stoica BA, Loane DJ, Wu J, Borroto C, Dorsey SG, Faden AI. Downregulation of miR-23a and miR-27a following experimental traumatic brain injury induces neuronal cell death through activation of proapoptotic Bcl-2 proteins. J Neurosci. 2014;34:10055–10071. doi: 10.1523/JNEUROSCI.1260-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang J, Chen L, Yang J, Ding J, Li S, Wu H, Zhang J, Fan Z, Dong W, Li X. MicroRNA-22 targeting CBP protects against myocardial ischemia-reperfusion injury through anti-apoptosis in rats. Mol Biol Rep. 2014;41:555–561. doi: 10.1007/s11033-013-2891-x. [DOI] [PubMed] [Google Scholar]
- 15.Zhang H, Tang J, Li C, Kong J, Wang J, Wu Y, Xu E, Lai M. MiR-22 regulates 5-FU sensitivity by inhibiting autophagy and promoting apoptosis in colorectal cancer cells. Cancer Lett. 2015;356:781–790. doi: 10.1016/j.canlet.2014.10.029. [DOI] [PubMed] [Google Scholar]
- 16.Liu L, Jiang Y, Zhang H, Greenlee AR, Yu R, Yang Q. miR-22 functions as a micro-oncogene in transformed human bronchial epithelial cells induced by anti-benzo[a] pyrene-7,8-diol-9,10-epoxide. Toxicol In Vitro. 2010;24:1168–1175. doi: 10.1016/j.tiv.2010.02.016. [DOI] [PubMed] [Google Scholar]
- 17.Gong QH, Wang Q, Shi JS, Huang XN, Liu Q, Ma H. Inhibition of caspases and intracellular free Ca2+ concentrations are involved in resveratrol protection against apoptosis in rat primary neuron cultures. Acta Pharmacol Sin. 2007;28:1724–1730. doi: 10.1111/j.1745-7254.2007.00666.x. [DOI] [PubMed] [Google Scholar]
- 18.Yu H, Wu M, Zhao P, Huang Y, Wang W, Yin W. Neuroprotective effects of viral overexpression of microRNA-22 in rat and cell models of cerebral ischemia-reperfusion injury. J Cell Biochem. 2015;116:233–241. doi: 10.1002/jcb.24960. [DOI] [PubMed] [Google Scholar]
- 19.Han Z, Chen F, Ge X, Tan J, Lei P, Zhang J. miR-21 alleviated apoptosis of cortical neurons through promoting PTEN-Akt signaling pathway in vitro after experimental traumatic brain injury. Brain Research. 2014;1582:12–20. doi: 10.1016/j.brainres.2014.07.045. [DOI] [PubMed] [Google Scholar]
- 20.Zhao Y, Luo P, Guo Q, Li S, Zhang L, Zhao M, Xu H, Yang Y, Poon W, Fei Z. Interactions between SIRT1 and MAPK/ERK regulate neuronal apoptosis induced by traumatic brain injury in vitro and in vivo. Exp Neurol. 2012;237:489–498. doi: 10.1016/j.expneurol.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 21.Jovicic A, Zaldivar Jolissaint JF, Moser R, Silva Santos Mde F, Luthi-Carter R. MicroRNA-22 (miR-22) overexpression is neuroprotective via general anti-apoptotic effects and may also target specific Huntington’s disease-related mechanisms. PLoS One. 2013;8:e54222. doi: 10.1371/journal.pone.0054222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang J, Yang Y, Yang T, Liu Y, Li A, Fu S, Wu M, Pan Z, Zhou W. microRNA-22, downregulated in hepatocellular carcinoma and correlated with prognosis, suppresses cell proliferation and tumourigenicity. Br J Cancer. 2010;103:1215–1220. doi: 10.1038/sj.bjc.6605895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fan TJ, Han LH, Cong RS, Liang J. Caspase Family Proteases and Apoptosis. Acta Biochim Biophys Sin (Shanghai) 2005;37:719–727. doi: 10.1111/j.1745-7270.2005.00108.x. [DOI] [PubMed] [Google Scholar]
- 24.Roy MJ, Vom A, Czabotar PE, Lessene G. Cell death and the mitochondria: therapeutic targeting of the BCL-2 family-driven pathway. Br J Pharmacol. 2014;171:1973–1987. doi: 10.1111/bph.12431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang H, Zhao PJ, Su D, Feng J, Ma SL. Paris saponin I induces apoptosis via increasing the Bax/Bcl-2 ratio and caspase-3 expression in gefitinib-resistant non-small cell lung cancer in vitro and in vivo. Mol Med Rep. 2014;9:2265–2272. doi: 10.3892/mmr.2014.2108. [DOI] [PubMed] [Google Scholar]
- 26.Del Poeta G, Venditti A, Del Principe MI, Maurillo L, Buccisano F, Tamburini A, Cox MC, Franchi A, Bruno A, Mazzone C, Panetta P, Suppo G, Masi M, Amadori S. Amount of spontaneous apoptosis detected by Bax/Bcl-2 ratio predicts outcome in acute myeloid leukemia (AML) Blood. 2003;101:2125–2131. doi: 10.1182/blood-2002-06-1714. [DOI] [PubMed] [Google Scholar]
- 27.Wu B, Wang X, Chi ZF, Hu R, Zhang R, Yang W, Liu ZG. Ursolic acid-induced apoptosis in K562 cells involving upregulation of PTEN gene expression and inactivation of the PI3K/Akt pathway. Arch Pharm Res. 2012;35:543–548. doi: 10.1007/s12272-012-0318-1. [DOI] [PubMed] [Google Scholar]
- 28.Stechschulte LA, Wuescher L, Marino JS, Hill JW, Eng C, Hinds TD Jr. Glucocorticoid receptor beta stimulates Akt1 growth pathway by attenuation of PTEN. J Biol Chem. 2014;289:17885–17894. doi: 10.1074/jbc.M113.544072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Omer AD, Janas MM, Novina CD. The chicken or the egg: microRNA-mediated regulation of mRNA translation or mRNA stability. Mol Cell. 2009;35:739–740. doi: 10.1016/j.molcel.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 30.Fabian MR, Mathonnet G, Sundermeier T, Mathys H, Zipprich JT, Svitkin YV, Rivas F, Jinek M, Wohlschlegel J, Doudna JA, Chen CY, Shyu AB, Yates JR 3rd, Hannon GJ, Filipowicz W, Duchaine TF, Sonenberg N. Mammalian miRNA RISC recruits CAF1 and PABP to affect PABPdependent deadenylation. Mol Cell. 2009;35:868–880. doi: 10.1016/j.molcel.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]