

Abstract
Chronic pain constitutes a challenge for the scientific community and a significant economic and social cost for modern societies. Given the failure of current drugs to effectively treat chronic pain, which are based on suppressing aberrant neuronal excitability, we propose in this review an integrated approach that views pain not solely originating from neuronal activation but also the result of a complex interaction between the nervous, immune, and endocrine systems. Pain assessment must also extend beyond measures of behavioural responses to noxious stimuli to a more developmentally informed assessment given the significant plasticity of the nociceptive system during the neonatal period. Finally integrating the concept of perinatal programming into the pain management field is a necessary step to develop and target interventions to reduce the suffering associated with chronic pain. We present clinical and animal findings from our laboratory (and others) demonstrating the importance of the microbial and relational environment in programming pain responsiveness later in life via action on hypothalamo-pituitary adrenal (HPA) axis activity, peripheral and central immune system, spinal and supraspinal mechanisms, and the autonomic nervous system.
Keywords: Pain, Psychoneuroimmunology, Neuroendocrinology, HPA axis, LPS
Background
Physiological pain plays an essential and primordial function in our survival. It signals potential damage to the body and produces a wide range of sophisticated actions to prevent this damage. The perception of noxious stimuli is typically initiated by the activation of peripheral receptors (i.e. nociceptors) which signal the spinal dorsal horn and supraspinal neurons of any potentially tissue-damaging stimuli. In pathological conditions, however, failure to sense pain such as in the case of congenital insensitivity often lead to self mutilation, bone fractures, joint deformities, amputations, and even early death, underscoring the importance of the physiological, protective role of nociceptive pain [1]. Chronic pain, the unbearable pain that persists long after an injury has healed, can drive the individual to immobility, psychological distress, depression, and disruption of family relationships, and potentially loss of employment. Chronic pain is common in developed countries. Prevalence rates of 16–22 % have been reported from epidemiological surveys [2–4]. One recent study lists chronic back pain (along with depression) in the top 10 causes of morbidity in over 180 countries [5]. Chronic pain incurs a multi-billion dollar cost to most developed western countries [6, 7]. Despite mounting evidence regarding the complexity of the nature of chronic pain there remains a general resistance [8] to the understanding that the nature of chronic pain is not primarily a matter of nociception (i.e. a noxious stimulus generating neural impulse), despite ample evidence of the multidimensional nature of chronic pain [9].
Pharmaceutical treatments, still our primary treatment modality, have a 25–30 % placebo rate, making therapeutic benefits difficult to establish. Some of the drugs such as the use of opioids (i.e. morphine), which can act on opioid-receptor-expressing neurons in brain areas that control pain (i.e. PAG) are effective but have a very narrow therapeutic window and in most of the time the dose needs to be limited to avoid side effects or analgesic tolerance which occurs upon repeated administration of opioids requiring increased dosage to maintain satisfactory level of pain control. Why in the 21st century is pain still a stubborn condition? What are the limitations of the current therapeutic approaches? Are we understanding and managing pain in its full complexity?
While our understanding of the complex mechanisms involved in the development and maintenance of chronic pain has increased dramatically in recent decades, treatments in various modalities continue to be based on a reductionist approach that understanding the precise nociceptive mechanisms that cause pain, will result in more targeted and hence more effective treatments. Given the myriad of neuroimmunoendocrine links now understood to modulate chronic pain, focusing on targeted specific mechanisms may be missing the bigger picture. Furthermore, given that pain has also a cognitive, aversive component there is a need to target the cognitive/emotional aspect of pain not only the sensory discriminative component.
From Galen to the gate-control theory developed by Melzack and Wall, pain has been viewed either as a separate, independent sensation, an emotion, or a result from activation of nociceptors, spinal and supraspinal pathways. This review proposes a shift in conceptualising the problem of chronic pain away from a dichotomous standpoint to an integrative view which captures both the sensory and the emotional-cognitive in a framework that emphasises the neuroendocrine to immune communication pathways and the relevance of early premorbid events as predictors.
Specifically we focus on the early life environment and its ability to program the organism’s physiological, affective and behavioural responses in later life [10–14]. Almost invariably, randomised control trials (RCTs) commence their assessment of pain by assessing the baseline values from the time the pain started. There is an implicit assumption, that prior to the onset of pain, “patients” are physiologically naïve and functionally equivalent—though there is some acknowledgement that their psychosocial history may differ. The thesis proposed here is that a neurodevelopmental understanding is required to account for the individual variability in response to the initiating noxious insult/stimulus as well as to subsequent treatments. We argue that not taking account of the premorbid physiological milieu may be limiting efforts to develop more tailored and more effective treatment interventions. In short, chronic pain patients may be differentially predisposed to recovering (or not recovering) from a physical injury, infection or trauma because of their premorbid psychoneuroimmunological status.
The traditional theory of pain
A brief historical overview
Aristotle (384–322 BC) considered pain to be an emotion. Later the Greek physician Galen (AD 130–201) using the experimental scientific method disagreed with Aristotle’s view and considered pain to be a sensation and the brain an organ for feeling. Avicenna (AD 980–1037) a renowned Persian philosopher, who is considered the father of modern medicine, argued that pain can be dissociated from touch. During the mid-1900s several studies performed by imminent Scottish and English neurologists demonstrated that hemi-section of the spinal cord resulted in a loss of pain sensation. The work by Blix and Goldcheider in 1884 and of Von Frey in 1897 demonstrated that different sensory experiences (i.e. pressure, cold, heat, pain) evoke the activation of different nerve endings. The terms noxious and nociception appears with Sherrington in 1906 who considered that noxious or painful events result in the activation of sense organs and this in turn produces pain [15]. In the 1930s, several studies by Bishop and Gasser demonstrated that noxious sensations are conveyed by two types of nerve fibers, namely Aδ and C [16]. It was with the development of the gate-control theory by Ronald Melzack and Patrick Wall in 1965 that the representation of pain as a unidirectional pathway from the site of injury to the sensation of pain was challenged. Instead pain was viewed as a result of supraspinal (brain) activity [17]. The development of new imaging techniques such as positron emission tomography (PET) and functional magnetic resonance imaging (fMRI) have revealed that pain results from activation of a number of brain regions such as the amygdala, insula or the anterior cingulate cortex.
Taken together, this brief historical outline illustrates the classical view that pain is an independent sensation, an emotion, a product of activation of nociceptors or spinal and supraspinal pathways. More recent conceptualisations of pain now view pain as a result of the complex interaction between the immune, nervous (both CNS and autonomic nervous system), and endocrine systems. Furthermore, a number of studies including findings from our laboratory clearly demonstrated that exposure to early life insults such as hindpaw inflammation, psychological/emotional or physical stress, as well as viral/bacterial infection can all lead to altered future pain responses [10, 18–24].
Overview of chronic pain
Chronic pain described as a continuing (unremitting) presence for periods longer than 3–6 months, has been defined as: “an unpleasant sensory and emotional experience associated with actual or potential tissue damage or described in terms of such damage” by the International Association of for the Study of Pain (IASP).
Our current understanding of chronic pain is that it falls into one of 4 categories—nociceptive, neuropathic, psychogenic or a combination of these. Nociceptive pain arises from tissue injury, which activates nociceptors (pain receptors) in the periphery that send signals to the central nervous system (CNS). Nociceptive pain can be inflammatory or musculoskeletal in origin. Neuropathic pain, arises from damage to nerves that results in an altered quality (e.g. sharp, burning) and intensity of pain signals. However in clinical practice, these divisions are not easily discriminated from each other and often overlap. Nevertheless, the diagnostic divisions assist with directing therapeutic options.
Indicative of the complexity of chronic pain is the phenomenon of sensitization. There are two types of sensitization. The first type refers to peripheral sensitization which occurs in response to the release of a plethora of inflammatory molecules such as histamine, prostaglandins, pro-inflammatory cytokines which sensitizes (or increase the excitability) of nociceptors by creating an “inflammatory soup” environment which enhances pain sensitivity by reducing the threshold of nociceptors activation. The second type is central sensitization which refers to exacerbation of pain sensation and can be caused by hyperexcitability of spinal dorsal horn neurons or a decrease in inhibition of spinal dorsal horn neurons. The two phenomena of hyperalgesia (an exaggerated response to a normally painful stimulus) and allodynia (an exaggerated response to a normally non-painful stimulus) are clinical markers used to detect the presence of sensitization. There is now accumulating evidence that chronic pain sensitivity is elevated across different modes (e.g. pressure, thermal, pricking) within chronic pain populations. This has led to the use of quantitative sensory testing (QST), that attempts to measure pain related parameters such as pain thresholds, temporal summation and pain duration using repeated stimuli in multiple locations on the body and with multiple modalities (e.g. thermal, chemical, mechanical) [25]. So to illustrate, pain threshold can be evaluated by measuring the time taken for the person to report the sensation of pain once a graduated stimulus (e.g. temperature/pressure) is applied.
DNIC (one type of QST) is considered to be a marker of endogenous (possibly opioid) analgesia, and reflects the capacity of the CNS to modulate (down-regulate) nociceptive signals [25]. DNIC comprises the inhibition of a nociceptive test stimulus when a distant, usually contralateral noxious conditioning stimulus has been pre-emptively applied. In other words the first painful stimulus “dulls” the intensity of the second (“test”) stimulus. This reduction in intensity of the second test stimulus, is understood to be mediated by bi-directional opioid-sensitive neurons in the spinal-medullary-spinal pathway [26]. This capacity has been found to be reduced in a range of idiopathic pain related conditions such as tempero-mandibular disorders, fibromyalgia, tension head-ache, migraine and irritable bowel syndrome [26] as well as in a healthy non-patient population [27].
However attention is drawn to the predictive capacity of DNIC in post-surgical pain, which suggests that at least part of the pain response is determined by physiological factors that precede the onset of pain. The significance of this should not be overlooked, as this observation suggests that pre-existing vulnerability to cope with the surgical-related trauma. Consistent with this line of evidence, are recent observations from imaging studies that indicate that functional connectivity between the medial prefrontal cortex, (mPFC), and nucleus accumbens, (NAc) are also predictive of those who progress from sub-acute (<4 months) to chronic (>6 months) lower back pain [28, 29]. Again note that the altered connectivity precedes the onset of acute pain, again implicating premorbid factors involved in the transition from (sub) acute to chronic pain. There has been a long-standing debate whether the features of chronic pain populations (e.g. demographics, education, gender, depression, sleeplessness, not coping, fear of pain, catastrophization, job satisfaction, helplessness) explain the acute to chronic pain transition. While these factors collectively account for 25 % of pain CP intensity, and 33 % of total disability [30], it is notable that for chronic back pain, the most common site of pain [2, 3], there are cognitive-emotional cortical regions that account for 70–80 % of the variance of pain intensity and duration [31]. In other words, higher order processing (including of emotional elements) appears to be more salient in the transition to and maintenance of chronic pain than is commonly appreciated. It is therefore of concern when treatment of chronic (low-back) pain is primarily focused on linking structural and mechanical causes (that predict only 17 % of the pain intensity variance and is unrelated to disability [31], to behavioural manifestations of the pain. This may be more easily remembered by the phrase—‘the gain in the pain happens mainly in the brain’.
Pain pathways
The conventional, traditional anatomical circuit of pain starts with the activation of nociceptors by multimodal noxious stimuli (i.e. thermal, physical, or chemical). There are two major classes of nociceptors. The first class includes thinly myelinated Aδ that are responsible for detecting well localized sharp, fast pain (speed of conduction: 5–30 m/s). The second category or C-fibers (unmyelinated; velocity less than 1 m/s) and belong to primary sensory neurons whose cell bodies are present in the dorsal root ganglia for the body or the trigeminal ganglion when innervating the face. Most Aδ fibres respond to heat as well as mechanical stimuli, whereas most C fibres are polymodal and respond to noxious thermal, mechanical, and chemical stimuli. Moreover, there are two main groups of C fibres: those that express the P2X3 purine receptor, the IB4-lectin-binding site and receptors for Glial Cell Derived Neurotrophic Factor (GDNF). This first group of fibres project exclusively to lamina II. The second group expresses peptides such as substance P and calcitonin gene related peptide (CGRP), but also expresses the receptor for nerve growth factor (NGF), neurotrophic tyrosine kinase receptor type 1 (TrkA). This group projects mainly to Lamina I. When activated, these peptidergic C fibres induce plasma extravasation and vasodilatation [30]. Many of the neurons in the marginal layer (Lamina I) respond exclusively to noxious stimuli and are called “nociceptive-specific neurons”, whereas some neurons respond to both noxious and non-noxious stimuli and are called “wide dynamic range” neurons. The substantia gelatinosa or Lamina II is made up exclusively of excitatory (glutamate) and inhibitory (gamma aminobutyric acid, GABA) interneurons. The nociceptive message, after being detected by peripheral nerves, is conveyed to neurons in the spinal cord that cross the spinothalamic tract (see Fig. 1) and make synapses in the thalamus. Once the message arrives at the somatosensory cortex, the conscious perception of pain occurs. The sensori-discriminative component of pain is integrated in the somatosensory cortex, whereas the emotional-affective component is coded by limbic structures such as the anterior cingulate cortex, amygdala, and insular cortex [31]. Other ascending pathways exist for the transmission of pain such as the one that initiates in the spinal cord and projects to the parabrachial nucleus, and then to the hypothalamus and amygdala; or the spinoreticular pathway that crosses the reticular formation in the medulla level, then thalamus and finally reaches the postcentral gyrus. In addition to these ascending pathways, the control of pain involves descending pathways also known as inhibitory descending control systems.
Fig. 1.
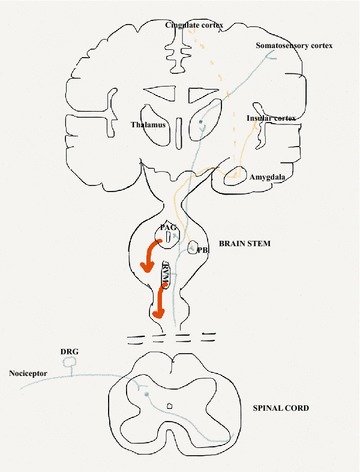
Anatomy of the pain pathways. Primary afferent nociceptors convey noxious information to neurons within the spinal dorsal horn of the spinal cord. These neurons project to the thalamus via the spinothalamic pathway and then to somatosensory cortex. This projection provides information about the location and nature of the stimulus (pathway in blue). The affective component is provided by projections to the cingulate and insular cortices via projections in the brainstem (parabrachial nucleus) and amygdala (pathway in orange). Descending inhibitory control pathways (Red line) originating in the PAG and RVM in the brainstem control the output of spinal nociceptive neurons in response to a noxious stimulus. PAG periaqueductal grey, PB parabrachial nucleus, RVM rostroventral medulla, DRG dorsal root ganglia
Figure 1 illustrates the complexity of the processing, as indicated by the number of tracts and regions involved. It would seem evident that there is no single anatomical region in the CNS that registers and processes nociception. This complexity would suggest processing far beyond that of mere auditing pain intensity and location. Also the involvement of the hypothalamus, thalamus, LC and parabrachial regions raises the possibility that all 3 stress-related neuroendocrine pathways listed above, being activated by pain. Finally activation of the prefrontal cortex (medial and lateral), the cingulate gyrus and insula cortex raises the distinct likelihood that higher order cognitive and emotional elements are involved with the processing of nociceptive stimuli.
In addition, recent studies using magnetic resonance spectroscopy have shown that there are differences in the neural processing between experimental pain within control subjects, and chronic pain in clinical populations. Afferent nociceptive information (via the thalamus) which projects to the somatosensory, insular and anterior cingulate cortices is more prominent in experimental pain conditions; by contrast projection to the prefrontal cortex (possibly via spinohypothalamic, spinoparabrachial and spinoreticular pathways) is more evident in chronic pain conditions [32]. Of note is that the prefrontal cortex (and thalamus) is associated with significant loss of grey matter in chronic back-pain patients [33]. What appears to be increasingly recognized is the importance of higher order processing of the nociceptive signals [34–36]. Therefore it is reasonable to examine conditions that have a pervasive and enduring impact upon the same higher order processing regions, prior to the onset of chronic pain. Childhood psychosocial stressors have such an impact upon adult neuroendocrine functioning and have been associated with the presence of chronic pain [37–42].
Why pain treatment is ineffective?
Sixty-plus years of intensive research in the pain field have led to the discovery of countless drugs including aspirin and pioneer findings such as the induction of analgesia by direct stimulating of the periaqueductal grey (PAG) in rodents [43] and humans [44]. Opioids are effective in attenuating the noxious stimulus. These molecules (whether endogenous or exogenous) exert their action through activation of opioid receptors which are μ, δ (DOR/OPDR), κ (KOR/OPRK1), and opioid-like receptors (ORL1/OPRL) [45] that are expressed peripherally by leukocytes or by primary sensory neurons but also can be expressed centrally in key nuclei involved in the descending pain inhibitory pathways such as PAG and rostroventral medulla (RVM). However the use of opioids in modulating pain has a downside, they can be addictive and enhance alcohol consumption. They can also induce opioid tolerance where repeated exposure to opioids decreases drug efficacy over time. The use of slow-release drug patches, local anaesthetics, or spinal epidurals, which can act either by blocking the transmission of noxious input to the brain or by attenuating the excitability of spinal and supraspinal neurons have brought relief to some patients but not all. Surprisingly drugs that were traditionally prescribed to control depression were shown to be effective in treating chronic pain and the same discovery applied for drugs that were originally designed to treat epilepsy and were found to relieve severe chronic pain. These drugs shifted the focus onto the brain. However despite the effectiveness of these drugs, treating chronic pain is still challenging for clinicians. We argue that one of the reasons resulting from this challenge is the complexity of the interaction between multiple physiological systems that modulate pain as well as the role of early life events in programming pain responses later in life. Early life exposure to adverse events whether it is of psychological, physiological, bacterial or viral nature has been shown to exert programming effects on the hypothalamus–pituitary–adrenal axis [10, 46–48], a key system that modulates the stress response to physiological or psychological stressor. Given that pain is an aversive, stressful experience, these early adverse events are also capable of altering future pain responses through action on the endocrine system. Below we present clinical and animal evidence that demonstrate the impact of early life insult on neuroendocrine and pain responses.
Perinatal exposure to adverse events alter the hypothalamus–pituitary–adrenal (HPA) axis later in life
The experience of stress, from an evolutionary perspective, plays a pivotal role in promoting survival of the organism. Perceiving an environmental stimulus as threatening and learning to avoid it is essential for survival. The HPA axis is the major neuroendocrine mediator of the stress response and is known to be susceptible to perinatal programming [49, 50]. The cascade of signalization in the HPA axis in response to stress begins with activation of a group of neurosecretory neurons in the paraventricular nucleus of the hypothalamus (PVN). These neurons secrete corticotropin-releasing hormone (CRH) and to a lesser extent arginine vasopressin (AVP) into the portal vessel system to reach the anterior pituitary where the synthesis of pro-opiomelanocortin (POMC) takes place and then processed to adrenocorticotropic hormone (ACTH) [51]. Subsequent increase in circulating ACTH then drives the synthesis of cortisol (in humans) and corticosterone (in rodents). Attenuation of the magnitude of the HPA stress response occurs through negative feedback of glucocorticoids on glucocorticoid (GR) and mineralocorticoid (MR) receptors expressed in neurons of the hippocampus, hypothalamus, and pituitary. This results in a negative feedback loop [52, 53] and maintains homeostasis. The HPA axis is therefore designed to provide a quick and appropriate response to stress by stimulating a relatively brief influx of glucocorticoids into the body. Moreover, this responsivity is determined, in part, by the relative affinity of glucocorticoids for MRs and GRs [54]. The HPA axis goes through a period of significant change and development during the perinatal period and exposure to adverse events during this period is known to produce long-term alterations in neuroendocrine function and behavior.
Prenatal studies
Human studies
A growing body of clinical literature has demonstrated that exposure to stress during the perinatal period is linked to adverse health outcomes later in life [55–58] including susceptibility to developing psychiatric disorders and cognitive impairments. This is of particular relevance given that in modern society we are increasingly exposed to pollutants and stress, and thus it is proposed that the propensity for dysregulation of immune and metabolic homeostasis is rising.
The intrauterine events can have significant lasting or even lifelong repercussions such as cardiovascular disease or diabetes type 2 [59]. This delayed insult to the organism’s integrity is referred to as programming and is associated with sensitive periods during gestation. Various components of the stress related system develop at different rates. For instance, ACTH has been detected as early as 7 weeks, while corticotropin-releasing hormone CRH is produced by the placenta and hypothalamus and appears at 12–13 weeks [60]. The significance of these developmental stages for maternal stress is still unknown, however it is clear that levels of maternal glucocorticoids above that required for normal maturation [61] can have a negative “programming” impact on the foetus [53, 62]. These effects include growth retardation, HPA dysregulation, behavioural alterations and glucose intolerance in the offspring. [63]. Exposure to stress during pregnancy has also been linked to altered endocrine responses later in life. Maternal anxiety, depression, or stress have been associated with increased basal responsivity of the HPA axis in the offspring of children at 6 months [64], 5 years [65], and 10 years of age [58]. Pregnant mothers subjected to psychosocial stress showed alterations in HPA axis activity as indicated by high level of ACTH and decreased serum cortisol level when compared to a prenatally non-stressed group [66]. It has also been noted that social stress and lack of support during pregnancy, can increase IL-6 levels, increase CRH levels and decrease IL-10 levels within the maternal circulation [67].
Animal studies
The HPA axis goes through a period of increased plasticity during the perinatal period. Whilst GR mRNA in the hypothalamus can be detected as early as gestational day (GD) 16 [68], MR mRNA is not detected until PND 2 [69]. Thus, exposure to stressors during this sensitive period of development can have long-term consequences on the HPA axis. Physiological levels of glucocorticoids play a crucial role in brain development. They are involved in axonal and dendritic remodeling, and can affect cell survival [70]. However, sustained increases or removal of glucocorticoids during development can permanently alter brain structure and function [49, 71]. For instance, administration of the glucocorticoid synthetic Dexamethasone in pregnant rats contributed to delay in the maturation of neurons, myelination, glia, and vasculature in the offspring, leading to altered neuronal cytoarchitecture, altered synaptogenesis, and attenuated levels of neurogenesis [72]. Furthermore, adult rats prenatally exposed to glucocorticoids displayed increased CRH levels in the central nucleus of the amygdala [73], a key region in the regulation of the emotional component of pain [74]. Exposure to a novel environment combined with a saline injection (mild stress) from GD14 to GD21 is also associated with elevated plasma levels of ACTH and corticosterone in adulthood. Moreover when offspring from prenatally stressed rats were exposed to an acute foot shock, they exhibited a greater amount of shock-induced freezing time than the control group [75].
Postnatal studies
Human studies
The hippocampus is a key regulator of the stress response having inputs into a variety of key loci in the brain that regulate behavioural and endocrine responses to stress [76, 77]. The hippocampus represents a major regulator of HPA axis activity. Activation of glucocorticoid (GR) and mineralocorticoid (MR) receptors by circulating glucocorticoids (e.g. corticosterone) provides a negative feedback, which serves a key role in maintaining normal functioning of HPA axis. A number of studies have shown that exposure to adverse life events such as poverty [78], abuse [79], or orphanage rearing [80] can lead to children with increased stress reactivity and dysregulated HPA function in adulthood. For instance, in an aged human population, long-term exposure to endogenous levels of glucocorticoids resulted in memory impairments and a significant decrease (14 %) in hippocampal volume [81]. Low birth weight combined with low maternal care contributed to smaller hippocampal volume in adulthood, leading to increased susceptibility to stress-related disorders in later life [82, 83]. Maternal emotional well-being can also affect the development of the hippocampus in children. Magnetic Resonance Imaging (MRI) studies revealed that children from mothers who experienced high level of anxiety during the last trimester of pregnancy exhibit slower growth of both hemispheres of the hippocampus over the first 6 months of postnatal life [77]. Decreased hippocampal volume has also been associated with being sexually abused at 3–5 and 11–13 years of age [84]. Parent–child interactions and the emotional state of the pregnant mother are also critical factors in determining HPA axis responsivity of the child later in life. There is increasing evidence that children from depressed mothers are at higher risk of increased HPA axis activity [78].
Animal studies
In rodents, the first two postnatal weeks constitute an important time window for HPA axis ontogeny whereby the HPA axis goes through a “stress hyporesponsive period” (SHRP) from PND 4 to PND 14 during which adrenal sensitivity to stressors is diminished and corticosterone concentrations are maintained at low levels [85]. Furthermore, the maturation of hypothalamic neurocircuits is not completed at birth and undergoes fine-tuning until the third postnatal week. Whilst the neuronal cell population in the hypothalamus is determined prior to birth, the formation of functional neuronal networks via axonal projections and synaptic connections occurs later during the postnatal period [86]. Adult rats challenged with LPS as neonates displayed increased CRH mRNA in the PVN, indicating decreased levels of negative feed-back and hence increased HPA axis activity [47]. Maternal care constitutes a major determinant of an individual’s development, shaping the phenotype by affecting brain structure and function and ultimately determining personality, physiology and behaviours. Maternal separation, an animal model of human child maltreatment, has been linked to increased density in CRH binding sites in the hippocampus, prefrontal cortex, amygdala, and hypothalamus, as measured post-infancy [48]. Daily handling from PND 2 to PND 8 resulted in down-regulation of hypothalamic CRH mRNA levels in PND 23 rats [46]. Our laboratory was recently able to demonstrate that exposure to the bacterial mimetic LPS at PNDs 3 and 5 can also alter HPA axis in a developmentally regulated way as indicated by increased circulating corticosterone at PNDs 7 and 22 but not 13 and decreased GR mRNA and increased MR mRNA in the hypothalamus of PND 22 rats following formalin injection [10]. Similar findings were reported by other researchers whereby LPS exposure during PNDs 3 and 5 resulted in decreased GR binding and density in the hippocampus, hypothalamus, and frontal cortex of adult rats following adrenalectomy [47]. Moreover, adult rats challenged with LPS as neonates displayed increased CRH mRNA in the PVN, indicating decreased levels of negative feed-back and hence increased HPA axis activity [47].
Taken together, animal and human studies suggest that exposure to stress during the perinatal period where key brain areas are still developing at the time of stress exposure, can have long-term consequences on HPA axis activity, leading to exaggerated responses to a stressful event encountered later in life. The effects on HPA axis depend on the nature of the stressor as well as the timing when it occurs (i.e. prenatal vs. postnatal).
Plasticity of the nociceptive system during the neonatal period
Nociceptive neural circuitries undergo significant changes during the early neonatal period, and therefore remain vulnerable to the effects of early insults. For instance, substance P, a specific marker of peptidergic C fibres, is present at birth suggesting the development of chemical properties of these fibres at an early stage. The peptidergic C fibers are a population of C fibers that release neuropeptides such as substance P and calcitonin-gene-related-peptide (CGRP) as well as the Trk A neurotrophin which binds to nerve growth factor (NGF) [31]. In the dorsal horn, substance P fibres are concentrated in Lamina I on day 1; in the next few days, the density in Lamina II is increased and by the end of the first week of development, the staining of the fibers is comparable to the one observed in adults [87]. Moreover, the non-peptidergic C fibres that express the IB4 isolectin cannot be detected in the synapse until PND 5 despite their presence in dorsal root ganglion from embryonic day 18. This delay suggests that these fibres form synapses later than peptidergic C-fibres. Finally, both neonate A-fibres and C fibres express µ-opioid receptors, although this expression becomes restricted to C fibres by PND 21 [88].
There is a functional maturation of spinal dorsal horn which is also developmentally regulated. Dorsal horn cells in deep laminae do not respond to C fibre input until PNDs 7–8. Similarly, specific C-fibre-evoked reflexes produced by mustard oil (a chemical skin irritant) do not occur until rat pups reach day 10–11 [89]. The activation of C fibres is unable to evoke spike activity in the spinal cord before the second postnatal week. Aδ fibres are completely unmyelinated until the pups reach 2–3 weeks [89]. In the mature state, Aβ fibres which convey non-noxious stimuli project into Laminae III to V [90], while nociceptive fibres terminate in the superficial laminae (Laminae I and II). However, in the immature state large myelinated fibres, which normally integrate non-painful stimuli such as touch, are found to project to Laminae I to V including Laminae II. In other words, fibres that normally convey non-noxious stimuli can integrate noxious information as well. At the supraspinal level, the inhibitory descending control system is not apparent until the second postnatal week. Interestingly, electrical stimulation of the PAG does not produce analgesia until at least PND 10 [91]. Of particular interest, a recent study showed that there is a developmental postnatal shift in the brainstem rostroventral medulla (RVM) control of spinal dorsal horn neuronal circuits from A fibre facilitation activity in young animals to inhibition of nociceptive C fibre input in the adult animal [92].
Taken together, these data demonstrate that the nociceptive system during early life presents an important “window of vulnerability” during which an early insult such as inflammation, bacterial infection or a polymodal stimulus (mechanical, chemical or thermal), may result in the disruption of the neuronal and chemical circuitry modulating pain, leading to altered behavioural and neuronal responses to noxious stimuli later in life.
Neonatal noxious stimuli alter future pain responses
Human studies
In contrast to the traditional view that argues neonates are unable to feel pain, it is now well acknowledged that neonates are neuroanatomically and functionally able to discriminate between noxious and innocuous stimuli as indicated by scalp electroencephalography and spectroscopy studies [93, 94]. Preterm infants (<37 weeks of gestation) are born during a critical period when the brain is still undergoing significant plasticity and development. Preterm infants admitted to the neonatal intensive care unit (NICU) are routinely exposed to stressful and invasive procedures (i.e., heel lance, chest tube insertion, tape removal, and nasogastric tube insertion) to maintain their survival [95]. These infants exhibit significant alterations in sensory and pain processing later in adulthood [96, 97]. For instance infants subjected to repeated heel lance develop hyperalgesia compared to non-injured controls [98]. A number of studies have demonstrated that exposure to such adverse events can contribute to altered brain neurocircuitry and processing, leading to poorer outcomes at school age [99–101]. For instance infants, children, and preadolescents born preterm were found to exhibit altered development in the cerebellum and reduced cerebellar volume [102]; reduced white matter integrity [103]; as well as significant deficits in executive function and memory [104].
Animal studies
Animal studies are in accordance with the above-mentioned clinical findings whereby full thickness skin removal (wound of 2 mm in diameter) in rats performed at birth results in hyperinnervation of the wounded area that persist long after the wound has healed (i.e. up to 12 weeks). This hyperinnervation involved both Aδ and C-fibers and paralleled a decrease in the threshold to mechanical stimuli applied to the injured area [105]. Neonatal hindpaw injury produces hyperalgesia in adulthood [20] and intrathecal administration of minocycline, which inhibits microglial activity, at the time of adult insult prevent this hyperalgesia [106]. Finally, adult rats treated with the persistent inflammatory-inducing agent, Compound Freund Adjuvant (CFA), at PND 1 displayed increased density of dorsal horn nociceptive primary afferents [107]. And electrophysiological recording of dorsal horn neuron from adult rats treated with CFA as neonates revealed enhanced evoked responses to brush and noxious pinch compared to untreated rats [107].
The mechanisms underlying the hyperalgesia following early life adverse events are numerous. The hyperalgesia can be mediated by peripheral nerve sprouting such as the innervation of nociceptors is increased as previously confirmed [89]. Also, the ensuing inflammation may contribute to enhanced dorsal horn central sensitization as Ruda et al. previously reported [107]. Furthermore, the hyperalgesia can result from altered descending inhibitory control systems and decreased opioidergic drive in the PAG. Finally and of particular importance to this review the altered pain responses can result from altered neuroimmune responses as described below.
Neuroimmune interface in pain
Traditionally implicated in defending the organism against microbial pathogens or “non-self”, the immune system is now well appreciated to play a critical role in pain modulation [108–110]. Following injury, immune cells release proinflammatory cytokines and other inflammatory molecules such as histamine, prostaglandins, eicosanoids and cytokines that sensitize nociceptors. For instance, nociceptors are known to be interleukin (IL)-1-β sensors and IL-1-β can directly activate nociceptors to generate action potentials and induce hyperalgesia [111]. Moreover, intraplantar injection of IL-1-β increase the discharge of spinal dorsal horn neurons to thermal and mechanical stimuli [112]. The contribution to pain hypersensitivity can be mediated by inflammatory mediators released by immune cells but also by immune cells infiltration per se. The release of cytokines contributes to the recruitment of granulocytes, monocytes and mast cells at the site of inflammation. An increasing body of evidence suggests that infiltration by macrophages and T-cells in the dorsal root ganglia following nerve injury [113, 114] as well as activation of microglial cells in the spinal cord that coincides with enhanced neuropathic pain [115]. Hyperalgesia following nerve injury is substantially reduced in mice lacking T cells [116–118] and following the inhibition of microglia by minocycline [119]. Infiltration of immune cells at the peripheral level is also important in contributing to pain modulation. Following injury, mast cells are first to infiltrate the site of inflammation and they can degranulate within minutes of inflammation leading to the release of histamine, prostaglandin, bradykinin and other inflammatory markers that participate in the vasodilation and nociceptors sensitization [120]. Interestingly, mast cell degranulation requires direct contact with nociceptors via molecule of adhesion (N-cadherin) [121]. In our laboratory, we have demonstrated that neonatal exposure to the bacterial mimetic lipopolysaccharide (LPS) produced hyperalgesia in preadolescent rats that coincided with enhanced mast cell degranulation [23]. In a recent clinical study, Di Nardo et al. showed that children suffering from Irritable Bowel Syndrome (IBS) exhibit an increased number of mast cells in the proximity to nerves in the ileal and colonic mucosa [122]. More importantly, the intensity of abdominal pain reported by these children was significantly correlated to the close proximity of mast cells to nerve endings in the gut wall [122].
Other events that can contribute to the induction of the immune system are exposure to viruses, lipopolysaccharide (LPS), cell wall of gram-negative bacteria, or fungal infections during both the neonatal and adult life. When exposed to such components, immune cells including monocytes, lymphocytes, leukocytes, macrophages, T cells and mast cells not only signal other components of the immune system (i.e. complement) but also the brain [123] to control the inflammatory response and to maintain it at homeostatic levels. This is made possible by the release of a plethora of pro-inflammatory cytokines including IL-1-β [124], IL-6 [125], IL-8 [126], IL-12 [127], interferon-α (IFN-α) and -δ (IFN-δ) [128] as well as tumor necrosis factor (TNF)-α [129, 130]. Since cytokines are large molecules that are unlikely to cross the blood brain barrier (BBB) [131], it has been postulated that they can act on the brain through different pathways. For instance, via the synthesis of prostaglandin E2 by brain microvessel endothelial cells [132]. Another possibility is through vagal afferences as subdiaphragmatic lesions of the visceral vagal afferents terminating in the nucleus tractus solitaris abolished the LPS or IL-1β-induced hyperalgesia [133]. Subdiaphragmatic lesions of the vagus nerve also abolishes expression of Fos-protein in the secondary projections of this nerve such as the paraventricular nucleus of the hypothalamus (PVN), central nucleus of the amygdala (CeA), and the bed nucleus of the stria terminalis following IP injection of LPS [134]. This indicates that systemic LPS requires an intact functional vagus nerve to induce neuronal activation of brain regions involved in neuroendocrine function such as the PVN. It has also been suggested that cytokines can reach the brain via areas devoid of BBB such as the organum vasculosum lamina terminalis (OVLT) [135] or the macrophage-like cells commonly known as circumventricular organs [136]. The brain production of IL-1β following peripheral inflammatory stimuli such as in the case of LPS administration can be first restricted to choroid plexus and circumventricular organs, and then to the brain side of the BBB by slow diffusion (via volume transmission) [137]. Finally cytokines can reach the brain through a saturable transport system, which although has been demonstrated to exist in mice but not rats [138, 139], its physiological relevance remains unclear.
All of these neuroimmune pathways ultimately lead to the production of cytokines by brain microglia and induce a set of physiological responses commonly known as “sickness behavior”, which include fever, malaise, lethargy, decreased appetite and libido [123, 140]. The sickness response is an adaptive mechanism assisting in the recovery process from acute viral/bacterial infections. For instance fever, induced by the action of the pyrogenic IL-1 on anterior hypothalamus, elevates the body’s temperature to assist the immune system in fighting infections by blocking the growth of microorganisms [141]. Fatigue helps conserve energy so much needed in order to fight infection. However, in some circumstances, this process can go seriously wrong such as in the case of uncontrolled, exaggerated and prolonged inflammatory response in the CNS. This maladaptive response can be exemplified in clinical settings of post-infectious chronic fatigue syndrome [142], irritable bowel syndrome [143], or post-viral depression [144]. Importantly, pain facilitation or hyperalgesia is also considered to be an integral part of sickness behavior [145, 146]. Proinflammatory cytokines released by immune cells are known to induce hyperalgesia when administered at the peripheral or central level. Among the many types of pro-inflammatory cytokines, IL-1β is the most characterized potent mechanical and thermal hyperalgesic agent when injected into peripheral tissue or the CNS [112, 133, 147, 148]. For instance, intracerebroventricular (ICV) injection of the recombinant human IL-1β (rhIL-1β) in rats significantly decreased paw lick latency to thermal noxious stimulus [148]. This thermal hyperalgesia was abolished by ICV administration of the IL-1β antagonist IL-1ra [148]. In our laboratory, we have demonstrated increased formalin-induced behavioural responses in LPS-treated adult rats that paralleled enhanced circulating IL-1β 1-h following formalin injection [23]. Additionally, adult rats treated with LPS at PND 3 and 5 displayed increased IL-1β in the hippocampus 1 h following formalin injection, an effect that coincided the increased formalin-induced nociception observed in adult LPS-treated rats. The neurocircuitry underlying central action of cytokines is not only restricted to the brain but can also involve the spinal cord by increasing the excitability of spinal dorsal horn neurons [149, 150]. As discussed previously, activation of microglia at the spinal level plays a critical role in producing this cytokine-induced hyperalgesia [151].
The impact of neonatal and adult LPS exposure on pain responses
Neonatal or adult exposure to the bacterial mimetic has been reported in both clinical and animal studies to produce increased pain sensitivity later in life. Boisse L et al. were the first to report that rats administered with LPS at PND 14 exhibited a significant decrease in withdrawal latency to thermal and mechanical stimuli in adulthood [152]. These adult rats displayed allodynia (a stimulus that is normally not painful is perceived as painful) and hyperalgesia (exaggerated responses to stimuli) in both tests of thermal (i.e. hot-plate test) and mechanical nociception (i.e. Von Frey test). Of particular interest, no significant differences were observed in nociceptive responses when a second LPS injection was administered in adulthood. This implies that a single LPS administration is enough to produce long-term alterations in nociceptive responses. Moreover, the thermal and mechanical hyperalgesia in neonatally LPS-treated rats was associated with upregulation of COX-2 protein level in the spinal cord [152], suggesting a potential role of prostaglandins in the hyperexcitability of spinal dorsal horn neurons in LPS-treated rats. We have replicated these findings such that rats exposed to LPS during PND 3 and 5 displayed increased formalin-induced nociceptive responses (i.e. flinching and licking) following formalin injection at PND 22 and PND 80–97 but not PND 7 or PND 13 [10, 23, 24]. This LPS-induced hyperalgesia was accompanied by peripheral and central neuroimmune and neuroendocrine changes as well as altered spinal and supraspinal neuronal responses [10, 23, 24]. More recently Hunter D and colleagues reconfirmed these findings showing that at 4 h and 8 h post LPS administration, PND 21 rats (PND0 as birth) displayed enhanced formalin-induced nociception during the late phase [153]. Adult LPS administration in rats is also reported to significantly decrease the tail-flick latency, which starts within few minutes of LPS injection and lasts for 60 min [154]. The LPS-induced hyperalgesia can be blocked following administration of IL1-ra, suggesting a critical role of IL-1 in inducing the LPS-induced hyperalgesia [155]. These animal studies are consistent with clinical findings showing that humans subjected to an IV injection of LPS exhibited enhanced flare, hyperalgesia, and allodynia in response to capsaicin administration [156] as well as lower pressure pain thresholds that correlated with enhanced circulating levels of IL-6 and TNF-α, an effect more pronounced in women [157]. This is consistent with a recent study, which demonstrated that IV injection of LPS produced a significant decrease in rectal and pressure pain thresholds [158]. This LPS-induced visceral hyperalgesia was accompanied by a significant increase in circulating pro-inflammatory cytokines as indicated by a peak of TNFα and IL6 1 and 2 h (respectively) following LPS administration [159].
Taken together, these animal and clinical findings suggest that far from the traditional view that posits that pain can solely be attributed to activation of neurons, the immune system via peripheral and central release of pro-inflammatory cytokines contribute significantly to pain modulation. Integrating the neuroimmune interface in pain modulation is thus a necessary step in order to find efficient treatments to treat chronic pain.
Neuroendocrine-immune crosstalk: Focus on IL-1β and implications for pain
During the late 80s, a series of elegant studies mainly carried out by Sapolsky and Besedovsky group have demonstrated that components of the immune system, namely cytokines and in particular IL-1β can directly activate the HPA axis leading to the release of glucocorticoids [160, 161]. In a clinical study on post-mortem tissue, immunohistochemistry staining using antiserum directed against human IL-1β revealed that the human hypothalamus is densely innervated by IL-1β fibers [161]. The densest innervations were found in the periventricular region that controls the anterior pituitary. IL-1β-immunoreactive fibers were also found in the arcuate nuclei and parvocellular part of the paraventricular nucleus [161]. In rats, intravenous injection of IL-1β induced cfos expression in corticotropin-releasing hormone (CRH) containing neurons in the paraventricular nucleus of the hypothalamus (PVN) [162]. Systemic or intracerebroventricular injection of IL-1β also induced the release of circulating corticosterone and adrenocorticotropic hormone (ACTH) [163, 164]. Moreover, immunoneutralization of IL-1β or CRH blocked the stimulatory effect of IL-1β on ACTH [164]. This response is primarily mediated by the release of CRH from the PVN [164]. Additionally, IL-1β has been reported to act directly on the anterior pituitary cells to release ACTH [165].
Adrenocorticoids such as cortisol in humans and corticosterone in rats are known for their immunosuppressive function [166]. However their exact contribution to pain modulation is still not clear and literature regarding the role of glucocorticoids in modulating pain is at best conflicting. It is not surprising that glucocorticoids play an important role in pain modulation since pain is an aversive stressful event and hence capable of activating the HPA axis. Whilst several clinical studies have reported hypocortisolemia in patients with fibromyalgia and chronic low back pain [167–169], others have found that corticosterone reduces the inflammation associated with pain [170, 171]. In our laboratory, we demonstrated that neonatal LPS exposure produced a developmentally regulated activation of HPA axis activity following a noxious stimulus (i.e. formalin injection) as indicated by increased plasma levels of corticosterone in PND 13 and 22 but not PND 7 rats [10]. Preadolescent rats neonatally treated with LPS also exhibited decreased GR mRNA transcripts in the hypothalamus compared to rats that had saline during the neonatal period [10]. This is in agreement with previous studies indicating that noxious stimuli including formalin injection are capable of activating the HPA axis [172–174]. These findings indicate that the relationships between peripheral HPA axis activity and pain modulation is complex. Further studies investigating the exact contribution of the HPA axis in pain modulation are needed.
Conclusion and final remarks
As human beings, our organism is very complex. Far from the traditional view that posits that every physiological system is self-regulated, every physiological system including the immune, endocrine, and nervous system are interconnected via well-organized and harmonious regulatory mechanisms. In fact, dysfunction in one physiological system will lead to dysfunction of another. The immune system communicates with the endocrine and nervous systems mainly via cytokines which can act directly on the brain to produce local neuroinflammation but also exert a direct action on the HPA axis to produce the release of cortisol. Given that the immune, endocrine, and nervous systems undergo significant plasticity during the perinatal period, exposure to adverse events during this critical window of development is susceptible to disturb the normal developmental trajectory of these systems and consequently disturb the highly organized neuroendocrine to immune network leading to long-term consequences including altered pain responses. The challenge now facing the scientific community and clinicians is how to translate the well-established concept of psychoneuroimmunology and psychoneuroendocrinology as well as the importance of early life events in programming future pain responses into therapeutic approaches to treat chronic pain. Instead of the reductionist approach, which consists of targeting neurons only, we can also target components of the endocrine system such as corticosterone blockers or components of the immune system such as blocking the inflammatory process via suppressing the activity of pro-inflammatory cytokines or enhancing the activity of anti-inflammatory cytokines (Fig. 2).
Fig. 2.
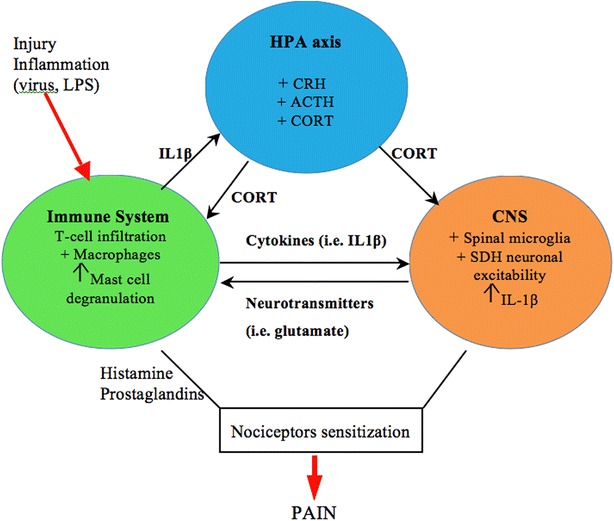
Neuroendocrine to immune communication in pain. Following a viral or bacterial infection, immune cells (i.e. T cells, macrophages, mast cells) are activated and infiltrate the site of inflammation. This results in the release of proinflammatory cytokines such as IL-1β. IL-1β then activates spinal microglia, produces the excitation of spinal dorsal horn (SDH) neurons and via activation of the HPA axis results in the release of corticosterone. Immune cells also release a plethora of inflammatory molecules including histamine and prostaglandins which results in the sensitization of nociceptors and increased pain sensitivity
Authors’ contributions
IZ, MDB, and DMH wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
All authors declare that they have no competing interests.
Contributor Information
I. Zouikr, Phone: +81484679779, Email: Ihssane.Zouikr@uon.edu.au
M. D. Bartholomeusz, Email: maurice.bartholomeusz@uon.edu.au
D. M. Hodgson, Email: deborah.hodgson@newcastle.edu.au
References
- 1.Reilly MM, Shy ME. Diagnosis and new treatments in genetic neuropathies. J Neurol Neurosurg Psychiatry. 2009;80:1304–1314. doi: 10.1136/jnnp.2008.158295. [DOI] [PubMed] [Google Scholar]
- 2.Blyth FM, March LM, Cousins MJ. Chronic pain-related disability and use of analgesia and health services in a Sydney community. Med J Aust. 2003;179:84–87. doi: 10.5694/j.1326-5377.2003.tb05441.x. [DOI] [PubMed] [Google Scholar]
- 3.Breivik H, Collett B, Ventafridda V, Cohen R, Gallacher D. Survey of chronic pain in Europe: prevalence, impact on daily life, and treatment. Eur J Pain. 2006;10:287–333. doi: 10.1016/j.ejpain.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 4.Blyth FM, March LM, Brnabic AJ, Jorm LR, Williamson M, Cousins MJ. Chronic pain in Australia: a prevalence study. Pain. 2001;89:127–134. doi: 10.1016/S0304-3959(00)00355-9. [DOI] [PubMed] [Google Scholar]
- 5.Vos T, Barber RM, Bell B, Bertozzi-Villa A, Biryukov S, Bolliger I, Charlson F, Davis A, Degenhardt L, Dicker D, et al. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;386:743–800. doi: 10.1016/S0140-6736(15)60692-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Leeuwen MT, Blyth FM, March LM, Nicholas MK, Cousins MJ. Chronic pain and reduced work effectiveness: the hidden cost to Australian employers. Eur J Pain. 2006;10:161–166. doi: 10.1016/j.ejpain.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 7.EFIC. Pain proposal improving the current and future management of chronic pain. A European consensus report. http://www.efic.org/userfiles/file/pain_proposal.pdf. 2010.
- 8.Goldberg JS. Revisiting the Cartesian model of pain. Med Hypotheses. 2008;70:1029–1033. doi: 10.1016/j.mehy.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 9.Chapman CR, Tuckett RP, Song CW. Pain and stress in a systems perspective: reciprocal neural, endocrine, and immune interactions. J Pain. 2008;9:122–145. doi: 10.1016/j.jpain.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zouikr I, Tadros MA, Barouei J, Beagley KW, Clifton VL, Callister RJ, Hodgson DM. Altered nociceptive, endocrine, and dorsal horn neuron responses in rats following a neonatal immune challenge. Psychoneuroendocrinology. 2014;41:1–12. doi: 10.1016/j.psyneuen.2013.11.016. [DOI] [PubMed] [Google Scholar]
- 11.Walker SM, Meredith-Middleton J, Cooke-Yarborough C, Fitzgerald M. Neonatal inflammation and primary afferent terminal plasticity in the rat dorsal horn. Pain. 2003;105:185–195. doi: 10.1016/S0304-3959(03)00201-X. [DOI] [PubMed] [Google Scholar]
- 12.Urakubo A, Jarskog LF, Lieberman JA, Gilmore JH. Prenatal exposure to maternal infection alters cytokine expression in the placenta, amniotic fluid, and fetal brain. Schizophr Res. 2001;47:27–36. doi: 10.1016/S0920-9964(00)00032-3. [DOI] [PubMed] [Google Scholar]
- 13.Taylor SE, Eisenberger NI, Saxbe D, Lehman BJ, Lieberman MD. Neural responses to emotional stimuli are associated with childhood family stress. Biol Psychiatry. 2006;60:296–301. doi: 10.1016/j.biopsych.2005.09.027. [DOI] [PubMed] [Google Scholar]
- 14.Gluckman PD, Hanson MA. Living with the past: evolution, development, and patterns of disease. Science. 2004;305:1733–1736. doi: 10.1126/science.1095292. [DOI] [PubMed] [Google Scholar]
- 15.Sherrington CS. The integrative action of the nervous system. Cambridge: Cambridge Univ Press; 1906. [Google Scholar]
- 16.Perl ER. Ideas about pain, a historical view. Nat Rev Neurosci. 2007;8:71–80. doi: 10.1038/nrn2042. [DOI] [PubMed] [Google Scholar]
- 17.Melzack R, Wall PD. The challenge of pain. London: Penguin books; 1982. [Google Scholar]
- 18.Sternberg WF, Ridgway CG. Effects of gestational stress and neonatal handling on pain, analgesia, and stress behavior of adult mice. Physiol Behav. 2003;78:375–383. doi: 10.1016/S0031-9384(03)00015-5. [DOI] [PubMed] [Google Scholar]
- 19.Ren K, Anseloni V, Zou SP, Wade EB, Novikova SI, Ennis M, Traub RJ, Gold MS, Dubner R, Lidow MS. Characterization of basal and re-inflammation-associated long-term alteration in pain responsivity following short-lasting neonatal local inflammatory insult. Pain. 2004;110:588–596. doi: 10.1016/j.pain.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 20.Walker SM, Tochiki KK, Fitzgerald M. Hindpaw incision in early life increases the hyperalgesic response to repeat surgical injury: critical period and dependence on initial afferent activity. Pain. 2009;147:99–106. doi: 10.1016/j.pain.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 21.Wang G, Ji Y, Lidow MS, Traub RJ. Neonatal hind paw injury alters processing of visceral and somatic nociceptive stimuli in the adult rat. J Pain. 2004;5:440–449. doi: 10.1016/j.jpain.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 22.Campbell EJ, Watters SM, Zouikr I, Hodgson DM, Dayas CV. Recruitment of hypothalamic orexin neurons after formalin injections in adult male rats exposed to a neonatal immune challenge. Front Neurosci. 2015;9:65. doi: 10.3389/fnins.2015.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zouikr I, Ahmed AF, Horvat JC, Beagley KW, Clifton VL, Ray A, Thorne RF, Jarnicki AG, Hansbro PM, Hodgson DM. Programming of formalin-induced nociception by neonatal LPS exposure: maintenance by peripheral and central neuroimmune activity. Brain Behav Immun. 2015;44:235–246. doi: 10.1016/j.bbi.2014.10.014. [DOI] [PubMed] [Google Scholar]
- 24.Zouikr I, James MH, Campbell EJ, Clifton VL, Beagley KW, Dayas CV, Hodgson DM. Altered formalin-induced pain and Fos induction in the periaqueductal grey of preadolescent rats following neonatal LPS exposure. PLoS One. 2014;9:e98382. doi: 10.1371/journal.pone.0098382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arendt-Nielsen L. Central sensitization in humans: assessment and pharmacology. Handb Exp Pharmacol. 2015;227:79–102. doi: 10.1007/978-3-662-46450-2_5. [DOI] [PubMed] [Google Scholar]
- 26.Yarnitsky D, Crispel Y, Eisenberg E, Granovsky Y, Ben-Nun A, Sprecher E, Best LA, Granot M. Prediction of chronic post-operative pain: pre-operative DNIC testing identifies patients at risk. Pain. 2008;138:22–28. doi: 10.1016/j.pain.2007.10.033. [DOI] [PubMed] [Google Scholar]
- 27.Edwards RR, Ness TJ, Weigent DA, Fillingim RB. Individual differences in diffuse noxious inhibitory controls (DNIC): association with clinical variables. Pain. 2003;106:427–437. doi: 10.1016/j.pain.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 28.Baliki MN, Petre B, Torbey S, Herrmann KM, Huang L, Schnitzer TJ, Fields HL, Apkarian AV. Corticostriatal functional connectivity predicts transition to chronic back pain. Nat Neurosci. 2012;15:1117–1119. doi: 10.1038/nn.3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mansour AR, Baliki MN, Huang L, Torbey S, Herrmann KM, Schnitzer TJ, Apkarian AV. Brain white matter structural properties predict transition to chronic pain. Pain. 2013;154:2160–2168. doi: 10.1016/j.pain.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hunt SP, Mantyh PW. The molecular dynamics of pain control. Nat Rev Neurosci. 2001;2:83–91. doi: 10.1038/35053509. [DOI] [PubMed] [Google Scholar]
- 31.Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Apkarian AV, Bushnell MC, Treede RD, Zubieta JK. Human brain mechanisms of pain perception and regulation in health and disease. Eur J Pain. 2005;9:463–484. doi: 10.1016/j.ejpain.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 33.Apkarian AV, Sosa Y, Sonty S, Levy RM, Harden RN, Parrish TB, Gitelman DR. Chronic back pain is associated with decreased prefrontal and thalamic gray matter density. J Neurosci. 2004;24:10410–10415. doi: 10.1523/JNEUROSCI.2541-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Apkarian AV, Baliki MN, Geha PY. Towards a theory of chronic pain. Prog Neurobiol. 2009;87:81–97. doi: 10.1016/j.pneurobio.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Geha PY, Baliki MN, Harden RN, Bauer WR, Parrish TB, Apkarian AV. The brain in chronic CRPS pain: abnormal gray-white matter interactions in emotional and autonomic regions. Neuron. 2008;60:570–581. doi: 10.1016/j.neuron.2008.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Metz AE, Yau HJ, Centeno MV, Apkarian AV, Martina M. Morphological and functional reorganization of rat medial prefrontal cortex in neuropathic pain. Proc Natl Acad Sci USA. 2009;106:2423–2428. doi: 10.1073/pnas.0809897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goldberg RT, Pachas WN, Keith D. Relationship between traumatic events in childhood and chronic pain. Disabil Rehabil. 1999;21:23–30. doi: 10.1080/096382899298061. [DOI] [PubMed] [Google Scholar]
- 38.McEwen BS. Plasticity of the hippocampus: adaptation to chronic stress and allostatic load. Ann NY Acad Sci. 2001;933:265–277. doi: 10.1111/j.1749-6632.2001.tb05830.x. [DOI] [PubMed] [Google Scholar]
- 39.Heim C, Ehlert U, Hanker JP, Hellhammer DH. Abuse-related posttraumatic stress disorder and alterations of the hypothalamic-pituitary-adrenal axis in women with chronic pelvic pain. Psychosom Med. 1998;60:309–318. doi: 10.1097/00006842-199805000-00017. [DOI] [PubMed] [Google Scholar]
- 40.De Bellis MD. Developmental traumatology: the psychobiological development of maltreated children and its implications for research, treatment, and policy. Dev Psychopathol. 2001;13:539–564. doi: 10.1017/S0954579401003078. [DOI] [PubMed] [Google Scholar]
- 41.Glaser D. Child abuse and neglect and the brain–a review. J Child Psychol Psychiatry. 2000;41:97–116. doi: 10.1017/S0021963099004990. [DOI] [PubMed] [Google Scholar]
- 42.De Bellis MD, Keshavan MS. Sex differences in brain maturation in maltreatment-related pediatric posttraumatic stress disorder. Neurosci Biobehav Rev. 2003;27:103–117. doi: 10.1016/S0149-7634(03)00013-7. [DOI] [PubMed] [Google Scholar]
- 43.Reynolds DV. Surgery in the rat during electrical analgesia induced by focal brain stimulation. Science. 1969;164:444–445. doi: 10.1126/science.164.3878.444. [DOI] [PubMed] [Google Scholar]
- 44.Hosobuchi Y, Adams JE, Linchitz R. Pain relief by electrical stimulation of the central gray matter in humans and its reversal by naloxone. Science. 1977;197:183–186. doi: 10.1126/science.301658. [DOI] [PubMed] [Google Scholar]
- 45.Eguchi M. Recent advances in selective opioid receptor agonists and antagonists. Med Res Rev. 2004;24:182–212. doi: 10.1002/med.10059. [DOI] [PubMed] [Google Scholar]
- 46.Avishai-Eliner S, Eghbal-Ahmadi M, Tabachnik E, Brunson KL, Baram TZ. Down-regulation of hypothalamic corticotropin-releasing hormone messenger ribonucleic acid (mRNA) precedes early-life experience-induced changes in hippocampal glucocorticoid receptor mRNA. Endocrinology. 2001;142:89–97. doi: 10.1210/endo.142.1.7917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shanks N, Larocque S, Meaney MJ. Neonatal endotoxin exposure alters the development of the hypothalamic-pituitary-adrenal axis: early illness and later responsivity to stress. J Neurosci. 1995;15:376–384. doi: 10.1523/JNEUROSCI.15-01-00376.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Anisman H, Zaharia MD, Meaney MJ, Merali Z. Do early-life events permanently alter behavioral and hormonal responses to stressors? Int J Dev Neurosci. 1998;16:149–164. doi: 10.1016/S0736-5748(98)00025-2. [DOI] [PubMed] [Google Scholar]
- 49.Matthews SG. Antenatal glucocorticoids and programming of the developing CNS. Pediatr Res. 2000;47:291–300. doi: 10.1203/00006450-200003000-00003. [DOI] [PubMed] [Google Scholar]
- 50.Matthews SG. Early programming of the hypothalamo-pituitary-adrenal axis. Trends Endocrinol Metab. 2002;13:373–380. doi: 10.1016/S1043-2760(02)00690-2. [DOI] [PubMed] [Google Scholar]
- 51.de Kloet ER, Joels M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci. 2005;6:463–475. doi: 10.1038/nrn1683. [DOI] [PubMed] [Google Scholar]
- 52.De Kloet ER, Reul JM. Feedback action and tonic influence of corticosteroids on brain function: a concept arising from the heterogeneity of brain receptor systems. Psychoneuroendocrinology. 1987;12:83–105. doi: 10.1016/0306-4530(87)90040-0. [DOI] [PubMed] [Google Scholar]
- 53.Welberg LA, Seckl JR. Prenatal stress, glucocorticoids and the programming of the brain. J Neuroendocrinol. 2001;13:113–128. doi: 10.1111/j.1365-2826.2001.00601.x. [DOI] [PubMed] [Google Scholar]
- 54.Lupien SJ, McEwen BS, Gunnar MR, Heim C. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat Rev Neurosci. 2009;10:434–445. doi: 10.1038/nrn2639. [DOI] [PubMed] [Google Scholar]
- 55.Maselko J, Kubzansky L, Lipsitt L, Buka SL. Mother’s affection at 8 months predicts emotional distress in adulthood. J Epidemiol Commun Health. 2011;65:621–625. doi: 10.1136/jech.2009.097873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mueller SC, Maheu FS, Dozier M, Peloso E, Mandell D, Leibenluft E, Pine DS, Ernst M. Early-life stress is associated with impairment in cognitive control in adolescence: an fMRI study. Neuropsychologia. 2010;48:3037–3044. doi: 10.1016/j.neuropsychologia.2010.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yehuda R, Engel SM, Brand SR, Seckl J, Marcus SM, Berkowitz GS. Transgenerational effects of posttraumatic stress disorder in babies of mothers exposed to the World Trade Center attacks during pregnancy. J Clin Endocrinol Metab. 2005;90:4115–4118. doi: 10.1210/jc.2005-0550. [DOI] [PubMed] [Google Scholar]
- 58.O’Connor TG, Ben-Shlomo Y, Heron J, Golding J, Adams D, Glover V. Prenatal anxiety predicts individual differences in cortisol in pre-adolescent children. Biol Psychiatry. 2005;58:211–217. doi: 10.1016/j.biopsych.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 59.Kajantie E. Fetal origins of stress-related adult disease. Ann NY Acad Sci. 2006;1083:11–27. doi: 10.1196/annals.1367.026. [DOI] [PubMed] [Google Scholar]
- 60.Meyer SE, Chrousos GP, Gold PW. Major depression and the stress system: a life span perspective. Dev Psychopathol. 2001;13:565–580. doi: 10.1017/S095457940100308X. [DOI] [PubMed] [Google Scholar]
- 61.Owen D, Andrews MH, Matthews SG. Maternal adversity, glucocorticoids and programming of neuroendocrine function and behaviour. Neurosci Biobehav Rev. 2005;29:209–226. doi: 10.1016/j.neubiorev.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 62.McMillen IC, Robinson JS. Developmental origins of the metabolic syndrome: prediction, plasticity, and programming. Physiol Rev. 2005;85:571–633. doi: 10.1152/physrev.00053.2003. [DOI] [PubMed] [Google Scholar]
- 63.Merlot E, Couret D, Otten W. Prenatal stress, fetal imprinting and immunity. Brain Behav Immun. 2008;22:42–51. doi: 10.1016/j.bbi.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 64.Lyons-Ruth K, Wolfe R, Lyubchik A. Depression and the parenting of young children: making the case for early preventive mental health services. Harv Rev Psychiatry. 2000;8:148–153. doi: 10.1080/hrp_8.3.148. [DOI] [PubMed] [Google Scholar]
- 65.Gutteling BM, de Weerth C, Buitelaar JK. Prenatal stress and children’s cortisol reaction to the first day of school. Psychoneuroendocrinology. 2005;30:541–549. doi: 10.1016/j.psyneuen.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 66.Entringer S, Kumsta R, Hellhammer DH, Wadhwa PD, Wust S. Prenatal exposure to maternal psychosocial stress and HPA axis regulation in young adults. Horm Behav. 2009;55:292–298. doi: 10.1016/j.yhbeh.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 67.Coussons-Read ME, Okun ML, Nettles CD. Psychosocial stress increases inflammatory markers and alters cytokine production across pregnancy. Brain Behav Immun. 2007;21:343–350. doi: 10.1016/j.bbi.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 68.Yi SJ, Masters JN, Baram TZ. Glucocorticoid receptor mRNA ontogeny in the fetal and postnatal rat forebrain. Mol Cell Neurosci. 1994;5:385–393. doi: 10.1006/mcne.1994.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.van Eekelen JA, Bohn MC, de Kloet ER. Postnatal ontogeny of mineralocorticoid and glucocorticoid receptor gene expression in regions of the rat tel- and diencephalon. Brain Res Dev Brain Res. 1991;61:33–43. doi: 10.1016/0165-3806(91)90111-U. [DOI] [PubMed] [Google Scholar]
- 70.Meyer JS. Early adrenalectomy stimulates subsequent growth and development of the rat brain. Exp Neurol. 1983;82:432–446. doi: 10.1016/0014-4886(83)90415-6. [DOI] [PubMed] [Google Scholar]
- 71.Fuxe K, Diaz R, Cintra A, Bhatnagar M, Tinner B, Gustafsson JA, Ogren SO, Agnati LF. On the role of glucocorticoid receptors in brain plasticity. Cell Mol Neurobiol. 1996;16:239–258. doi: 10.1007/BF02088179. [DOI] [PubMed] [Google Scholar]
- 72.Seckl JR. Glucocorticoids, developmental ‘programming’ and the risk of affective dysfunction. Prog Brain Res. 2008;167:17–34. doi: 10.1016/S0079-6123(07)67002-2. [DOI] [PubMed] [Google Scholar]
- 73.Cratty MS, Ward HE, Johnson EA, Azzaro AJ, Birkle DL. Prenatal stress increases corticotropin-releasing factor (CRF) content and release in rat amygdala minces. Brain Res. 1995;675:297–302. doi: 10.1016/0006-8993(95)00087-7. [DOI] [PubMed] [Google Scholar]
- 74.Bushnell MC, Ceko M, Low LA. Cognitive and emotional control of pain and its disruption in chronic pain. Nat Rev Neurosci. 2013;14:502–511. doi: 10.1038/nrn3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Griffin WC, 3rd, Skinner HD, Salm AK, Birkle DL. Mild prenatal stress in rats is associated with enhanced conditioned fear. Physiol Behav. 2003;79:209–215. doi: 10.1016/S0031-9384(03)00097-0. [DOI] [PubMed] [Google Scholar]
- 76.Pruessner JC, Dedovic K, Khalili-Mahani N, Engert V, Pruessner M, Buss C, Renwick R, Dagher A, Meaney MJ, Lupien S. Deactivation of the limbic system during acute psychosocial stress: evidence from positron emission tomography and functional magnetic resonance imaging studies. Biol Psychiatry. 2008;63:234–240. doi: 10.1016/j.biopsych.2007.04.041. [DOI] [PubMed] [Google Scholar]
- 77.Qiu A, Rifkin-Graboi A, Chen H, Chong YS, Kwek K, Gluckman PD, Fortier MV, Meaney MJ. Maternal anxiety and infants’ hippocampal development: timing matters. Transl Psychiatry. 2013;3:e306. doi: 10.1038/tp.2013.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lupien SJ, King S, Meaney MJ, McEwen BS. Child’s stress hormone levels correlate with mother’s socioeconomic status and depressive state. Biol Psychiatry. 2000;48:976–980. doi: 10.1016/S0006-3223(00)00965-3. [DOI] [PubMed] [Google Scholar]
- 79.Tarullo AR, Gunnar MR. Child maltreatment and the developing HPA axis. Horm Behav. 2006;50:632–639. doi: 10.1016/j.yhbeh.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 80.Gunnar MR, Morison SJ, Chisholm K, Schuder M. Salivary cortisol levels in children adopted from romanian orphanages. Dev Psychopathol. 2001;13:611–628. doi: 10.1017/S095457940100311X. [DOI] [PubMed] [Google Scholar]
- 81.Lupien SJ, Fiocco A, Wan N, Maheu F, Lord C, Schramek T, Tu MT. Stress hormones and human memory function across the lifespan. Psychoneuroendocrinology. 2005;30:225–242. doi: 10.1016/j.psyneuen.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 82.Buss C, Lord C, Wadiwalla M, Hellhammer DH, Lupien SJ, Meaney MJ, Pruessner JC. Maternal care modulates the relationship between prenatal risk and hippocampal volume in women but not in men. J Neurosci. 2007;27:2592–2595. doi: 10.1523/JNEUROSCI.3252-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gilbertson MW, Shenton ME, Ciszewski A, Kasai K, Lasko NB, Orr SP, Pitman RK. Smaller hippocampal volume predicts pathologic vulnerability to psychological trauma. Nat Neurosci. 2002;5:1242–1247. doi: 10.1038/nn958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Andersen SL, Tomada A, Vincow ES, Valente E, Polcari A, Teicher MH. Preliminary evidence for sensitive periods in the effect of childhood sexual abuse on regional brain development. J Neuropsychiatry Clin Neurosci. 2008;20:292–301. doi: 10.1176/jnp.2008.20.3.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Levine S. The ontogeny of the hypothalamic-pituitary-adrenal axis. The influence of maternal factors. Ann NY Acad Sci. 1994;746:275–288. doi: 10.1111/j.1749-6632.1994.tb39245.x. [DOI] [PubMed] [Google Scholar]
- 86.Bouret SG, Draper SJ, Simerly RB. Formation of projection pathways from the arcuate nucleus of the hypothalamus to hypothalamic regions implicated in the neural control of feeding behavior in mice. J Neurosci. 2004;24:2797–2805. doi: 10.1523/JNEUROSCI.5369-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fitzgerald M, Gibson S. The postnatal physiological and neurochemical development of peripheral sensory C fibres. Neuroscience. 1984;13:933–944. doi: 10.1016/0306-4522(84)90107-6. [DOI] [PubMed] [Google Scholar]
- 88.Nandi R, Beacham D, Middleton J, Koltzenburg M, Howard RF, Fitzgerald M. The functional expression of mu opioid receptors on sensory neurons is developmentally regulated; morphine analgesia is less selective in the neonate. Pain. 2004;111:38–50. doi: 10.1016/j.pain.2004.05.025. [DOI] [PubMed] [Google Scholar]
- 89.Fitzgerald M. The post-natal development of cutaneous afferent fibre input and receptive field organization in the rat dorsal horn. J Physiol. 1985;364:1–18. doi: 10.1113/jphysiol.1985.sp015725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fitzgerald M, Butcher T, Shortland P. Developmental changes in the laminar termination of A fibre cutaneous sensory afferents in the rat spinal cord dorsal horn. J Comp Neurol. 1994;348:225–233. doi: 10.1002/cne.903480205. [DOI] [PubMed] [Google Scholar]
- 91.Boucher T, Jennings E, Fitzgerald M. The onset of diffuse noxious inhibitory controls in postnatal rat pups: a C-Fos study. Neurosci Lett. 1998;257:9–12. doi: 10.1016/S0304-3940(98)00779-4. [DOI] [PubMed] [Google Scholar]
- 92.Koch SC, Fitzgerald M. The selectivity of rostroventral medulla descending control of spinal sensory inputs shifts postnatally from A fibre to C fibre evoked activity. J Physiol. 2014;592:1535–1544. doi: 10.1113/jphysiol.2013.267518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Slater R, Worley A, Fabrizi L, Roberts S, Meek J, Boyd S, Fitzgerald M. Evoked potentials generated by noxious stimulation in the human infant brain. Eur J Pain. 2010;14:321–326. doi: 10.1016/j.ejpain.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 94.Fabrizi L, Slater R, Worley A, Meek J, Boyd S, Olhede S, Fitzgerald M. A shift in sensory processing that enables the developing human brain to discriminate touch from pain. Curr Biol. 2011;21:1552–1558. doi: 10.1016/j.cub.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Carbajal R, Rousset A, Danan C, Coquery S, Nolent P, Ducrocq S, Saizou C, Lapillonne A, Granier M, Durand P, et al. Epidemiology and treatment of painful procedures in neonates in intensive care units. JAMA. 2008;300:60–70. doi: 10.1001/jama.300.1.60. [DOI] [PubMed] [Google Scholar]
- 96.Hohmeister J, Demirakca S, Zohsel K, Flor H, Hermann C. Responses to pain in school-aged children with experience in a neonatal intensive care unit: cognitive aspects and maternal influences. Eur J Pain. 2009;13:94–101. doi: 10.1016/j.ejpain.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 97.Walker SM, Franck LS, Fitzgerald M, Myles J, Stocks J, Marlow N. Long-term impact of neonatal intensive care and surgery on somatosensory perception in children born extremely preterm. Pain. 2009;141:79–87. doi: 10.1016/j.pain.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 98.Taddio A, Shah V, Gilbert-MacLeod C, Katz J. Conditioning and hyperalgesia in newborns exposed to repeated heel lances. JAMA. 2002;288:857–861. doi: 10.1001/jama.288.7.857. [DOI] [PubMed] [Google Scholar]
- 99.Petrie Thomas JH, Whitfield MF, Oberlander TF, Synnes AR, Grunau RE. Focused attention, heart rate deceleration, and cognitive development in preterm and full-term infants. Dev Psychobiol. 2012;54:383–400. doi: 10.1002/dev.20597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Brummelte S, Grunau RE, Synnes AR, Whitfield MF, Petrie-Thomas J. Declining cognitive development from 8 to 18 months in preterm children predicts persisting higher parenting stress. Early Hum Dev. 2011;87:273–280. doi: 10.1016/j.earlhumdev.2011.01.030. [DOI] [PubMed] [Google Scholar]
- 101.Synnes AR, Anson S, Arkesteijn A, Butt A, Grunau RE, Rogers M, Whitfield MF. School entry age outcomes for infants with birth weight </= 800 grams. J Pediatr. 2010;157(989–994):e981. doi: 10.1016/j.jpeds.2010.06.016. [DOI] [PubMed] [Google Scholar]
- 102.Ranger M, Zwicker JG, Chau CM, Park MT, Chakravarthy MM, Poskitt K, Miller SP, Bjornson BH, Tam EW, Chau V, et al. Neonatal pain and infection relate to smaller cerebellum in very preterm children at school age. J Pediatr. 2015;167(2):292–298. doi: 10.1016/j.jpeds.2015.04.055. [DOI] [PubMed] [Google Scholar]
- 103.Eikenes L, Lohaugen GC, Brubakk AM, Skranes J, Haberg AK. Young adults born preterm with very low birth weight demonstrate widespread white matter alterations on brain DTI. Neuroimage. 2011;54:1774–1785. doi: 10.1016/j.neuroimage.2010.10.037. [DOI] [PubMed] [Google Scholar]
- 104.Luu TM, Ment L, Allan W, Schneider K, Vohr BR. Executive and memory function in adolescents born very preterm. Pediatrics. 2011;127:e639–e646. doi: 10.1542/peds.2010-1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Reynolds M. Long-term sensory hyperinnervation following neonatal skin wounds. J Comp Neurol. 1995;358:487–498. doi: 10.1002/cne.903580403. [DOI] [PubMed] [Google Scholar]
- 106.Beggs S, Currie G, Salter MW, Fitzgerald M, Walker SM. Priming of adult pain responses by neonatal pain experience: maintenance by central neuroimmune activity. Brain. 2012;135:404–417. doi: 10.1093/brain/awr288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ruda MA, Ling QD, Hohmann AG, Peng YB, Tachibana T. Altered nociceptive neuronal circuits after neonatal peripheral inflammation. Science. 2000;289:628–631. doi: 10.1126/science.289.5479.628. [DOI] [PubMed] [Google Scholar]
- 108.Grace PM, Hutchinson MR, Maier SF, Watkins LR. Pathological pain and the neuroimmune interface. Nat Rev Immunol. 2014;14:217–231. doi: 10.1038/nri3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Marchand F, Perretti M, McMahon SB. Role of the immune system in chronic pain. Nat Rev Neurosci. 2005;6:521–532. doi: 10.1038/nrn1700. [DOI] [PubMed] [Google Scholar]
- 110.Ren K, Dubner R. Interactions between the immune and nervous systems in pain. Nat Med. 2010;16:1267–1276. doi: 10.1038/nm.2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Binshtok AM, Wang H, Zimmermann K, Amaya F, Vardeh D, Shi L, Brenner GJ, Ji RR, Bean BP, Woolf CJ, Samad TA. Nociceptors are interleukin-1beta sensors. J Neurosci. 2008;28:14062–14073. doi: 10.1523/JNEUROSCI.3795-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Fukuoka H, Kawatani M, Hisamitsu T, Takeshige C. Cutaneous hyperalgesia induced by peripheral injection of interleukin-1 beta in the rat. Brain Res. 1994;657:133–140. doi: 10.1016/0006-8993(94)90960-1. [DOI] [PubMed] [Google Scholar]
- 113.Cui JG, Holmin S, Mathiesen T, Meyerson BA, Linderoth B. Possible role of inflammatory mediators in tactile hypersensitivity in rat models of mononeuropathy. Pain. 2000;88:239–248. doi: 10.1016/S0304-3959(00)00331-6. [DOI] [PubMed] [Google Scholar]
- 114.Hu P, McLachlan EM. Macrophage and lymphocyte invasion of dorsal root ganglia after peripheral nerve lesions in the rat. Neuroscience. 2002;112:23–38. doi: 10.1016/S0306-4522(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 115.Tsuda M, Inoue K, Salter MW. Neuropathic pain and spinal microglia: a big problem from molecules in “small” glia. Trends Neurosci. 2005;28:101–107. doi: 10.1016/j.tins.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 116.Costigan M, Moss A, Latremoliere A, Johnston C, Verma-Gandhu M, Herbert TA, Barrett L, Brenner GJ, Vardeh D, Woolf CJ, Fitzgerald M. T-cell infiltration and signaling in the adult dorsal spinal cord is a major contributor to neuropathic pain-like hypersensitivity. J Neurosci. 2009;29:14415–14422. doi: 10.1523/JNEUROSCI.4569-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Moalem G, Xu K, Yu L. T lymphocytes play a role in neuropathic pain following peripheral nerve injury in rats. Neuroscience. 2004;129:767–777. doi: 10.1016/j.neuroscience.2004.08.035. [DOI] [PubMed] [Google Scholar]
- 118.Cao L, DeLeo JA. CNS-infiltrating CD4+ T lymphocytes contribute to murine spinal nerve transection-induced neuropathic pain. Eur J Immunol. 2008;38:448–458. doi: 10.1002/eji.200737485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hua XY, Svensson CI, Matsui T, Fitzsimmons B, Yaksh TL, Webb M. Intrathecal minocycline attenuates peripheral inflammation-induced hyperalgesia by inhibiting p38 MAPK in spinal microglia. Eur J Neurosci. 2005;22:2431–2440. doi: 10.1111/j.1460-9568.2005.04451.x. [DOI] [PubMed] [Google Scholar]
- 120.Lawrence T, Willoughby DA, Gilroy DW. Anti-inflammatory lipid mediators and insights into the resolution of inflammation. Nat Rev Immunol. 2002;2:787–795. doi: 10.1038/nri915. [DOI] [PubMed] [Google Scholar]
- 121.Folgueras AR, Valdes-Sanchez T, Llano E, Menendez L, Baamonde A, Denlinger BL, Belmonte C, Juarez L, Lastra A, Garcia-Suarez O, et al. Metalloproteinase MT5-MMP is an essential modulator of neuro-immune interactions in thermal pain stimulation. Proc Natl Acad Sci USA. 2009;106:16451–16456. doi: 10.1073/pnas.0908507106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Di Nardo G, Barbara G, Cucchiara S, Cremon C, Shulman RJ, Isoldi S, Zecchi L, Drago L, Oliva S, Saulle R, et al. Neuroimmune interactions at different intestinal sites are related to abdominal pain symptoms in children with IBS. Neurogastroenterol Motil. 2014;26:196–204. doi: 10.1111/nmo.12250. [DOI] [PubMed] [Google Scholar]
- 123.Dantzer R. Cytokine-induced sickness behavior: where do we stand? Brain Behav Immun. 2001;15:7–24. doi: 10.1006/brbi.2000.0613. [DOI] [PubMed] [Google Scholar]
- 124.Dinarello CA. Interleukin-1 and the pathogenesis of the acute-phase response. N Engl J Med. 1984;311:1413–1418. doi: 10.1056/NEJM198411293112205. [DOI] [PubMed] [Google Scholar]
- 125.Kaplanski G, Marin V, Montero-Julian F, Mantovani A, Farnarier C. IL-6: a regulator of the transition from neutrophil to monocyte recruitment during inflammation. Trends Immunol. 2003;24:25–29. doi: 10.1016/S1471-4906(02)00013-3. [DOI] [PubMed] [Google Scholar]
- 126.Middleton J, Neil S, Wintle J, Clark-Lewis I, Moore H, Lam C, Auer M, Hub E, Rot A. Transcytosis and surface presentation of IL-8 by venular endothelial cells. Cell. 1997;91:385–395. doi: 10.1016/S0092-8674(00)80422-5. [DOI] [PubMed] [Google Scholar]
- 127.Wysocka M, Robertson S, Riemann H, Caamano J, Hunter C, Mackiewicz A, Montaner LJ, Trinchieri G, Karp CL. IL-12 suppression during experimental endotoxin tolerance: dendritic cell loss and macrophage hyporesponsiveness. J Immunol. 2001;166:7504–7513. doi: 10.4049/jimmunol.166.12.7504. [DOI] [PubMed] [Google Scholar]
- 128.Ronni T, Sareneva T, Pirhonen J, Julkunen I. Activation of IFN-alpha, IFN-gamma, MxA, and IFN regulatory factor 1 genes in influenza A virus-infected human peripheral blood mononuclear cells. J Immunol. 1995;154:2764–2774. [PubMed] [Google Scholar]
- 129.Beutler B, Cerami A. Cachectin and tumour necrosis factor as two sides of the same biological coin. Nature. 1986;320:584–588. doi: 10.1038/320584a0. [DOI] [PubMed] [Google Scholar]
- 130.Beutler B, Cerami A. Cachectin (tumor necrosis factor): a macrophage hormone governing cellular metabolism and inflammatory response. Endocr Rev. 1988;9:57–66. doi: 10.1210/edrv-9-1-57. [DOI] [PubMed] [Google Scholar]
- 131.Banks WA, Erickson MA. The blood-brain barrier and immune function and dysfunction. Neurobiol Dis. 2010;37:26–32. doi: 10.1016/j.nbd.2009.07.031. [DOI] [PubMed] [Google Scholar]
- 132.Yamagata K, Matsumura K, Inoue W, Shiraki T, Suzuki K, Yasuda S, Sugiura H, Cao C, Watanabe Y, Kobayashi S. Coexpression of microsomal-type prostaglandin E synthase with cyclooxygenase-2 in brain endothelial cells of rats during endotoxin-induced fever. J Neurosci. 2001;21:2669–2677. doi: 10.1523/JNEUROSCI.21-08-02669.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Watkins LR, Wiertelak EP, Goehler LE, Smith KP, Martin D, Maier SF. Characterization of cytokine-induced hyperalgesia. Brain Res. 1994;654:15–26. doi: 10.1016/0006-8993(94)91566-0. [DOI] [PubMed] [Google Scholar]
- 134.Wan W, Wetmore L, Sorensen CM, Greenberg AH, Nance DM. Neural and biochemical mediators of endotoxin and stress-induced c-fos expression in the rat brain. Brain Res Bull. 1994;34:7–14. doi: 10.1016/0361-9230(94)90179-1. [DOI] [PubMed] [Google Scholar]
- 135.Katsuura G, Arimura A, Koves K, Gottschall PE. Involvement of organum vasculosum of lamina terminalis and preoptic area in interleukin 1 beta-induced ACTH release. Am J Physiol. 1990;258:E163–E171. doi: 10.1152/ajpendo.1990.258.1.E163. [DOI] [PubMed] [Google Scholar]
- 136.Maness LM, Kastin AJ, Banks WA. Relative contributions of a CVO and the microvascular bed to delivery of blood-borne IL-1alpha to the brain. Am J Physiol. 1998;275:E207–E212. doi: 10.1152/ajpendo.1998.275.2.E207. [DOI] [PubMed] [Google Scholar]
- 137.Konsman JP, Kelley K, Dantzer R. Temporal and spatial relationships between lipopolysaccharide-induced expression of Fos, interleukin-1beta and inducible nitric oxide synthase in rat brain. Neuroscience. 1999;89:535–548. doi: 10.1016/S0306-4522(98)00368-6. [DOI] [PubMed] [Google Scholar]
- 138.Banks WA, Kastin AJ, Ehrensing CA. Blood-borne interleukin-1 alpha is transported across the endothelial blood-spinal cord barrier of mice. J Physiol. 1994;479(Pt 2):257–264. doi: 10.1113/jphysiol.1994.sp020293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Banks WA, Niehoff ML, Zalcman SS. Permeability of the mouse blood-brain barrier to murine interleukin-2: predominance of a saturable efflux system. Brain Behav Immun. 2004;18:434–442. doi: 10.1016/j.bbi.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 140.Dantzer R. Cytokine-induced sickness behaviour: a neuroimmune response to activation of innate immunity. Eur J Pharmacol. 2004;500:399–411. doi: 10.1016/j.ejphar.2004.07.040. [DOI] [PubMed] [Google Scholar]
- 141.Kluger MJ, Kozak W, Conn CA, Leon LR, Soszynski D. Role of fever in disease. Ann NY Acad Sci. 1998;856:224–233. doi: 10.1111/j.1749-6632.1998.tb08329.x. [DOI] [PubMed] [Google Scholar]
- 142.Katz BZ, Jason LA. Chronic fatigue syndrome following infections in adolescents. Curr Opin Pediatr. 2013;25:95–102. doi: 10.1097/MOP.0b013e32835c1108. [DOI] [PubMed] [Google Scholar]
- 143.Spiller R, Garsed K. Postinfectious irritable bowel syndrome. Gastroenterology. 2009;136:1979–1988. doi: 10.1053/j.gastro.2009.02.074. [DOI] [PubMed] [Google Scholar]
- 144.Owen BM, Eccleston D, Ferrier IN, Young AH. Raised levels of plasma interleukin-1beta in major and postviral depression. Acta Psychiatr Scand. 2001;103:226–228. doi: 10.1034/j.1600-0447.2001.00162.x. [DOI] [PubMed] [Google Scholar]
- 145.Watkins LR, Maier SF, Goehler LE. Immune activation: the role of pro-inflammatory cytokines in inflammation, illness responses and pathological pain states. Pain. 1995;63:289–302. doi: 10.1016/0304-3959(95)00186-7. [DOI] [PubMed] [Google Scholar]
- 146.Watkins LR, Maier SF. The pain of being sick: implications of immune-to-brain communication for understanding pain. Annu Rev Psychol. 2000;51:29–57. doi: 10.1146/annurev.psych.51.1.29. [DOI] [PubMed] [Google Scholar]
- 147.Ferreira SH, Lorenzetti BB, Bristow AF, Poole S. Interleukin-1 beta as a potent hyperalgesic agent antagonized by a tripeptide analogue. Nature. 1988;334:698–700. doi: 10.1038/334698a0. [DOI] [PubMed] [Google Scholar]
- 148.Oka T, Aou S, Hori T. Intracerebroventricular injection of interleukin-1 beta induces hyperalgesia in rats. Brain Res. 1993;624:61–68. doi: 10.1016/0006-8993(93)90060-Z. [DOI] [PubMed] [Google Scholar]
- 149.Reeve AJ, Patel S, Fox A, Walker K, Urban L. Intrathecally administered endotoxin or cytokines produce allodynia, hyperalgesia and changes in spinal cord neuronal responses to nociceptive stimuli in the rat. Eur J Pain. 2000;4:247–257. doi: 10.1053/eujp.2000.0177. [DOI] [PubMed] [Google Scholar]
- 150.Sung CS, Wen ZH, Chang WK, Ho ST, Tsai SK, Chang YC, Wong CS. Intrathecal interleukin-1beta administration induces thermal hyperalgesia by activating inducible nitric oxide synthase expression in the rat spinal cord. Brain Res. 2004;1015:145–153. doi: 10.1016/j.brainres.2004.04.068. [DOI] [PubMed] [Google Scholar]
- 151.Watkins LR, Martin D, Ulrich P, Tracey KJ, Maier SF. Evidence for the involvement of spinal cord glia in subcutaneous formalin induced hyperalgesia in the rat. Pain. 1997;71:225–235. doi: 10.1016/S0304-3959(97)03369-1. [DOI] [PubMed] [Google Scholar]
- 152.Boisse L, Spencer SJ, Mouihate A, Vergnolle N, Pittman QJ. Neonatal immune challenge alters nociception in the adult rat. Pain. 2005;119:133–141. doi: 10.1016/j.pain.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 153.Hunter D, Chai C, Barr GA. Effects of COX inhibition and LPS on formalin induced pain in the infant rat. Dev Neurobiol. 2015;75(10):1068–1079. doi: 10.1002/dneu.22230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Mason P. Lipopolysaccharide induces fever and decreases tail flick latency in awake rats. Neurosci Lett. 1993;154:134–136. doi: 10.1016/0304-3940(93)90189-R. [DOI] [PubMed] [Google Scholar]
- 155.Maier SF, Wiertelak EP, Martin D, Watkins LR. Interleukin-1 mediates the behavioral hyperalgesia produced by lithium chloride and endotoxin. Brain Res. 1993;623:321–324. doi: 10.1016/0006-8993(93)91446-Y. [DOI] [PubMed] [Google Scholar]
- 156.Hutchinson MR, Buijs M, Tuke J, Kwok YH, Gentgall M, Williams D, Rolan P. Low-dose endotoxin potentiates capsaicin-induced pain in man: evidence for a pain neuroimmune connection. Brain Behav Immun. 2013;30:3–11. doi: 10.1016/j.bbi.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 157.Karshikoff B, Lekander M, Soop A, Lindstedt F, Ingvar M, Kosek E, Hoglund CO, Axelsson J. Modality and sex differences in pain sensitivity during human endotoxemia. Brain Behav Immun. 2015;46:35–43. doi: 10.1016/j.bbi.2014.11.014. [DOI] [PubMed] [Google Scholar]
- 158.Wegner A, Elsenbruch S, Rebernik L, Roderigo T, Engelbrecht E, Jager M, Engler H, Schedlowski M, Benson S. Inflammation-induced pain sensitization in men and women: does sex matter in experimental endotoxemia? Pain. 2015;156(10):1954–1964. doi: 10.1097/j.pain.0000000000000256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Benson S, Kattoor J, Wegner A, Hammes F, Reidick D, Grigoleit JS, Engler H, Oberbeck R, Schedlowski M, Elsenbruch S. Acute experimental endotoxemia induces visceral hypersensitivity and altered pain evaluation in healthy humans. Pain. 2012;153:794–799. doi: 10.1016/j.pain.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 160.Besedovsky H, del Rey A, Sorkin E, Dinarello CA. Immunoregulatory feedback between interleukin-1 and glucocorticoid hormones. Science. 1986;233:652–654. doi: 10.1126/science.3014662. [DOI] [PubMed] [Google Scholar]
- 161.Sapolsky R, Rivier C, Yamamoto G, Plotsky P, Vale W. Interleukin-1 stimulates the secretion of hypothalamic corticotropin-releasing factor. Science. 1987;238:522–524. doi: 10.1126/science.2821621. [DOI] [PubMed] [Google Scholar]
- 162.Veening JG, van der Meer MJ, Joosten H, Hermus AR, Rijnnkels CE, Geeraedts LM, Sweep CG. Intravenous administration of interleukin-1 beta induces Fos-like immunoreactivity in corticotropin-releasing hormone neurons in the paraventricular hypothalamic nucleus of the rat. J Chem Neuroanat. 1993;6:391–397. doi: 10.1016/0891-0618(93)90013-T. [DOI] [PubMed] [Google Scholar]
- 163.Uehara A, Gottschall PE, Dahl RR, Arimura A. Stimulation of ACTH release by human interleukin-1 beta, but not by interleukin-1 alpha, in conscious, freely-moving rats. Biochem Biophys Res Commun. 1987;146:1286–1290. doi: 10.1016/0006-291X(87)90788-1. [DOI] [PubMed] [Google Scholar]
- 164.Berkenbosch F, van Oers J, del Rey A, Tilders F, Besedovsky H. Corticotropin-releasing factor-producing neurons in the rat activated by interleukin-1. Science. 1987;238:524–526. doi: 10.1126/science.2443979. [DOI] [PubMed] [Google Scholar]
- 165.Lumpkin MD. The regulation of ACTH secretion by IL-1. Science. 1987;238:452–454. doi: 10.1126/science.2821618. [DOI] [PubMed] [Google Scholar]
- 166.Munck A, Guyre PM, Holbrook NJ. Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocr Rev. 1984;5:25–44. doi: 10.1210/edrv-5-1-25. [DOI] [PubMed] [Google Scholar]
- 167.Griep EN, Boersma JW, Lentjes EG, Prins AP, van der Korst JK, de Kloet ER. Function of the hypothalamic-pituitary-adrenal axis in patients with fibromyalgia and low back pain. J Rheumatol. 1998;25:1374–1381. [PubMed] [Google Scholar]
- 168.Muhtz C, Rodriguez-Raecke R, Hinkelmann K, Moeller-Bertram T, Kiefer F, Wiedemann K, May A, Otte C. Cortisol response to experimental pain in patients with chronic low back pain and patients with major depression. Pain Med. 2013;14:498–503. doi: 10.1111/j.1526-4637.2012.01514.x. [DOI] [PubMed] [Google Scholar]
- 169.Riva R, Mork PJ, Westgaard RH, Ro M, Lundberg U. Fibromyalgia syndrome is associated with hypocortisolism. Int J Behav Med. 2010;17:223–233. doi: 10.1007/s12529-010-9097-6. [DOI] [PubMed] [Google Scholar]
- 170.Buritova J, Honore P, Chapman V, Besson JM. Enhanced effects of co-administered dexamethasone and diclofenac on inflammatory pain processing and associated spinal c-Fos expression in the rat. Pain. 1996;64:559–568. doi: 10.1016/0304-3959(95)00167-0. [DOI] [PubMed] [Google Scholar]
- 171.Dray A, Bevan S. Inflammation and hyperalgesia: highlighting the team effort. Trends Pharmacol Sci. 1993;14:287–290. doi: 10.1016/0165-6147(93)90041-H. [DOI] [PubMed] [Google Scholar]
- 172.Newsome HH, Rose JC. The response of human adrenocorticotrophic hormone and growth hormone to surgical stress. J Clin Endocrinol Metab. 1971;33:481–487. doi: 10.1210/jcem-33-3-481. [DOI] [PubMed] [Google Scholar]
- 173.Aloisi AM, Albonetti ME, Muscettola M, Facchinetti F, Tanganelli C, Carli G. Effects of formalin-induced pain on ACTH, beta-endorphin, corticosterone and interleukin-6 plasma levels in rats. Neuroendocrinology. 1995;62:13–18. doi: 10.1159/000126983. [DOI] [PubMed] [Google Scholar]
- 174.Harbuz MS, Lightman SL. Stress and the hypothalamo-pituitary-adrenal axis: acute, chronic and immunological activation. J Endocrinol. 1992;134:327–339. doi: 10.1677/joe.0.1340327. [DOI] [PubMed] [Google Scholar]