

Lead In
Quinones are common stoichiometric reagents in organic chemistry. High potential para-quinones, such as DDQ and chloranil, are widely used and typically promote hydride abstraction. In recent years, many catalytic applications of these methods have been achieved by using transition metals, electrochemistry or O2 to regenerate the oxidized quinone in situ. Complementary studies have led to the development of a different class of quinones that resemble the ortho-quinone cofactors in Copper Amine Oxidases and mediate efficient and selective aerobic and/or electrochemical dehydrogenation of amines. The latter reactions typically proceed via electrophilic transamination and/or addition-elimination reaction mechanisms, rather than hydride abstraction pathways. The collective observations show that the quinone structure has a significant influence on the reaction mechanism and have important implications for the development of new quinone reagents and quinone-catalyzed transformations.
Keywords: Quinones, oxidation, dehydrogenation, DDQ, chloranil, amine oxidase
1. Introduction
The selective oxidation of organic compounds is a prominent challenge in the chemical industry. Considerable efforts have focused on the development of transition metal reagents and catalysts to accomplish such reactions, but redox-active organic molecules also serve as robust and efficient (co)catalysts for oxidative transformations. Nitroxyl radicals are among the most broadly used classes of organic oxidation catalysts. For example, 2,2,6,6-tetramethylpiperidine N-oxyl (TEMPO) [Eq. (1)] and its derivatives promote a range of oxidative transformations, perhaps most prominently, alcohol oxidation reactions.[ 1 ] Additionally, pthalimide N-oxyl (PINO) – generated in situ from N-hydroxyphthalimide (NHPI) – is widely employed in aerobic autoxidation reactions [Eq. (2)].[2] Both reagents are amenable to industrial scale oxidation processes.[3] The use of organic (co)catalysts provides access to reaction mechanisms that are often distinct from those mediated by transition-metal catalysts, leading to complementary reactivity, selectivity, and reaction conditions. Selective oxidation reactions promoted by organic catalysts and co-catalysts represent promising targets for future development.
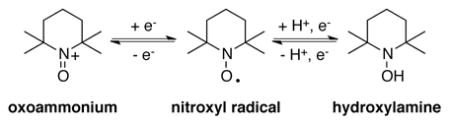 |
(1) |
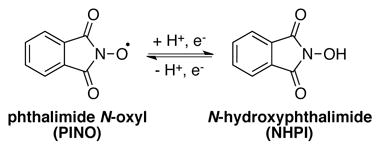 |
(2) |
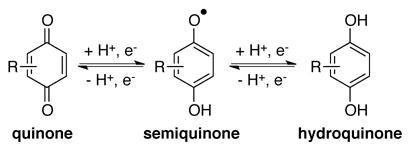 |
(3) |
Quinones are important redox-active organic molecules that have applications in diverse redox processes, including in the manufacture of industrial chemicals, in oxidation reactions for organic synthesis, and as electron carriers, antioxidants and cofactors in biological processes.[4] Like nitroxyl radicals, quinones feature three readily-accessible oxidation states, fully-oxidized quinone, one electron-reduced semiquinone, and two electron-reduced hydroquinone [Eq. (3)], and they are capable of mediating both closed- and open-shell redox processes. Relative to nitroxyl radicals, however, quinones are more commonly used as stoichiometric reagents, with less extensive use as catalysts for the oxidation of organic molecules.
Redox cycling between oxidized and reduced quinone species forms the basis for the anthraquinone-mediated industrial synthesis of hydrogen peroxide.[5] Hydrogen peroxide is produced in near quantitative yields by the stoichiometric autoxidation of 2-alkyl-9,10-anthrahydroquinone (Scheme 1, autoxidation step). The resulting oxidized anthraquinone co-product is subsequently reduced in a separate step via catalytic hydrogenation (Scheme 1, hydrogenation step). This sequence enables the net conversion of molecular oxygen and hydrogen into hydrogen peroxide (Scheme 1, net reaction). Over 95% of the world supply of hydrogen peroxide is made using this quinone-mediated process. While the incompatibility of the oxidation and reduction steps requires sequential operation of the two stoichiometric half reactions in this case, use of a quinone mediator common to both half reactions suggests broader potential for engaging quinones as catalysts in redox processes.
Scheme 1.
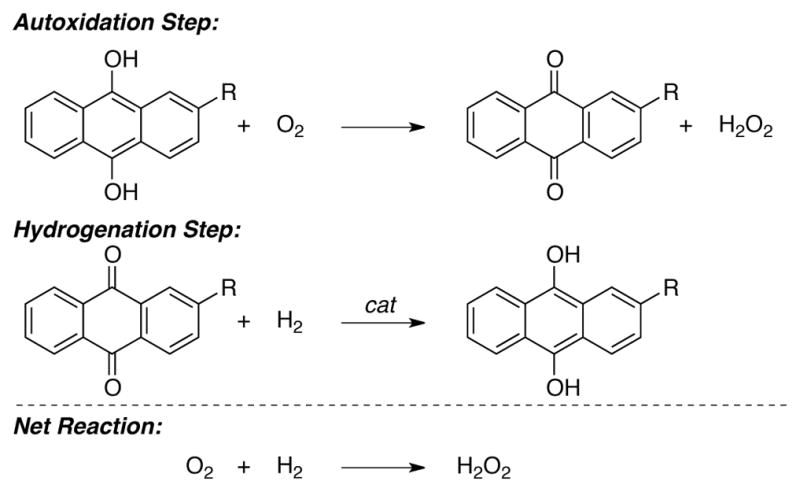
The anthraquinone oxidation (AO) process for industrial synthesis of H2O2, consisting of the sequential autoxidation and hydrogenation of a quinone mediator.
Nature provides a framework for the use of catalytic quinones as redox shuttles in oxidative transformations: plastoquinone and ubiqinone act as electron carriers in the photosynthetic and mitochondrial electron transport chains (ETCs), respectively. In the mitochondrial ETC, a series of cofactors shuttle electrons from a sacrificial reductant (NADH) to molecular oxygen. The reduction of molecular oxygen to water ultimately provides the thermodynamic driving force for synthesis of ATP via oxidative phosphorylation. Conceptually related synthetic “electron transfer mediators” or ETMs often feature quinones as catalytic redox shuttles in metal-catalyzed aerobic oxidation reactions, such as Pd-catalyzed 1,4-diacetoxylation of cyclohexadiene (Scheme 2).[6] Substrate oxidation first occurs via a transition metal-mediated step, and a quinone (commonly, benzoquinone[7] or a related derivative[8]) acts as a redox shuttle to transfer protons and electrons from the transition metal catalyst to a metal-macrocycle co-catalyst LM, such as [Co(salophen)] (salophen = N,N-salicylidene phenylenediamine), [Fe(pc)] (pc = phthalocyanine), or [Co(tpp)] (tpp = 5,10,15,20-tetraphenylporphyrin). Finally, the metal macrocycle co-catalyst is re-oxidized by molecular oxygen, the terminal oxidant. The series of coupled catalytic cycles are proposed to account for the efficient reactivity in these systems. In addition to serving as a redox shuttle between Pd and metal-macrocycle cocatalysts, benzoquinone additives may also promote oxidatively-induced reductive elimination of the substrate from PdII.[9] Applications of quinones as redox shuttles/ETMs are the subject of an extensive recent review by Piera and Bäckvall.[10] While not revisited here, many of the fundamental principles associated with quinone co-catalyzed reactions of the type in Scheme 2 may be exploited in oxidation reactions involving direct oxidation of the organic molecule by a quinone catalyst.
Scheme 2.
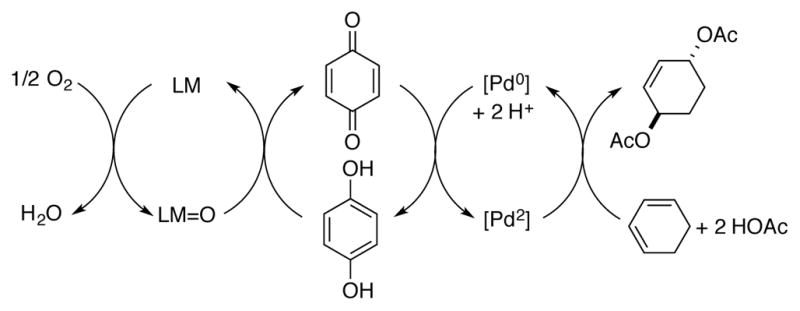
Use of quinones as redox shuttles in organic synthesis, represented here in Pd-catalyzed aerobic diacetoxylation of cyclohexadiene, involving Pd, quinone, and metal-macrocycle (LM)-coupled catalytic cycles.[6]
The present review highlights recent progress in the development of quinone-catalyzed oxidations of organic substrates, specifically, reactions involving direct reaction of the organic substrate with the quinone catalyst.[11] The quinone catalysts may be divided into two general families: high-potential quinones, such as 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) and chloranil; and o-quinone catalysts reminiscent of the o-quinone cofactors found in Copper Amine Oxidases (CAOs) and related enzymes.
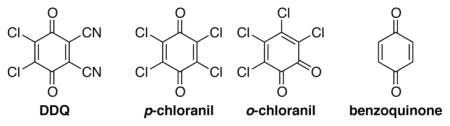
DDQ is the most widely used high-potential quinone, and it commonly mediates hydride transfer reactions. DDQ has found broad utility as a stoichiometric oxidant in the functionalization of activated C–H bonds and the dehydrogenation of saturated C–C, C–O and C–N bonds, including in several process-scale pharmaceutical syntheses.[12,13] The cost and toxicity of DDQ underlies recent efforts to develop methods that employ catalytic quantities of DDQ in combination with alternate stoichiometric oxidants. These efforts are the focus of Section 2 of this review. This section begins with an overview of the types of transformation and reaction mechanisms promoted by high-potential quinones, such as DDQ and chloranil. This context provides a foundation for consideration of methods that have been developed with these quinones as catalysts. Reactions promoted by catalytic DDQ closely parallel those accessible with stoichiometric DDQ, although applications that benefit from or are only effective with catalytic DDQ have also been identified.
A second family of quinone catalysts resemble the o-quinone cofactors found in enzymatic oxidases and dehydrogenases. These bioinspired quinones typically have lower reduction potentials than DDQ or chloranil, and they react via different mechanisms. Consequently, different substrate scope and reactivity patterns are observed relative to high-potential quinones. Section 3 begins with an overview of copper amine oxidases and related naturally occurring quinoenzymes, highlighting the oxidation mechanisms employed by these enzymes. This context provides the basis for consideration of synthetic o-quinone catalysts and their applications to aerobic and electrochemical dehydrogenation of saturated C–N bond in amines, including nitrogen heterocycles.
2. DDQ-Catalyzed Oxidations of Organic Substrates
2.1. Mechanistic Insights from Stoichiometric DDQ-Mediated Transformations
High potential quinones, notably DDQ and chloranil, are important stoichiometric reagents for the oxidation of organic compounds.[4,14] The synthetic applications of DDQ fit into several categories, many of which feature quinone-mediated hydride abstraction from substrate. This mechanism is thought to proceed through the formation of a quinone-substrate charge-transfer complex (Scheme 3).[15,16] Substrate oxidation subsequently occurs via hydride transfer from substrate to quinone, forming an ion-paired product.[17] The specific mechanism of hydride abstraction by DDQ and/or chloranil has been the subject of some controversy. Depending on the substrate, net hydride abstraction may be initiated by single-electron transfer[18] or hydrogen-atom transfer;[19] however, polar reaction mechanisms involving direct hydride transfer[20,21] are typically favored.[22]
Scheme 3.

Hydride-transfer-initiated reaction pathways that accounts for the majority of known DDQ-mediated oxidation/dehydrogenation reactions.
The substrate-cation/DDQH− ion pair formed upon hydride transfer can then undergo a range of subsequent chemical steps (Scheme 3). Deprotonation of the substrate by DDQH− can afford the corresponding dehydrogenated product,[23,24] as featured in the formation of (hetero)aromatic compounds from unsaturated precursors,[25] aldehydes and ketones from activated alcohols,[26] and oxocarbenium and iminium species from ethers and amines, respectively.[27,28] Alternatively, a carbocationic intermediate can undergo intramolecular rearrangement (for example, a Wagner-Meerwein rearrangement[29]) or be subject to intra- or intermolecular addition reactions, leading to oxidative C–H functionalization products.[30] Collapse of the ion pair may also occur, leading to (typically undesired) quinol ether products.[31,32] These types of hydride-abstraction-initiated reactions account for the majority of DDQ-mediated transformations in the literature.
A second mechanistic pathway has been proposed for the DDQ-mediated dehydrogenation of ketones and silyl enol ethers to α,β-unsaturated ketones.[33,34] In this case, an “electrophilic” reaction pathway is proposed, rather than a hydride-abstraction pathway.[35] Mayr and coworkers have provided evidence for competing electrophilic and electron-transfer (ET) pathways in the reaction of DDQ with silyl enol ethers and silyl ketene acetals.[36] In the stoichiometric reaction of DDQ with 1-trimethylsilyloxy-cyclohexene, two adducts are observed: a C–C linked adduct resulting from reversible conjugate addition of the silyl enol ether to the quinone, and a C–O linked quinol ether resulting from irreversible single-electron transfer and radical coupling (Scheme 4). The ratio of C–C and C–O product formation is a consequence of the reaction conditions (solvent, temperature, concentration) as well as the identity of the quinone and reactant.[37] Productive reaction of the C–C adducts obtained from polar addition of the nucleophile to the quinone is typically achieved at elevated temperatures and results in the formation of dehydrogenation products. The C–O linked quinol ether products, in this case initiated by ET pathways, are not competent intermediates en route to substrate dehydrogenation.
Scheme 4.

An electrophilic/polar pathway proposed for DDQ-mediated dehydrogenation of ketones and silyl enol ethers, together with an unproductive, competing electron-transfer (ET) pathway leading to a C–O coupled adduct. Scheme adapted from ref 36.
The mechanistic studies summarized here have several important implications for understanding the reactivity of high potential quinones and for subsequent catalytic reaction development. First, while DDQ is a strong thermodynamic oxidant, a reaction mechanism involving direct hydride abstraction limits dehydrogenation reactions to substrates containing quite activated C–H bonds (e.g., benzylic, allylic). Second, electrophilic reaction mechanisms involving DDQ may lead to different reaction products relative to oxidation reactions initiated by electron or hydride transfer. Studies by Mayr and coworkers highlight the potential generality of the electrophilic pathway and the involvement of covalent substrate/quinone intermediates in dehydrogenation reactions mediated by a range of different quinones.[37] This mechanistic pathway provides kinetic access to reactions that may not be possible via hydride abstraction pathways.
2.2 DDQ- and Chloranil-Catalyzed Reactions
Despite the versatility of DDQ as a stoichiometric reagent, there are numerous limitations to the use of DDQ on large scale. Issues include its relatively high toxicity (LD50 82 mg/kg rat) and high cost (>$500/mol), environmental hazards associated with water-mediated liberation of HCN, and challenges in removing DDQH2 from reaction products. DDQ exhibits unique reactivity relative to chloranil and other quinones, however, and the use of alternative, more-benign reagents is not always possible. Therefore, in spite of its limitations, DDQ continues to be an important reagent in the process-scale synthesis of pharmaceutical intermediates and drug candidates.[12,13,35] A widely cited example is the process scale synthesis of Finasteride by Merck.[35] In this case, it was possible to lower the process costs by isolating the hydroquinone, DDQH2, from the aqueous waste stream (96% recovery) and treating it with HNO3/AcOH in a subsequent step to regenerate DDQ (75% yield).[35,38]
Transition Metal Salts as Stoichiometric Terminal Oxidant
The considerations noted above have provided motivation to develop methods capable of using catalytic amounts of DDQ or choranil in quinone-mediated reactions. Catalytic quantities of DDQ can be used by employing an alternative stoichiometric reagent such as FeCl3, Mn(OAc)3, PbO2, or MnO2 as the terminal oxidant (Scheme 5). Though these reactions are catalytic in DDQ, catalyst loadings remain rather high (10–20 mol%), and a large excess of the terminal oxidant is typically employed.
Scheme 5.
A generic representation of quinone-catalyzed substrate oxidation employing stoichiometric transition metal as the terminal oxidant.
Cacchi and coworkers reported the first DDQ-catalyzed transformation in 1978, showing that the oxidation of allylic alcohols to α,β-unsaturated ketones could be accomplished using 10 mol% DDQ in the presence of 30 mol% periodic acid under slightly acidic,[39] biphasic conditions at room temperature (Scheme 6).[40]
Scheme 6.
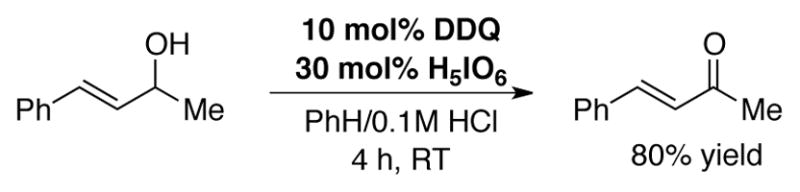
Catalytic oxidation of activated alcohols using catalytic DDQ with periodic acid under biphasic conditions.[40]
Helquist and coworkers subsequently demonstrated similar reactivity with Mn(OAc)3 as the stoichiometric oxidant. With 20 mol% DDQ and 6.0 equiv Mn(OAc)3, allylic alcohols and electron-rich benzylic alcohols are oxidized to the corresponding aldehydes and ketones under mild conditions (Scheme 7A).[41] Good chemoselectivity for allylic alcohols over benzylic alcohols was observed (Scheme 7B).
Scheme 7.
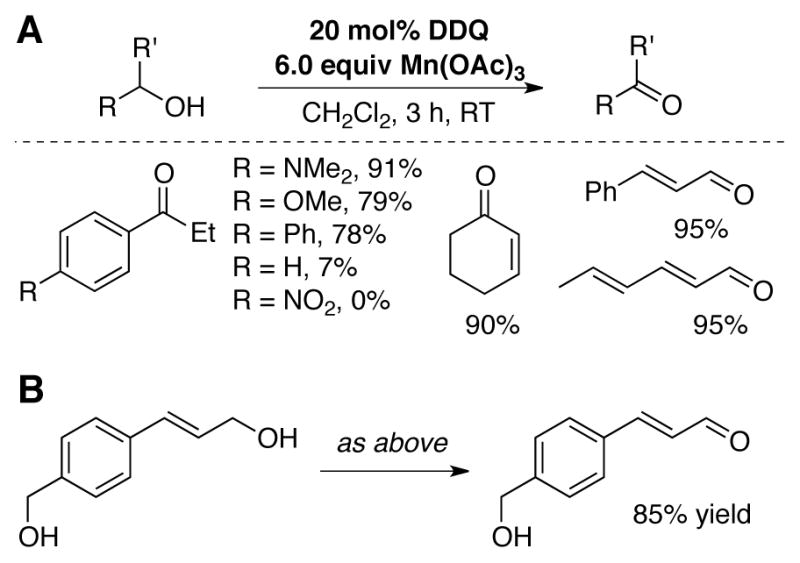
(A) Oxidation of activated alcohols using catalytic DDQ with Mn(OAc)3 as the terminal oxidant. (B) Selective oxidation of allylic alcohols over benzylic alcohols.[41]
Chandrasekhar reported the deprotection of PMB (4-methoxybenzyl) and DMB (3,4-dimethoxybenzyl) ethers using 10 mol% DDQ in combination with 3.0 equiv FeCl3 under biphasic conditions (Scheme 8A).[42] While substrate scope is limited, these catalytic conditions allow for the same selective removal of PMB ether protecting groups as with stoichiometric application of DDQ. To avoid incompatibility with acid-sensitive substrates, Sharma reported neutral conditions for PMB ether deprotection using 10 mol% DDQ and 3.0 equiv Mn(OAc)3 in CH2Cl2 at room temperature (Scheme 8B).[43]
Scheme 8.
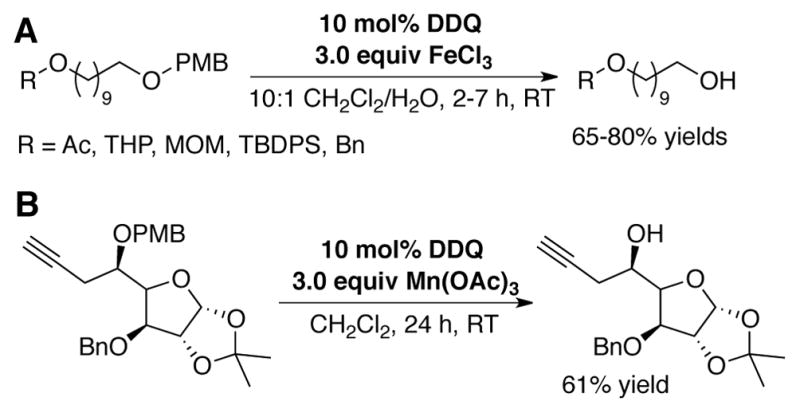
Catalytic deprotection of PMB ethers using DDQ in combination with (A) 3.0 equiv FeCl3, or (B) 3.0 equiv Mn(OAc)3 as the (super)stoichiometric terminal oxidant.[42]
Floreancig and coworkers have reported several innovative examples of DDQ-mediated oxidative C-C bond forming reactions, including intramolecular dehydrogenative-coupling of allylic ethers to form tetrahydropyranone (Scheme 9A).[28g–o] In connection with this effort, Floreancig and Liu developed conditions for carrying out some of these reactions catalytically (15–20 mol% DDQ) with either excess PbO2 or MnO2 (Scheme 9B).[44]
Scheme 9.

(A) Stoichometric DDQ-mediated oxidative intramolecular synthesis of tetrahydropyranone derivatives,[28k] and (B) subsequently-developed catalytic conditions for similar transformations.[44]
The versatility of these catalytic conditions was demonstrated in a series of other applications, including PMB ether deprotection (Scheme 10A), arene and heteroarene dehydrogenation reactions (Scheme 10B and C, respectively), and a cross-dehydrogenative coupling reaction of isochroman with acetophenone (Scheme 10D), originally developed as a stoichiometric DDQ-mediated reaction by Li (vida supra).[28c]
Scheme 10.
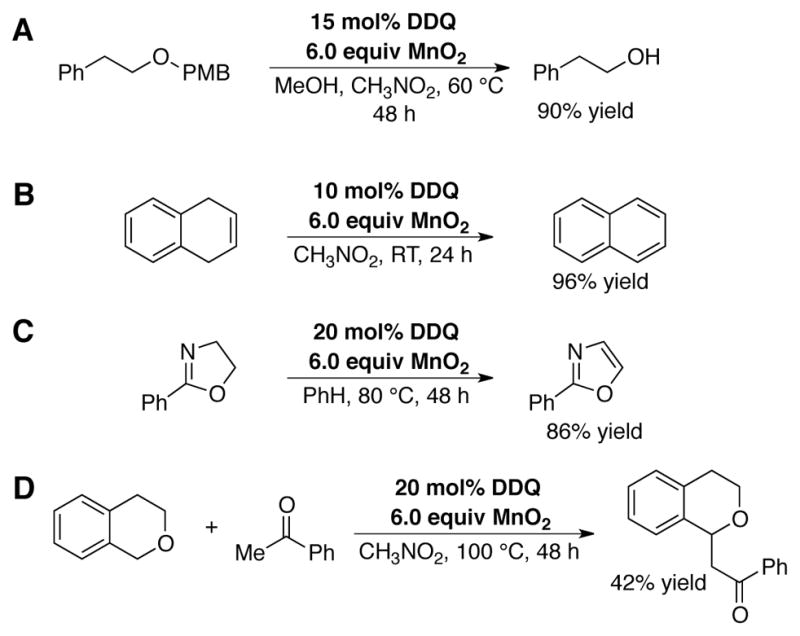
Examples of DDQ-catalyzed transformations with MnO2 as the terminal oxidant: (A) PMB deprotection, (B) arene and (C) heteroarene dehydrogenation reactions, as well as (D) CDC reaction of isochroman with acetophenone, based on reaction conditions developed for tetrahydropyranone synthesis.[44]
Ghosh and coworkers disclosed a similar DDQ-catalyzed synthesis of tetrahydropyran derivatives, based on a stoichiometric DDQ-mediated transformation developed in their efforts towards (−)-Zampanolide (Scheme 11A).[45] A variety of substituted tetrahydropyran derivatives were obtained using 20 mol% DDQ, 2.0 equiv PPTS, 2.0 equiv ceric ammonium nitrate (CAN), and 4Å MS in MeCN at −38 °C (Scheme 11B).[46]
Scheme 11.

(A) Stoichiometric DDQ-mediated oxidative cyclization reaction towards the synthesis of Zampanolide,[45] and (B) subsequently developed DDQ-catalyzed conditions employing 2.0 equiv CAN as terminal oxidant.[46]
A DDQ-catalyzed method for the oxidative C–O coupling of diaryl methane C–H bonds with carboxylic acids was reported by Lei and coworkers. Good yields of cross-coupled products could be obtained by using 20 mol% DDQ and 5.0 equiv MnO2 in dichloroethane at 100 °C (Scheme 12).[47]
Scheme 12.
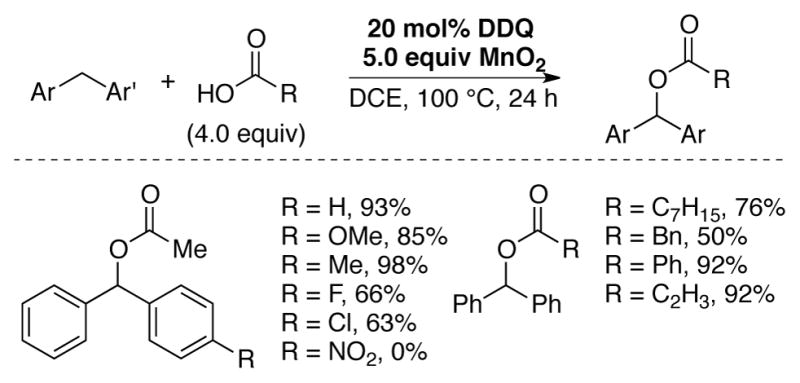
DDQ-catalyzed oxidative C-O coupling of diarylmethane C–H bonds employing MnO2 as the terminal oxidant.[47]
Muramatsu and Nakano reported a DDQ-catalyzed cross-dehydrogenative coupling reaction, again based on an earlier transformation employing stoichiometric DDQ (Scheme 13A).[48] New C–C bonds are formed from oxidation of isochroman or tetrahydroisoquinoline substrates followed by addition of aryl Grignard reagents. Catalytic DDQ (20 mol%) was employed with 1.0 equiv of [bis(trifluoroacetoxy)iodo]-benzene (PIFA) as the stoichiometric oxidant (Scheme 13B).[49]
Scheme 13.
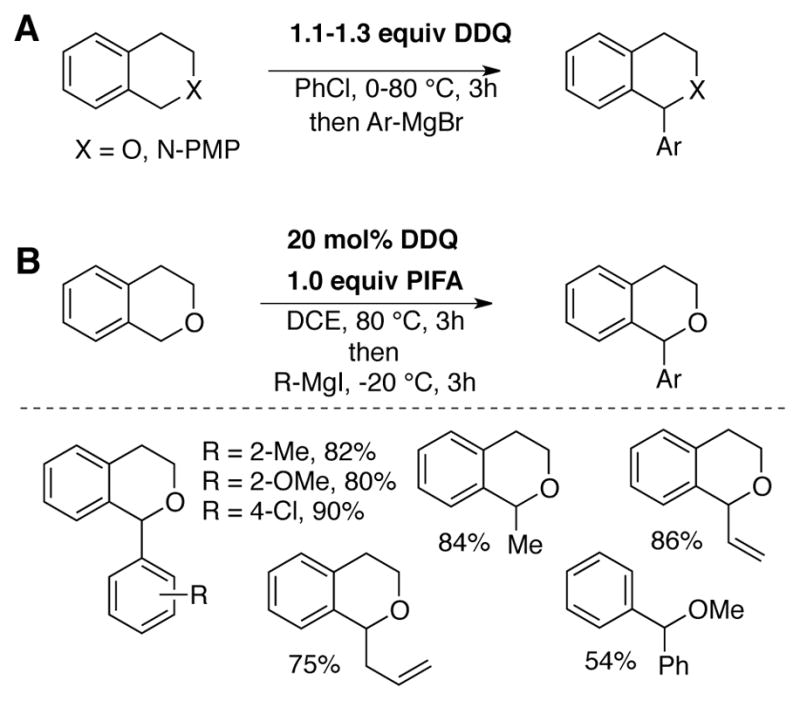
Cross-dehydrogenative coupling of activated C–H bonds with aryl Grignard reagents using (A) stoichiometric DDQ,[48] or (B) catalytic DDQ with [bis(trifluoroacetoxy)iodo]-benzene (PIFA) as stoichiometric oxidant.[49]
Electrochemical regeneration of DDQ
Electrolysis has been used as a method for regeneration of DDQ following a reaction,[50] and in situ electrochemical oxidation of DDQH2 could provide a means to achieve catalytic applications of DDQ (Scheme 14). Redox mediators have been widely studied for electrochemical oxidation of organic substrates,[51] but applications in which DDQ has been used as the electrochemical mediator are still rare.
Scheme 14.
Generic representation of electrochemical quinone-catalyzed oxidation of organic substrates.
In the course of synthetic studies on the euglobal family of natural products, Chiba and coworkers found that stoichiometric DDQ (2.0 equiv) could promote a Diels-Alder reaction between grandinol (and related model compounds, such as 5) and pinene derivatives (Scheme 15A).[52] Under these conditions, however, one of the desired reaction partners, α–phellandrene, undergoes an undesired Diels-Alder reaction with DDQ in near-quantitative yield and no formation of desired product (Scheme 15B). The researchers subsequently developed an electrocatalytic method, involving DDQ-catalyzed oxidation of 5 to an intermediate quinone methide species at a polytetrafluoroethylene (PTFE)-fiber coated working electrode at 0.70 V (NHE) in Et4NOTs (50 mM in MeNO2). The steady-state concentration of DDQ is kept low under these conditions, and the desired Diels-Alder reaction of the quinone methide intermediate with α-phellandrene or α- or β-pinene generates the desired euglobal analog in excellent yield (Scheme 15C).[53] Six euglobal natural products were prepared by using this electrochemical approach. This work elegantly shows how catalytic conditions may be used to overcome synthetic limitations that may be encountered when using a reactive stoichiometric reagent (DDQ).
Scheme 15.

Stoichiometric DDQ-promoted Diels-Alder reaction of compound 5 leads to desired product with (A) β-pinene, but not with (B) α-phellandrene as the reaction partner.[52] (C) Reaction with α-phellandrene is successful when electrochemical catalytic DDQ conditions are employed.[53]
Crabtree and coworkers have reported conditions for electrochemical regeneration of DDQ in the context of “virtual hydrogen storage” research. With 15 mol% DDQ in MeCN (0.5 M NaClO4) at room temperature, N-phenylbenzylidene could be obtained from N-phenylbenzylamine in 95% yield (Scheme 16) after a 6 h controlled potential electrolysis at 0.964 V NHE.[16] Separately, Utley and Rosenberg employed DDQ as an electrocatalyst for the benzylic oxidation of electron-rich 2-alkylnaphthalenes to the corresponding benzylic ethers and ketones.[32f, 54]
Scheme 16.
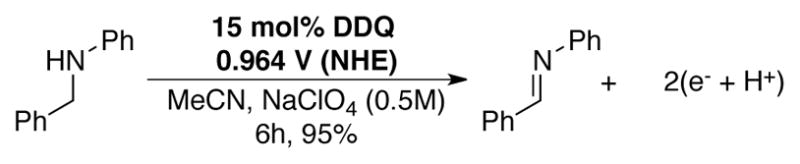
Electrochemical DDQ-catalyzed dehydrogenation of C–N bonds by controlled potential electrolysis at 0.964 V.[16]
Aerobic regeneration of DDQ/quinones
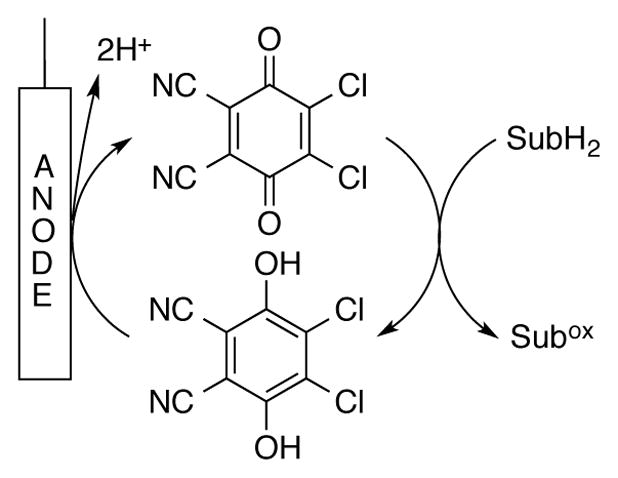
Molecular oxygen is an ideal terminal oxidant, and quinone-mediated oxidations of organic molecules are well-suited for “oxidase”-type aerobic oxidation reactions (cf. Scheme 2).[55] In contrast to the facile autoxidation of low-potential quinones (cf. Scheme 1), direct aerobic oxidation of high-potential hydroquinones is typically not feasible.[56] Nevertheless, several strategies have been identified for mediating aerobic oxidation of hydroquinone species by using a heterogeneous co-catalyst or a soluble co-catalytic electron-transfer mediator (ETM, Scheme 17A), such as a polyoxometalate or “NOx” source, the latter of which contributes to a NO/NO2 redox cycle (Scheme 17B).
Scheme 17.
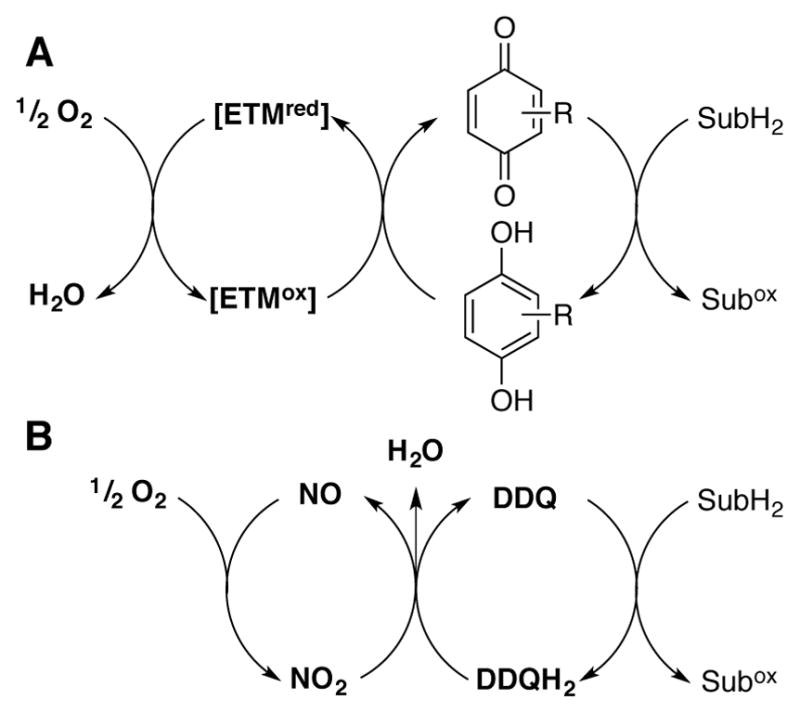
A generic representation of aerobic quinone-catalyzed substrate oxidation employing (A) an electron transfer mediator (ETM) or (B) NO/NO2 redox couple to facilitate hydroquinone reoxidation.
Aerobic oxidation of hydroquinones to quinones, either uncatalyzed (autoxidation)[5] or using catalysts such as metal macrocycles[57] and various catecholase mimics,[58] have been studied previously, but examples of such studies with high potential quinones, such as DDQ or chloranil, are rare. Miyamura, Kobayashi and coworkers reported the aerobic oxidation of a wide variety of hydroquinone derivatives with heterogeneous, polymer-incarcerated Au[59] (PI Au) and Pt[60] (PI Pt) nanoclusters. Reaction conditions are mild, proceeding with low catalyst loadings at room temperature in CHCl3/H2O under 1 atm O2 (Scheme 18). Of particular note is the oxidation of tetrachlorohydroquinone to p-choranil in 99% yield at room temperature within 3 h using 1 mol% PI-Pt catalyst.
Scheme 18.
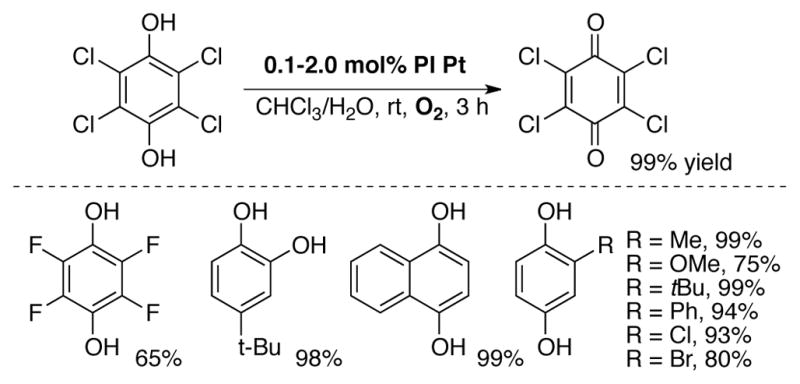
Aerobic oxidation of tetrachlorohydroquinone and other hydroquinone derivatives to corresponding quinones using polymer incarcerated Pt (PI Pt) nanoclusters.[60]
A subsequent study demonstrated that a catalyst system composed of catalytic o-chloranil and co-catalytic hybrid organic/inorganic platinum nanocluster catalyst (HB Pt) can be employed in the oxidation of organic substrates using molecular oxygen as the terminal oxidant.[61] Efficient oxidation of Hantzsch-type dihydropyridines to substituted pyridines was accomplished using 5–10 mol% o-chloranil in combination with 0.5–1.0 mol% HB Pt in CH2Cl2/H2O solvent at room temperature under 1 atm O2 (Scheme 19A). Dehydrogenation of 2-methylindoline to 2-methylindole was also accomplished with this catalyst system (Scheme 19B), and efficient PMB ether deprotection was demonstrated under modified reaction conditions consisting of 1 mol% oxidation-resistant polymer-incarcerated Pt catalyst (RPI Pt) and 10 mol% o-chloranil in DCE/H2O at 100 °C (Scheme 19C). When stoichiometric chloranil (3.0 equiv) was used in the oxidation of tetrahydroquinoline derivative 6, a substrate/chloranil-derived ketal adduct 8 was obtained as the major product. Under catalytic conditions, however, the desired oxygenated compound 7 was obtained in 88% yield (Scheme 20).[61]
Scheme 19.

Aerobic o-chloranil-catalyzed dehydrogenation of (A) dihydropyridines to pyridines and (B) 2-methylindoline to 2-methylindole using co-catalytic hybrid organic/inorganic platinum nanocluster catalyst (HB Pt), and (C) PMB ether deprotection under similar conditions employing oxidation-resistant polymer-incarcerated Pt co-catalyst (RPI Pt).[61]
Scheme 20.

Stoichiometric oxidation of tetrahydroquinoline derivative 6, with 3.0 eq chloranil leads to substrate/chloranil ketal adduct 8. Desired product 7 can be obtained under previously-developed catalytic conditions.[61]
Polyoxometalates (also called heteropolyacids) are effective co-catalysts for the aerobic regeneration of high potential quinones. Neumann and coworkers reported an aerobic oxidation of allylic and benzylic alcohols using 5 mol% o-chloranil with 1.5 mol% Na5PV2Mo10O40 at 90 °C in H2O/decalin under 1 atm O2.[62]
In 1994, Kochi and coworkers demonstrated quantitative aerobic oxidation of hydroquinone to benzoquinone using 1 mol% NO2 in CH2Cl2 at −10 °C under 1 atm O2 (Scheme 21).[63] A range of substituted quinones were generated in this manner.[64] It wasn’t until 2008, however, that the NO/NO2 redox couple was used in combination with DDQ to catalyze the aerobic oxidation of an organic molecule (cf Scheme 17B). Xu and coworkers used a catalyst system consisting of 5 mol% DDQ and 5 mol% NaNO2 under 1.3 MPa O2 at 120 °C to dehydrogenate dihydroanthracene to anthracene in >99% yield in 8 h (Scheme 22).[65]
Scheme 21.
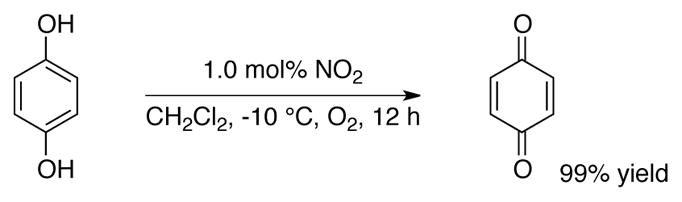
NO2-catalyzed aerobic oxidation of hydroquinones to quinones.[63]
Scheme 22.
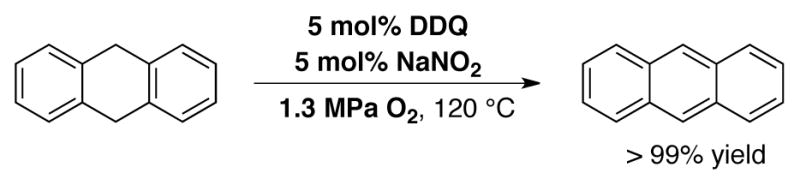
DDQ-catalyzed aerobic dehydrogenation of dihydroanthracene employing co-catalytic NaNO2 at elevated temperatures and pressures.[65]
The observation that a NO/NO2 redox couple enables aerobic DDQ-catalyzed oxidation of organic molecules is quite significant. Traditional electron-transfer mediators (ETMs), which couple the oxidation of hydroquinone to the reduction of O2 to H2O2, are typically not effective with DDQ because the reduction potential of DDQ/DDQH2 (E° = 0.750 V NHE) is higher than the O2/H2O2 reduction potential (0.670 V NHE). O2 reduction by NO, however, proceeds via 4e− reduction of O2 without formation of H2O2, and NO2 is a sufficiently strong oxidant to promote oxidation of DDQH2 to DDQ.
Mo, Hu and coworkers have reported a similar catalyst system for the oxidation of activated alcohols, using 5 mol% DDQ and 5 mol% tBuNO2 in dichloroethane at 80 °C under 0.2 MPa O2 pressure. Excellent yields of the corresponding aldehydes and ketones were obtained (Scheme 23A).[66] Under modified reaction conditions (ethylene glycol diethyl ether as solvent, 120–140 °C), the authors found that methyl aryl ethers are converted to the corresponding benzylic aldehydes (Scheme 23B), and PMB ethers undergo selective deprotection (Scheme 23C).
Scheme 23.
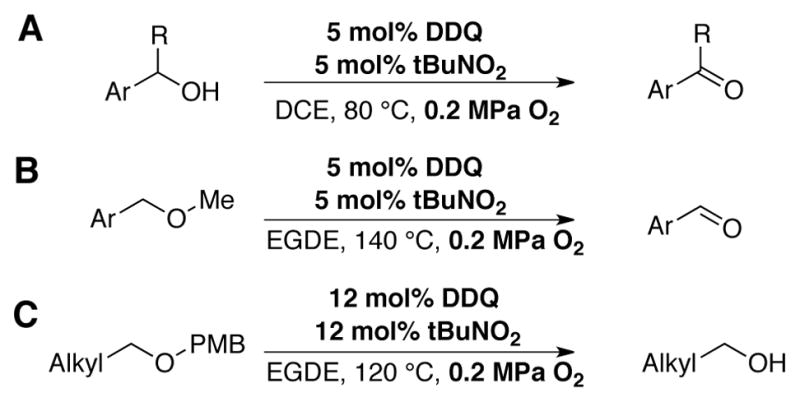
DDQ-catalyzed aerobic oxidation of (A) alcohols and (B) ethers, and (C) selective deprotection of PMB using tert-butylnitrite as a co-catalyst.[66]
Gao and colleagues reported a DDQ-catalyzed (1–20 mol%) method for the aerobic oxidation of allylic and benzylic alcohols to corresponding aldehydes.[67] The reaction uses 10 mol% NaNO2 and is carried out in a CH2Cl2/AcOH solvent mixture at ambient temperatures under an O2 balloon (Scheme 24). Reactions could also proceed in good yield under air. DDQ-catalyzed aerobic alcohol oxidation has been applied to lignin depolymerization using a NOx co-catalyst system,[68,69] and other aerobic alcohol oxidation reactions have been reported in which additional cocatalysts, such as TEMPO[70a] or N-bromosuccinimide,[70b] are used in combination with DDQ and NOx.
Scheme 24.
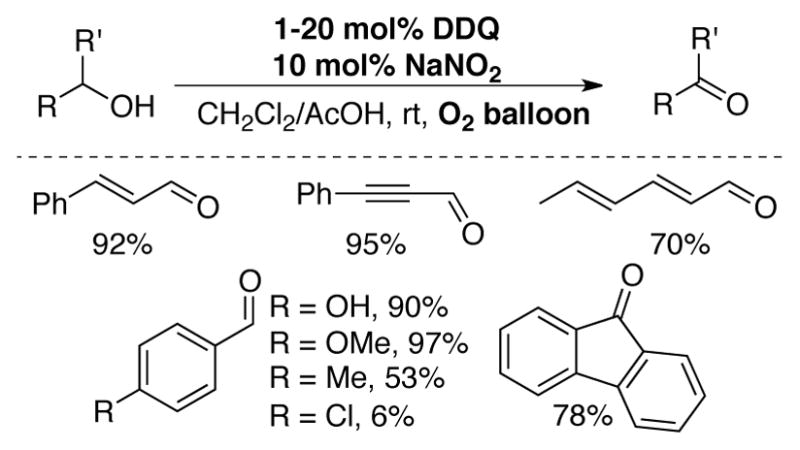
DDQ-catalyzed aerobic oxidation of activated alcohols to aldehydes employing co-catalytic NaNO2 under mild conditions.[67]
Shen, Hu and colleagues then reported a modified method for aerobic DDQ-catalyzed deprotection of PMB ethers that the corresponding alcohol in excellent yield.[71] Conditions resembled those developed by Gao for oxidation of activated alcohols (5 mol% DDQ, 5 mol% NaNO2), but were carried out at 100 °C in chlorobenzene (which represent somewhat milder conditions that those shown in Scheme 23). Moody then reported even milder conditions for PMB ether deprotection in AcOH as the solvent, employing 1.5–5 mol% DDQ, 3–10 mol% NaNO2 at room temperature under 1 atm O2 (balloon).[72]
Yan and coworkers reported aerobic DDQ-catalyzed oxidative coupling of diarylpropenes with 1,3-diketones.[73] A range of new C–C coupled products were obtained in good-to-excellent yields using 1 mol% DDQ and 10 mol% NaNO2 in MeNO2/HCO2H at room temperature under O2 balloon (Scheme 25).
Scheme 25.

DDQ-catalyzed aerobic C-C coupling of diarylpropenes and 1,3-dicarbonyls employing co-catalytic NaNO2.[73]
Aerobic DDQ-catalyzed transformations have also been promoted by using AIBN[74] and Fe(pc)[75] co-catalysts as well.
3. Bioinspired o-Quinone-Catalyzed Oxidation of Amines
3.1. Enzymatic Context
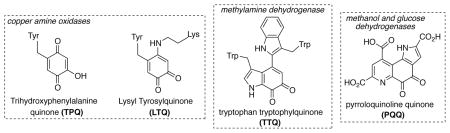
Quinones play an important role as cofactors in the enzymatic oxidation of organic substrates. Several families of “quinoenzymes”[ 76] are known, and include copper amine oxidases (containing active-site cofactors TPQ[77] and LTQ[78]), methylamine dehydrogenase (TTQ[79]), and methanol and glucose dehydrogenases (PQQ).
Copper amine oxidases (CAOs) convert primary amines to aldehydes using molecular oxygen (Scheme 26A).[80] The copper present in the active site of these enzymes reacts with oxygen and mediates post-translational modification of an active-site tyrosine residue to generate the o-quinone cofactor (i.e., TPQ or LTQ).[81] Oxidation of the amine substrate is mediated by the quinone without direct involvement of the Cu center. Two mechanisms were initially proposed for substrate oxidation by the quinone cofactor: a “transamination” mechanism and an “addition-elimination” mechanism (Schemes 26B and 26C). In the transamination mechanism, substrate oxidation occurs via initial condensation of primary amine with the quinone cofactor to give an imine adduct, 9.[82] Imine 9 undergoes tautomerization via net prototropic rearrangement to give a second imine species, 10. Hydrolysis of the imine in 10 liberates the aldehyde product and affords the reduced aminohydroquinone cofactor, 11. Aerobic reoxidation of 11 to an iminoquinone species 12, followed by transamination with another equivalent of amine, closes the catalytic cycle (Scheme 26B). In the addition-elimination mechanism, substrate oxidation occurs via an initially formed hemiaminal adduct 13, which directly liberates an aldimine product via a pericyclic mechanism together with formation of the reduced hydroquinone cofactor 14 (Scheme 26C).
Scheme 26.

Copper amine oxidases carry out (A) the aerobic oxidation of primary amines in vivo. Two initially-proposed substrate oxidation mechanisms, the (B) transamination mechanism, and (C) addition-elimination mechanism. Scheme adapted from ref 116.
Extensive mechanistic studies demonstrated that amine oxidation in these enzymes proceeds through a transamination mechanism. In connection with mechanistic studies of CAO and methylamine dehydrogenase quinoenzymes, Klinman,[83] Sayre,[84] Itoh[85] and others developed biomimetic model quinones that promote selective aerobic oxidation of primary amines to imines and aldehydes in the absence of the enzymes (Scheme 27). Like the native enzymes, substrate oxidation by these model quinones was proposed to proceed through a transamination mechanism.
Scheme 27.

Aerobic primary amine oxidation catalyzed by model quinone cofactors (A) TBHBQ, developed by Klinman,[83] (B) Piv-TPQ, developed by Sayre,[84] and (C)Me-TTQ, developed by Itoh.[85]
Itoh and coworkers further examined mechanistic features of the C–H bond-cleaving step in the tryptophan tryptophylquinone (TTQ) model quinone Me-TTQ.[85a] Kinetic studies, including observation of large KIEs (7.8 – 9.2), provided evidence for two competing C–H bond cleavage processes: a slow “spontaneous” intramolecular prototropic rearrangement, k1; and a faster bimolecular "base-catalyzed” rearrangement step, k1′ (Scheme 28). These findings suggest a possible distinction between these comparatively low potential biomimetic o-quinone catalysts and the high-potential quinones, such as DDQ, described above: the former induce a deprotonation-type C–H bond cleavage, while the latter promote C–H bond cleavage via hydride-transfer.
Scheme 28.

Mechanistic proposal for C-H bond breaking step in biomimetic model quinone- catalyzed aerobic oxidation of primary amines to imines, involving competing intramolecular (“spontaneous”) and intermolecular (“base-catalyzed”) C-H cleavage steps.[85a]
Another quinone cofactor, pyrroloquinoline quinone (PQQ) is found in bacterial methane and glucose dehydrogenases. Biochemical studies of the PQQ cofactor established a complementary mechanism for substrate oxidation.[86] In this case an “addition-elimination” mechanism is supported, whereby substrate oxidation (typically an alcohol, such as methanol) occurs from a hemiacetal intermediate (Scheme 29).[87]
Scheme 29.

Proposed mechanism of alcohol oxidation mediated by cofactor PQQ.
In model studies, Itoh, Fukuzumi and coworkers showed that the oxidation of low molecular weight alcohols such as ethanol and methanol are oxidized to the corresponding aldehydes using the trimethyl ester of PQQ (Scheme 30).[88, 89] PQQ is not regenerated by O2 in nature, but the synthetic model is capable of using molecular oxygen as the terminal oxidant. The thermodynamic potential of PQQ-3OMe is low, estimated at 0.05 V (vs. NHE in MeCN; cf. free PQQ −0.05 V vs. NHE in DMF).[96] These values contrast DDQ, which has a significantly higher reduction potential (0.750 V vs. NHE in MeCN).[90] Nevertheless, DDQ has not been shown to oxidize methanol, and many DDQ-mediated substrate oxidations actually take place in methanol as solvent. These observations together with the efficient dehydrogenation of methanol by PQQ clearly show that quinone reactivity is not controlled solely by its reduction potential. This conclusion has important implications for the design of new quinone catalysts for organic oxidation reactions.
Scheme 30.

Biomimetic, aerobic oxidation of methanol and ethanol catalyzed by PQQ-3OMe.[88]
Unlike other quinone cofactors, PQQ is not covalently bound to the enzyme. The glucose dehydrogenase apoenzyme can be reconsitituted with simplified PQQ derivatives (related to 1,7- and 4,7-phenanthroline quinones) to give an active enzyme, albeit with somewhat lower activity.[91] In addition, Bruice has carried out a series of mechanistic studies of non-enzymatic alcohol and amine oxidation using PQQ and simplified PQQ analogs such as di-decarboxy-PQQ and 1,10-, 1,7-, and 4,7-phenanthroline-derived quinones (Scheme 31A).[92,93]
Scheme 31.

(A) PQQ and model compounds, didecarboxy PQQ and phenanthroline-derived quinones. (B) Adduct formation is observed in the reaction of 4,7-phenanthroline-1,10-dione with excess morpholine, implicating the transamination mechanism.[93] (C) Different reaction mechanisms lead to different reduction products, detected by end-product analysis.
In a series of mechanistic studies, Bruice showed that the stoichiometric reduction of phenanthroline-derived o-quinones by primary amines (cyclohexylamine and glycine) results in the formation of aminohydroquinone products, consistent with a transamination mechanism.[93] The secondary amine morpholine reacts more slowly and forms a substrate-quinone adduct as the predominant product (Scheme 31B), also consistent with a transamination-type mechanism. Oxidation of the tertiary amine N,N-dimethylbenzylamine is also significantly slower than primary amine oxidation, and leads to the formation of hydroquinone as well as benzaldehyde and formaldehyde products. The stoichiometric oxidation of p-methylbenzylalcohol was also tested with these phenanthroline-derived quinones. No reaction was observed after 7 days at 60 °C with 1,10- and 1,7-phenanthrolinedione, and only 6% yield was observed using 4,7-phenanthrolinedione. In contrast, 90% yield was obtained when DDQ was employed as the oxidant under otherwise identical conditions. Across each of these substrate classes, Bruice found that quinones containing a nitrogen atom adjacent to the quinone carbonyls (e.g., 1,7- and 4,7-phenanthrolinedione) mediate substrate oxidation more rapidly that 1,10-phenanthrolinedione, despite nearly identical electrochemical potential.
The phenanthroline-derived quinones studied by Bruice share a number of physical and chemical similarities with PQQ, including similar electrochemical potentials (cf. Scheme 31A), facile formation of covalent adducts with water, methanol, and acetone, and ability to mediated stoichiometric oxidation of simple organic substrates.[94–96] In contrast, while secondary and tertiary amines react with phenanthroline-derived quinones, they are not substrates for the didecarboxy-PQQ model compound. End-product analysis of the reaction of didecarboxy-PQQ with primary amines reveals the formation of hydroquinone, rather than aminohydroquinone (cf. Scheme 31C).[97] These findings are consistent with the proposal by Itoh, Ohshiro, and coworkers that oxidation of primary amines by PQQ itself proceeds through an “addition-elimination”-like mechanism.[98] This proposal was supported by kinetic evidence, in addition to the observation of mixtures of aminohydroquinone (transamination product) and hydroquinone (addition-elimination product) species isolated at the end of model reactions.[99]
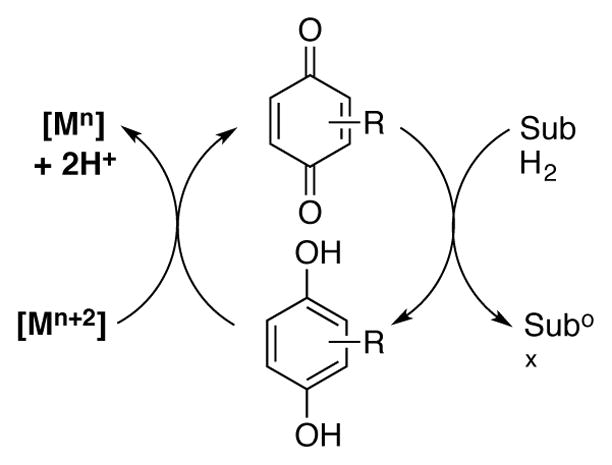
3.2 o-Quinone Catalyzed C–N Bond Dehydrogenation Reactions
The early mechanistic and model studies of enzymatic quinones discussed above have provided the basis for a number of bioinspired quinone-mediated transformations in recent years. The majority of these studies have focused on o-quinones that resemble the enzymatic quinones and are distinct from DDQ and other high-potential p-quinones described in Section 2. Perhaps not surprisingly, many applications of the o-quinones have targeted amine dehydrogenations.[100]
Even before the quinone cofactors were identified in amine oxidase enzymes, Corey demonstrated that branched primary amines could be oxidized with stoichiometric 3,5-di-tert-butyl-o-quinone in MeOH to give a tautomeric imine adduct.[101] Upon hydrolysis, ketone products were obtained in excellent yields (Scheme 32).[102] Unbranched primary amines were not effective substrates, as they generated benzoxazole products instead.[103]
Scheme 32.

Stoichiometric oxidation of branched primary amines to ketones using 3,5-di-tert-butyl-o-quinone. Oxazole adducts are obtained when unbranched primary amines are used as substrates.[101]
Independent attempts to regenerate 3,5-di-tert-butylquinone from the reduced aminophenol by O2 led to the dimeric species 15 (Scheme 33A), while electrochemical and chromate regeneration under acidic conditions gave the quinone in 64% and 56% yields, respectively (Scheme 33B).[104]
Scheme 33.
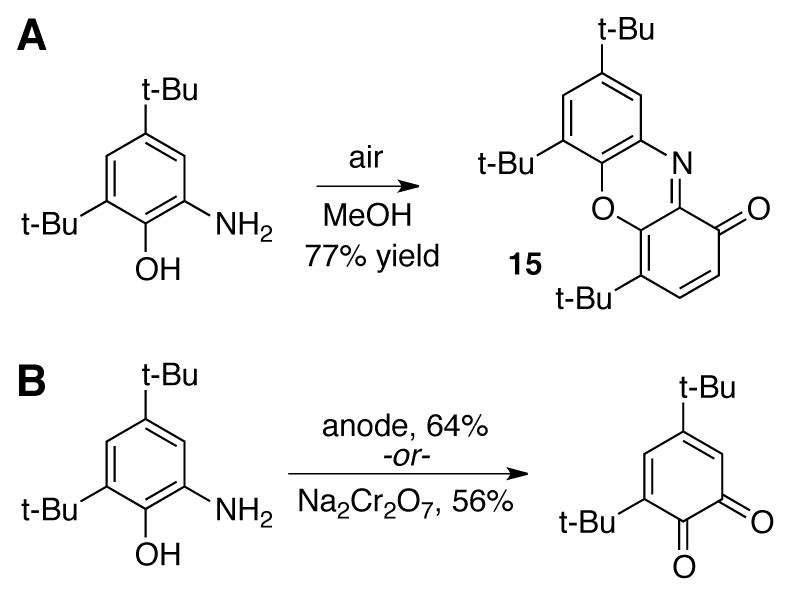
(A) Aerobic oxidation of aminohydroquinone results in formation of dimeric species 15, (B) and additional approaches to regenerate quinone from reduced aminohydroquinone.[104]
Catalytic aerobic oxidation of primary amines to ketones and aldehydes was achieved by Itoh under aqueous micellar conditions using 1 mol% of the quinone PQQ and 10 mol% hexadecyltrimethylammonium bromide (CTAB) in pH 9–10 solution under ambient air at room temperature (Scheme 34).[105, 106]
Scheme 34.
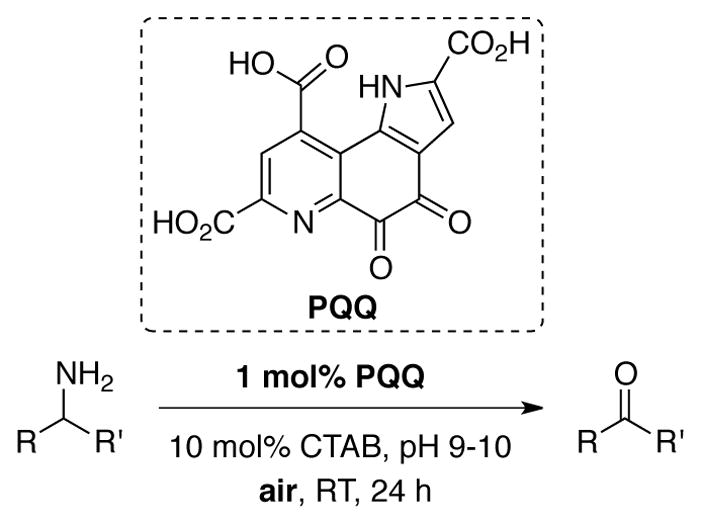
Catalytic oxidation of primary amines to aldehydes and ketones using PQQ promoted by micellar conditions.[105]
Largeron and Fleury developed o-quinones Q1red and Q2red as catalysts for the electrochemical oxidation of primary amines to imines.[107] Controlled potential electrolysis of benzylic or aliphatic primary amines was accomplished by using 2 mol% of precatalyst Q1red or Q2red at 0.60 V vs. SCE with a Pt anode in MeOH at room temperature (Scheme 35A). Good catalyst turnover numbers could be obtained, but the products had to be isolated as dinitrophenylhydrazones. To address this limitation, Largeron and coworkers generated cross-coupled imine compounds by carrying out the oxidation of benzylamine in the presence of a second, less-readily oxidized amine. Following electrolysis, the Pt anode is replaced with a Hg pool cathode. Electrolysis at −1.6 V for 1 h reduced the cross-coupled imines to secondary amines, which could be isolated after workup (Scheme 35B).[108]
Scheme 35.

(A) Electrochemical oxidation of amines to imines[107] and (B) secondary amines, [108] by CAO mimics Q1red and Q2red. (C) When CuI is added as a co-catalyst, the aerobic oxidation of primary amines is also achieved.[109]
Largeron and Fleury later showed that the Q2red catalyst is effective in aerobic oxidation reactions. Primary amines undergo efficient aerobic oxidation at room temperature under ambient air in the presence of 2 mol% Q2red and 0.2 mol% CuI(MeSal) (MeSal = methylsalicylate) as a co-catalyst.[109] Dimeric and cross-coupled imine products were obtained in excellent yields (Scheme 35C).
Wendlandt and Stahl have demonstrated the aerobic oxidation of a diversity of benzylic amines to the corresponding secondary imines using TBHBQ,[110,111] a model quinone initially developed by Mure and Klinman[83] (Scheme 36). Selective cross-coupling products could be obtained by running the reaction in the presence of a second, unactivated (and non-reactive) amine. The homocoupled benzylamine-derived imine is formed at early stages of the latter reactions; however, imine exchange under the reaction conditions enables full oxidation the benzylamine and selective formation of the cross-coupled product.
Scheme 36.

Aerobic oxidation of primary amines to imines using biomimetic o-quinone catalyst TBHBQ.[110]
Unbranched amines are readily oxidized by Q1red, Q2red, and TBHBQ, but these catalysts react poorly with α-branched substrates. Luo and coworkers recently reported that branched benzylic primary amines are readily dehydrogenated to imines by 4-methoxy-5-tert-butyl-o-quinone, Q3.[112] Excellent yields of dimeric imine products were obtained using 10 mol% Q3 at room temperature under an O2 atmosphere (Scheme 37).
Scheme 37.

Aerobic oxidation of a-branched primary amines to imines using a biomimetic o-quinone catalyst, Q3. [112]
Q1red, Q2red, and TBHBQ are also not effective for dehydrogenation of secondary and tertiary amines. Nor are primary alcohols oxidized under the reaction conditions. These limitations are beneficial for chemoselective oxidation reactions, but they also limit the scope. The exquisite selectivity for primary amines can be rationalized by the transamination mechanism, in which substrate oxidation involves formation of an imine adduct. Secondary amines have been shown to be mechanism-based inhibitors of quinones such as TBHBQ.[113] In these cases, the iminium adducts are precursors to irreversible modification of the catalyst (Scheme 38).
Scheme 38.

Irreversible pyrrolation of TBHBQ catalyst via transamination-type mechanism is observed when 3-pyrroline substrates are used.[113]
In principle, quinone-catalyzed dehydrogenation of secondary and tertiary amines would be possible if the reaction proceeded by an addition-elimination, rather than a transamination, mechanism (cf. Scheme 26). Thus, a shift between these two mechanistic pathways has important implications for reaction scope.
Kobayashi and coworkers reported a method for the aerobic dehydrogenation of amines with 0.5 mol% of a block co-polymer-incarcerated Pt/Ir alloy catalyst in combination with a 15–60 mol% catechol as a co-catalyst (Scheme 39A and 39B).[114] C–H cleavage of the amine was proposed to be mediated by the Pt/Ir nanocluster, but o-quinone was proposed to play a crucial role in substrate activation. A hemiaminal intermediate, reminiscent of the addition-elimination mechanism, was proposed (Scheme 39C). Similar reactivity was reported recently by Doris and coworkers, who showed that Rh nanoparticles supported on carbon nanotubes (Rh-CNT) are efficient co-catalysts with 4-tert-butyl-o-quinone in the aerobic dehydrogenations of N-heterocycles.[115]
Scheme 39.

(A) Dehydrogenation of C-N bonds using Pt/Ir alloy incarcerated coblock polymer catalyst, (B) used in combination with co-catalytic catechol additives. (C) A mechanism is proposed wherein substrate oxidation by Pt involved quinone-substrate hemiaminal intermediates.[114]
The first definitive evidence for an addition-elimination pathway was reported by Wendlandt and Stahl using the PQQ model, 1,10-phenanthroline-5,6-dione (phd). This catalyst was shown to mediate aerobic dehydrogenation of secondary amines and a diverse range of N-heterocycles in the presence of co-catalytic quantities of ZnI2 and pyridinium p-toluenesulfonic acid (PPTS) (Scheme 40). The hemiaminal intermediate shown in Scheme 41C was observed and characterized by NMR spectroscopy.[116]
Scheme 40.

(A) Conditions for the aerobic phd-catalyzed oxidation of a variety of secondary amines, (B). (C) An “addition-elimination” mechanism is proposed involving a hemiaminal intermediate.[116]
Scheme 41.

Dehydrogenation of tetrahydroquinolines to quinolines under ambient conditions with co-catalyst system consisting of [Ru(phd)3](PF6)2 and [Co(salophen)].
ZnI2 plays two roles in this catalytic reaction. Coordination of phenanthroline nitrogen atoms to Zn2+ activates the quinone and enhances the rate of substrate oxidation. In addition, the iodide counterions serve as redox-active co-catalysts to promote aerobic turnover of the quinone catalyst (Scheme 40C). The origin of the shift in mechanism from transamination to addition-elimination is not fully understood, but this mechanism provides the basis for the significantly improved substrate scope, specifically allowing dehydrogenation of a broad range of secondary amine-based heterocycles.
Wendlandt and Stahl leveraged the modularity of catalyst system to develop improved phd-based catalysts. For example, dehydrogenation of 1,2,3,4-tetrahydroquinolines to quinolines was not effective with the phd/ZnI2 catalyst, but a broad range of quinoline products were obtained by using a [Ru(phd)3](PF6)2 catalyst in combination with [Co(salophen)] as a co-catalyst (Scheme 41).[117] Changing the co-catalyst from I−/I3− to [Co(salophen)] exhibits a marked improvement in reaction rate (Scheme 42), and allowed the reaction to be carried out using ambient air rather than pure O2. The [Co(salophen)] has been widely used as a co-catalyst in aerobic redox chains, such as Pd-catalyzed oxidation reactions that use benzoquinone as a co-catalyst.[6–9]
Scheme 42.

Influence of different Lewis acid promoters and co-catalysts on the quinone-catalyzed aerobic dehydrogenation of tetrahydroquinoline to quinoline by phd and phd-coordinated Ru complexes. Scheme adapted from ref 117.
PQQ and related phenanthroline-derived quinones, including phd and metal-phd coordination complexes have seen extensive application in the aerobic and electrochemical oxidation of NADH to NAD+[118,119] for synthetic enzymatic transformations,[120] as well as in applications as biosensors.[121,122]
4. Summary and Outlook
This review has focused on two rather distinct classes of quinones and their use as catalysts for selective oxidation of organic molecules. High potential quinones, such as DDQ and chloranil, often operate via hydride-abstraction mechanisms and, therefore, typically react with electron-rich substrates capable of stabilizing carbocationic intermediates. Examples of single-electron transfer and addition-elimination reactions have also been implicated with these quinones. DDQ and related high-potential quinones are usually used as stoichiometric reagents, but significant progress has been made in the development of catalytic methods in which the quinone is used in combination with a more-desirable terminal oxidant. Applications include methods compatible with O2 as the stoichiomentric oxidant, and the collective findings have important implications for large-scale implementation of DDQ-based oxidation reactions.
The second class of quinones consists of o-quinone catalysts inspired by cofactors in enzymes that mediate amine dehydrogenations. This family of quinones has been shown to be compatible with electrochemical and aerobic catalytic turnover. Mechanistic studies show that these quinones engage in electrophilic reaction pathways such as transamination and addition-elimination mechanisms, which are quite different from the pathway commonly featured with DDQ and the high-potential quinones. While the applications emphasize amine oxidation reactions, the synthetic scope has been expanded well beyond that known from the enzymes. The differences include dehydrogenation of diverse heterocyclic secondary amines, and arise primarily the development of catalysts that operate via an addition-elimination rather than the enzymatic transamination mechanism.
The bioinspired o-quinones have significantly lower reduction potentials than DDQ or chloranil, but their unique mechanisms introduce kinetically accessible pathways not readily available to the more-oxidizing p-quinones (cf. Scheme 29). These observations hint towards a broader, yet still largely unexplored opportunity to achieve catalyst control within quinone-mediated transformations. The reactivity differences between DDQ and chloranil are typically rationalized on the basis of their differential reduction potentials, but many of the findings discussed herein and elaborated elsewhere in the literature[37,85a] suggest that reactivities observed across diverse quinone structure types do not exhibit simple linear-free-energy correlations. Instead, reactivity is closely linked to the reaction pathways accessible to the different quinone structures. The ability to exploit different reaction pathways will benefit from closely coupling reaction development efforts with mechanistic studies of new catalytic reactions.
Finally, the results summarized herein highlight the ability to use quinones as catalysts, rather than stoichiometric reagents. The use and/or development of new co-catalysts that enable efficient catalytic turnover with O2 as the terminal oxidant has important implications for “green” large-scale applications of quinone-catalyzed oxidation of organic molecules.
Acknowledgments
We thank the NIH (R01-GM100143) for financial support of our work on catalytic dehydrogenation reactions.
Biographies
Shannon S. Stahl is a Professor of Chemistry at the University of Wisconsin-Madison, where he began his independent career in 1999. His research group specializes in catalysis, with an emphasis on aerobic oxidation reactions and their applications in organic chemistry. He was an undergraduate at the University of Illinois at Urbana-Champaign, and subsequently attended Caltech (Ph.D., 1997), where he was an NSF predoctoral fellow with Prof. John Bercaw. From 1997–1999, he was an NSF postdoctoral fellow with Prof. Stephen Lippard at MIT.
Alison E. Wendlandt received her B.S. in Chemistry from the University of Chicago in 2007. She began her doctoral studies at the University of Wisconsin – Madison in 2010, under the supervision of Prof. Shannon S. Stahl. She completed her Ph.D. in 2015, and is currently a postdoctoral fellow at Harvard University working with Prof. Eric Jacobsen.
References
- 1.For selected reviews on the applications of nitroxyl radicals with and without metal co-catalysts, see: Nooy AEJd, Besemer AC, Bekkum Hv. Synthesis. 1996;1996:1153–1176.Cao Q, Dornan LM, Rogan L, Hughes NL, Muldoon MJ. Chem Commun. 2014;50:4524–4543. doi: 10.1039/c3cc47081d.Wertz S, Studer A. Green Chemistry. 2013;15:3116–3134.Tebben L, Studer A. Angew Chem Int Ed. 2011;50:5034–5068. doi: 10.1002/anie.201002547.Sheldon RA, Arends IWCE. J Mol Cat A: Chem. 2006;251:200–214.Sheldon RA, Arends IWCE. Adv Synth Catal. 2004;346:1051–1071.Bobbitt JM, Brückner C, Merbouh N. Org React. 2009;74:106–132.Ryland BL, Stahl SS. Angew Chem Int Ed. 2014;53:8824–8838. doi: 10.1002/anie.201403110.Miles KC, Stahl SS. Aldrichimica Acta. 2015;48:8–10.
- 2.For reviews focusing on synthetic applications of N-hydroxyphthalimide (NHPI)/Phthalimide N-oxyl (PINO), see: Recupero F, Punta C. Chem Rev. 2007;107:3800–3842. doi: 10.1021/cr040170k.Melone L, Punta C. Beilstein J Org Chem. 2013;9:1296–1310. doi: 10.3762/bjoc.9.146.Galli C, Gentili P, Lanzalunga O. Angew Chem Int Ed. 2008;47:4790–4796. doi: 10.1002/anie.200704292.Coseri S. Catalysis Reviews. 2009;51:218–292.Ishii Y, Sakaguchi S. Catal Today. 2006;117:105–113.Ishii Y, Sakaguchi S, Iwahama T. Adv Synth Catal. 2001;343:393–427.
- 3.a) Ciriminna R, Pagliaro M. Org Process Res Dev. 2010;14:245–251. [Google Scholar]; b) Iwahama T, Syojyo K, Sakaguchi S, Ishii Y. Org Process Res Dev. 1998;2:255–260. [Google Scholar]; c) Ishii Y, Kajikawa Y. 7,015,356. US Patent. (Daicel Chem. Ind), issued Mar 21, 2006.
- 4.The Chemistry of the Quinonoid Compounds. Wiley-Interscience; New York: 1974 and 1988. [Google Scholar]; Buckle DR, Collier SJ, McLaws MD. Encyclopedia of Reagents for Organic Synthesis. John Wiley & Sons, Ltd; 2001. [Google Scholar]
- 5.a) Goor G, Glenneberg J, Jacobi S. Ullmann’s Encyclopedia of Industrial Chemistry. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim: 2012. Hydrogen Peroxide. [Google Scholar]; b) Eul W, Moeller A, Steiner N. Hydrogen Peroxide. In: Seidel A, Bickford M, editors. Kirk-Othmer Encyclopedia of Chemical Technology. John Wiley & Sons, Inc; 2000. [Google Scholar]; c) Jones CW, Clark JH, editors. Applications of Hydrogen Peroxide and Derivatives. The Royal Society of Chemistry; 1999. [Google Scholar]
- 6.a) Bäckvall JE, Awasthi AK, Renko ZD. J Am Chem Soc. 1987;109:4750–4752. [Google Scholar]; b) Bäckvall JE, Hopkins RB, Grennberg H, Mader M, Awasthi AK. J Am Chem Soc. 1990;112:5160–5166. [Google Scholar]
- 7.a) Bäckvall JE, Hopkins RB. Tetrahedron Lett. 1988;29:2885–2888. [Google Scholar]; b) Bäckvall JE, Chowdhury RL, Karlsson U. J Chem Soc, Chem Commun. 1991:473–475. [Google Scholar]; c) Yokota T, Fujibayashi S, Nishiyama Y, Sakaguchi S, Ishii Y. J Mol Cat A: Chem. 1996;114:113–122. [Google Scholar]; d) Grennberg H, Bergstad K, Bäckvall JE. J Mol Cat A: Chem. 1996;113:355–358. [Google Scholar]; e) Bergstad K, Grennberg H, Bäckvall JE. Organometallics. 1998;17:45–50. [Google Scholar]; f) Wöltinger J, Bäckvall JE, Zsigmond Á. Chem – Eur J. 1999;5:1460–1467. [Google Scholar]; g) Piera J, Närhi K, Bäckvall JE. Angew Chem Int Ed. 2006;45:6914–6917. doi: 10.1002/anie.200602421. [DOI] [PubMed] [Google Scholar]; h) Volla CMR, Bäckvall JE. Angew Chem Int Ed. 2013;52:14209–14213. doi: 10.1002/anie.201308448. [DOI] [PubMed] [Google Scholar]
- 8.a) Wang GZ, Andreasson U, Bäckvall JE. J Chem Soc, Chem Commun. 1994:1037–1038. [Google Scholar]; b) Csjernyik G, Éll AH, Fadini L, Pugin B, Bäckvall J-E. J Org Chem. 2002;67:1657–1662. doi: 10.1021/jo0163750. [DOI] [PubMed] [Google Scholar]; c) Samec JSM, Éll AH, Bäckvall JE. Chem –Eur J. 2005;11:2327–2334. doi: 10.1002/chem.200401082. [DOI] [PubMed] [Google Scholar]
- 9.For a leading reference, see: Campbell AN, White PB, Guzei IA, Stahl SS. J Am Chem Soc. 2010;132:15116–15119. doi: 10.1021/ja105829t.
- 10.Piera J, Bäckvall J-E. Angew Chem Int Ed. 2008;47:3506–3523. doi: 10.1002/anie.200700604. [DOI] [PubMed] [Google Scholar]
- 11.We limit our discussion to reactions of quinones in their ground-state. Photoexcited quinones have been demonstrated to be extremely powerful oxidants with unique reactivity. For a recent review and a prominent specific example, see: Lerch S, Unkel LN, Wienefeld P, Brasholz M. Synlett. 2014;25:2673–2680.Ohkubo K, Fujimoto A, Fukuzumi S. J Am Chem Soc. 2013;135:5368–5371. doi: 10.1021/ja402303k.
- 12.For selected examples involving the use of (super)-stoichiometric DDQ, see: Merschaert A, Boquel P, Van Hoeck JP, Gorissen H, Borghese A, Bonnier B, Mockel A, Napora F. Org Process Res Dev. 2006;10:776–783.Sharma PK, Kolchinski A, Shea HA, Nair JJ, Gou Y, Romanczyk LJ, Schmitz HH. Org Process Res Dev. 2007;11:422–430.Mickel SJ, Sedelmeier GH, Niederer D, Schuerch F, Seger M, Schreiner K, Daeffler R, Osmani A, Bixel D, Loiseleur O, Cercus J, Stettler H, Schaer K, Gamboni R, Bach A, Chen GP, Chen W, Geng P, Lee GT, Loeser E, McKenna J, Kinder FR, Konigsberger K, Prasad K, Ramsey TM, Reel N, Repič O, Rogers L, Shieh WC, Wang RM, Waykole L, Xue S, Florence G, Paterson I. Org Process Res Dev. 2003;8:113–121.
- 13.For an example involving the use of stoichiometric chloranil, see: Armitage M, Bret G, Choudary BM, Kingswood M, Loft M, Moore S, Smith S, Urquhart MWJ. Org Process Res Dev. 2012;16:1626–1634.
- 14.Walker D, Hiebert JD. Chem Rev. 1967;67:153–195. doi: 10.1021/cr60246a002. [DOI] [PubMed] [Google Scholar]
- 15.This charge transfer complex is thought to be a true intermediate – rather than an off-cycle species – by the observation of negative reaction entropy. See: Yamamoto S, Sakurai T, Yingjin L, Sueishi Y. Phys Chem Chem Phys. 1999;1:833–837.Fukuzumi S, Ohkubo K, Tokuda Y, Suenobu T. J Am Chem Soc. 2000;122:4286–4294.. See also: Zaman KM, Yamamoto S, Nishimura N, Maruta J, Fukuzumi S. J Am Chem Soc. 1994;116:12099–12100.
- 16.Charge transfer complex formation is also supported by theoretical studies. For example: Luca OR, Wang T, Konezny SJ, Batista VS, Crabtree RH. New J Chem. 2011;35:998–999.
- 17.Lemaire M, Guy A, Imbert D, Guette J-P. J Chem Soc, Chem Commun. 1986:741–742. [Google Scholar]
- 18.Jung HH, Floreancig PE. Tetrahedron. 2009;65:10830–10836. doi: 10.1016/j.tet.2009.10.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.For selected mechanistic studies proposing related HAT-based processes, see: Höfler C, Rüchardt C. Liebigs Annalen. 1996;1996:183–188.Rüchardt C, Gerst M, Ebenhoch J. Angew Chem Int Ed. 1997;36:1406–1430.
- 20.Trost BM. J Am Chem Soc. 1967;89:1847–1851. doi: 10.1021/ja00984a017. [DOI] [PubMed] [Google Scholar]
- 21.Mayr has suggested that in the case of C–H hydride donors reaction occurs are the carbonyl oxygen of DDQ, forming a new O–H bond, while with Sn-H and B-H donors, a new C-H bond is formed by reaction at C5: Guo X, Zipse H, Mayr H. J Am Chem Soc. 2014;136:13863–13873. doi: 10.1021/ja507598y.
- 22.For mechanistic studies on the dehydrogenation of hydroaromatic substrates which suggest that direct hydride abstraction is not feasible, see: Mueller P, Rocek J. J Am Chem Soc. 1972;94:2716–2719.Wurche F, Sicking W, Sustmann R, Klärner FG, Rüchardt C. Chem – Eur J. 2004;10:2707–2721. doi: 10.1002/chem.200305686.
- 23.a) Braude EA, Jackman LM, Linstead RP. J Chem Soc. 1954:3548–3563. [Google Scholar]; b) Braude EA, Jackman LM, Linstead RP. J Chem Soc. 1954:3564–3568. [Google Scholar]; c) Braude EA, Brook AG, Linstead RP. J Chem Soc. 1954:3569–3574. [Google Scholar]; d) Braude EA, Linstead RP, Wooldridge KR. J Chem Soc. 1956:3070–3074. [Google Scholar]; e) Barnard JR, Jackman LM. J Chem Soc. 1960:3110–3115. [Google Scholar]; f) Braude EA, Jackman LM, Linstead RP, Shannon JS. J Chem Soc. 1960:3116–3122. [Google Scholar]; g) Braude EA, Jackman LM, Linstead RP, Lowe G. J Chem Soc. 1960:3133–3138. [Google Scholar]
- 24.a) Paukstat R, Brock M, Heesing A. Chem Ber. 1985;118:2579–2592. [Google Scholar]; b) Dost N. Recl Trav Chim Pays-Bas. 1952;71:857–868. [Google Scholar]; c) Müller P, Joly D. Helv Chim Acta. 1983;66:1110–1118. [Google Scholar]
- 25.For representative examples of DDQ-mediated synthesis of arenes and heteroarenes, see: Fu PP, Harvey RG. Chem Rev. 1978;78:317–361., and references therein. Hilt G, Danz M. Synthesis. 2008;2008:2257–2263.Pla D, Marchal A, Olsen CA, Albericio F, Álvarez M. J Org Chem. 2005;70:8231–8234. doi: 10.1021/jo051083a.Ohmura T, Masuda K, Takase I, Suginome M. J Am Chem Soc. 2009;131:16624–16625. doi: 10.1021/ja907170p.Ploypradith P, Petchmanee T, Sahakitpichan P, Litvinas ND, Ruchirawat S. J Org Chem. 2006;71:9440–9448. doi: 10.1021/jo061810h.Manning JR, Davies HML. J Am Chem Soc. 2008;130:8602–8603. doi: 10.1021/ja803139k.Sun J, Xia EY, Zhang LL, Yan CG. Eur J Org Chem. 2009;2009:5247–5254.Wurz RP, Charette AB. Org Lett. 2005;7:2313–2316. doi: 10.1021/ol050442l.Shimizu M, Takahashi A, Kawai S. Org Lett. 2006;8:3585–3587. doi: 10.1021/ol0615634.Haldar P, Barman G, Ray JK. Tetrahedron. 2007;63:3049–3056.Bellur E, Freifeld I, Langer P. Tetrahedron Lett. 2005;46:2185–2187.Taylor EC, Macor JE. Tetrahedron Lett. 1986;27:431–432.
- 26.For representative examples, see: Peng K, Chen F, She X, Yang C, Cui Y, Pan X. Tetrahedron Lett. 2005;46:1217–1220.Steenkamp JA, Mouton CHL, Ferreira D. Tetrahedron. 1991;47:6705–6716.Mori K, Koseki K. Tetrahedron. 1988;44:6013–6020.Becker HD, Bjoerk A, Adler E. J Org Chem. 1980;45:1596–1600.Ohki A, Nishiguchi T, Fukuzumi K. Tetrahedron. 1979;35:1737–1743.Brown DR, Turner AB. J Chem Soc, Perkin Trans 2. 1975:1307–1309.McKittrick BA, Ganem B. J Org Chem. 1985;50:5897–5898.
- 27.For selected examples of PMB ether deprotections, and related transformations, see: Oikawa Y, Yoshioka T, Yonemitsu O. Tetrahedron Lett. 1982;23:885–888.Oikawa Y, Tanaka T, Horita K, Yoshioka T, Yonemitsu O. Tetrahedron Lett. 1984;25:5393–5396.Rahim MA, Matsumura S, Toshima K. Tetrahedron Lett. 2005;46:7307–7309.Vatèle JM. Tetrahedron. 2002;58:5689–5698.Sharma GVM, Rakesh Tetrahedron Lett. 2001;42:5571–5573.Yadav JS, Chandrasekhar S, Sumithra G, Kache R. Tetrahedron Lett. 1996;37:6603–6606.Jarowicki K, Kocienski P. J Chem Soc, Perkin Trans 1. 2001:2109–2135.Horita K, Yoshioka T, Tanaka T, Oikawa Y, Yonemitsu O. Tetrahedron. 1986;42:3021–3028.Tanaka, Oikawa Y, Hamada T, Yonemitsu O. Chem Pharm Bull. 1987;35:2209–2218.
- 28.For selected examples of cross-dehydrogenative coupling-type reactions, see: Ying BP, Trogden BG, Kohlman DT, Liang SX, Xu YC. Organic Letters. 2004;6:1523–1526. doi: 10.1021/ol036314j.Li Z, Li H, Guo X, Cao L, Yu R, Li H, Pan S. Organic Letters. 2008;10:803–805. doi: 10.1021/ol702934k.Zhang Y, Li CJ. J Am Chem Soc. 2006;128:4242–4243. doi: 10.1021/ja060050p.Zhang Y, Li CJ. Angew Chem Int Ed. 2006;45:1949–1952. doi: 10.1002/anie.200503255.Tsang ASK, Todd MH. Tetrahedron Letters. 2009;50:1199–1202.Tsang ASK, Jensen P, Hook JM, Hashmi ASK, Todd MH. Pure and Applied Chemistry. 2011;83:655–665.Liu L, Floreancig PE. Angew Chem Int Ed. 2010;49:3069–3072. doi: 10.1002/anie.201000033.Liu L, Floreancig PE. Angew Chem Int Ed. 2010;49:5894–5897. doi: 10.1002/anie.201002281.Peh G, Floreancig PE. Org Lett. 2012;14:5614–5617. doi: 10.1021/ol302744t.Tu W, Floreancig PE. Angew Chem Int Ed. 2009;48:4567–4571. doi: 10.1002/anie.200901489.Tu W, Liu L, Floreancig PE. Angew Chem Int Ed. 2008;47:4184–4187. doi: 10.1002/anie.200706002.Brizgys GJ, Jung HH, Floreancig PE. Chem Sci. 2012;3:438–442.Liu L, Floreancig PE. Org Lett. 2009;11:3152–3155. doi: 10.1021/ol901188q.Cui Y, Floreancig PE. Org Lett. 2012;14:1720–1723. doi: 10.1021/ol3002877.Han X, Floreancig PE. Org Lett. 2012;14:3808–3811. doi: 10.1021/ol301720u.Yu B, Jiang T, Li J, Su Y, Pan X, She X. Org Lett. 2009;11:3442–3445. doi: 10.1021/ol901291w.
- 29.Braude EA, Jackman LM, Linstead RP, Lowe G. J Chem Soc. 1960:3123–3132. [Google Scholar]
- 30.For representative examples of oxidative functionalization of C–H bonds, see: Aubry S, Pellet-Rostaing S, Lemaire M. Eur J Org Chem. 2007:5212–5225.Marcantoni E, Petrini M, Profeta R. Tetrahedron Lett. 2004;45:2133–2136.Lehr RE, Kole PL, Tschappat KD. Tetrahedron Lett. 1986;27:1649–1652.Oikawa Y, Yonemitsu O. J Org Chem. 1977;42:1213–1216.Tomioka K, Kubota Y, Koga K. J Chem Soc, Chem Commun. 1989:1622–1624.Tomioka K, Kubota Y, Koga K. Tetrahedron. 1993;49:1891–1900.Cheng D, Bao W. J Org Chem. 2008;73:6881–6883. doi: 10.1021/jo8010039.Cheng D, Bao W. Adv Synth Catal. 2008;350:1263–1266.Benfatti F, Capdevila MG, Zoli L, Benedetto E, Cozzi PG. Chem Commun. 2009:5919–5921. doi: 10.1039/b910185c.Li Y, Bao W. Adv Synth Catal. 2009;351:865–868.Jin J, Li Y, Wang Z-j, Qian W-x, Bao W-l. Eur J Org Chem. 2010;2010:1235–1238.Damu GLV, Jon Paul Selvam J, Venkata Rao C, Venkateswarlu Y. Tetrahedron Lett. 2009;50:6154–6158.Ramesh D, Ramulu U, Rajaram S, Prabhakar P, Venkateswarlu Y. Tetrahedron Lett. 2010;51:4898–4903.
- 31.Crabtree and Batista have reported the isolation of a similar quinol ether formed between DDQ and toluene, and report its subsequent transformations: Batista VS, Crabtree RH, Konezny SJ, Luca OR, Praetorius JM. New J Chem. 2012;36:1141–1144.
- 32.The formation of covalent quinone/substrate adducts have been observed in many instances, however typically as undesired side-products rather than as intermediates. For selected examples, see ref 29 and: Kiefer EF, Lutz FE. J Org Chem. 1972;37:1519–1522.Foster R, Horman I. J Chem Soc B: Phys Org. 1966:1049–1053.Lutz FE, Kiefer EF. J Chem Soc D: Chem, Commun. 1970:1722b–1723.Lemaire M, Doussot J, Guy A. Chemistry Lett. 1988;17:1581–1584.Wallace DJ, Gibb AD, Cottrell IF, Kennedy DJ, Brands KMJ, Dolling UH. Synthesis. 2001:1784–1789.Utley JHP, Rozenberg GG. J Appl Electrochem. 2003;33:525–532.
- 33.Bhattacharya A, DiMichele LM, Dolling UH, Grabowski EJJ, Grenda VJ. J Org Chem. 1989;54:6118–6120.Matsuura S, Iinuma M, Ishikawa K, Kagei K. Chem Pharm Bull. 1978;26:305–306.Jung ME, Pan YG, Rathke MW, Sullivan DF, Woodbury RP. J Org Chem. 1977;42:3961–3963.Ryu I, Murai S, Hatayama Y, Sonoda N. Tetrahedron Lett. 1978;19:3455–3458.Fevig TL, Elliott RL, Curran DP. J Am Chem Soc. 1988;110:5064–5067.Fleming I, Paterson I. Synthesis. 1979;1979:736–738.. See related: Heathcock CH, Mahaim C, Schlecht MF, Utawanit T. J Org Chem. 1984;49:3264–3274.
- 34.Extensive historical effort was directed towards the dehydrogenation of steroids. See ref 14a and references therein. For selected examples, see: Turner AB, Ringold HJ. J Chem Soc C: Organic. 1967:1720–1730.Westerhof P, Hartog J. Recueil Trav Chim Pays-Bas. 1965;84:918–931.Bagli JF, Morand PF, Wiesner K, Gaudry R. Tetrahedron Lett. 1964;5:387–389.Pradhan SK, Ringold HJ. J Org Chem. 1964;29:601–604.Edwards JA, Calzada MC, Ibáñez LC, Rivera MEC, Urquiza R, Cardona L, Orr JC, Bowers A. J Org Chem. 1964;29:3481–3486.Edwards JA, Orr C, Bowers A. J Org Chem. 1962;27:3378–3384.
- 35.Bhattacharya A, DiMichele LM, Dolling UH, Douglas AW, Grabowski EJJ. J Am Chem Soc. 1988;110:3318–3319.. For detailed discussion, see Williams JM. The Art of Process Chemistry. Wiley-VCH Verlag GmbH & Co KGaA; 2010. pp. 77–115.
- 36.Guo X, Mayr H. J Am Chem Soc. 2013;135:12377–12387. doi: 10.1021/ja405890d.For earlier studies, see: Fukuzumi S, Fujita M, Matsubayashi G, Otera J. Chem Lett. 1993:1451–1454.
- 37.Guo X, Mayr H. J Am Chem Soc. 2014;136:11499–11512. doi: 10.1021/ja505613b. [DOI] [PubMed] [Google Scholar]
- 38.For selected examples of methods for the regeneration of DDQ from DDQH2, see: Walker D, Waugh TD. J Org Chem. 1965;30:3240–3240.Newman MS, Khanna VK. Org Prep Proc Int. 1985;17:422–423.Kim KH, Grunewald GL. Org Prep Proc Int. 1976;8:141–143.Scott JW, Parrish DR, Bizzarro FT. Org Prep Proc Int. 1977;9:91–94.
- 39.While DDQ decomposes in water, it is stable in aqueous mineral acids: see ref 38a.
- 40.Cacchi S, La Torre F, Paolucci G. Synthesis. 1978;1978:848–849. [Google Scholar]
- 41.Cosner CC, Cabrera PJ, Byrd KM, Thomas AMA, Helquist P. Org Lett. 2011;13:2071–2073. doi: 10.1021/ol200441g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chandrasekhar S, Sumithra G, Yadav JS. Tetrahedron Lett. 1996;37:1645–1646. [Google Scholar]
- 43.Sharma GVM, Lavanya B, Mahalingam AK, Krishna PR. Tetrahedron Lett. 2000;41:10323–10326. [Google Scholar]
- 44.Liu L, Floreancig PE. Org Lett. 2010;12:4686–4689. doi: 10.1021/ol102078v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ghosh AK, Cheng X. Org Lett. 2011;13:4108–4111. doi: 10.1021/ol201626h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ghosh AK, Cheng X. Tetrahedron Lett. 2012;53:2568–2570. doi: 10.1016/j.tetlet.2012.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yi H, Liu Q, Liu J, Zeng Z, Yang Y, Lei A. ChemSusChem. 2012;5:2143–2146. doi: 10.1002/cssc.201200458. [DOI] [PubMed] [Google Scholar]
- 48.Muramatsu W, Nakano K, Li C-J. Org Lett. 2013;15:3650–3653. doi: 10.1021/ol401534g. [DOI] [PubMed] [Google Scholar]
- 49.Muramatsu W, Nakano K. Org Lett. 2014;16:2042–2045. doi: 10.1021/ol5006399. [DOI] [PubMed] [Google Scholar]
- 50.Brinker UH, Tyner M, III, Jones WM. Synthesis. 1975;1975:671. [Google Scholar]
- 51.Francke R, Little RD. Chem Soc Rev. 2014;43:2492–2521. doi: 10.1039/c3cs60464k.. See also: Steckhan E. Angew Chem Int Ed. 1986;25:683–701.Steckhan E. In: Electrochemistry I. Steckhan E, editor. Vol. 142. Springer; Berlin Heidelberg: 1987. pp. 1–69.
- 52.Chiba K, Arakawa T, Tada M. Chem Commun. 1996:1763–17. [Google Scholar]
- 53.Chiba K, Arakawa T, Tada M. J Chem Soc, Perkin Trans 1. 1998:2939–2942. [Google Scholar]
- 54.Utley JHP, Rozenberg GG. Tetrahedron. 2002;58:5251–5265. [Google Scholar]
- 55.For a discussion of synthetic oxidase-type aerobic oxidation reactions, see ref. 10 and the following leading references: Stahl SS. Angew Chem Int Ed. 2004;43:3400–3420. doi: 10.1002/anie.200300630.Stahl SS. Science. 2005;309:1824–1826. doi: 10.1126/science.1114666.Wendlandt AE, Suess AM, Stahl SS. Angew Chem Int Ed. 2011;50:11062–11087. doi: 10.1002/anie.201103945.
- 56.For an example proposing the direct aerobic reoxidation of p-chloranil, see: Fan Y, An Z, Pan X, Liu X, Bao X. Chem Commun. 2009:7488–7490. doi: 10.1039/b915412d.
- 57.For selected examples, see: Sakamoto T, Yonehara H, Pac C. J Org Chem. 1997;62:3194–3199. doi: 10.1021/jo961377j.Sain B, Murthy PS, Rao TV, Rao TSRP, Joshi GC. Tetrahedron Lett. 1994;35:5083–5084.Sakai K, Tsubomura T, Matsumoto K. Inorg Chim Acta. 1995;234:157–161.Hwang DR, Hwang DR, Chu CY, Wang SK, Uang BJ. Synlett. 1999;1999:77–78.Fujibayashi S, Nakayama K, Nishiyama Y, Ishii Y. Chemistry Lett. 1994;23:1345–1348.Kim S, Kim D, Park J. Adv Synth Catal. 2009;351:2573–2578.
- 58.For selected examples, see: (Sakamoto H, Funabiki T, Yoshida S, Tarama K. Bull Chem Soc Jpn. 1979;52:2760–2765.Tsuruya S, Yanai S, Masai M. Inorg Chem. 1986;25:141–146. doi: 10.1021/ic00227a041.Sakata K, Kikutake T, Shigaki Y, Hashimoto M, Iyehara Ogawa H, Kato Y. Inorganica Chimica Acta. 1988;144:1–3.Simándi LI, Simándi TM, May Z, Besenyei G. Coord Chem Rev. 2003;245:85–93.
- 59.Miyamura H, Shiramizu M, Matsubara R, Kobayashi S. Chemistry Lett. 2008;37:360–361. [Google Scholar]
- 60.Miyamura H, Shiramizu M, Matsubara R, Kobayashi S. Angew Chem Int Ed. 2008;47:8093–8095. doi: 10.1002/anie.200802192. [DOI] [PubMed] [Google Scholar]
- 61.Miyamura H, Maehata K, Kobayashi S. Chem Commun. 2010;46:8052–8054. doi: 10.1039/c0cc02865g. [DOI] [PubMed] [Google Scholar]
- 62.Neumann R, Khenkin AM, Vigdergauz I. Chem – Eur J. 2000;6:875–882. doi: 10.1002/(sici)1521-3765(20000303)6:5<875::aid-chem875>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 63.a) Bosch E, Rathore R, Kochi JK. J Org Chem. 1994;59:2529–2536. [Google Scholar]; b) Rathore R, Bosch E, Kochi JK. Tetrahedron Lett. 1994;35:1335–1338. [Google Scholar]
- 64.NO/NO2 redox couples have also been used to promote the aerobic reoxidation of hydroquinones to quinones in Pd-catalyzed oxidation reactions. See: An Z, Pan X, Liu X, Han X, Bao X. J Am Chem Soc. 2006;128:16028–16029. doi: 10.1021/ja0647912.Zhang G, Xie X, Wang Y, Wen X, Zhao Y, Ding C. Org Biomol Chem. 2013;11:2947–2950. doi: 10.1039/c3ob40277k.
- 65.a) Zhang W, Ma H, Zhou L, Sun Z, Du Z, Miao H, Xu J. Molecules. 2008;13:3236–3245. doi: 10.3390/molecules13123236. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang W, Ma H, Zhou L, Miao H, Xu J. Chin J Catal. 2009;30:86–88. [Google Scholar]
- 66.Shen Z, Dai J, Xiong J, He X, Mo W, Hu B, Sun N, Hu X. Adv Synth Catal. 2011;353:3031–3038. [Google Scholar]
- 67.Wang L, Li J, Yang H, Lv Y, Gao S. J Org Chem. 2011;77:790–794. doi: 10.1021/jo202301s. [DOI] [PubMed] [Google Scholar]
- 68.Lancefield CS, Ojo OS, Tran F, Westwood NJ. Angew Chem Int Ed. 2015;54:258–262. doi: 10.1002/anie.201409408. [DOI] [PubMed] [Google Scholar]
- 69.For related earlier methods for lignin oxidation using photochemical DDQ/NOx and AcNH-TEMPO/NOx catalyst systems, see: Walsh K, Sneddon HF, Moody CJ. Org Lett. 2014;16:5224–5227. doi: 10.1021/ol502664f.Rahimi A, Azarpira A, Kim H, Ralph J, Stahl SS. J Am Chem Soc. 2013;135:6415–6418. doi: 10.1021/ja401793n.
- 70.a) Qiu C, Jin L, Huang Z, Tang Z, Lei A, Shen Z, Sun N, Mo W, Hu B, Hu X. ChemCatChem. 2012;4:76–80. [Google Scholar]; b) Tong X, Sun Y, Yan Y, Luo X, Liu J, Wu Z. J Mol Catal A: Chem. 2014;391:1–6. [Google Scholar]
- 71.Shen Z, Sheng L, Zhang X, Mo W, Hu B, Sun N, Hu X. Tetrahedron Lett. 2013;54:1579–1583. [Google Scholar]
- 72.Walsh K, Sneddon HF, Moody CJ. Tetrahedron. 2014;70:7380–7387. [Google Scholar]
- 73.Cheng D, Yuan K, Xu X, Yan J. Tetrahedron Lett. 2015;56:1641–1644. [Google Scholar]
- 74.Prabhu and coworkers reported cross-dehydrogenative coupling reactions to form C–C and C–P bonds with more-traditional N-aryl tetrahydroisoquinoline substrates by using catalytic quantities of DDQ and AIBN under aerobic conditions. See: Alagiri K, Devadig P, Prabhu KR. Chem – Eur J. 2012;18:5160–5164. doi: 10.1002/chem.201200100.
- 75.Dehydrogenation reactions to afford porphyrins have been carried out with catalytic DDQ or p-chloranil in combination with Fe(pc) co-catalysts under aerobic conditions. See: Ravikanth M, Achim C, Tyhonas JS, Münck E, Lindsey JS. J Porphyrins Phthalocyanines. 1997;1:385–394.Lindsey JS, MacCrum KA, Tyhonas JS, Chuang YY. J Org Chem. 1994;59:579–587.Zaidi SHH, Fico RM, Lindsey JS. Org Proc Res Dev. 2006;10:118–134.
- 76.Anthony C. Biochem J. 1996;320:697–711. doi: 10.1042/bj3200697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Janes S, Mu D, Wemmer D, Smith A, Kaur S, Maltby D, Burlingame A, Klinman J. Science. 1990;248:981–987. doi: 10.1126/science.2111581. [DOI] [PubMed] [Google Scholar]
- 78.Wang SX, Mure M, Medzihradszky KF, Burlingame AL, Brown DE, Dooley DM, Smith AJ, Kagan HM, Klinman JP. Science. 1996;273:1078–1084. doi: 10.1126/science.273.5278.1078. [DOI] [PubMed] [Google Scholar]
- 79.McIntire W, Wemmer D, Chistoserdov A, Lidstrom M. Science. 1991;252:817–824. doi: 10.1126/science.2028257. [DOI] [PubMed] [Google Scholar]
- 80.In contrast to CAOs, which are aerobic enzymes in nature, TTQ and PQQ use the enzymes Amicyanin, Heme C, Cytochrome C and Ubiquinone as the terminal electron acceptors for substrate oxidation. Gray KA, Davidson VL, Knaff DB. J Biol Chem. 1988;263:13987–13990.Chen L, Durley R, Mathews F, Davidson V. Science. 1994;264:86–90. doi: 10.1126/science.8140419.
- 81.For an authoritative review on o-quinone cofactor self-processing, see: Klinman JP, Bonnot F. Chem Rev. 2014;114:4343–4365. doi: 10.1021/cr400475g.
- 82.a) Mure M, Mills SA, Klinman JP. Biochem. 2002;41:9269–9278. doi: 10.1021/bi020246b. [DOI] [PubMed] [Google Scholar]; b) Mure M. Acc Chem Res. 2004;37:131–139. doi: 10.1021/ar9703342. [DOI] [PubMed] [Google Scholar]
- 83.a) Mure M, Klinman JP. J Am Chem Soc. 1995;117:8698–8706. [Google Scholar]; b) Mure M, Klinman JP. J Am Chem Soc. 1995;117:8707–8718. [Google Scholar]
- 84.a) Lee Y, Sayre LM. J Am Chem Soc. 1995;117:3096–3105. [Google Scholar]; b) Lee Y, Sayre LM. J Am Chem Soc. 1995;117:11823–11828. [Google Scholar]; c) Ling KQ, Kim J, Sayre LM. J Am Chem Soc. 2001;123:9606–9611. doi: 10.1021/ja011141j. [DOI] [PubMed] [Google Scholar]
- 85.a) Itoh S, Takada N, Haranou S, Ando T, Komatsu M, Ohshiro Y, Fukuzumi S. J Org Chem. 1996;61:8967–8974. doi: 10.1021/jo961705f. [DOI] [PubMed] [Google Scholar]; b) Itoh S, Takada N, Ando T, Haranou S, Huang X, Uenoyama Y, Ohshiro Y, Komatsu M, Fukuzumi S. J Org Chem. 1997;62:5898–5907. doi: 10.1021/jo961705f. [DOI] [PubMed] [Google Scholar]; c) Itoh S, Taniguchi M, Fukuzumi S. Chem Commun. 2000:329–330. [Google Scholar]; d) Itoh S, Ogino M, Haranou S, Terasaka T, Ando T, Komatsu M, Ohshiro Y, Fukuzumi S, Kano K. J Am Chem Soc. 1995;117:1485–1493. [Google Scholar]
- 86.a) Davidson VL. In: In Advances in Protein Chemistry. Judith P, Klinman JED, editors. Vol. 58. Academic Press; 2001. p. 95. [DOI] [PubMed] [Google Scholar]; b) Anthony C, Ghosh M, Blake CC. Biochem J. 1994;304:665–674. doi: 10.1042/bj3040665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.For selected mechanistic studies: Forrest HS, Salisbury SA, Kilty CG. Biochem Biophys Res Commun. 1980;97:248–251. doi: 10.1016/s0006-291x(80)80161-6.Itoh S, Kawakami H, Fukuzumi S. Biochem. 1998;37:6562–6571. doi: 10.1021/bi9800092.Frank J, van Krimpen SH, Verwiel PEJ, Jongejan JA, Mulder AC, Duine JA. Eur J Biochem. 1989;184:187–195. doi: 10.1111/j.1432-1033.1989.tb15006.x.. For computational studies: Zheng YJ, Bruice TC. Proc Natl Acad Sci. 1997;94:11881–11886. doi: 10.1073/pnas.94.22.11881.Andres J, Moliner V, Domingo LR, Picher MT, Krechl J. J Am Chem Soc. 1995;117:8807–8815.Andrés J, Moliner V, Krechl J. Bioorg Chem. 1994;22:58–71.Oubrie A. Biochim Biophys Acta - Proteins and Proteomics. 2003;1647:143. doi: 10.1016/s1570-9639(03)00087-6.
- 88.Itoh S, Kawakami H, Fukuzumi S. J Am Chem Soc. 1997;119:439–440.. See also: Itoh S, Kawakami H, Fukuzumi S. Biochem. 1998;37:6562–6571. doi: 10.1021/bi9800092.
- 89.Itoh, Ohshiro and coworkers additionally demonstrated the PQQ-mediated oxidation of simple sugars to the corresponding carboxylic acids (eg glucose to gluconic acid), modeling the activity of PQQ quinoenzyme glucose dehydrogenase: Itoh S, Mure M, Ohshiro Y. J Chem Soc, Chem Commun. 1987:1580–1581.
- 90.Sasaki K, Kashimura T, Ohura M, Ohsaki Y, Ohta N. J Electrochem Soc. 1990;137:2437–2443. [Google Scholar]
- 91.Duine JA, Frank Jzn J, Verwiel PEJ. Eur J Biochem. 1980;108:187–192. doi: 10.1111/j.1432-1033.1980.tb04711.x. [DOI] [PubMed] [Google Scholar]
- 92.Prior to the discovery of Topaquinone it was thought that PQQ was the cofactor associated with the copper amine oxidase family of enzymes. Accordingly PQQ was studied in the context of amine oxidation as well as for alcohol oxidation.
- 93.Eckert TS, Bruice TC. J Am Chem Soc. 1983;105:4431–4441. [Google Scholar]
- 94.Dekker RH, Duine JA, Frank J, Verwiel JPEJ, Westerling J. Eur J Biochem. 1982;125:69–73. doi: 10.1111/j.1432-1033.1982.tb06652.x. [DOI] [PubMed] [Google Scholar]
- 95.Sleath PR, Noar JB, Eberlein GA, Bruice TC. J Am Chem Soc. 1985;107:3328–3338. [Google Scholar]
- 96.Eckert TS, Bruice TC, Gainor JA, Weinreb SM. Proc Natl Acad Sci. 1982;79:2533–2536. doi: 10.1073/pnas.79.8.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rodriguez EJ, Bruice TC. J Am Chem Soc. 1989;111:7947–7956. [Google Scholar]
- 98.a) Itoh S, Kitamura Y, Ohshiro Y, Agawa T. Bull Chem Soc Jpn. 1986;59:1907–1910. [Google Scholar]; b) Ohshiro Y, Itoh S. Bioorg Chem. 1991;19:169–189. [Google Scholar]
- 99.Reaction end-product analyses of this kind are complicated by redox scrambling of the quinone and hydroquinone species, and are therefore difficult to interpret. Interconversion of aminoquinol and quinol catalyzed by the presence of even a small amount of quinone occurs even under anaerobic conditions, and the ratios of products are found to depend significantly on reaction conditions. For additional examples, see refs 84b, 97b, and 98b.
- 100.Largeron M, Fleury M-B. Science. 2013;339:43–44. doi: 10.1126/science.1232220. [DOI] [PubMed] [Google Scholar]
- 101.Corey EJ, Achiwa K. J Am Chem Soc. 1969;91:1429–1432. [Google Scholar]
- 102.Wanner and Koomen employed this quinone-mediated amine oxidation method to prepare diverse alkaloid natural products, including anabasine, lupinamine, and sparteine: Wanner MJ, Koomen G-J. J Org Chem. 1996;61:5581–5586.
- 103.Similar benzoxazole adducts have been observed as decomposition products in enzymatic systems: Lee Y, Shepard E, Smith J, Dooley DM, Sayre LM. Biochem. 2000;40:822–829. doi: 10.1021/bi002118y.
- 104.Klein RFX, Bargas LM, Horak V. J Org Chem. 1988;53:5994–5998. [Google Scholar]
- 105.Ohshiro Y, Itoh S, Kurokawa K, Kato J, Hirao T, Agawa T. Tetrahedron Lett. 1983;24:3465–3468. [Google Scholar]
- 106.In a subsequent report, Itoh and coworkers extended this reaction to the oxidative decarboxylation of amino acids and oxidative dealdolation of β-hydroxy amino acids to give aldehydes. See: Itoh S, Kato N, Ohshiro Y, Agawa T. Tetrahedron Lett. 1984;25:4753–4756.Mure M, Suzuki A, Itoh S, Ohshiro Y. J Chem Soc, Chem Commun. 1990:1608–1611.. Aerobic PQQ-catalyzed oxidation of thiols to disulfide is also achieved under similar conditions: Itoh S, Kato N, Ohshiro Y, Agawa T. Chem Lett. 1985;14:135–136.
- 107.a) Largeron M, Fleury M-B. J Org Chem. 2000;65:8874–8881. doi: 10.1021/jo000478l. [DOI] [PubMed] [Google Scholar]; b) Largeron M, Neudorffer A, Fleury M-B. Angew Chem Int Ed. 2003;42:1026–1029. doi: 10.1002/anie.200390263. [DOI] [PubMed] [Google Scholar]; c) Largeron M, Chiaroni A, Fleury M-B. Chem – Eur J. 2008;14:996–1003. doi: 10.1002/chem.200700876. [DOI] [PubMed] [Google Scholar]; d) Largeron M, Fleury M-B, Strolin Benedetti M. Org Biomol Chem. 2010;8:3796–3800. doi: 10.1039/c004501b. [DOI] [PubMed] [Google Scholar]
- 108.a) Largeron M, Fleury M-B. Org Lett. 2009;11:883–886. doi: 10.1021/ol802885b. [DOI] [PubMed] [Google Scholar]; b) Largeron M. Electrochimica Acta. 2009;54:5109–5115. [Google Scholar]
- 109.Largeron M, Fleury M-B. Angew Chem Int Ed. 2012;51:5409–5412. doi: 10.1002/anie.201200587. [DOI] [PubMed] [Google Scholar]
- 110.Wendlandt AE, Stahl SS. Org Lett. 2012;14:2850–2853. doi: 10.1021/ol301095j. [DOI] [PubMed] [Google Scholar]
- 111.Yamamoto K. Encyclopedia of Reagents for Organic Synthesis. John Wiley & Sons, Ltd; 2001. [Google Scholar]
- 112.Qin Y, Zhang L, Lv J, Luo S, Cheng J-P. Org Lett. 2015;17:1469–1472. doi: 10.1021/acs.orglett.5b00351. [DOI] [PubMed] [Google Scholar]
- 113.Lee Y, Huang H, Sayre LM. J Am Chem Soc. 1996;118:7241–7242.Lee Y, Ling KQ, Lu X, Silverman RB, Shepard EM, Dooley DM, Sayre LM. J Am Chem Soc. 2002;124:12135–12143. doi: 10.1021/ja0205434.Zhang Y, Ran C, Zhou G, Sayre LM. Bioorg Med Chem. 2007;15:1868–1877. doi: 10.1016/j.bmc.2006.11.025.. See also ref 93.
- 114.a) Yuan H, Yoo W-J, Miyamura H, Kobayashi S. J Am Chem Soc. 2012;134:13970–13973. doi: 10.1021/ja306934b. [DOI] [PubMed] [Google Scholar]; b) Yuan H, Yoo W-J, Miyamura H, Kobayashi S. Adv Synth Catal. 2012;354:2899–2904. [Google Scholar]
- 115.Jawale DV, Gravel E, Shah N, Dauvois V, Li H, Namboothiri INN, Doris E. Chem – Eur J. 2015;21:7039–7042. doi: 10.1002/chem.201500148. [DOI] [PubMed] [Google Scholar]
- 116.Wendlandt AE, Stahl SS. J Am Chem Soc. 2014;136:506–512. doi: 10.1021/ja411692v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wendlandt AE, Stahl SS. J Am Chem Soc. 2014;136:11910–11913. doi: 10.1021/ja506546w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.For selected reports of NADH oxidation, see: Goss CA, Abruna HD. Inorg Chem. 1985;24:4263–4267.Wu Q, Maskus M, Pariente F, Tobalina F, Fernández VM, Lorenzo E, Abruña HD. Anal Chem. 1996;68:3688–3696.Hilt G, Jarbawi T, Heineman WR, Steckhan E. Chem – Eur J. 1997;3:79–88.Pinczewska A, Sosna M, Bloodworth S, Kilburn JD, Bartlett PN. J Am Chem Soc. 2012;134:18022–18033. doi: 10.1021/ja307390x.Yokoyama K, Ueda Y, Nakamura N, Ohno H. Chemistry Lett. 2005;34:1282–1283.Catalin Popescu I, Domínguez E, Narváez A, Pavlov V, Katakis I. J Electroanal Chem. 1999;464:208–214.
- 119.O-quinones as a class have been recognized as particularly effective for electrocatalytic NADH oxidation. For examples, see: Tse DC-S, Kuwana T. Anal Chem. 1978;50:1315–1318.Jaegfeldt H, Kuwana T, Johansson G. J Am Chem Soc. 1983;105:1805–1814.Jaegfeldt H, Torstensson ABC, Gorton LGO, Johansson G. Anal Chem. 1981;53:1979–1982.Huck H, Schmidt H-L. Angew Chem Int Ed. 1981;20:402–403.
- 120.For selected examples, see: Itoh S, Kinugawa M, Mita N, Ohshiro Y. J Chem Soc, Chem Commun. 1989:694–695.Rivera N, Colón Y, Guadalupe AR. Bioelectrochem Bioenergetics. 1994;34:169–175.Itoh S, Mita N, Ohshiro Y. Chem Lett. 1990;19:1949–1952.Hilt G, Lewall B, Montero G, Utley JHP, Steckhan E. Liebigs Annalen. 1997;1997:2289–2296.Hilt G, Steckhan E. J Chem Soc, Chem Commun. 1993:1706–1707.
- 121.For a review of biosensors based on NAD(P)-dependent dehydrogenase enzymes: Lobo MJ, Miranda AJ, Tuñón P. Electroanalysis. 1997;9:191–202.
- 122.For selected examples, see: Santiago MEB, Daniel GA, David A, Casañas B, Hernández G, Guadalupe AR, Colón JL. Electroanalysis. 2010;22:1097–1105.Santiago MEB, Vélez MM, Borrero S, Díaz A, Casillas CA, Hofmann C, Guadalupe AR, Colón JL. Electroanalysis. 2006;18:559–572. doi: 10.1002/elan.200503432.Lorenzo E, Pariente F, Hernàndez L, Tobalina F, Darder M, Wu Q, Maskus M, Abruña HD. Biosensors and Bioelectronics. 1998;13:319–332. doi: 10.1016/s0956-5663(97)00138-3.Tobalina F, Pariente F, Hernández L, Abruña HD, Lorenzo E. Analytica Chimica Acta. 1999;395:17–26.Hedenmo M, Narvaez A, Dominguez E, Katakis I. Analyst. 1996;121:1891–1895.